A todas las personas que con
Texto completo
Figure




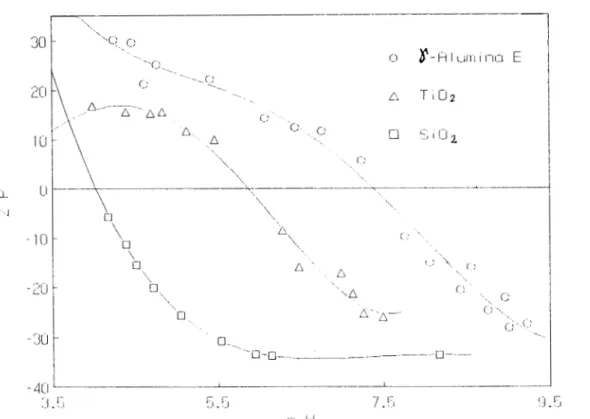



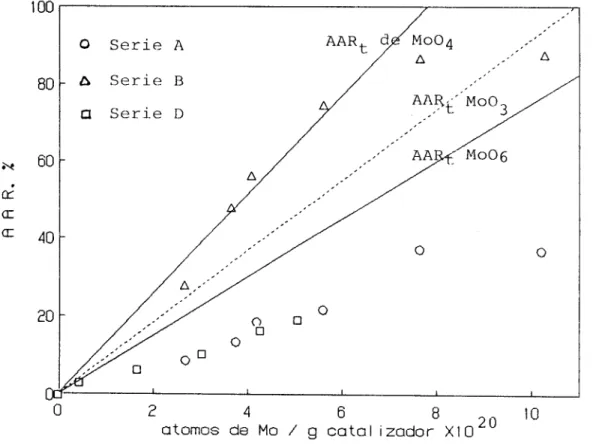
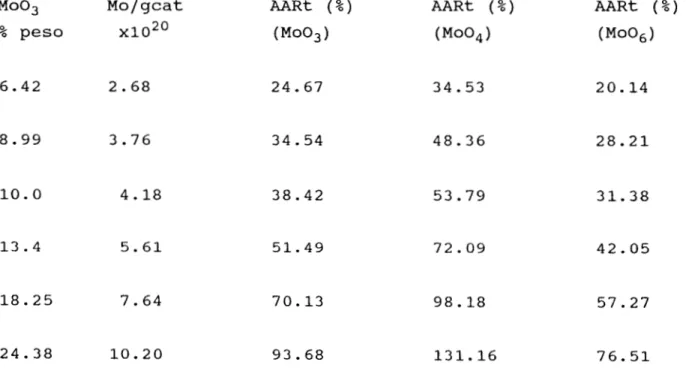
Documento similar
En cuarto lugar, se establecen unos medios para la actuación de re- fuerzo de la Cohesión (conducción y coordinación de las políticas eco- nómicas nacionales, políticas y acciones
b) El Tribunal Constitucional se encuadra dentro de una organiza- ción jurídico constitucional que asume la supremacía de los dere- chos fundamentales y que reconoce la separación
If certification of devices under the MDR has not been finalised before expiry of the Directive’s certificate, and where the device does not present an unacceptable risk to health
In addition to the requirements set out in Chapter VII MDR, also other MDR requirements should apply to ‘legacy devices’, provided that those requirements
The notified body that issued the AIMDD or MDD certificate may confirm in writing (after having reviewed manufacturer’s description of the (proposed) change) that the
que hasta que llegue el tiempo en que su regia planta ; | pise el hispano suelo... que hasta que el
Para ello, trabajaremos con una colección de cartas redactadas desde allí, impresa en Évora en 1598 y otros documentos jesuitas: el Sumario de las cosas de Japón (1583),
Entre nosotros anda un escritor de cosas de filología, paisano de Costa, que no deja de tener ingenio y garbo; pero cuyas obras tienen de todo menos de ciencia, y aun
