PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS
PROGRAMA DE DOCTORADO EN CIENCIAS BIOLOGICAS
ASOCIACIÓN DE LAS MUTACIONES SOMÁTICAS DE LOS GENES NKX2.5 Y GATA4 EN PACIENTES CON CARIOPATÍAS CONGÉNITAS ESPORÁDICAS
Marleny Salazar Salazar
TESIS
presentada como requisito parcial para optar al título de
Doctora en Ciencias Biológicas
ASOCIACIÓN DE LAS MUTACIONES SOMÁTICAS DE LOS GENES NKX2.5 Y GATA4 EN PACIENTES CON CARIOPATÍAS CONGÉNITAS ESPORÁDICAS
Marleny Salazar Salazar
TESIS
presentado como requisito parcial para optar al título de
Doctora en Ciencias Biológicas
Director
Doctor Jaime Eduardo Bernal Villegas
Director Instituto de Genética
Pontificia Universidad Javeriana
Pontificia Universidad Javeriana
Facultad de Ciencias
Bogotá
“La Universidad no se hace responsable de los conceptos emitidos por sus alumnos en sus trabajos de tesis. Solo velará porque no se publique nada contrario al dogma y a la moral católica y por que las tesis no contengan ataques personales contra persona alguna, antes bien se vea en ellas el anhelo de buscar la verdad y la justicia”
ASOCIACIÓN DE LAS MUTACIONES SOMÁTICAS DE LOS GENES NKX2.5 Y GATA4 EN PACIENTES CON CARIOPATÍAS CONGÉNITAS ESPORÁDICAS
Marleny Salazar Salazar
________________________________ ______________________________
Jaime Eduardo Bernal V, MD., PhD Jurado
Director
_____________________________ ________________________________
Jurado Jurado
_____________________________ ________________________________
ASOCIACIÓN DE LAS MUTACIONES SOMÁTICAS DE LOS GENES NKX2.5 Y GATA4 EN PACIENTES CON CARIOPATÍAS CONGÉNITAS ESPORÁDICAS
Marleny Salazar Salazar
________________________________ _______________________________
Ingrid Schuler García Manuel Antonio Franco Cortés
Decana Académica Director
AGRADECIMIENTOS
Mis sinceros agradecimientos a las instituciones y personas que aportaron sus conocimientos en la elaboración del trabajo de grado.
La Universidad del Quindío por su apoyo económico y académico para la realización de mis estudios de Doctorado
La Universidad del Rosario y al Laboratorio de Biología Molecular y Celular por la capacitación y colaboración económica en la ejecución de la primera fase del proyecto. A las Doctoras: Sandra Ramírez PhD en Ciencias, Decana de la Facultad de Ciencias, Victoría Villegas Msc en Ciencias, Heidi Matteus MD Msc en Genética, por la capacitación y acompañamiento en el desarrollo del proyecto.
Clínica Shaio especialmente a los Doctores Sonia Pachón, Federico Núnéz y Victor Caicedo en la toma de muestras de los pacientes del estudio. A la señora Stella Acevedo por su valiosa colaboración
Instituto Css-Mendel de Roma Italia por la capacitación, aporte académico y económico al proyecto, especialmente a los Doctores: Bruno Dalapicolla Director del Instituto Mendel, Alessandro DeLuca y Federica Consolini por su dedicación y trabajo en equipo.
Doctor Jaime Bernal Director del Instituto de Genética Pontificia Universidad Javeriana tutor del proyecto por sus aportes.
Doctora Magdalena María Martínez Agüero PhD en Ciencias por sus valiosos aportes en el proyecto
Doctora Diana Torres López PhD en Ciencias por su acompañamiento y valiosos aportes durante el desarrollo del proyecto.
Doctora Nelsy Loango y Doctor Ailan Farid Arenas por su colaboración y acompañamiento
Doctor Rafael Correa por su colaboración
Doctor Ignacio Zarante por sus enseñanzas durante mis estudios de Doctorado.
Dedicatoria
A mi madre que siempre estuvo a mi lado para ofrecerme su apoyo incondicional para seguir adelante con mis metas propuestas.
TABLA DE CONTENIDO
Contenido
INDICE DE ANEXOS………14
INDICE DE ABREVIATURAS ... 13
2. JUSTIFICACIÓN ... 19
3. ANTECEDENTES BIBLIOGRÁFICOS ... 21
3.1EMBRIOLOGÍA DEL CORAZÓN EN HUMANOS ... 21
3.1.1 Fase premorfogenética de la cardiogénesis ... 21
3.1.2 Fase morfogenética de la cardiogénesis ... 22
3.2 Proceso de torsión y rotación del tubo cardíaco. ... 23
3.3. INICIO DE LA TABICACIÓN DEL CORAZÓN. ... 24
3.3.1 Looping del tubo cardiaco: ... 24
3.4 FORMACIÓN DE LAS CÁMARAS CARDÍACAS: ... 25
3.4.1 Formación de los atrios ... 26
3.4.2 Formación de los ventrículos ... 27
3.4.3 Formación del tronco arterial ... 28
3.4.4 Formación del septum y del sistema venoso ... 29
3.4.5 Formación del septo auricular ... 29
3.5 Formación del septum atrioventricular ... 31
3.4.6 Formación del septum ventricular ... 32
3.5 Formación del sistema de conducción ... 33
3.5 Formación del nódulo sinoauricular ... 35
4. CARDIOPATÍA CONGÉNITA ... 36
4.1 DEFECTO CONGÉNITO CARDÍACO ... 36
4.2 CARDIOPATÍAS NO CIANOSANTES ... 36
4.2.1 Cardiopatía con cortocircuito de izquierda a derecha ... 37
4.2.2 Cardiopatías obstructivas izquierdas ... 37
4.2.3 Cardiopatías obstructivas derechas no cianosantes ... 37
4.3 CARDIOPATÍAS CIANOSANTES ... 38
4.3.1 Comunicación Interauricular (CIA) ... 38
4.4COMUNICACIÓN INTERVENTRICULAR (CIV) ... 41
4.5 DEFECTOS DEL CANAL ATRIOVENTRICULAR: ... 42
4.5.1 Clasificación ... 44
4.5.2 Defectos asociados ... 44
4.6 COARTACIÓN DE LA AORTA (COA) ... 45
4.6.1 Definición ... 45
4.6.2 Anomalías asociadas ... 45
4.9 TRANSPOSICIÓN DE GRANDES ARTERIAS (TGA) ... 48
4.9.1 Clasificación: ... 49
4.10 TETRALOGÍA DE FALLOT: ... 50
4.10.1 Defectos asociados: ... 51
5 EPIDEMIOLOGIA DE LA CARDIOPATIA CONGÉNITA Y MONITOREO ... 52
5.1 ETIOLOGÍA ... 54
5.2 FACTORES DE RIESGO DE RECURRENCIA ... 56
5.3 EPIDEMIOLOGÍA DE LAS MALFORMACIONES CARDIACAS EN SUR AMÉRICA ... 58
5.4 FRECUENCIA DE MALFORMACIONES CARDIACAS EN COLOMBIA. ... 59
6 EVOLUCIÓN DEL DESARROLLO DEL CORAZÓN ... 60
6.1 ONTOGENIA... 60
6.2 DESARROLLO DEL CORAZÓN EN INVERTEBRADOS ... 61
6.2.1 Drosophila melanogaster ... 61
6.2.2 Genes en el desarrollo del corazón en Drosophila ... 62
6.3 CORDADOS PRIMITIVOS ... 63
6.3.1 Tunicados ... 63
6.3.2 Amphioxus ... 65
6.4 VERTEBRADOS ... 65
6.4.1 Peces ... 66
6.4.2 Anfibios ... 67
6.4.3 Reptiles ... 69
6.4.4 Aves... 70
6.4.5 Mamíferos ... 71
6.5 FACTORES DE TRANSCRIPCIÓN EN LA CARDIOGÉNESIS ... 74
6.5.1 Factor de transcripción Nkx2.5... 75
6.5.2 Proteína GATA4 ... 78
6.5.3 Factor de transcripción TBX ... 80
6.5.4 Familia MEF2 ... 81
6.5.5 Microdeleción 22q11 ... 82
7.1. OBJETIVO GENERAL ... 85
7.2. OBJETIVOS ESPECÍFICOS ... 85
7.3. HIPÓTESIS ... 86
8. MATERIALES Y MÉTODOS ... 87
8.1. METODOLOGÍA PROPUESTA ... 87
8.1.1. Diseño del estudio ... 87
Es un estudio descriptivo realizado desde marzo de 2008 a marzo de 2009. ... 87
8.2 TOMA DE MUESTRAS ... 89
8.2.1 Entrevista y variables demográficas. ... 90
8.2.2 Criterios de exclusión: ... 90
8.2.3 Criterios de inclusión: ... 90
8.3 EXTRACCIÓN DE ADN DE SANGRE ... 90
8.4.1 Preparación de primers para PCR ... 91
8.4.2 Reacción en cadena de la polimerasa (PCR) ... 92
8.4.3 Análisis electroforético:... 94
8.5 ANÁLISIS DE LAS MUTACIONES ... 95
8.5.1 Análisis de DHPLC ... 95
8.5.2 Análisis de secuencia del protocolo de PCR ... 98
8.5.3 PCR de secuencia ... 98
8.5.4 Purificación de la secuencia de reacciones y análisis de electroferogramas ... 99
8.5.5 .Secuenciación ... 99
9 RESULTADOS... 102
9.2 ANÁLISIS DE LOS GENES NKX2.5 Y GATA4 ... 103
9.3 ANÁLISIS MUTACIONAL... 104
9.4 ANÁLISIS DE LA MUTACIÓN EN EL GEN NKX2.5 ... 105
9.4.1 Análisis de secuencias del exón 1.1 del gen NKX2.5... 105
9.4.2 Análisis de las secuencias del exón 1.2 del gen NKX2.5 ... 109
9.4.3 Análisis de las secuencias del exón 2.1 del gen NKX2.5 ... 110
9.4.4 Análisis de las secuencias del exón 2.2 del gen NKX2.5 ... 111
9.5 ANÁLISIS DE LAS SECUENCIAS DEL GEN GATA4 ... 113
9.5.1 Análisis de las secuencias del exón 1.1del gen GATA4 ... 114
9.5.2 Análisis de las secuencias del exón 1.2 del gen GATA 4 ... 114
9.5.3 Análisis de las secuencias del exón 1.3 del gen GATA4 ... 115
9.5.4 Análisis de las secuencias del exón 2 del gen GATA4 ... 116
9.5.5 Análisis de las secuencias del exón 4 del gen GATA4 ... 117
9.5.6 Análisis de las secuencias del exón 5 del gen GATA4 ... 118
9.5.7 Análisis de las secuencias del exón 6 del gen GATA4 ... 120
9.6 ANÁLISIS DE LAS VARIANTES ENCONTRADAS EN LOS GENES NKX2.5 Y GATA4 ... 121
9.7 ANÁLISIS DE LAS MUESTRAS DE SANGRE DE LOS PACIENTES DE NÁPOLES (ITALIA) ... 122
10 DISCUSIÓN ... 125
10.1 MICRODELECIÓN 22Q11 ... 125
10.2 GENES NKX2.5 Y GATA4……… 129
11 . CONCLUSIONES ... 133
12 RECOMENDACIONES ... 134
INFORME DE CONSENTIMIENTO INFORMADO PARA PARTICIPAR EN EL PROYECTO DE INVESTIGACIÓN: ... 160
ANEXOS ... 163
TÉCNICA DE EXTRACCIÓN DE DNA – PROBE... 164
3. CENTRIFUGAR A 3000 RPM DURANTE 8 MINUTOS SIN FRENO. ... 164
INDICE DE ANEXOS
INDICE DE ABREVIATURAS
ADN Acido Desoxirribonucleico
AHF Campo Secundario del Corazón
ANF Factor Natiurético Atrial
AP Arteria Pulmonar
AI Aurícula Izquierda
AD Aurícula Derecha
A.V Auriculoventricular
AVV Válvula Atrio-Ventricular
BMP Proteínas Morfogenéticas Óseas
C.A.V Canal Auriculoventricular
C.C Cardiopatías Congénitas
CIA Comunicación atrial
CIV Comunicación interventricular
CoA Coartación de la Aorta
DAP Ductus Arterioso Persistente
DVA Drenaje Venoso Anómalo
EA Estenosis Aórtica
EP Estenosis Pulmonar
FGF Factores de Crecimiento de Fibroblastos
FGF8 Factor de Crecimiento de Fibroblastos 8
GATA4 Factor de trascripción de la Familia GATA
IAA Interrupción del Arco Aórtico
IM Insuficiencia Mitral
IV- VI Ventrículo Izquierdo
LA-AI Atrio Izquierdo
LV-VI Ventrículo Izquierdo
MEF Factor Enhancer Miocitos
MSA Membrana Sub-Aórtica
NKX2.5 Factor de Trascripción de la Familia NK
NSL Dominio para la Localización Nuclear
PHF Campo Primario del Corazón
SA Saco Aórtico
SA SinoAuricular
SVIH Síndrome del Corazón Hipoplásico
TA Truncus Arterioso
TAD Dominio de Transactivación
TBX5 Factor de Tascripción de la Caja T
TC Troncocono
TF Tetralogía de Fallot
TGA Transposición de Grandes Arterias
TGFß Miembros de la Familia de Factores de Crecimiento,),
TGFB Factor de Crecimiento de Trofoblasto B
VCS Vena Cava Superior
ASOCIACIÓN DE LAS MUTACIONES SOMÁTICAS DE LOS GENES NKX2.5 Y GATA4 DE PACIENTES CON CARDIOPATIA CONGENITA ESPORÁDICA
Resumen
Los defectos cardiacos son las malformaciones congénitas más frecuentes, con una incidencia que se ha estimado entre 1-2% nacidos vivos y en el 10% de los fetos abortados espontáneamente Estos tienen una etiología multifactorial en donde se suman la predisposición genética y los factores ambientales. El estudio molecular del desarrollo cardíaco embrionario ha identificado dos factores de transcripción particulares, NKX2.5 y GATA4, que tienen un papel fundamental en la morfogénesis
cardíaca. Para la identificación de las mutaciones somáticas de los genes NKX2.5 y
NKX2.5 AND GATA4 SOMATIC MUTATION ASSOCIATION IN PATIENTS WITH SPORADIC CONGENITAL CADIOPATHY
Abstract
Cardiac defects are the most frequent congenital malformations, with an incidence estimated between 1-2% newborns and 10% in stillborn. Their etiology is multifactorial and might be attributed to genetic predispositions and environmental factors. In embryological studies two key transcriptional factors have been associated with cardiac morphogenesis: NKX2.5 and GATA4. NKX2.5 and GATA4 somatic mutations were identified and compared in DNA sequences obtained from affected cardiac biopsies, normal tissue and peripheral blood in patients with congenital cardiopathy. 54 patients with congenital cardiopathy were analyzed. DNA was extracted from
peripheral blood and tissue. NKX.25 and GATA4 were amplified by PCR, visualized
1. INTRODUCCIÓN
Los defectos cardiacos son las malformaciones congénitas más frecuentes, con una incidencia que se ha estimado entre 1-2% nacidos vivos y en el 10% de los fetos abortados espontáneamente (Hoffman, 1995; Hoffman y Kaplan, 2002)
La cardiopatía congénita (CC) es una de las principales causas de muerte infantil, y la alta susceptibilidad humana a desarrollar estas anomalías pueden deberse en parte a la complejidad del desarrollo embrionario, debido a la participación de varios eventos moleculares y morfogenéticos. La etiología de la cardiopatía congénita en la mayoría de los casos es desconocida y pueden derivarse de factores genéticos, ambientales y de la interacción de estos. Se ha podido identificar que un 10% de las cardiopatías congénitas son causados por alteraciones cromosómicas y el 90% restante tiene un
origen multifactorial (Lu Icardo et al., 2002).
En las últimas décadas, se ha observado un aumento gradual en el conocimiento sobre las causas genéticas de enfermedades humanas. El estudio de las cardiopatías congénitas (CC), ha permitido la identificación de algunos genes cuyas mutaciones son responsables o predisponen la aparición de estas enfermedades en los seres humanos.
Las condiciones de las cardiopatías congénitas son genéticamente heterogéneas, en parte debido a mutaciones en genes como NKX2.5 y GATA4, los cuales son factores de trascripción que juegan un papel fundamental en la morfogénesis cardiaca. El interés de estos dos genes en particular se ha incrementado a raíz de la identificación de mutaciones en los mismos pacientes con cardiopatías congénitas
Mutaciones en el gen NKX2.5 han sido identificados en grupos familiares y en personas que presentan una enfermedad del corazón congénita esporádica no sindrómica (con o sin bloqueo atrioventricular), incluyendo comunicación
Mutaciones en el gen GATA4 se presentan predominantemente en pacientes con familiares que tienen defectos del tabique cardiaco. La familia de los factores de transcripción GATA desempeña un importante papel en la diferenciación, crecimiento y supervivencia de diferentes tipos de células (Molkentin 2000, Weiss 1995). Seis miembros de la familia GATA se han identificado en vertebrados, los cuales contienen dos dominios con dedos de zinc (A/T) GATA (A/G) mediante la interacción de cofactores GATA1, 2 y 3 que son los primeros linajes hematopoyéticos que se traducen (Weiss y Orkin 1995) y GATA4, 5 y 6 se expresan en el mesodermo, endodermo y los tejidos derivados como el corazón, hígado, pulmón y tracto digestivo (Molkentin 2000). GATA4 regula la expresión de genes que son críticos para la contracción del corazón, así como la expresión de otros factores de la transcripción tales como NKX2.5, Hand2 y MEF2C (factor potenciador de los miocitos (McFadden
et al., 2000, Dodou et al., 2004)
Las mutaciones heterocigotas en GATA4 también se asocian con cardiopatías
congénitas, en los seres humanos (Garg et al., 2003). Teniendo en cuenta que los
pacientes con defectos del tabique y defectos conotruncales pueden compartir una base genética común, es posible que los pacientes con otros tipos de enfermedades
del corazón también presenten mutaciones en el gen GATA4. (Garg et al., 2003).
2. JUSTIFICACIÓN
Las enfermedades cardiacas congénitas y las arritmias cardiacas son causas frecuentes de mortalidad y morbilidad en niños de todas las edades, especialmente durante el periodo perinatal. Las anomalías cardiovasculares representan la clase más común de defectos al nacer (García 2004), con una incidencia que se ha estimado entre 1-2% nacidos vivos y en el 10% de los fetos abortados espontáneamente (Hoffman, 1995; Hoffman y Kaplan, 2002)
Las malformaciones congénitas cardiacas tienen una etiología multifactorial en donde se suman la predisposición genética y los factores ambientales (Friedman, 1998, Flores 1999; Hoffman 2002, Benson 2002, Burn 2002).
La alta tasa de incidencia de defectos cardiovasculares en niños genera un elevado costo para las familias, el sistema de salud y la sociedad en general. La incidencia de las cardiopatías congénitas esta probablemente subestimada debido a la corta
estancia del neonato en el hospital y a que no se contemplan los defectos letales in
útero, los que presentan cardiopatías leves que pasan inadvertidos, a veces hasta la
edad adulta, o que se resuelven de manera espontánea y que solo llegan a ser detectados por cianosis o por soplo cardiaco. A pesar del cuidado médico las cardiopatías congénitas siguen siendo una causa importante de mortalidad infantil y neonatal. Muchos de los abortos espontáneos y muerte en los primeros días de vida son debidos a cardiopatías congénitas aisladas que podrían ser evitadas con un diagnóstico precoz.
Los elevados costos quirúrgicos de cardiopatías congénitas se encuentran en un promedio de US$10.450 Las cardiopatías cianóticas, dentro de la cual se encuentra el defecto septal atrial aportan costos entre US$21780 por paciente.
(http://www.medicalworldtourism.com/en/prices.htm)
asesoría genética. Algunas de las búsquedas genéticas más prometedoras en cardiopatías congénitas son a nivel molecular y están enfocadas a mapear y determinar la función de los genes. Esta búsqueda pretende brindar conocimiento acerca del conocimiento cardiaco normal y los procesos fisiopatologicos que causan la enfermedad, ya que solamente conociendo como se expresan los genes normalmente y cómo los cambios o mutaciones causan defectos congénitos, será posible prevenir estos defectos cardiacos.
Las malformaciones pleitrópicas pueden ser el resultado de mutaciones discretas en factores de transcripción específicos y proteínas claves en el desarrollo embrionario y
la morfogénesis cardiaca. (Benson et al., 1999, Benson 2000, Robles et al., 2005)
Factores de trascripción como GATA4, NKX2.5 son genes que se expresan tempano durante el desarrollo embrionario y la morfogénesis cardiaca, y su expresión es crucial para la activación de otros genes que participan en el desarrollo del corazón. Mutaciones específicas en cada uno de estos genes se traducen severas anormalidades cardiacas como defectos septales y defectos de conducción, lo que pone de relieve el papel crítico que desempeñan estos genes en el trastorno del
desarrollo cardiaco (Bruneau et al., 2001, Garg et al., 2000).
3. ANTECEDENTES BIBLIOGRÁFICOS
3.1 Embriología del Corazón en Humanos
Un rasgo primitivo del desarrollo del corazón es su morfología simétrica. Los primordios del sistema cardiovascular se originan como clúster o pares simétricos de células mesenquimáticas en el mesodermo celómico. Inicialmente localizado en la región cefálica y dorsal del embrión, aparecen temprano para migrar alrededor de la membrana bucofaríngea para la formación del intestino anterior. La cardiogénesis es un proceso secuencial, progresivo, ininterrumpido e irreversible. Se divide en dos fases:
3.1.1 Fase premorfogenética de la cardiogénesis
El sistema circulatorio es indispensable para sustentar la embriogénesis, provee de nutrientes y oxígeno al organismo en desarrollo. De igual manera está implicado en la eliminación de desechos y sustancias tóxicas.
En esta fase premorfogenética no existe una estructura anatómica que identifique al corazón, sólo están presentes estructuras que paulatinamente están determinadas para transformarse en miocardio. La etapa más temprana en las que han detectado células con esta característica es la blástula de pollo, similar al embrión humano de 7±1 día de gestación. En esta etapa se han descrito dos preáreas cardiogénicas, ubicadas en el epiblasto, a cada lado de la mitad posterior de la línea primitiva (Rosenquist y Haan 1996), cuyas células han iniciado la especificación para
transformarse en miocardio (Montgomery et al., 1994). Durante la gastrulación las
células precárdicas migran a través de la línea primitiva hasta establecerse en el mesodermo y formar las áreas cardiógenas (15 ±1 día). Estos dos grupos celulares de forma oval se encuentran uno a cada lado de la línea primitiva, a la altura del Nodo de Hensen. En este caso, las células ya están especificadas y determinadas para
formar miocardio y endocardio (Rawles 1943).
endodermo de la región cefálica del embrión, comienzan a doblarse y forman el pliegue cefálico, que consta de la placa neural y el intestino portal anterior. Al mismo tiempo, el mesodermo se separa en dos capas, la somatopleura y la esplacnopleura; en esta última se encuentran células cardiogénicas, formando una creciente cardiogénica, que tiene forma de herradura. Simultáneamente, las células de la creciente cardiogénica comienzan a expresar genes característicos del miocardio
como los NKX2-5 y GATA4. (Laverriere et al., 1994).
NKX2-5 es considerado el gen maestro de la cardiogénesis (Srivastava y Olson 2000,
Lints et al., 1993), debido a su actividad en el mesodermo pericárdico y estructuras en
las que se transforma es fundamental para la expresión de MEF2 y GATA4, que a su vez participan en el control de la trascripción de proteínas contráctiles propias del músculo cardíaco.
3.1.2 Fase morfogenética de la cardiogénesis
Esta fase se divide en tres períodos: Corazón en tubo recto, proceso de torsión y rotación del tubo cardíaco e inicio de la tabicación del corazón
3.1.2.1 Formación del Tubo del Corazón
Los tejidos mesodérmicos dan origen al corazón primario y esto se hace evidente cuando el embrión está proceso conocido como gastrulación. En los humanos, ocurre en la tercera semana de desarrollo
3.1.2.2 Corazón en Tubo Recto
El corazón en esta etapa está constituido por las siguientes cuatro cavidades cardíacas primitivas: Bulbo aórtico = arterias pulmonar y aorta, Bulbos cordis = ventrículo derecho, Ventrículo primitivo=ventrículo izquierdo, Atrios primitivos derecho
e izquierdo = atrios definitivos (Salazar et al., 2006). El corazón en tubo recto, se
región anatómicamente izquierda. Ambos segmentos están separados por dos surcos relativamente profundos, denominados derecho e izquierdo.
3.2 Proceso de torsión y rotación del tubo cardíaco.
Los segmentos cardíacos primitivos inicialmente están en serie y siguen una dirección caudo-cefálica, sin embargo, como consecuencia del proceso de torsión y rotación del tubo cardíaco, poco a poco cambian de posición, hasta adquirir la polaridad derecha-izquierda. Este proceso sucede en tres etapas: Asa en C, Asa en S
y Asa avanzada. (Salazar et al., 2006).
3.2.1 Asa en C.
Esta etapa ocurre en el día 23±1 día, por la incorporación de células en los extremos caudal y cefálico. Se mantiene en el plano frontal, pero comienza a torcerse hacia la derecha, siendo ésta la primera manifestación de la asimetría del organismo. Simultáneamente, aparecen tres segmentos nuevos, dando como resultado un corazón con cinco segmentos cardíacos primitivos, que en sentido caudo-cefálico son: los atrios primitivos derecho e izquierdo, (región caudal de la rama caudal del asa); el primordio de la región trabeculada del ventrículo izquierdo (región cefálica de la rama caudal del asa); el primordio de la región trabeculada del ventrículo derecho (rama cefálica del asa) y el primordio de los tractos de salida (segmento proximal del
tracto de salida embrionario, llamado cono).(Salazar et al., 2006).
3.2.2 Asa en S.
El embrión empieza a flexionarse a nivel craneal y cervical, afectando la torsión y rotación del corazón (23±1 día). Simultáneamente, el asa cardiaca se coloca en el plano sagital y toma la forma se S. La curvatura mayor se vuelve ventral y la menor dorsal, también aparece el seno venoso (Anselmi y De la Cruz 1998).
3.2.3 Asa Avanzada.
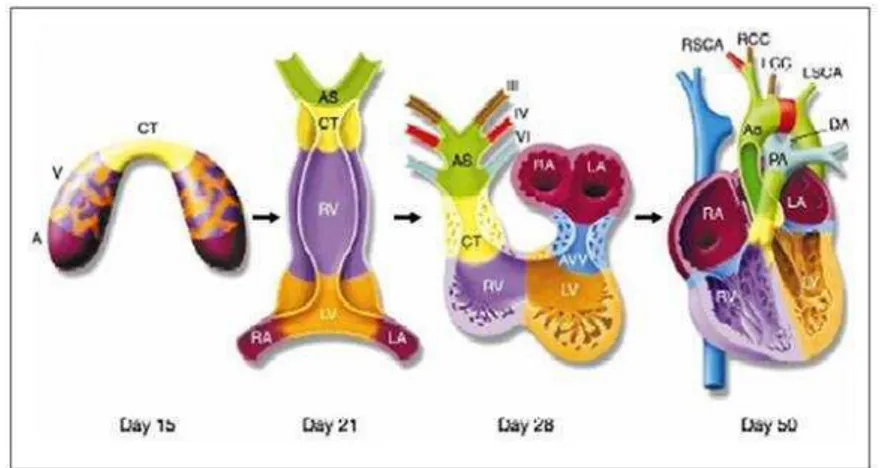
embrionario y empieza a formarse su segmento distal. La curvatura mayor cambia de posición y constituye el ápice ventricular (Figura 1).
Figura 1. Representación esquemática de la rotación del tubo cardiaco primitivo, para dar origen a las cuatro cámaras.
AS: saco aórtico; TC: troncocono; RV: ventrículo derecho; LV: ventrículo izquierdo; RA: atrio derecho; LA: atrio izquierdo; AVV: válvula atrio-ventricular
3.3. Inicio de la tabicación del corazón.
Inicia en el día 29, una vez que la región ventricular se ha colocado en posición caudal y la atrial es dorso-cefálica. Casi inmediatamente aparece el septum cardíaco primitivo, constituido por tres elementos: septum primum que separa los atrios, cojines ventral (superior) y dorsal (inferior) del canal atrioventricular y septum
interventricular primitivo, divide la región trabeculada de los ventrículos (De La Cruz et
al., 1983).
3.3.1 Looping del tubo cardiaco:
liberado el tubo se dobla hacia la derecha proceso conocido como looping del tubo del corazón, considerado como la primera evidencia de asimetría dentro del embrión,
aunque el canal atrioventricular está formado en forma asimétrica (Nonaka et al.,
2002).
Cuando se ha formado el looping ventricular, éste tiene componentes de tracto de entrada y de salida. El tracto de salida a su vez, alimenta las arterias que surgen del saco de la aorta que se extienden hacia los arcos faríngeos, los cuales se observan
en el día 25 del desarrollo. (Hiruma et al., 2002) (Figura 2).
Figura 2. Microfotografía electrónica de un embrión de ratón de 12 somitas.
Se observa el looping derecho del corazón (LV) parte apical del ventrículo formación del canal atrio-ventricular y desarrollo del looping del segmento atrial (tomado de: Hirumaet al., 2002)
3.4 Formación de las cámaras cardíacas:
del atrio. La sangre que fluye a través del componente auricular del tubo debe atravesar la totalidad del looping ventricular para alcanzar el tracto de salida. En esta etapa, se forma una constricción que marca el sitio del agujero interventricular primario, que se ha formado entre la entrada y la salida del agujero interventricular, que se convertirán en los ventrículos izquierdo y derecho respectivamente. En el tubo del corazón se producen importantes cambios con el fin de separar los componentes
en forma correcta (Christoffels et al., 2000).
Figura 3. Corte sagital de un embrión humano en estado 14 de Carnegie.
Se observa el desarrollo del ventrículo en comunicación con los componentes atriales del corazón, las almohadillas endocárdicas atrioventriculares y la vena pulmonar. (Tomado de Christoffels et al., 2000)
3.4.1 Formación de los atrios
siempre mantiene la unión atrioloventricular izquierda dentro de sus paredes, separados del atrio principal. El componente auricular principal del conducto del corazón ha crecido a ambos lados del tracto de salida para formar los apéndices auriculares, que son la primera diferencia morfológica de los lados derecho e izquierdo del atrio principal que está bajo el control del gen Pitx2. (Harvey 2002).
El seno venoso sistémico se incorpora dentro de la aurícula derecha en formación, que se insinúa entre el apéndice y el tabique primario en desarrollo. El apéndice se continúa distalmente con el vestíbulo de la aurícula derecha, formado por la incorporación de la musculatura del canal atrioventricular en el desarrollo de los atrios. Con el desarrollo del atrio derecho parte del canal atrioventricular se incorpora
dentro del atrio izquierdo, como el vestíbulo de la válvula mitral (Kim, et al., 2001).
Los componentes de la vena pulmonar inicialmente son insignificantes, pero después de la tabicación el componente venoso se expande para formar el techo de la aurícula izquierda y finalmente producir el arreglo definitivo de cuatro orificios
venosos (Kelly et al., 2001).
3.4.2 Formación de los ventrículos
Los ventrículos derivan del looping ventricular. Inicialmente parte del tubo primario se formó por el tallo del tubo en forma de Y; derivado desde la parte primaria distal de la creciente cardiaca. En este estadio toda la sangre desde el segmento atrial debe pasar a través de dos partes del tubo primario del corazón hasta alcanzar el tracto de salida.
Después del looping el tubo del corazón presenta dos curvaturas una externa y otra interna. La parte interna de la curvatura del tubo da lugar al desarrollo de la parte apical del ventrículo izquierdo y la parte externa da origen al componente apical del ventrículo derecho (Davis 2002).
en desarrollo. (Lamers et al., 1992). En este estadio el miocardio rodea el foramen interventricular y se denomina Foramen Oval que modela los segmentos ventriculares al interior del tubo con uniones en el canal atrioventricular proximal y distal del tracto de salida. Este modelamiento permite la separación de los atrios y la división del tracto de salida y ser compartida con los componentes apicales de los ventrículos izquierdo y derecho. Este intercambio requiere la expansión del canal atrioventricular
(Kim et al., 2001). La formación final del ventrículo izquierdo requiere que la parte
media proximal del tracto de salida sea transferido por encima del ventrículo derecho en desarrollo para formar el vestíbulo de la aorta, dejando el resto del tracto de salida como el infundíbulo subpulmonar. Esto requiere que se produzcan cambios en el
tracto de salida (Davis 1927, Lamers et al., 2002) (Figura 4).
Figura 4. Componentes del atrio y ventrículos (Davis CL 2002)
3.4.3 Formación del tronco arterial
aorta, que es inicialmente una estructura con paredes exclusivamente de miocardio, y en la parte proximal están separadas por curvas características. Debido a procesos aún no determinados, las paredes distales de salida cambian rápidamente el fenotipo de miocardio a uno arterial. Análogo a estos cambios el tubo que inicialmente era solitario es reemplazado por porciones intra-pericardiacas de la aorta ascendente y el tronco pulmonar. La parte proximal que pierde el fenotipo de miocardio se separa en dos componentes, la parte arterial y sus senos constituidos por la arteria proximal a la curva que marca el sitio de formación de las uniones sinutubulares. La parte proximal del tracto de salida es separada por la fusión de las almohadillas endocárdicas o cojinetes endocárdicos, y se forma nuevo endocardio dentro de ellas para producir la parte media del infundíbulo subpulmonar, que conserva su origen en el ventrículo
derecho (Van den Hoff et al., 1999.) Al mismo tiempo, la parte subaórtica del
segmento de salida divide el ventrículo izquierdo, por la fusión de los cojines de la cresta del tabique interventricular muscular, y el miocardio en la curvatura interna del corazón, que con el tiempo desaparece para permitir la continuidad fibrosa de las
valvas de las válvulas aortica y mitral en el techo del ventrículo izquierdo (Moorman et
al., 2003)
3.4.4 Formación del septum y del sistema venoso
El sistema venoso existe desde el inicio del sistema circulatorio. En embriones humanos en el estadio 12 de Carnegie las venas ya se han unido con el componente principal de la aurícula del tubo del corazón primario. El cuerno del seno izquierdo se incorpora dentro del surco auriculoventricular izquierdo en desarrollo, pasando por debajo del mesocardio posterior. El mesocardio constituye la vena portal pulmonar que entra al atrio izquierdo (Allwork y Anderson 1979). Así mismo, el sistema venoso tributario esta reorientado como un preludio para entrar en aurícula iniciando la septación atrial
3.4.5 Formación del septo auricular
Junto con la reorientación del sistema venoso tributario se desarrolla la vena pulmonar como una nueva estructura que utiliza el mesocardio posterior para llegar a
cambios en la topografía venosa, el atrio primario del corazón se divide en componentes izquierdo y derecho, cada uno con un apéndice en el lado parietal del
cuerpo de la aurícula (Moorman et al., 2003); ésta estructura le confiere especificidad
morfológica a los atrios. La septación se lleva a cabo en varios pasos: las dos almohadillas endocárdicas son producidas en la unión del canal auriculoventricular común del tubo del corazón (Figura 5), mientras que el tabique auricular crece como una porción muscular desde el techo de la aurícula; ésta placa muscular se desarrolla dentro de la cavidad atrial entre el sistema pulmonar y venoso que tienen su origen en región primaria de la aurícula izquierda, caracterizado por la expresión del gen PITX2.
Figura 5. Microfotografía donde se muestra el canal atrioventricular y el desarrollo del tubo del corazón en un ratón de 10 días.
(Moorman 2003)
La parte del atrio con características de lado izquierdo se extiende hacia la derecha
junto a la válvula del sistema venoso izquierdo (Franco et al., 2000). Con el desarrollo
superior del tabique muscular primario se abre para formar el foramen interauricular o el “ostium secundum”.
Después de formadas estas estructuras, las venas pulmonares izquierda y derecha se separan e incorporan en sus orificios como estructuras separadas en el techo de la aurícula izquierda, formando un tabique usualmente descrito como “septum secundum”; y los pliegues del colgajo de la valva limitan con el foramen oval que se
cierra en la vida post-natal (Robert et al., 2003)
3.5 Formación del septum atrioventricular
Antes de la formación del septum atrial, se han desarrollado dos masas de tejido mesenquimal en la parte superior e inferior entre el canal atrioventricular, denominadas almohadillas endocárdicas, y dividen el canal en componentes izquierdo y derecho. Estas almohadillas se adhieren a la cresta del septum ventricular muscular que forma los componentes trabeculares apicales desarrollando los
ventrículos izquierdo y derecho (Robert et al., 2003). Esta parte del tabique muscular
se desarrolla con el aumento de tamaño del ventrículo derecho para la posterior división del canal atrioventricular, que se expande hacia la derecha y hacia la parte inferior de las almohadillas atrioventriculares.
La fusión con la almohadilla inferior forma el borde supero-posterior del foramen interventricular embrionario. Esta parte del foramen se ha convertido en una estructura que proporciona el vestíbulo de la parte sub-aórtica del tracto de salida del ventrículo izquierdo. Los orificios tricúspide y mitral también incrementan su tamaño, que en un estadio posterior completan la septación ventricular. Para la separación del canal atrioventricular se forma un anillo fibro-adiposo que expande las uniones atrioventriculares que son ocupadas por las almohadillas atrioventriculares (Wessels
et al., 1996).
La fusión del componente del septo ventricular membranoso con el componente auriculoventricular, éste último derivado de la fusión de las almohadillas auriculoventriculares, se cierran en el foramen interventricular embrionario formando
3.4.6 Formación del septum ventricular
La septación de los ventrículos está coordinada por la formación de las válvulas auriculoventriculares y la septación de los tractos de salida. Una vez que los canales auriculoventriculares, los ventrículos y los tractos de salida cardiaco están alineados correctamente, todo está preparado para que se formen los siguientes pasos de la morfogénesis cardiaca: la tabicación ventricular, la división de los tractos de salida de la aorta ascendente, tronco pulmonar y desarrollo de las válvulas cardiacas. A finales de la cuarta semana aparece un tabique ventricular muscular que separa los dos ventrículos; al mismo tiempo el miocardio empieza a engrosarse y aparecen trabéculas o crestas miocárdicas en la superficie interna de ambos ventrículos (Larsen 2002).
Otro aspecto importante en la septación, es la separación de los orificios de las valvas mitral y tricúspide, el desarrollo del ventrículo derecho y la formación del anillo fibro-adiposo atrioventricular. El componente ventricular derecho, el cual forma el revestimiento atrioventricular, conserva su posición en la cresta del septum ventricular recién formado (Figura 6). Un segundo componente muscular se incorpora dentro del septo ventricular definitivo derivado del tabique muscular (Wenink 1981) o del septum
del canal atrioventricular (Van Praagh et al., 1989) Para el cierre del foramen
interventricular deben separarse completamente los ventrículos derecho e izquierdo recién formados, y la expansión del canal atrioventricular que existe entre la parte derecha del ventrículo y la aurícula. Después de la expansión del canal el ventrículo derecho continúa con el desarrollo del tracto de salida y los componentes de salida sub-aórticos se forman por la fusión de las almohadillas proximales o crestas que se
encuentran dentro del segmento de salida del tubo primario del corazón (Anderson et
Figura 6. Plano axial de un embrión humano en el estado 22 de Carnegie 22.
Se observa septación completa. En líneas amarillas punteadas se observa el septo atrioventricular recién formado. (Anderson 2003)
La septación ventricular concluye con el cierre del foramen ventricular. Este orificio tiene un límite inferior fijo: el borde libre del tabique ventricular, pero los límites superiores cambian a medida que se realiza el desplazamiento hacia la línea media del orificio atrioventricular y la migración bulbar. Antes de iniciarse este movimiento el orificio atrioventricular está delimitado por arriba por el espolón atrioventricular, en una fase intermedia está limitada hacia arriba por el espolón bulboauricular, hacia atrás por los cojinetes endocárdicos, al momento de cerrarse, tiene por límite superior el espolón bulboventricular. El cierre del foramen se efectúa por: a) crecimiento concéntrico del borde libre del tabique ventricular, b) crecimiento del espolón bulboventricular y c) tejido fibroso que prolifera desde atrás hacia los cojinetes endocárdicos.
(ChuaquiB.
http://escuela.med.puc.cl/publ/AnatomiaPatologica/01Cardiovascular/1malformacione s.html)
3.5 Formación del sistema de conducción
izquierda son derivadas desde el canal atrioventricular del tubo primario del corazón
(Kim et al., 2000).
Las células especializadas en el sistema de conducción del corazón están formadas por diferentes células cardiacas. Los genes NKX2.5 y TBX5 juegan un papel
importante en la formación y función del sistema de conducción (Gourdine et al.,
1999). Mutaciones en estos genes ocasionan defectos en el nodo AV (Kasandra 2001)
El sistema de conducción, el nódulo y el haz auriculoventricular se derivan de células en las paredes del seno venoso y el conducto atrioventricular. Inicialmente el músculo de la aurícula y ventrículo es continuo. La aurícula primitiva actúa como el marcapasos temporal del corazón, pero el seno venoso se hace cargo de esta función en poco tiempo. El nódulo sino-auricular (SA) se desarrolla a lo largo de la quinta semana. En un principio se encuentra en la pared derecha del seno venoso, pero se incorpora a la pared de la aurícula derecha con éste.
El nodo SA se localiza en la parte superior de la aurícula derecha, cerca de la entrada de la Vena Cava Superior (VCS) .Tras la incorporación del seno venoso, las células de su pared izquierda aparecen en la base del tabique ínterauricular inmediatamente delante de la desembocadura del seno coronario. Junto con las células de la región atrioventricular (AV), forman el nódulo y haz AV, situados inmediatamente encima de los cojinetes endocárdicos. Las fibras que surgen del haz AV pasan de la aurícula hacia el ventrículo y se dividen en las ramas derecha e izquierda del haz. Estas
ramas se distribuyen por todo el miocardio ventricular. (Kim et al., 2000)
Figura 7. Sistema de conducción eléctrica del corazón.
Las señales eléctricas del corazón viajan a través de vías (amarillo). Esto estimula la contracción de las cavidades superiores (aurículas) e inferiores (ventrículos).
3.5 Formación del nódulo sinoauricular
4. CARDIOPATÍA CONGÉNITA
Las cardiopatías congénitas (CC) son defectos estructurales o funcionales del corazón. Comprende la malformación cardíaca, la cardiomiopatía y la arritmia cardiaca. Las malformaciones congénitas constituyen la segunda causa de muerte en niño menores de un año en Colombia (Departamento Administrativo Nacional de
Estadística 2006 (http://www.dane.gov.co/), y fueron responsables del 20.8% de las
muertes en Colombia, según lo reportado por el estudio realizado por Zarante et al.,
2010 en tres ciudades de Colombia, identificaron en 52.744 nacimientos de los cuales
1.650 (3,12%) presentaron algún tipo de malformación congénita. Las CC se pueden
clasificar en función de la lesión anatómica y patológica o al mecanismo por el que se originan durante el desarrollo embrionario. Los recientes avances en genética están permitiendo la clasificación de algunas enfermedades cardiacas, basadas en procesos moleculares que muestran un alto grado de heterogeneidad genética (fenotipos similares que surgen de mutaciones en diferentes loci). (Sadler y Lagman 2005)
4.1 Defecto congénito cardíaco
Se define como una anormalidad en la estructura y/o función del corazón en el recién nacido, establecida durante la gestación. Se calcula que el 8% de las malformaciones cardiacas obedecen a factores genéticos, 2% a causas ambientales y una gran mayoría a la interacción compleja de influencias genéticas, ambientales y causas multifactoriales (Villagrá 2004). Los ejemplos clásicos de teratógenos cardiovasculares ambientales comprenden el virus de la rubéola y la talidomida, aunque existen otros como difenilhidantoína, litio, alcohol y muchos otros
compuestos.También, las enfermedades maternas como la diabetes
insulinodependientes, la hipertensión y el lupus eritematoso, se asocian también con defectos cardíacos. (Sadler y Lagman 2005)
4.2 Cardiopatías no cianosantes
cardiopatías obstructivas del corazón izquierdo, y otras menos frecuentes como la insuficiencia valvular y la cardiopatía obstructiva derecha no cianótica.
4.2.1 Cardiopatía con cortocircuito de izquierda a derecha
Constituye el grupo más numeroso de cardiopatías congénitas, alcanzando alrededor del 50% de ellas. El cortocircuito de izquierda a derecha puede ocurrir: a nivel auricular, como en la comunicación interauricular (CIA) y el drenaje venoso anómalo parcial (DVA); a nivel ventricular, como en la comunicación interventricular (CIV), a nivel auricular y ventricular, como la comunicación ventricular o canal atrio-ventricular (CAV); o a nivel de grandes arterias, como en el ductus arterioso persistente (DAP) y en la ventana aorta-pulmonar. La alteración fisiopatológica que define a este grupo de cardiopatías es el paso de sangre oxigenada desde el lado izquierdo del corazón (aurícula izquierda, ventrículo izquierdo, o aorta) hacia el lado derecho de éste (aurícula derecha, ventrículo derecho o arteria pulmonar) y la sangre que recircula por los pulmones no entra a la circulación arterial sistémica periférica. Las consecuencias fisiopatológicas y clínicas del cortocircuito van a depender de la
magnitud y del nivel anatómico en que ocurre.
4.2.2 Cardiopatías obstructivas izquierdas
Son todas aquellas que impiden o dificultan el normal flujo sanguíneo a través del lado izquierdo del corazón desde las venas pulmonares hasta la aorta torácica. La obstrucción al flujo sanguíneo en el lado izquierdo del corazón ocurre frecuentemente a nivel de la salida ventricular, obstrucción que puede ser total, como en la atresia aórtica, o parcial, lo que se denomina estenosis.
4.2.3 Cardiopatías obstructivas derechas no cianosantes
4.3 Cardiopatías cianosantes
Constituyen un grupo heterogéneo siendo la característica más común la presencia del cortocircuito de derecha a izquierda, con la consiguiente hipoxemia, manifestada clínicamente por la cianosis marcada de piel y mucosas (Heuser 2007)
4.3.1 Comunicación Interauricular (CIA)
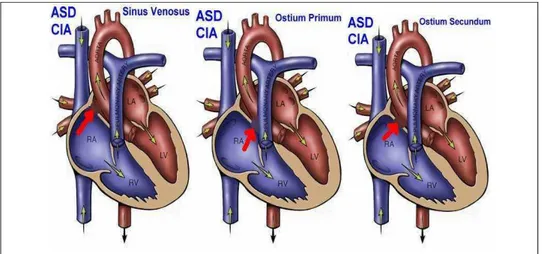
Los defectos inter-atriales fueron descritos por primera vez por Rokitanski en 1875; sin embargo, el cuadro clínico no fue descrito sino hasta 1941 por Bedfor (DE Papp and Parkinson Bedfor, 1991) Estas descripciones patológicas, se conocen desde la época del médico griego Claudio Galeno en el siglo II y fue muy bien descrita por Fawset en 1900 (Fawcett y Blachford 1900). El CIA es un defecto del tabique interauricular que comunica las dos aurículas, corresponde al 25-30% de las cardiopatías congénitas como defecto aislado (Heusser 2007) Estos defectos pueden estar localizados en cualquier sitio del tabique interauricular, siendo el más frecuente
la región del foramen oval, alrededor del 80% (Figura 8) (Martínez et al 1999,
Friedman 1998)
Figura 8. Esquema de los diferentes tipos de CIA.
A. Tipo ostium secundum o foramen oval. B. Tipo seno venoso superior. C. Tipo seno coronario. D. Tipo seno coronario E. Tipo ostium primum. Figura tomada
D A
C E
4.3.1.1 CLASIFICACIÓN
4.3.1.2 CIA tipo ostium secundum o tipo foramen oval
Este tipo de defecto se debe casi siempre a un ostium secumdum demasiado amplio, pero también puede deberse a una hipoplasia del septum secundum. Se localizan en la zona del foramen oval, (Figura 9). .Son las más frecuentes en adultos con un 80%
(Martinez et al., 1999, Friedman 1998). A nivel molecular Gruber y Ebstein (2004) en
una revisión de la literatura listan los siguientes genes implicados en la comunicación
interauricular (CIA): Agpt, Cited2, Fgf8, Nr2f2, Rxrα, Zfpm2, Tbx5, Gata4 y Nkx2.5.
Estos tres últimos han sido caracterizados en modelos animales y en humanos. (Gruber y Epstein 2004).
En 1998 Schott y colaboradores (Schott et al., 1998) describieron por primera vez tres
mutaciones del gen NKX2.5 (locus 5q35) Identificaron 4 familias con alteraciones de
patrón autosómico dominante que incluían CIA y/o alteraciones del ritmo como bloqueos atrio-ventriculares de 1º 2º y 3º grado (en algunos individuos progresaba con la edad) y muerte súbita. El defecto fue localizado electrofisiológicamente en el nodo aurículo-ventricular (AV) (Figura 7)
4.3.2.2. Defectos tipo ostium primum:
Situado en la parte baja del septo, muy próxima a las válvulas atrio-ventriculares, por encima de las dos valvas A-V, a la cual se asocia una hendidura en la valva anterior mitral capaz de producir una insuficiencia mitral. Una forma más completa de esta lesión es el canal atrioventricular (asociado al Síndrome de Down), asociada a una comunicación interventricular. (Figura 9).Constituye el 30% de todos los defectos septales atriales (Valdez Cruz L, Cayré R 1998)
los cojinetes y c) con solo fisuras incompletas de los velos septales tricuspideo (Chuaqui 2009)
Figura 9. Formación el setum Primum y septum secundum (Chuaqui 2009.
(http://escuela.med.puc.cl/pub/anatomiapatologica/01Cardiovascular/malformaciones.
htlm))
4.3.1.3 Defecto septal auricular tipo seno venoso:
Figura 10. Esquemas representativos de defectos septales atriales.
(http://www.medicalecho.net/IMAGES/4EYCIAHEART_F.htm)
4.4 Comunicación interventricular (CIV)
El defecto del septum interventricular es la anomalía cardíaca más común encontrada
en niños. Las manifestaciones de la anormalidad tienen rangos que van desde un soplo cardíaco de tono alto en un niño asintomático hasta signos severos de falla cardíaca en un niño con defecto de gran tamaño. (Texto de cardiología Sociedad
Colombiana de Cardiología y Cirugía Cardiovascular 2007). El defecto del septum
interventricular se ha estimado en un rango aproximado de 1,5 a 2 por cada 1.000 nacidos vivos, y en el reporte de Atlanta en 1980, se consideró que la incidencia era de 2.6 en 1.000 nacidos vivos. Tennesse y Norway encontraron una mayor incidencia: 5,6 y 5,7 por 1.000 nacidos vivos. (Gumbiner y Atsuyoshi T. 1998)
la localización del defecto. (Figura11) Las consecuencias hemodinámicas por el cortocircuito de izquierda a derecha son: a) sobrecarga del volumen de las cavidades ventriculares que producen dilatación y aumento de la presión diastólica, b) hiperflujo pulmonar tiende a aumentar la presión en el lecho vascular, alterando la relación de esta presión con el espacio intersticial pulmonar y c) compromiso del gasto cardiaco
sistémico. (Texto de cardiología Sociedad Colombiana de Cardiología y Cirugía
Cardiovascular 2007)
Figura 11. Esquema donde se observa la relación de un corazón normal y con
defecto septal ventricular(Nucleus Medical Art, Inc. 2009).
4.5 Defectos del canal atrioventricular:
El defecto septal atrioventricular (CAV) corresponde a un 4-5% de las personas con enfermedad cardíaca congénita. Recientes estudios reportan una incidencia de 0,19 por cada mil nacidos vivos. La forma completa es la más común y la transicional, la más rara. La relación entre hombres y mujeres para la forma completa del canal atrioventricular (CAV) es de 1:1. No hay un incremento de la enfermedad basado en
El desarrollo anormal de los cojines endocárdicos ocasiona defectos interauriculares e interventriculares y alteraciones en las valvas septales mitral y tricuspídea (Figura 12). En la forma parcial de defecto atrioventricular (AV) hay una fusión incompleta
de los cojines endocárdicos superior e inferior, resultando en una hendidura de la
válvula mitral anterior y en defecto del septum interauricular del tipo ostiumprimum.
En contraste, la forma completa de este defecto es asociada con falta de fusión entre los cojines superior e inferior, consecuentemente con división de la válvula mitral anterior en dos componentes separados, la valva anterior y posterior (Texto de
cardiología Sociedad Colombiana de Cardiología y Cirugía Cardiovascular 2007).
Figura 12. Esquemas representativos de un corazón normal y con defecto cardiaco atrioventricular
4.5.1 Clasificación
4.5.1.1 Canal Atrioventricular CAV Parcial
El CAV parcial se caracteriza por tener el anillo mitral y el tricuspídeo separados por los siguientes rasgos anatómicos que se pueden presentar en forma aislada o en varias combinaciones:
- Comunicación interauricular tipo ostium primum.
- Comunicación interventricular del tracto de entrada.
- Hendidura de la valva mitral anterior.
- Hendidura de la valva septal tricuspídea.
4.5.1.2 Canal Atrioventricular Completo (CAV)
El canal AV completo tiene como características un defecto septal grande, que compromete interauricular e interventricular una válvula auriculoventricular (AV) común que conecta ambas aurículas con ambos ventrículos. A la vez, el canal AV completo se ha subclasificado de acuerdo con la relación anatómica entre la valva
anterior y el septum interventricular. Esta clasificación fue descrita por Rastelli y cols
(Rastelli et al., 1999).
4.5.2 Defectos asociados
Los defectos parciales y completos AV están asociados con formas severas y complejas de enfermedades cardíacas congénitas, incluyendo síndrome de heterotaxia, Tetralogía de Fallot, doble salida del ventrículo derecho, anomalía total del retorno venoso pulmonar y transposición de grandes arterias.
Los pacientes con canal AV completo a menudo tienen una comunicación
interauricular tipo ostium secundum. El patrón de ductus arterioso se ha visto en un
4.6 Coartación de la aorta (CoA)
4.6.1 Definición
El término coartación de la aorta se refiere a un estrechamiento de la arteria aorta que causa una obstrucción al flujo aórtico (Rudolph AM 2001), el ventrículo izquierdo tiene que bombear más fuerte para impulsar la sangre por la válvula. El esfuerzo excesivo puede agrandar el ventrículo izquierdo, lo cual puede dar lugar a una
insuficiencia cardíaca (Figura 13) (Carl et al., 2000)
La ocurrencia de coartación de aorta es de 0,2 a 0,6 por 1.000 recién nacidos vivos y representa de la quinta a la octava forma más común de cardiopatía congénita (Fernández y Manrique 2007)
Figura 13. Esquema que representa estenosis valvular aórtica.
(http://www.texasheartinstitute.org/HIC/Topics_Esp/Cond/coarct_sp.cfm)
4.6.2 Anomalías asociadas
Las más frecuentes son las anomalías de la válvula aórtica, especialmente la bicúspide oscilando entre un 15% a 85% de acuerdo a los diferentes estudios.
restricción de las márgenes libres de la valva anterior, posición anormal de los músculos papilares, válvula mitral en paracaídas. Se observa asociación de coartación en cardiopatías complejas como: el canal atrio-ventricular, transposición de grandes arterias y truncus arterioso (Morris y McNamara 1998). El 13% de los pacientes con coartación de la aorta presentan aneurismas intracraneales.
4.7 Interrupción del arco aórtico (IAA):
La interrupción del arco aórtico (IAA) es una cardiopatía congénita severa y rara, definida como la falta de continuidad luminal y anatómica entre dos segmentos del arco aórtico (Weinberg 2001, Morris y McNamara DG 1998)
Es una cardiopatía poco frecuente y ocasiona un cuadro clínico severo de insuficiencia cardiaca. Engloba dos lesiones: a) Una comunicación interventricular, y b) Una interrupción de la aorta a nivel del nacimiento de los vasos que llevan sangre a la cabeza y a los brazos Esta zona se llama arco aórtico, de ahí el nombre de interrupción del arco aórtico (IAA). A través de la Comunicación Interventricular (CIV) pasa sangre oxigenada del ventrículo izquierdo (VI), al ventrículo derecho (VD), mezclándose con la sangre no oxigenada que proviene de la aurícula derecha (AD), y que regresa al pulmón a oxigenarse estando ya previamente oxigenada. Por otro lado del VI sale sangre hacia la aorta de la que nacen los vasos que llevan sangre al brazo derecho, cerebro, brazo izquierdo, no prolongándose como es habitual con la aorta descendente que lleva la sangre a la mitad inferior del cuerpo. La aorta descendente recibe sangre mezclada de la arteria pulmonar a través del ductus.
Figura 14. Esquema do
(http://www.cardiopatia
4.8 Ductus
El ductus arterioso per distal del arco aórtico presencia es necesaria hacia la aorta descen permeable, se localiza su diámetro puede ser circula el 70% del gasto
El ductusarterioso pers
recién nacidos vivos, s edad gestacional del p posible encontrarlo en
un 60% (Zachman et a
el 9 y 12% de las car ciudades localizadas a
La presencia del ductu derecha entre la aorta
donde se observa: IIA, CIA, CIV, y DAP
tiascongenitas.net/pinta_htmlbd_n_interaotxt.ht
tus Arterioso Persistente (DAP)
persistente es una estructura vascular que co co con la región proximal de la arteria pulmo ria en la vida fetal para desviar la sangre de endente; durante este período se denomina za justo entre las arterias pulmonares, tiene mo ser mayor que el de cada arteria pulmonar, y
sto cardíaco fetal.
ersistente tiene una incidencia que varía entre , siendo su frecuencia inversamente proporcio
l paciente. Así, en recién nacidos entre 32 y n un 20% de los casos y en menores de 30 s
t al., 1984). Se estima que como lesión aislada
ardiopatías congénitas, incrementándose has a más de 2.500 m sobre el nivel del mar. (Miao
ctus arterioso persistente permite un cortocircu
ta descendente y la arteria pulmonar izquierda,
t.htm)
comunica la porción onar izquierda. Su del tronco pulmonar
ina ductus arterioso
morfología tubular y , ya que por su luz
tre 1/2.500 y 1/5.000 cional al peso y a la 2 y 36 semanas es 0 semanas hasta en da representa entre asta un 20% en las iao et al., 1988)
flujo sanguíneo pulmonar y, por consiguiente, el retorno venoso hacia la aurícula izquierda.
Lo anterior incrementa la precarga del ventrículo izquierdo en grado variable según el
tamaño del ductus y la resistencia vascular pulmonar. La sobrecarga volumétrica
induce dilatación progresiva de la pared ventricular y activa los mecanismos neurohumorales del eje renina-angiotensina aldosterona.
En los pacientes portadores de ductus pequeños, el incremento del flujo sanguíneo
pulmonar es mínimo o imperceptible, por lo tanto, ellos son asintomáticos. A medida que aumenta el tamaño aparecen los signos de sobrecarga volumétrica de la aurícula y el ventrículo izquierdo y, por consiguiente, los signos de insuficiencia cardíaca congestiva compensada, inicialmente, y descompensada si el paciente no recibe tratamiento. (Stapper 2007)
4.9 Transposición de Grandes Arterias (TGA)
La transposición de grandes arterias es un defecto cardíaco congénito en el cual la relación de las grandes arterias está inversa al igual que la conexión ventrículo-arterial, es decir, la aorta está conectada totalmente o en gran parte al ventrículo derecho y la arteria pulmonar se conecta totalmente o en gran parte al ventrículo
izquierdo, la aorta se encuentra anterior a la arteria pulmonar. (Díaz et al., 2003,
Kirklin y Barratt-Boyes 1993)
La TGA es la cardiopatía cianógena más común en la infancia, le corresponde entre el 5% y el 9,9% del total de las cardiopatías congénitas en los países desarrollados (Talner 1980, Yuh y Reitz 2002)
través de un ductus arterioso, una comunicación interauricular o una comunicación interventricular, que permitirá el paso de la sangre oxigenada a la circulación sistémica y, a su vez, lograr la oxigenación de la sangre venosa sistémica.
El paciente con TGA tiene mayor tendencia a desarrollar enfermedad vascular pulmonar y especialmente cuando se asocia a la comunicación interventricular. Las causas de este fenómeno pueden estar asociadas a la saturación de oxígeno, el contenido de CO2 y el pH arterial pulmonar (Backer 2003).
Se han encontrado cambios severos de la circulación pulmonar a edades tan tempranas como los 2 meses de edad, y aproximadamente en el 80% al año de edad
en los pacientes con CIV a diferencia de los que tienen un septum interventricular
íntegro (Díaz et al., 2003, Vélez et al., 1999) (Figura 15)
Figura 15. Representación esquemática de transposición de Grandes Vasos
(www.rush.edu/spanish/speds/cardiac/tga.html)
4.9.1 Clasificación:
4.9.1.1 Tipo I
56% de los casos y en el 92% de los casos es evidente en el primer día de vida. El diagnóstico temprano en este caso es crítico.
4.9.1.2 Tipo II.
Transposición de grandes arterias con comunicación interventricular amplia (cortocircuito amplio) y flujo pulmonar aumentado En este caso puede no haber signos ni síntomas de enfermedad cardíaca con excepción de cianosis leve que se incrementa con el llanto. El cuadro de insuficiencia cardíaca congestiva se presenta en el transcurso de las primeras 2 a 6 semanas de vida, y se manifiesta por presencia de soplo cardíaco que puede ser más intenso con el transcurso de los días
4.9.1.3 Tipo III.
Transposición de grandes arterias con: CIV, obstrucción al tracto de salida ventricular izquierdo (estenosis subpulmonar) y flujo pulmonar restringido. Este grupo representa el 5%-8% de los neonatos con transposición. Los hallazgos clínicos son similares a los pacientes con tetralogía de Fallot, con cianosis extrema desde el nacimiento.
4.9.1.4 Tipo IV
Transposición de grandes arterias con CIV y flujo pulmonar restringido secundario a hipertensión arterial pulmonar severa. La hipertensión arterial pulmonar se hace presente después del nacimiento, con incremento de la cianosis. Puede no existir soplo cardíaco o solo un soplo sistólico eyectivo suave con la progresión de la hipertensión arterial pulmonar. (Vélez y Echeverry 2007)
4.10 Tetralogía de Fallot:
Figura 16. Esquema donde se observa Tetralogía de Fallot.
(www.monografias.com/trabajos63/desarrollo.car.)
4.10.1 Defectos asociados: — Foramen oval: 83%.
— Arco aórtico derecho: 25%.
— Persistencia de vena cava superior izquierda: 11%.
— Arteria subclavia izquierda aberrante: 10%.
— Anomalía de implantación de las arterias coronarias: 5% (9-10).
Otros defectos asociados son la presencia de canal atrioventricular, múltiples comunicaciones interventriculares, conexión venosa pulmonar anómala, enfermedad
de Ebstein, etc. El ductusarterioso está ausente en un 30% y esto es más común en
5 EPIDEMIOLOGIA DE LA CARDIOPATIA CONGÉNITA Y MONITOREO
Los defectos cardiacos son las malformaciones congénitas más frecuentes, con una incidencia que se ha estimado entre 1-2% en nacidos vivos y en el 10% de los fetos abortados espontáneamente (Hoffman, 1995; Hoffman y Kaplan, 2002). Las diferencias en la tasa de los distintos estudios se deben, en parte, a los diferentes
criterios de registro y de diagnóstico, así como a la época de estudio (Cloarec et al.,
1999, Hoffman et al., 1990, Pradat et al., 2003). Se ha observado un aumento
aparente de la incidencia de las cardiopatías congénitas en los trabajos más recientes, especialmente de las cardiopatías más leves, como la comunicación interatrial (CIA) y la comunicación interventricular (CIV), permaneciendo constante la prevalencia de las más severas, como la transposición de las grandes arterias (TGA) o el síndrome de corazón izquierdo hioplásico (SVIH. Este incremento se debe, al menos en parte, a una mejora en las técnicas de diagnóstico, fundamentalmente el Eco-Doppler, capaz de detectar defectos septales interventriculares muy pequeñas
(Cloarec S. et al 1999, Botto et al., 2003), que con frecuencia se cierran
espontáneamente en los primeros meses. En el trabajo de Cloarec y cols, la prevalencia de las cardiopatías congénitas disminuía de 9.8 a 5.3 por 1000 si se excluían los CIVs musculares de diámetro inferior a los 3 mm., que representaban el 70,2 % de todas las CIVs. La prevalencia de las cardiopatías también varía con la edad de la población estudiada, habiéndose estimado en un 8 por 1000 antes del
primer año de vida y en un 12 por 1000 antes de los 16 años (Martin et al., 1998). En
el sexo masculino hay un ligero predominio, en las obstrucciones al tracto de salida
del ventrículo izquierdo (Samánek et al., 1990, Guia et al., 2002).
TABLA 1. Síndromes Malformaticos con Afectación Cardiáca
Hernia diafragmática Atresia duodenal
Atresia de esófago y Fístula traqueo-esofágica Ano imperforado
Asociación VACTERL Asociación CHARGE
S. de Ivemark (Heterotaxia)Onfalocele Pentalogía de Cantrell y Ectopia Cordis Agenesia renal (S. de Potter) S. de Goldenhar
Agenesia del cuerpo calloso
TABLA 2. Cromosomopatías más comunes con Afectación Cardiaca
Visibles con técnicas convencionales
Cromosomopatía Porcentaje Defecto Cardiaco Trisomía 21 (S. de Down) 50 % CIV, CIA Trisomía 13 (S. de Pattau) >90% CIV, DAP, Valvulopatías
Trisomía 18 (S. de Edwards) >90 % CIV, DAP, Valvulopatías
45 X0 (S. de Turner) 25 % CoA, EP, EA, Otras 4p- ((S. de Wolff) 40 % DSV, DSA, DAP 13q- 50 % DSV
18q- 50 % DSV, DSA 5p- (Cri du chat) 25 % DAP Síndromes de microdelección
22q11 (CATCH-22) 75 % Malformaciones Troncoconales
12q22 (Noonan) > 50% EP, Miocard. Hipertrófica 7q11.23 (Villiams-Beuren) 75% EA Supra., EP Perif. 12q (Holt-Oram) 75 % CIA, CIV, Trastornos conducción
20p (Alagille) 85 % EP, TF
Abreviaturas: CIA: Comunicación interauricular. CIV: Comunicación interventricular. DAP: Ductus arterioso permeable.EA: Estenosis aórtica. EP: Estenosis pulmonar. TF: Tetralogía de Fallot
La mortalidad por cardiopatía congénita en niños menores de 1 año supone algo más de 1/3 de las muertes por anomalías congénitas y alrededor de 1/10 de todas las
muertes en ese periodo de la vida (Rosano et al., 2000). La mortalidad por esta causa