PREDICCIÓN ESTRUCTURAL Y FUNCIONAL DE LA PROTEÍNA HIPOTÉTICA PA2481 DE Pseudomonas Aeruginosa PAO1
DAVID ALBERTO DÍAZ DE LOS RIOS
PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS BÁSICAS
DEPARTAMENTO DE BIOLOGÍA BOGOTÁ D. C.
PREDICCIÓN ESTRUCTURAL Y FUNCIONAL DE LA PROTEÍNA HIPOTÉTICA PA2481 DE Pseudomonas Aeruginosa PAO1
DAVID ALBERTO DÍAZ DE LOS RIOS
__________________________ _____________________________ INGRID SCHULER PHD ANDREA FORERO RUÍZ DECANA ACADEMICA DIRECTORA
PREDICCIÓN ESTRUCTURAL Y FUNCIONAL DE LA PROTEÍNA HIPOTÉTICA PA2481 DE Pseudomonas Aeruginosa PAO1
DAVID ALBERTO DÍAZ DE LOS RIOS
_____________________________ __________________________ JANNETH GONZÁLEZ GEORGE BARRETO
NOTA DE ADVERTENCIA
"La Universidad no se hace responsable por los conceptos emitidos por sus alumnos en sus trabajos de tesis. Solo velará por que no se publique nada contrario al dogma y a la moral católica y por qué las tesis no contengan ataques personales contra persona alguna, antes bien se vea en ellas el anhelo de buscar la verdad y la justicia".
Resumen
El género Pseudomonas sp., es un grupo muy diverso de bacilos Gram negativos aerobios, ubicuos y que causa más infecciones oportunistas en seres humanos, como otitis externa, foliculitis de tina y fibrosis quística, entre otras. Específicamente, la Pseudomonas aeruginosa
PAO1, es un microorganismo con gran capacidad infecciosa en diferentes organismos y
resistencia a sustancias antibióticas y desinfectantes. Puesto que los antígenos son los encargados de producir esta resistencia, y la mayoría de estos son proteínas, el objetivo de este trabajo fue predecir computacionalmente la estructura tridimensional y función de la proteína hipotética PA2481 de Pseudomonas aeruginosa, determinando su relación en la resistencia antibiótica de esta bacteria. Para esto, primero se realizó el análisis computacional de la estructura primaria, utilizando los servidores PROSITE, PFAM, BLAST, PROTPARAM, GLOBPLOT, y PROTSCALE; luego, se obtuvo la estructura secundaria por el consenso de los algoritmos SOPM, PREDATOR, DPM, DSC, y GOR4; y por último la estructura tridimensional de la proteína se obtuvo mediante el servidor I-TASSER y se evaluó su conformación estereoquímica con la herramienta STRUCTURE ASSESSMENT. El modelo final fue visualizado con el programa PyMOL. En el presente trabajo se presenta la estructura tridimensional de PA2481 y se identifican dos dominios correspondientes al citocromo c, encontrándose 2 regiones desordenadas en el domino más significativo. Se determina que la proteína presenta características propias de los citocromo c oxidasa, cbb3, subunidad III, y que su función posiblemente está relacionada con la cadena trasportadora de electrones y el bombeo de protones para la generación de ATP en Pseudomonas aeruginosa. La predicción estructural y funcional de PA2481 permite entender mejor el papel de esta proteína en la fisiología de esta bacteria.
Palabras clave: Fibrosis quística, modelamiento estructural, otitis externa, Pseudomonas aeruginosa, Proteína hipotética.
Introducción
y animales (Hardalo & Edberg 1997). Como consecuencia de su resistencia a los antibióticos y a los desinfectantes Pseudomonas .aeruginosa es una bacteria que se caracteriza por ser uno de los principales patógenos oportunistas en humanos, provocando serias complicaciones causadas por infecciones en pacientes particularmente susceptibles como personas con deficiencias del sistema inmunológico, víctimas de quemaduras de la piel, pacientes cateterizados los cuales sufren infecciones del tracto urinario y en pacientes con respiradores provocando neumonía nosocomial. También es la causa predominante de la morbilidad y la mortalidad en pacientes con fibrosis quística colonizando los pulmones (Stover et al. 2000).
del genoma de P. aeruginosa cepa PAO1, el cual se destacó por su diversa capacidad metabólica y gran tamaño con 6.3 millones de pares de bases, siendo el mayor genoma bacterial secuenciado, sin embargo, cerca del 30 al 40% de los genes no tienen asignada una función. Además, en los estudios de anotación de genes, una gran fracción de marcos de lectura abierta están etiquetados como proteínas hipotéticas conservadas y muchas de estas proteínas hipotéticas se encuentran en más de una especie bacteriana (Dunham 2000). Para la asignación de funciones a nuevas proteínas, la homología basada en anotaciones de genes ha sido el estándar durante las últimas décadas, pues infiere características moleculares mediante la trasferencia de información de proteínas caracterizadas experimentalmente (Roberts 2004).
El conocimiento de la secuencia completa del genoma incluyendo las proteínas hipotéticas y los procesos de codificación, proporcionan una gran cantidad de información para el descubrimiento y explotación de nuevos blancos de antibióticos, y la esperanza para el desarrollo de estrategias más efectivas para el tratamiento de las infecciones oportunistas potencialmente mortales causadas por P. aeruginosa en los seres humanos (Stover et al. 2000).
Por lo tanto en este estudio se realiza la predicción de la estructura tridimensional, y se realiza una aproximación funcional de la proteína hipotética PA2481 de Pseudomonas aeruginosa PAO1 a través de diferentes herramientas bioinformáticas y softwares, para determinar si está implicada en la resistencia antibiótica de este microorganismo, con el fin de entender mejor la fisiología de esta bacteria.
Justificación y Planteamiento del problema
De acuerdo a esto es importante determinar cuáles proteínas están implicadas en estos mecanismos de resistencia, determinando su estructura y función, para poder desarrollar estrategias efectivas contra esta bacteria.
Por otro lado, los avances en las técnicas de secuenciación de proteínas en los últimos años hace que se almacenen gran cantidad de secuencias en bases de datos, pertenecientes a genomas completos de diversas bacterias, arqueas y eucariotas, dentro de los cuales se encuentra la secuencia completa del genoma de P. aeruginosa cepa PAO1. Sin embargo, cerca del 30 al 40% de los genes no tienen asignada una función. Además, en su secuencia génica se encuentran gran cantidad de proteínas hipotéticas conservadas y muchas de estas se encuentran en más de una especie bacteriana (Dunham, 2000).
El conocimiento de las estructuras tridimensionales de las proteínas hipotéticas y su función, tanto los procesos de codificación, proporcionan una gran cantidad de información para el descubrimiento y explotación de nuevos blancos de antibióticos, y la esperanza para el desarrollo de estrategias más efectivas para el tratamiento de las infecciones oportunistas potencialmente mortales causadas por P. aeruginosa en los seres humanos (Stover et al. 2000).
Partiendo de la secuencia de residuos de aminoácidos de la proteína hipotética PA2481 de Pseudomonas aeruginosa PAO1, en el presente trabajo se realizará una predicción de su estructura secundaria y terciaria y, con el resultado obtenido, se buscará una aproximación funcional para determinar si está implicada en la resistencia antibiótica de este microorganismo.
Marco teórico
Pseudomonas aeruginosa es una bacteria Gram-negativa que se caracteriza por su versatilidad ambiental, y su habilidad para causar enfermedades en individuos particularmente susceptibles, desarrollando resistencia a los antibióticos. La complicación más seria causada por este microorganismo es la fibrosis quística, una infección del tracto respiratorio. Los pacientes con cáncer, al igual que algunas otras personas con deficiencias del sistema inmunológico, sufren graves infecciones por este microorganismo (Winsor et al. 2011).
una amplia gama de compuestos orgánicos como fuentes de alimento. En su secuencia génica se encuentran genes implicados en la locomoción, apego, transporte y utilización de nutrientes, flujo de salida de los antibióticos, y los sistemas de detección involucrados en la respuesta al estrés y cambio ambiental (Winsor et al. 2011).
Pseudomonas aeruginosa PAO1 es la cepa más comúnmente utilizada en la investigación de este microorganismo como patógeno. La cepa PAO1, es un derivado de la cepa original aislada PAO, que se ha distribuido en todo el mundo para laboratorios y colecciones de cepas. En las últimas décadas han surgido fenotipos discordantes de PAO1 sublíneas. Estas difieren en su capacidad para hacer frente a la limitación de nutrientes y su virulencia, por ejemplo, en un modelo murino de infección aguda de vías respiratorias (Klockgether et al. 2010).
Los mecanismos de resistencia antibiótica que presentan las bacterias Gram negativas como Pseudomonas aeruginosa, es consecuencia de una combinación de mecanismos inherentes a la especie y otros adquiridos (Gómez 2005). En términos generales, los principales mecanismos de resistencia antibiótica en Pseudomonas aeruginosa son: 1) modificación y desactivación del antibiótico por hidrólisis mediada por enzimas; 2) disminución de la permeabilidad del antibiótico a través de la membrana externa debido a la disminución en la expresión de porinas; 3) aumento de la expulsión del antibiótico mediada por la activación de las bombas de flujo, y 4) modificación o mutación del sitio blanco del antibiótico (Suarez et al. 2006).
1996), PSI-BLAST (Altschul et al. 1997), HMM (Soding 2005), MulPSSM (Gowri et al. 2006), los cuales son conocidos para identificar relaciones de secuencias distantes en el espacio.
Cuando las proteínas presentan una alta similitud en su secuencia y estructura, frecuentemente también muestran una buena similaridad funcional (Roy et al. 2009). Estas relaciones funcionales entre proteínas ya han sido estudiadas (Chothia & Lesk 1986; Russell & Barton 1994; Wood & Pearson 1999; Wilson et al. 2000). Los umbrales de similitud de secuencias para la transferencia de anotaciones funcionales también han sido propuestas (Wilson et al. 2000; Todd et al. 2001; Tian & Skolnick. 2003).
A parte de inferir la función a través de proteínas homologas con estructura y función conocida, se puede inferir la función por medio de la búsqueda de patrones o motivos, y utilizando procedimientos de coincidencias de patrones, sin embargo este método solo es aplicable cuando se conoce la estructura de las proteínas (Via & Helmer 2004).
Las proteínas presentan dominios que se definen como unidades autónomas de plegado con independencia evolutiva y funcional (Doolittle 1995). La búsqueda de estos dominios en bases de datos es más efectiva en comparación con la secuencia completa (George y Heriga. 2002). Entre las bases de datos de dominios estructurales de las proteínas disponibles en la actualidad se encuentran SCOP (Clasificación Estructural de Proteínas) (Murzin et al.1995), CATH (Clase, Arquitectura, Topología y homología) (Orengo et al. 2003), FSSP (Familias de Proteínas Estructuralmente Similares) (Holm & Sander 1996) y PFAM (Protein Database Familias) (Bateman et al. 2002). En las bases de datos CATH y FSSP las proteínas se agrupan en función de los resultados de los algoritmos de comparación estructural y no reflejan ninguna similitud funcional o una relación evolutiva. Los resultados de PFAM están derivados de los alineamientos de secuencias de proteínas y de perfiles HMMs (Roy et al. 2009).
Objetivo General
Objetivos específicos
1. Predecir in silico la estructura secundaria y terciaria de la proteína hipotética PA2481 de Pseudomonas aeruginosa PA01.
2. Predecir la función de la proteína hipotética PA2481 de Pseudomonas aeruginosa PA01 con relación a su posible participación en los mecanismos de resistencia que presenta este microorganismo.
Metodología
Análisis computacional de la estructura primaria de PA2481
El análisis de la secuencia de PA2481 de 291 residuos de aminoácidos con función y estructura desconocida se realizó a través de diversas herramientas bioinformáticas y bases de datos disponibles en la red. La secuencia de la proteína hipotética PA2481 (Acc. No. AAG05869.1) fue obtenida a través del GenBank en el Centro Nacional de Información para la Biotecnología (NCBI) (http://www.ncbi.nlm.nih.gov/). La búsqueda de similitud con otras secuencias reportadas se realizó por medio de BLAST disponible en NCBI.
Para la caracterización fisicoquímica de la proteína, se determinó el punto isoeléctrico, peso molecular, coeficiente de extinción (Gill & Von Hippel 1989), índice de inestabilidad (Guruprasad et al. 1990), índice alifático (Ikai 1980) y promedio general de la hidropatía (Kyte & Doolittle 1982), mediante la herramienta PROTPARAM (Gasteiger et al. 2005) del portal de recursos bioinformaticos EXPASY (http://EXPASY.org/tools), la cual realiza estos cálculos basándose en la composición de aminoácidos y en el residuo N-terminal. En cuanto al análisis de hidrofobicidad se utilizó la herramienta PROTSCALE y el algoritmo de Kyte y Doolittle del servidor EXPASY Proteomic tools, basándose en las propiedades físicas y químicas de los aminoácidos que componen la proteína. La predicción de las regiones globulares y regiones desordenadas o no estructurales de la proteína fueron determinadas con GLOBPLOT (Imer & Cavas 2009).
Predicción de la estructura secundaria
Para predecir la estructura secundaria de la proteína hipotética PA2481 se utilizó el servidor NSP@ (Combet et al. 2000), en donde se usó el consenso de cinco algoritmos, dentro de los cuales se seleccionaron dos métodos fundamentados en el uso de parámetros de probabilidad determinados por las frecuencias relativas de las apariciones de cada aminoácido en cada tipo de estructura secundaria (SOPM y PREDATOR) y tres métodos basados en probabilidad de inferencia bayesiana (GOR4, DPM y DSC).
Predicción de la estructura tridimensional
Resultados y Discusión
Análisis computacional de la estructura primaria de PA2481
Al realizar el alineamiento local de secuencias con BLAST, se identificaron 6 proteínas con una secuencia de aminoácidos similar en un 99% y 98% a la secuencia de PA2481 (Tabla 1). Puesto que la identidad de cada proteína se basa en el orden especifico de cada uno de sus aminoácidos (Claverie & Notredame 2007), el porcentaje de identidad de la proteína PA2481 con cada una de las 6 proteínas identificadas, permite inferir que estas son probablemente homologas y casi idénticas en la conformación de su estructura tridimensional y por consiguiente en su función. Teniendo en cuenta que las proteínas son hipotéticas, no presentan una estructura ni función definida a pesar de presentar anotaciones en el NCBI con respecto a dominios conservados correspondientes al citocromo c, lo cual es un buen indicio para predecir su posible función biológica.
TABLA 1. Resultados del alineamiento local con BLAST para la búsqueda de similitud con secuencias reportadas.
Descripción Id secuencia Max
identidad Proteína hipotética PACG_01397 [Pseudomonas aeruginosa
C3719]
ref|ZP_04928804.1| 99%
Proteína hipotéticaPaerPAb_13161 [Pseudomonas aeruginosa PAb1]
ref|ZP_06878575.1| 99%
Proteína hipotéticaPA14_32540 [Pseudomonas aeruginosa UCBPP-PA14]
ref|YP_790756.1| 99%
Proteína hipotéticaHMPREF1030_01871 [Pseudomonas sp. 2_1_26]
ref|ZP_09052785.1| 98%
Proteína hipotéticaG655_12625 [Pseudomonas aeruginosa B136-33]
ref|YP_007709370.1| 98%
Proteína hipotéticaPSPA7_2757 [Pseudomonas aeruginosa PA7]
TABLA 2. Resultados encontrados a partir de la secuencia de aminoácidos de la proteína PA2481 con las herramientas PROSITE y PFAM.
Programa Descripción Aminoácidos Puntaje
PROSITE Dominio Citocromo c 165 a 250 9.309
PROSITE Sitio de unión covalente al grupo hemo 178 y 181
PROSITE Hierro (hemo 1 ligando axial) 182
PROSITE Dominio Citocromo c 44 a 141 6.95
PROSITE Sitio de unión covalente al grupo hemo 70 y 73
PROSITE Hierro (hemo 1 ligando axial) 74
PFAM Dominio de Citocromo c oxidasa, cbb3, subunidad III
165 a 246 42.6
PFAM Dominio Citocromo c 46 a 135 11
En las proteínas pertenecientes a la familia de los Citocromos c, el grupo hemo se une covalentemente por enlaces tioeter a dos residuos de cisteína conservados localizados en el centro del Citocromo c. Los Citocromos c, cumplen un papel importante en la transferencia de electrones (Lee 2009); además, en los centros de los Citocromos c también se encuentran sitios activos de muchas enzimas que cumplen un papel importante en la apoptosis de células eucariotas (Martinou 2000). En las estructuras conocidas de los Citocromos de tipo c, hay cerca de 6 clases que varían en sus pliegues (Allen 2003).
Las propiedades fisicoquímicas obtenidas mediante PROTPARAM (Tabla 3), permitieron determinar el peso molecular de PA2481 el cual coincide con el peso molecular de la cadena c del citocromo cbb3 oxidasa, subunidad III, reportado por Zufferey et al. (1996) el cual es de 31 kDa. El punto isoeléctrico es donde las cargas positivas y negativas se igualan anulando la existencia de movimiento en un campo eléctrico, representando el pH de la proteína PA2481, el cual podría presentar un mínimo de solubilidad en ensayos experimentales facilitando su aislamiento en un campo eléctrico (Werner 2007). En cuanto al tiempo de vida de PA2481, este hace referencia al tiempo (in vitro) que tarda una proteína en desaparecer una vez es sintetizada por la célula. De acuerdo al tiempo de vida de la proteína PA2481, se puede determinar que el residuo N terminal corresponde a metionina, serina o alanina, treonina, valina o lisina, pues estos aminoácidos son específicos para proteínas con tiempo de vida superiores a 20 horas, ya que se ha demostrado que el último aminoácido de la cadena determina la sobrevivencia de la proteína (Bachmair et al. 1986). Experimentalmente también se puede determinar el residuo N terminal para saber con exactitud cuál aminoácido de los anteriores mencionados es el N terminal (Jinling et al. 2011).
TABLA 3. Propiedades fisicoquímicas de PA2481 encontradas mediante PROTPARAM
Peso molecular (Da) 31249.4
Índice de estabilidad 41.06
Punto isoeléctrico 8.56
Tiempo de vida (horas) 30
Índice alifático 71.51
Promedio general de la hidropatía (GRAVY) 0.271
(k/mol) que se usa para transferir un segmento de secuencia de longitud definida de un medio hidrofóbico a un medio hidrofílico (Lieberman et al. 2009).
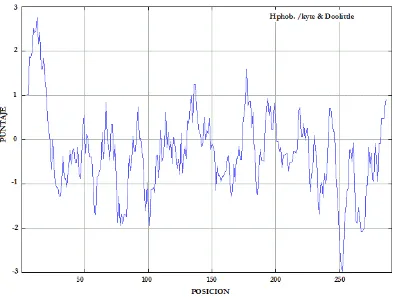
Los resultados arrojados por ProtScale (Figura 1), permiten identificar las regiones hidrofóbicas e hidrofílicas de la proteína PA2481, en donde se observa el puntaje en función del residuo. Los picos más altos indican regiones hidrofóbicas y los más bajos cercanos a -2 indican regiones hidrofílicas. Utilizando una ventana de 9, se determinó que PA2481 es una proteína altamente hidrofílica, es decir gran parte de su estructura se encuentra en contacto con un medio acuoso, lo que coincide con los resultados del promedio general de la hidropatía. PA2481 también presenta 3 regiones hidrofóbicas que podrían estar en contacto con la parte interna de la membrana celular.
Figura 1. Análisis hidropático de la proteína PA2481 con base en los valores de Kyte y Doolittle.
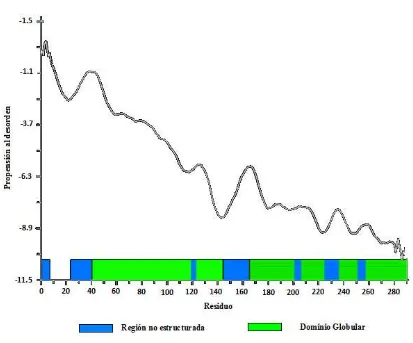
Las regiones desordenadas reveladas por GLOBPLOT (Figura 2) muestran 7 regiones con desordenes intrínsecos entre la posición de aminoácidos del 1-7, 24-40, 119-123, 145-165, 201-206, 225-236 y 251-257, las cuales están posiblemente involucradas en funciones importantes de Pseudomonas aeruginosa. Además, 2 de las 7 regiones desordenadas (residuos 201-206 y
Figura 2. Análisis de GLOBPLOT para la proteína PA2481. Las regiones con desordenes intrínsecos corresponden a las regiones de color azul, mientras que las regiones globulares están en verde.
Predicción de la estructura secundaria
[image:19.612.100.513.63.406.2]aminoácido haga parte de una estructura secundaria considerando que los aminoácidos adyacentes adquieran la misma estructura, mientras que los algoritmos GORIV, DSC, y DPM utilizan conjuntos de entrenamiento cuyos elementos son estructuras resueltas para identificar secuencias motivo comunes asociadas a disposiciones particulares de estructuras secundarias (Barreto et al. 2012).
[image:20.612.99.506.156.589.2]De acuerdo con lo anterior, la estructura secundaria de PA2481 revelo la existencia de 8 hélices α entre los residuos de aminoácidos: 5-14, 42-57, 104-113, 129-141, 171-179, 215-222, 239-247 y 263-269 representados en un 29.90%; 11 bucles entre los aminoácidos: 17-41, 57-62, 65-94, 118-128, 142-165, 182-189, 197-213, 225-238, 248-261, 270-276 y 278-284 con un 60.82%; y hebras no plegadas representadas con tan solo el 4.12%. La prevalencia de conformación en bucle hace a estas moléculas más dúctiles y se explica por la necesidad funcional de establecer fácilmente interacciones con proteínas (Sáenz et al. 2011).
Predicción de la Estructura Tridimensional
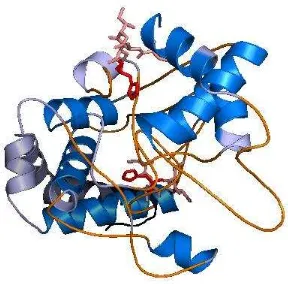
Figura 4. Modelo tridimensional de PA2481 generado por I-TASSER y visualizado con PyMOL. Las hélices mostradas de color azul agua marina y los bucles mostrados en naranja, corresponden a las regiones donde hay coincidencia con la predicción de la estructura secundaria. Las regiones en color salmón mostradas en “sticks” corresponden a los motivos de unión covalente al grupo hemo el cual se presenta en color rojo.
un buen acercamiento a la estructura real de la proteína PA2481, además el modelo coincide en gran parte con la predicción de la estructura secundaria predicha anteriormente.
Conclusiones
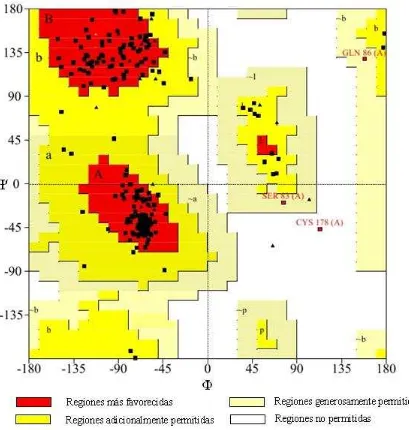
El modelo tridimensional predicho constituye un buen acercamiento a la posible estructura real de la proteína PA2481, pues este coincide en gran parte con la estructura secundaria predicha y además se aseguró su correcta conformación por medio de la evaluación geométrica (Grafico Ramachandran).A través de las diferentes herramientas bioinformáticas usadas en el presente estudio, se determinó que la proteína hipotética PA2481 de Pseudomonas aeruginosa PAO1 pertenece a la superfamilia de los Citocromos c, presentando las características más significativas de este tipo de proteínas como los dos sitios de unión covalente al grupo hemo. Esto indica que posiblemente PA2481 participa en procesos biológicos metabólicos celulares, de óxido-reducción, en la cadena trasportadora de electrones y en la generación de precursores de metabolitos y energía, contribuyendo con los mecanismos de resistencia antibiótica de esta bacteria, por el bombeo de este tipo de sustancias al exterior, lo cual requiere ATP, o por procesos de óxido-reducción que llevan a su degradación.
Referencias
Allen J.W, Daltrop O, Stevens J.M, Ferguson S.J.(2003). C-type cytochromes: diverse structures and biogenesis Systems pose evolutionary problems. Philios. Trans. R. Soc. Lond, B, Biol. Sci. 358:255-266.
Altschul, S. F. and Gish, W. (1996). Local alignment statistics. Methods Enzymol. 266, 460-480.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389-3402.
Arnold K, Bordoli L, Kopp J, Schwede T. (2006). The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 22,195-201.
Bachmair. A, D. Finley and A. Varshavsky. (1986). In vivo half-life of a protein is a function of its amino-terminal residue. Science. Vol. 234. No 4773. Pp. 179-186.
Barreto Leidy Viviana, George Emilio Barreto, Ludis Morales, Orlando Emilio Acevedo, Janneth Gonzales Santos. (2012). Proteína LIC10494 de Leptospira interrogans serovarcopenhageni: modelo estructural y regiones funcionales asociadas. Universitas Scientiarum. Vol. 17. No 1:16-27
Barker P.D., Ferguson S.J. (1999). Still a puzzle: why is covalently attached in c-Type cytochromes?. Structure 7: 281-290
BakerD, A. Sali A.(2001). Protein structure prediction and structural genomics. Science 294, 93-96.
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. and Sonnhammer, E. L. (2002). The PFAM protein families database. Nucleic Acids Res. 30, 276-280.
Buschmann. S, Warkentin. E, Xie. H, Langer. J.D, Ermier. U, Michel. H. (2010). The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science. Jul16; 329(5989):327-30.
Chothia, C. and Lesk, A. M. (1986). The relation between the divergence of sequence and structure in proteins. EMBO Journal. 5, 823-826.
Claverie J, Notredame, C. Bioinformatics for Dummies. (2007). Segunda Edición. Wiley Publishing, Inc. Indianapolis., Indiana. 457 p.
Combet C., Blanchet C., Geourjon C, Deléage G. (2000). NPS@: Network Protein Sequence Analysis TIBS. Vol. 25, No 3 [291]:147-150.
Delano WL. 2008. The PyMOL Molecular Graphics System. Delano Scientific Homepage.
Doolittle, R. F. (1995). The multiplicity of domains in proteins. Annu. Rev. Biochem. 64, 287-314.
Dunham, I. (2000) Genomics-the new rock and roll? Trends Genet. 16, 456–461 Finn R. D, et al. (2012). The PFAM protein families database. Nucleic Acid Research.
Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. (2005). Protein Identification and Analysis Tools on the EXPASY Server; (In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press. pp. 571-607
George, R. A. and Heriga, J. (2002). Protein domain identification and improved sequence similarity searching using PSI-BLAST. Proteins 48, 672-681.
Gill C. Staney and Von Hippel. P.H. (1989). Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry 182, 319-326.
Gómez Carlos Andres, Leal Aura Lucia, Pérez María de Jesús, Navarrete Myriam Lucia. (2005). Mecanismos de Resistencia en Pseudomonas aeruginosa: Entendiendo a un poderoso enemigo. Revista Facultad de Medicina Universidad Nacional de Colombia. Vol. 53. No 1. 27-34.
Gowri, V. S., Krishnadev, O., Swamy, C. S. and Srinivasan, N. (2006). MulPSSM: a database of multiple position-specificnscoring matrices of protein domain families. Nucleic Acid Res. 34, D243-D246
Guruprasad K., Reddy BV. Pandit MVV. (1990). Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Dec; 4(2):155-61.
Hardalo, C. & Edberg, S. C. (1997). Pseudomonas aeruginosa: assessment of risk from drinking water. Crit. Rev. Microbiol. 23, 47–75
Hieter P, Boguski M. (1997). "Functional Genomics: It's All How You Read It". Science 278: 601- 602.
Holm, L. and Sander, C. (1996). The FSSP database-fold classification based on structure alignment of proteins Nucleic Acids Res. 24, 206-209.
Ikai AJ. (1980). Thermostability and aliphatic index of globular proteins. Biochem. Dec; 88(6):1895-8. [PMID: 7462208]
Imer O, Cavas L. (2009). The Bioinformatics Tools for the Estimation of Disordered. Biomedical engineering Meeting; 1-4.
KlockgetherJ., Munder A., Neugebauer J., Colin F., et al. 2010.Genome Diversity of Pseudomonas aeruginosa PAO1 Laboratory Strains. Journal of Bacteriology. Vol 192.n 4. 1113-1121.
Kyte J &Doolittle RF. (1982). A simple method for displaying the hydropathic character of a protein. Journal Molecular Biology. May 5; 157(1):105-32.
Lee, B. (2009). Physical properties of cytochromes c: 1. role of cytochrome c' as a nitric oxide carrier 2. Folding of cytochrome c. State University of New York at Albany). ProQuest Dissertations and Theses, 109. Retrieved from http://search.proquest.com/docview/305097001?accountid=13250. (305097001).
Lieberman M, Marks AD, Smith CM. Marks. (2009). basic medical biochemistry: a clinical approach. Tercera edición. Wolters Kluwer Health. China. 997 p.
Makino S, Ewing TJ, Kuntz ID. (1999). Dream++: Flexible docking program for virtual combinatorial libraries. Journal Comput-Aided Mol Des 13:513–532
Martinou J.-C., Desagher S., Antonsson B. (2000) .Cytochrome c release from mitochondria: all or nothing. Nat.Cell Biol. 2:E41-E43.
Murzin, A., Brenner., S. E., Hubbard, T. and Chothia, C. (1995). SCOP: a structural classification of proteins database for the investigation of sequences and structures. Journal Molecular Biology. 247, 536-540.
Orengo, C. A., Pearl, F. M. and Thornton, J. M. (2003). The CATH domain structure database. Methods Biochem. Anal. 44, 249-271.
Pearson, W. R. (2000). Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 1 32, 185-219.
Pollastri G, Martin AJM, Mooney C, Vullo A. (2007). Accurate prediction of protein secondary structure and solvent accessibility by consensus combiners of sequence and structure information. BMC Bioinformatics. 8, 201.
Roberts RJ (2004) Identifying Protein Function—A Call for Community Action. PLoSBiol 2(3): e42
Roy Ambrish, Alper Kucukural, Yang Zhang. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. April; 5(4):725-738. Roy A, Narayanas wamy Srinivasan, Venkatraman S. Gowri. (2009). Molecular and
Domains: Implications for Function Annotation Based on Homology Searches and Structural Genomics. In Silico Biology 9. S41–S55.
Russell, R. B. and Barton, G. J. (1994). Structural features can be unconserved in proteins with similar folds. An analysis of side-chain to side-chain contacts secondary structure and accessibility. J. Mol. Biol. 244, 332-350.
Sáenz-Suarez. Homero, Leonardo Rene Lareo, Carlos Oribio-quinto, Juan Martínez-Mendoza, Aura Chavez-Zobel. (2011). Predicción computacional de estructura terciaria de las proteínas humanas Hsp27, αB-cristalina y HspB8. Universitas Scientiarum. Vol. 16. No 1: 15-28.
Sigrist CJA, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N (2010). .PROSITE, a protein domain database for functional characterization and annotation.Nucleic Acids Res. 38(Database issue) 161.
Soding, J. (2005). Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951-960.
Skolnick J, JS Fetrow, A Kolinski. (2000) Structural genomics and its importance for gene function analysis. Nature Biotechnologic 18:283–287.
Stover, K.C. et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1: an opportunistic pathogen. Nature. 406: 959-964.
Suarez, C., Kattan J, Guzmán A, Villegas V. (2006). Mecanismos de resistencia a carbapenemsen P. aeruginosa, Acinetobacter y Enterobacteriaceae y estrategias para su prevención y control. Infect. 2006, vol.10, n.2, pp. 85-93. ISSN 0123-9392.
Terwilliger TC, Waldo G, Peat TS, Newman JM, Chu K, Berendzen J. (1998).Class-directed structure determination: foundation for a protein structure initiative. Protein Sci Sep; 7(9):1851-1856.
Todd, A. E., Orengo, C. A. and Thornton, J. M. (2001). Evolution of function in protein superfamilies, from a structural perspective. Journal Molecular Biology.307, 1113-1143. Tian, W. and Skolnick, J. (2003). How well is enzyme function conserved as a function of
Tribelli, Paula María. (2011). Influencia del regulador global Anr en la fisiología de Pseudomonas extremaustralis, una bacteria productora de polihidroxibutirato. Tesis Doctoral. Facultad de Ciencias Exactas y Naturales Universidad de Buenos Aires, p 1, 14. Tosatto S, Battistutta R (2007). TAP score: torsion angle propensity normalization
applied to local protein structure evaluation. BMC Bioinformatics.8, 155.
Uversky VN, Radivojac P, Lakoucheva LM, Obradovic Z, Dunker AK. (2007). Prediction of intrinsic disorder and its use in functional proteomics. Methods in Molecular Biology.408(1): 69-92.
Uversky VN, Dunker AK. (2010). Understanding protein non-folding. Biochimica et Biophysical Acta. 1804(6): 1231-1264.
Via, A. and Helmer, Citterich, M. (2004). A structural study for the optimization of functional motifs encoded in protein sequences. BMC Bioinformatics 5, 50.
Werner MS. (2007). Biología: Fundamentos para Medicina y Ciencias de la Vida. Reverté. Barcelona, España. 661p.
Wilson, C. A., Kreychman, J. and Gerstein, M. (2000). Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J. Mol. Biol. 297, 233-249.
Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS (2011). Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. Jan; 39(Database issue):D596-600. PubMed.
Wood, T. C. and Pearson, W. R. (1999). Evolution of protein sequences and structures. J. Mol. Biol. 291, 977-995.
Recursos electrónicos
Centro Nacional de Información para la Biotecnología (NCBI) http://www.ncbi.nlm.nih.gov/
Protein Data Bank (PDB) http://www.rcsb.org/pdb/home/home.do