UNIVERSIDAD AUTÓNOMA METROPOLITANA-IZTAPALAPA
JCB1-
DEPARTAMENTO DE ClUíMlCA
J
ESTUDIOS DE FLUORESCENCIA Y DICROíSMO CIRCULAR EN PROTEASAS TIÓLICAS HOMÓLOGAS
T E S I S
que para obtener el grado de
/MAESTRO EN CIENCIAS
( ClUíMiCA )
PRESENTA
J
EMMA ADRIANA URBY SILVA
Y'.
Casa abierta al tiempo
UNIVERSIDAD AUTONOMA METROPOLITANA
DEPARTAMENTO DE QU~MICA
ESTUDIOS DE FLUORESCENCIA Y DICRO~SMO CIRCULAR EN PROTEASAS TIÓLICAS HOM~LOGAS
Q. Emma Adrianu Urby
Silva
UNIDAD IZTAPALAPA
Este trabajo se llevó a cabo bajo la asesoría del Doctor Andrés Hernández
J
Arana (Departamento de Química, UAM-lztapalapa) y del M. en C. Gerard0AGRADECIMIENTOS
Muchas personas han hecho posible la realización de este trrabajo y a ellas
quiero expresar m i más profundo agradecimiento.
AI Dr. Andrés Hernández Arana, y al M.en C. Gerard0 Muñoz Hernández por
la dirección de esta tesis y s u valiosa ayuda durante mis estudios de posgrado.
AI Dr. Arturo Rojo Dominguez por la revisión de la tesis, sus valiosos
comentarios y por aceptar ser parte del jurado en el examen de grado.
A mis amigos del laboratorio de Biofisicoquímica por su apoyo y sincera
amistad sin la cual esta tesis n o hubiera sido posible realizar.
Agradesco el apoyo económico del Consejo Nacional de Ciencia y Tecnología
CONTENIDO
I. I N T R O D U C C I ~ N
2. PROTEASAS TldLlCAS
2.1 Objetivos
2.2 Obtención de las proteasas
2.2.1 Materiales
2.2.2 Aislamiento y purificación de las enzimas
2.2.3 Purificación
3. TÉCNICAS EXPERIMENTALES
3.1 Actividad Optica
3.2 Luminiscencia
3.2.1 Fluoróforos
3.2.2 Efecto del p H
3.3 Preparación de las muestras
3.4 Espectros de fluorescencia
3.5 Espectros de Dicroísmo Circular
4. RESULTADOS
4.1 Dicroísmo Circular
4.2 Fluorescencia
5. CONCLUSIONES
1 3 11 11 11 12 13 14 14 18 19 19 20 20 22 25 25 36 44
1.
INTRODUCCION
Dentro de todas las biomoléculas los ácidos nucleicos y las proteínas son las más importantes; su importancia se debe al hecho de que ambos tipos de moléculas son "informativas", es decir, almacenan en su estructura química las instrucciones que determinan
Io
que ocurre en la célula, dichas instrucciones son necesarias para el ensamblaje de las proteínas de la célula. Cada proteína (única por su estructura, y en consecuencia por sus funciones) participa en los procesos que demarcan la individualidad de la célula y del organismo entero. Las funciones que desempeñan las proteínas son muchas, pero las de mayor importancia son las que actúan como enzimas o catalizadores biológicos, ya que prácticamente todas las reacciones que se efectúan en la célula viva necesitan una enzima, y casi todas requieren que ésta sea específica. Sin embargo, las proteínas no sólo catalizan los procesos metabólicos importantes de un organismo directamente (como en el caso de muchas enzimas o inmunoglobulinas) sino tambien indirectamente, al participar en la producción de otras moléculas de interes biológico como el ADN, ARN, Iípidos u otras proteínas. Por ejemplo; si se conoce la secuencia del ADN de un organismo individual se podrían leer (potencialmente) las secuencias de residuos aminoácidos de todas las proteínas enese
organismo; y a partir del conocimiento del comportamiento de estas proteínas podrían calcularse, en principio, todos los comportamientos biológicos del organismo y por consiguiente su manipulación.Con estos antecedentes es comprensible el creciente interés a nivel mundial por estudiar a este tipo de macromoléculas y a la fecha es posible el diseño de nuevas proteínas".". Hasta donde se sabe, la función biológica de una proteína está determinada por su estructura tridimensional global; ésta depende directamente de la secuencia de las unidades monoméricas o estructura primaria; de aquí resulta obvia la importancia de determinar la secuencia total de una proteína. Sin embargo,
el
metodo actual de secuenciación (degradación secuencia1 de Edman) se limita entre 20 y 40 ciclos; la secuencia más grande obtenida hasta el momento para una sola cadena polipeptídica consta de 1 O21 aminoácidos. Mas aún, suponiendo quepudiera determinarse la secuencia exacta de una macromolécula (como el ADN o
una proteína), otro problema al que se enfrenta la biología molecular es determinar
la forma en que dicha secuencia debe ordenarse en la estructura tridimensional
específica que determine su función biológica.
Las proteínas c o n funciones similares tienen secuencias semejantes
(homólogas); los grados de variación son mínimos, aunque en algunos casos llegan
a ser considerables. Las comparaciones secuenciales de tales proteínas ayudan a
determinar cuál o cuales partes del péptido son cruciales para s u intervención en
ciertos tipos de reacciones. Por otro lado, si se determinan las características
generales de u n grupo de proteínas homólogas, es posible, en principio, aplicarlo
a proteínas mutantes c o n función específica.
El presentre trabajo se encuadra dentro del proyecto de caracterización de
la familia de enzimas denominada proteasas sulfhidrílicasvegetales, particularmente tres diferentes tipos obtenidas a partir del látex de papaya: papaína, quimopapaína
y proteinasa
R;
así como tambien la bromelaína obtenida del tallo de la piña, laactinidina obtenida a partir del fruto llamado k i w i y la ficina del higo. Dicho
proyecto se esta llevando a cabo en el Área de Biofisicoquímica de la UAM-I.
2. PROTEASAS
TIÓLICAS
Las proteasas o enzimas proteolíticas es un grupo de enzimas que atacan el
enlace peptídico de proteínas y péptidos; cuando este ataque se realiza en u n
enlace peptídico del interior de la cadena se les conoce como proteinasas. Este tipo
de enzimas pueden ser encontradas tanto en plantas como en animales. U n grupo
particular de esta clase de enzimas son las proteasas tiólicas o cisteínicas, cuya
actividad proteolítica depende de u n grupo sulfhidrilo de u n residuo de cisteína. Se
encuentran distribuídas ampliamente a través de los sistemas vivos, ya que pueden
encontrarse tanto en animales y plantas así como en bacterias llevando a cabo una
gran variedad de funciones aún no completamente conocidas. Se tiene evidencia
de que la mayor parte de ellas pertenecen a una familia de enzirnas similares, tanto
estructural como f ~ n c i o n a l m e n t e ' ~ ' . En cuanto a s u reactividad, se ha mostrado que
el sistema cisteína-histidina es una característica común de este tipo de enzimas
tanto en plantas como en a n i r n a l e ~ ' ~ ~ ~ . ' ) . AI analizar la especificidad y forma de
unión c o n el sustrato de algunas de estas enzimas, los resultados m u e ~ t r a n ' ~ . ~ , ~ ' que
comparten estructuras c o n sitios activos y mecanismos de acción similares.
Las proteasas tiólicas más conocidas y utilizadas son las obtenidas a partir
de fuentes vegetales. La razón de ello es que estas enzimas se obtienen fácilmente,
en gran cantidad y tienen muchas aplicaciones; en particular, la papaína, que es
empleada en procesos tan diversos como la clarificación de la cerveza, el
ablandamiento de carne y como auxiliar digestivo"".
Existe una gran cantidad de proteasas cisteínicas vegetales caracterizadas
por sus pesos moleculares y puntos isoeléctricos los cuales son m u y variables
reflejando, probablemente, el entorno biológico diferente en los cuales operan
(Tabla i). El papel biológico de muchas proteasas n o se conoce c o n precisión; se
cree que su función puede ser la de proteger al fruto contra el ataque de insectos;
sin embargo, el porqué se producen multiples enzimas c o n propiedades m u y
similares en una planta es una pregunta que aún n o es posible responder.
En algunos casos, se pueden aislar diferentes enzimas de una misma fuente, como en el caso del látex de papaya (Carica papaya) del cuál al menos cuatro
distintas enzimas con actividad proteolítica (papaína, quimopapaína proteinasa
Q
y proteinasafi) han sido
caracterizadas"'~'2~'3~'4'.
E stas formas múltiples obtenidas de una misma fuente no son simples productos de degradación, ya que existen diferencias claramente establecidas en sus propiedades físicas y en la secuencia de aminoácidos (algunas de estas diferencias se mostrarán posteriormente). Todas ellas guardan una estrecha semejanza u homología en su secuencia de aminoácidos, con una cantidad de residuos idénticos que se encuentran en el intervalo de 40 a 90% '15).Actualmente se han caracterizado por difracción de rayos X las estructuras tridimensionales de tres proteasas tiólicas vegetales: papaína(16', a~tinidina"~', y proteinasa Q('*I; mientras que se tienen secuenciadas solamente siete: papaína, actinidina, proteinasa 0 , quimopapaína, bromelaína, papaya peptidasa IV y a i e ~ r a í n a " ~ - ~ ~ '
.
De la observación de la arquitectura de las tres primeras, se sabe que están integradas por dos dominios estructurales de aproximadamente 1 O0residuos de aminoácidos cada uno, lo que corresponde a cerca de 200 residuos por cadena polipeptídica. También se conoce que el sitio activo, localizado en la hendidura interdominio, se encuentra integrado por tres aminoácidos: una cisteína en el primer dominio y de una histidina y una asparagina en el segundo dominio. En esta tercia de aminoácidos. el grupo sulfhidrilo de la cisteína interacciona fuertemente con el anillo imidazol de la h i ~ t i d i n a " ~ ' , el cual a su vez se encuentra estabilizado por el carbonilo de la cadena lateral de la asparagina.
PAPAINA
Muchos de los trabajos en papaína han sido realizados utilizando la enzima preparada del látex seco. El estudio de la papaína se extendió después del informe de Kimmel y Smith en 1954'261 quienes la obtuvieron por una modificación al procedimiento que Balls y Lineweaver (1 939)'"' habían utilizado para preparar papaína cristalina del látex de papaya. Esta modificación permitía la preparación de cristales de papaína parcialmente activa. Actualmente existen métodos de preparación de papaína completamente activa conteniendo un sistema interactivo nucleofílico tiol-imidazol por molécula de proteína'28'.
La papaína es una enzima proteolítica cuyo peso molecular es de 23,350 Da con punto isoeléctrico de 8.75'7.38'. Uno de los dominios esta constituído predominantemente por u-hélices, mientras que el otro está formado básicamente de hojas p antiparalelas. El sitio activo se localiza entre los dos dominios con uno de los residuos catalíticamente importantes, la cisteína 25, en uno de los dominios, y con la histidina 159 en el otro dominio.
QUI M OPAPAl NA
La quimopapaína es la proteinasa cisteínica más abundante del látex de papaya, constituyendo casi con el 30%; fué aislada por primera vez por Jansen y Balls en 1941, generalmente se considera como heterogenea dadas las multiples formas de esta enzima que han sido separadas por cromatografía de intercambio c a t i ó n i ~ o ' ~ . ~ ~ ' ; está formada por una sola cadena polipeptídica con masas moleculares que van de 24000 a 24400 Da para cada uno de los cuatro
componentes
identificado^'^^'.
Como la papaína, la quimopapaína es una tiolenzima que requiere de una cisteína o de cianuro para su activación y es inhibida por reactivos que protegen ai grupo tioV7'. La papaína y la quimopapaína tienen un considerable grado de similitud en su especificidad por el s u ~ t r a t o ' ~ ' ~ . En contraste con la papaína, la quimopapaína es muy estable a pH á ~ i d o ' ~ ~ ~ ~ ~ ' , además de tener dos grupos tiol libres siendo enzimáticamente activo sólo uno de ellos '14.321.PROTEINASA
f2
El extracto acuoso del látex de papaya contiene la enzima proteinasa
f2
comoun componente en menor cantidad. Esta proteasa se encuentra como coeluyente
de la quimopapaína cuando es separada en sus fracciones por cromatografía de
intercambio catiónico, debido a que es la proteasa más es la última en ser
eluída de la columna al incrementar el gradiente salino (Fig.1 a). La proteinasa
f2
esel tercer constituyente proteolítico del látex de papaya y se caracteriza por su
extrema basicidad y u n punto isoeléctrico alto (Tabla I); fue aislada primero por
S ~ h a c k " ~ ' y ha sido llamada papaya proteinasa A, papaya peptidasa 11, papaya
proteinasa 111 y papaya proteinasa 0. Tiene una masa molecular de 24 O00 Da. La
actividad de la proteinasa
f2
es similar a la papaína aunque c o n diferencias sutilesen especificidad y estabilidad. Tiene una estructura similar a la de la papaha'"'
pero con una inserción adicional de cuatro residuos y c o n otras diferencias en la
conformación de la cadena principal resultado de sustituciones de prolina. El sitio
activo (cisteína 25, histidina 1 5 9 y asparagina 179, 1 7 5 en papaína) está en
posición idéntica en ambas proteasas.
Una comparación de la secuencia de aminoácidos de la papaína, la
quimopapaína y la proteinasa 0 muestra la gran similitud que existe entre ellas
(Tabla 11). La quimopapaína contiene 21 8 residuos de aminoácidos; de éstos, 1 2 6
son idénticos a los de la papaína (58.9%) y 1 4 1 (65%) a la proteinasa
Q,
incluyendo los tres enlaces disulfuro y las cisteínas requeridas para la actividad
enzimática; es decir, todos los aminoácidos involucrados en el mecanismo catalítico
son conservados a excepción de algunos residuos del sitio de enlace c o n el
sustrato, por lo que es razonable asumir que la quimopapaína y la proteinasa
f2
pertenecen a la misma familia de enzimas de la papaína'19.20.35'; esta idea ha sido
confirmada en el caso de la proteinasa
f2,
ya que s u estructura tridimensional esm u y semejante a la de la papaína, como se mencionó antes. Además, los espectros
de dicroísmo circular en el ultravioleta lejano muestran diferencias
mínima^'^^.^'',
esto indica que la conformación en solución de estas dos enzimas es prácticamente
idéntica; sin embargo, el espectro de la quimopapaína difiere considerablemente de
los de las otras dos e n z i m a ~ ' ~ ~ ' lo cual plantea una interrogante sobre las
CaracterÍsticas estructurales que son responsables de tales
difereiicias
espectralesCOMPOSICION DE AMINOÁCIDOS DE SIETE PROTEASAS Ti6LiCAS
SEO. CDOE Ai. Arg Lsn Asp Cyq tin Giu t h y H i s i i e LEU Lys Met ?he P r o Ser lhi l r p l y r V a l k x C l x Yo11
...
T A B L A
I 1
5 10 15 20 A
PAP -1le-Pro~G1u-Tyr-Val~Asp-Trp-Arg~G1n-Lys-Gly-Ala~Val~Thr-Pro-Val-Lys~Asn~Gln~Gly~Ser~Cys-Gly-Sfr-Cys QUI -Tyr-Pro~Glu-Ser-I1e~Asp-Trp-Arg~Ala~Lys-Gly-Ala~Val~Thr-Pro-Val-Lys-Asn~Gln~Gly~Ala~Cys~G1y-SEr-Cys
PPQ -Leu-Pro~Glu-Asn-Val~Asp-Trp-Arg~Lys~Lys-Gly-Ala~Val~Thr-Pro-Val-Arg-His~Gln~Gly~Ser~Cys~Gly-SEr-Cys
70 35 40 45 3
PAP -Trp~Ala-Phe-Ser-Ala~Val-Val-Thr~Ile~Glu-Gly-Ile~Ile~Lys-Ile-Arg-Thr-G1y-Asn~Leu~Asn~G1u~Tyr~S~r-Glu Q ü i -Trp-Ala~Phe-Ser-Thr~Ile~Ala-Thr-Val~Glu-Gly-Ile-Asn-Lys~Ile~Val-Thr-Gly-Asn-Leu-Leu-Glu-Leu~S~~r~Glu
PPQ -TrD-Ala-Phe-Ser-Ala-Val~Ala-Thr-Val~Glu-Gly-Ile-Asn-Lys~Ile~Arg-Thr~Gly-Lys-Leu-Val-G1u-Leu~Ser~Glu
55 60 65 70 15
PAP -Gln-Glu~Leu-Leu-Asp-Cys~Asp-Arq-Arg-Ser~Tyr-Gly-Cys-Asn-Gly~Gly~Tyr~Pro~Trp-Ser-Ala-Leu-Gln-Leu~Val QUI ~Gln-Glu-Leu~Val-Asp-Cys~Asp-Lys-His-Ser~Tyr~Gly-Cys-Lys-Gly~Gly~Tyr~G1n~Thr~Thr-Ser-Leu-Gln-T~~r-Val
PPQ ~Gln~Glu-Leu-Val~Asp-Cys-G1u~Arg~Arg-Ser-His-Gly~Cys~Lys-Gly-Gly-Tyr-Pro-Pro-Tyr~Ala~Leu~Glu~Tyr-Val
80 85 90 95 _-Loa
PAP -Ala-Gln~Tyr-Gly-Ile-His~Tyr-Arg-Asn-Thr~Tyr~Pro-Tyr-G1u-Gly-Val-Gln~Arg~Tyr~Cys~Arg~Ser~Arg-Glu-Lys QUI ~Ala-Asn-Asn~Gly-Val-His~Thr~Ser-Lys-Val-Tyr-Pro~Tyr~Gln-Ala-Lys-Gln-Tyr-Lys-Cys-Arg-Ala~Thr~A~;p-Lys
PPQ ~Ala-Lys~Asn~Gly-Ile-His~Leu~Arg-Ser-Lys-Tyr-Pro~Tyr~Lys~Ala-Lys-Gln-G1y-Thr-Cys-Arg-Ala-Lys~G~n~Val
105 110 115 120 v5
PAP -Gly~Pro-Tyr-Ala-Ala~Lys-Thr-Asp-Gly~Val~Arg~Gln-Val-Gln-Pro-Tyr-Asn~Glu~Gly~Ala~Leu~Leu~Tyr~St?r-Ile QUI ~Pro-Gly-Pro~Lys-Val-Lys-I1e~Thr-Gly-Tyr-Lys-Arg~Val~Pro~Ser-Asn-Cys-G1u-Thr-Ser-Phe-Leu-G1y~Aa~Leu
PPQ ~Gly-Gly-Pro~Ile-Val-Lys-Thr~Ser~Gly-Val-Gly-Arg~Val~Gln~Pro-Asn-Asn-G1u-Gly-Asn-Leu-Leu-Asn-A~la~Ile
130 135 140 145 2
PAP -Ala~Asn~Gln-Pro-Val-Ser~Val-Val-Leu-G1u-Ala~Ala~Gly~Lys-Asp-Phe-Gln-Leu-?yr-Arg-Gly-Gly-Ile~Plie~Val
QUI -Ala-Asn~Gln-Pro-Leu-Ser~Val-Leu-Val-Glu-Ala~Gly~Gly~Lys-Pro-Phe-Gln-Leu-Tyr-Lys-Ser-Gly-Val~Plie~Asp
PPQ ~Ala-Lys-Gln~Pro-Val-Ser-Val~Val~Val~Glu-Ser-Lys-Gly-Arg~Pro~Phe~G1n~Leu~Tyr-Lys-Gly-Gly-Ile-Phe-Glu
I55 160 165 170 175
PAP -Gly~Pro~Cys-Gly-Asn-Lys~Val-Asp-His-Ala-Val~Ala~Ala~Val~Gly-Tyr-Gly-Pro-. , , - , . .-Asn-Tyr-Ile
QUI -Gly-Pro~Cys-Gly-Thr-Lys~Leu~Asp-His-Ala-Val-Thr~Ala~Val~Gly~Tyr-Gly~Thr-Ser-Asp-Gly-Lys-Asn-T:ir~Ile
PPQ ~Gly-Pro-Cys~Gly-Thr-Lys-Val~Asp~His-Ala-Val-Thr-Ala-Val~G1y~Tyr~Gly~Lys-Ser-Gly-Gly-Lys-Gly-T:ír~Ile
18Q u 5 190 195 2nn
PAP ~Leu~I1e-Lys-Asn-Ser~Trp-Gly-Thr-GlyTrpGly-Glu-Asn-Gly-Tyr-11e-Arg~Ile~Lys~Arg-Gly-Thr-Gly-A:;n-Ser QUI ~Ile-Ile-Lys~Asn-Ser-Trp-Gly~Pro-Asn-Trp-Gly-G1u~Lys~Gly~Tyr~Met~Arg~Leu-Lys-Arg-Gln-Ser-Gly~A;n~Ser
PPQ ~Leu-Ile-Lys~Asn-Ser-Trp~Gly~Thr-Ala-Trp-Gly-Glu~Lys~Gly~Tyr-Ile-Arg-11e-Lys-Arg-Ala-Pro~G1y~Ain~Ser
705 21 o 21 5
PAP ~Tyr~Gly-Val-Cys~Gly~Leu-Tyr-Thr-SerSerPheTyrPro-Val-Lys-Asn
11
Ibl Tiempo
~ , g 1 l a ) Crmwtograma de separaci9n de quimopapaína ( 0 y proteinasa (2 (11)
2.1 OBJETIVOS
El presente trabajo tiene como objetivo fundamental la determinación, en forma
comparativa, de algunas caractrísticas espectroscópicas de las tres proteasas de
Caricapapaya. En particular se estudiará la fluorescencia y el dicroísmo circular en
la región donde absorben los residuos aromáticos ( 2 5 0 - 3 2 0 nm), buscando
establecer las semejanzas o diferencias que existen en el microambiente de los
residuos de tirosina y triptofano contenidos en las enzimas mencionadas.
2.2 OBTENCIÓN DE LAS PROTEíNAS
2.2.1. Materiales
Para la separación de la quimopapaína y la proteinasa 0 así como la
purificación de la papaína se utilizaron como materiales de partida lotes con las
siguientes características:
-Quimopapaína parcialmente purificada, libre de papaína y lisozima. Lote
64F8045 (EC.3.4.22.6) de Sigma Chemical Co. Esta preparación se utilizó para
obtener quimopapaína y proteinasa 0.
-Papaína doblemente cristalizada. Lote 11 3F8135 (EC:3.4.22.2) de Sigma
Chemical Co.
La concentración de la papaína fue determinada (de acuerdo c o n Glazer y
Smithi7’) por absorción Óptica a 280 nm, empleando u n coeficiente de extinción de
2 . 5 0 ml/(crn.mg)’” y similarmente para las otras dos proteasas utilizando un coeficiente de 1.83 ml/(cm.mg) 137.481
.
2.2.2. Aislamiento y purificación de las enzimas.
Con el fin de asegurar que los resultados obtenidos correspondan
únicamente a la proteína estudiada, es necesario purificar la enzima comercial por
medio de cromatografía líquida de alta resolución (HPLC). En este trabajo se utilizó
una columna TSK-SP5PW de intercambio catiónico (0.75x7.5 cm) instalada en un
cromatógrafo Varian 5020B.
Dado que las proteínas, así como los ácidos nucleicos, los aminoácidos y
péptidos son polielectrolitos, pueden ser separados por medio de cromatografía de
intercambio iónico. En este tipo de cromatografía, la fase estacionaria se constituye por partículas que se modificaron químicamente, de m o d o que presenten grupos
iónicos de carga opuesta a la de la muestra problema. Así, se hablará de
cromatografía de intercambio catiónico si el relleno de la columna tiene naturaleza
aniónica y la muestra problema de naturaleza catiónica; y se hablará de cromatografía de intercambio aniónico si el relleno tiene naturaleza catiónica y la
muestra problema es a n i ó n i ~ a ' ~ ~ ' .
AI llegar la muestra problema al interior de la columna, ésta, que tiene
afinidad por los iones de la muestra (de signo contrario) los retiene a través de la
formación de uniones electrostáticas. Mientras mayor sea la carga y el número de
uniones de los iones, mayor será la afinidad; luego, u n aumento en la fuerza iónican
al incrementar el gradiente salino, será capaz de liberar a las moléculas del soluto
de esta retención y arrastrarlas a lo largo de la columna. Cuando las sustancias que
se van a separar se mueven lentamente en comparación al líquido eluyente se
obtiene el cromatograma mejor hasta cierto límite.
2.2.3. Purificación
Aproximadamente 8 mg/ml de quimopapaína comercial parcialmente
purificada se disolvieron en un amortiguador de fosfatos (NaH,PO, /Na,HPO,) con
una concentración 0 . 0 5 M, p H 7.0. Los componentes de la proteína son eluídos de
la columna aplicando como gradiente salino lineal de NaCl en u n intervalo de 0.0
a 0.4M. Los componentes de la proteína son eluídos de la columna a una velocidad
de flujo de 0.5 ml/min, empleando un programa de gradiente bifásico de NaCI: de
0.0 a 0.4M de NaCl (pendiente de 0.015 M/ml) y de 0.4 a 0.8 M de NaCl
(pendiente de 0.04 M/ml). (Las condiciones cromatográficas experimentales más
adecuadas para la separación fueron establecidas por Solís Mendiola en s u tesis de
M a e ~ t r í a ' ~ ~ ' ) . La papaína fue purificada por este procedimiento, solo que únicamente
se inyecta una solución proteica de 4mg/ml, a p H 4.0, debido al límite de
solubilidad de la proteína a pH neutro. Dado que éstas proteínas son enzimas
proteolíticas que se autohidrolizan fácilmente, los sitios activos fueron previamente
bloqueados; para esto, primero se activaron con mercaptoetanol O. 1 M durante 30
minutos y luego se bloquearon con yodoacetamida O. 1 2 M dejando reaccionar
durante 60 min. Una vez bloqueadas se realizó la separación de la quimopapaína
y la proteinasa 0 y la purificación de la papaína.
A fin de eliminar las sales, las muestras fueron dializadas contra agua
después de ser separadas en HPLC. Los cromatogramas obtenidos se muestran en
la Fig.1.
3. TECNICAS EXPERIMENTALES
3. I . ACTIVIDAD OPTICA
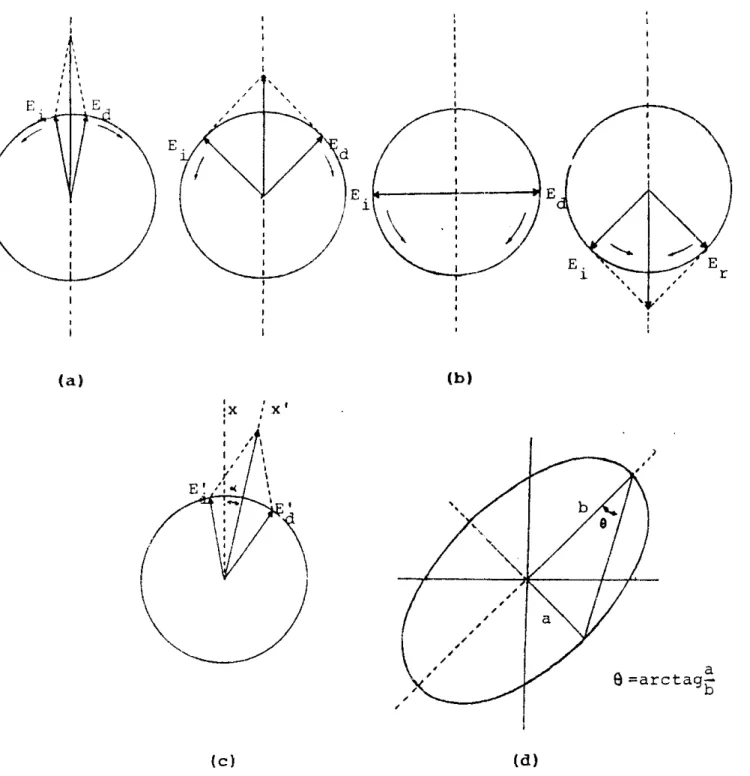
Casi todas las moléculas sintetizadas por organismos vivientes son ópticamente activas (pueden modificar las características de la luz polarizada que incide sobre ellas). La actividad óptica de pequeñas moléculas se incrementa conforme disminuye la simetría (particularmente por la presencia de átomos de carbono asimétricos y del efecto de éstos sobre cualquier cromóforo cercano) manifestándose através de cuatro fenómenos: rotación óptica, elipticidad, dicroísmo circular y birrefringencia circular.
Supongamos que el haz incidente es plano polarizado (el cual es generado por el paso de luz no polarizada através de un polarizador) cuyo vector eléctrico es
la resultante de la suma de dos vectores que rotan a la misma velocidad pero en sentido opuesto (Fig. 2a). Cuando estos vectores se encuentren en la misma dirección y sentido se reforzarán y la resultante, en el plano vertical, será máximo (positivo o negativo) mientras que cuando se encuentren en la misma dirección pero en sentido opuesto, la resultante será cero y la emisión será una onda senoidal en el plano vertical (el plano de polarización) (Fig. 2b).
Los dos componentes son llamados rayos circularmente polarizados a la izquierda y a la derecha y describen una hélice circular en el sentido y en contrasentido de las manecillas del reloj. Estos componentes no son una abstracción, ya que la luz circularmente polarizada puede ser generada en la práctica, como ocurre en un espectropolarímetro.
Ahora, en un medio ópticamente activo el índice de refracción de la luz circularmente polarizada a la izquierda es diferente a la del otro componente y cuando emerge de la muestra cada uno será aún circularmente polarizado, pero con diferente fase (Fig. 2c). La resultante estará ahora en un ángulo con el plano vertical: el ángulo de rotación
o.
Así el plano de polarización es rotado en un medioópticamente activo; éste es el fenómeno conocido como rotación óptica, y es una medida de la diferencia entre los índices de refracción para la luz circularmente polarizada a la izquierda y a la derecha.
Dado que en un medio ópticamente activo los índices de refracción asociados a cualquier transición son diferentes para los rayos circularmente polarizados a la izquierda y a la derecha, no es de sorprender el encontrar que esta diferencia en la interacción del medio con la luz se extienda tambien a la absorción, es decir, la luz circularmente polarizada a la izquierda y a la derecha se absorbe con diferente intensidad (A,#A,). Este fenómeno es llamado dicroísmo circular (DC),
y puede ser medido directamente. La diferencia de absorción (A,- A,) en una banda
puede ser positiva (efecto Cotton positivo) o negativa (efecto Cotton negati~o)‘~’’. El efecto es pequeño (una diferencia del 1
YO
es considerada como una señal grande de DC). Debemos de notar que si una de las componentes circulares es absorbida preferencialmente los dos vectores rotantes serán de longitud desigual. El efecto puede ser fácilmente visto ya que la resultante describe una elipse. La elipticidad de la luz después de pasar através del medio es así una medida alternativa del DC, y se utiliza cornunmente para de~cribirlo‘~”. Los valores de DC se reportan en AE donde, por convención, AE = E,- E,, siendo €,y E, los coeficientes de extinciónmolares para la radiación circularmente polarizada a la izquierda y a la derecha, respectivamente. Es posible relacionar la elipticidad con AE mediante la siguiente ecuación:
IOI =
3300
AEPara una solución protéica típica, en la región del ultravioleta lejano (1 80- 250 nm), se manejan concentraciones del orden de 0.2 mg/ml en celdas de 0.1 cm de recorrido óptico, mientras que en el ultravioleta cercano (250-350 nm) comúnmente se utilizan concentraciones de 2 mg/ml con celdas de 1 c m de recorrido Óptico. Cabe aclarar que para calcular los valores de [O] se utiliza la concentración de residuos de aminoácidos, dado que son los enlaces de amida los responsables de las señales de DC en esta región’58’; así, el valor de elipticidad reportado es el promedio por residuo.
En principio hay tres caminos por los cuales se obtiene actividad Óptica al
tratar con una proteína nativa. En primer lugar, la cadena polipeptídica está hecha
de residuos en la conformación L y la actividad Óptica está presente en virtud de
éstos. Segundo, un arreglo ordenado de residuos en espiral como es una u-hélice,
crean una nueva base para la actividad óptica y tercero, la distribución asimétrica
de cargas o dipolos cerca de un cromóforo que resulta de la estructura terciaria
rígida, puede tambien provocar actividad Óptica. Debemos dejar claro que la
actividad óptica de una cadena lateral bajo la influencia de u n solo carbono u es
imperceptible para fines prácticos, es decir, en u n polipéptido de cadena aleatoria
(una proteína completamente desnaturalizada, por ejemplo), los efectos Cotton de
las cadenas laterales de tirosina y triptofano son apenas observables y pueden ser
despreciados en comparación con la actividad óptica de los cromóforos del
p é p t i d ~ ' ~ ' . ~ * ' .
Inicialmente los estudios de DC en proteínas se llevaban a cabo a longitudes
de onda menores de 250nm, en parte, porque estas bandas son mucho más
intensas que en la región comprendida entre 2 5 0 a 330 nm (UV cercano).
Actualmente la disponibilidad de espectropolarimetros de DC mejorados ha
permitido extensos estudios de bandas de DC en proteínas. Estas bandas son
debidas principalmente a residuos de tirosina, triptofano, fenilalanina, cisteína y
ciertos grupos prostéticos. Las bandas de DC en el U V cercano reflejan la
estructura terciaria, mientras que las bandas en el U V lejano usualmente indican
estructura secundaria de acuerdo con Adler (et al.), Sears y Beychok'53,54'.
Fig. 2 al Luz plano polarizada dividida
en
sus dos componentes circularmente polarizados (derecho e izquierdo)encuentra en u11 plano
la rotación y e l plano de polarizacidn
b) La suma de los dos componentes de luz circularmente polarizada se c) Resultante después de atravesar U I medio ópticamente activo mostrando
3.2. LUMINISCENCIA
Cuando un material es expuesto a una fuente de radiación, ultravioleta o
visible, algunos de los iones o moléculas que lo constituyen pueden absorber parte
de esta radiación produciéndose en ellos transiciones electrónicas entre el estado
base y algún estado excitado. El ión o molécula en estado excitado regresa a s u
estado base utilizando diversos canales para disipar el exceso de energía; en
particular, una parte se disipa en forma de calor (vibraciones) hacia los iones y
moléculas vecinas y la parte restante puede ser liberada en forma de fotones, este
último fenómeno se conoce como luminiscencia.
La luminiscencia se ha clasificado de acuerdo c o n la duración de la emisión
después de que se interrumpe la excitación. Cuando la excitación se suspende,
existe siempre un decaimiento exponencial de la luz emitida. Cuando el tiempo para
que la intensidad inicial de emisión decaiga a l / e (donde "e" es la base del
logaritmo natural) de s u valor original es del orden de 1 O-3 seg. o menor, el proceso
luminiscente se denomina fluorescencia. Pero si este tiempo es de segundos, o
aún, horas, entonces el fenómeno recibe el nombre de fosforescencia.
Como podemos observar de lo dicho anteriormente, la luminiscencia de una
molécula dependerá, en general, fuertemente de las interacciones c o n el medio en
que esté inmerso; así, si las interacciones son m u y débiles la longitud de onda de
la emisión difiere m u y poco de la longitud de onda de excitación, mientras que para
las interacciones fuertes la especie química en estado excitado cede una cantidad
apreciable de energía en forma de calor hacia sus alrededores y esto traerá como
consecuencia la emisión de fotones con longitud de onda mayor a la de excitación;
incluso, eventualmente toda la energía se puede disipar en forma de calor; en tales
casos se dice que se presenta una transcición no-radiativa. Así la luminiscencia de
un emisor nos proporciona, cualitativamente, información acerca de la interacción
de éste c o n sus alrededores inmediatos.
3.2.1 Fluoróforos
Las moléculas que presentan fluorescencia observable se les llama
fluoróforos. Muchos compuestos aromáticos presentan fluorescencia como la
riboflavina, los aminoácidos tirosina, triptofano y fenilalanina o moléculas con
dobles enlaces como la vitamina A (retinol). Sin embargo, n o todas las moléculas
fluorescen eficientemente. Las bases nucleicas, por ejemplo, son particularmente no fluorescentes, mientras que una variedad de heterociclos similares muestran
fuertes emisiones. Las porfirinas son intensamente fluorescentes, mientras que sus
complejos c o n hierro (hemos) n o fluorescen. La causa de estas variaciones es que
algunas moléculas pueden perder su energía de excitación no-radiativamente. Sólo
aquellas especies en las cuales el proceso no-radiativo es lento, emiten
f ~ e r t e m e n t e ' ~ ~ ' .
3.2.2 Efecto del pH
Aparentemente pequeños cambios en el ambiente pueden tener también
efectos en la f l u o r e ~ c e n c i a ' ~ ~ ~ . Un ejemplo de esto es el cambio en el pH.A menudo
los espectros de la forma ácida y básica de una molécula son m u y diferentes.
Algunas veces se sigue el cambio de la emisión al pasar de una forma a otra; otras
veces solo una de las dos formas emite significativamente a cualquier longitud de
onda. Esto n o es de sorprender, ya que la protonación puede fácilmente afectar el
sistema 17 electrónico de una molécula.
Los estados excitados de las moléculas participan tambien en el equilibrio
ácido-base, pero a menudo exhiben valores de pKa que difieren por muchas
unidades de p H de las del estado base.
Las cadenas laterales aromáticas son responsables de la mayor parte de la
absorción ultravioleta y las propiedades fluorescentes de la proteína parámetros que
son pruebas Útiles de la estructura de una proteína'55'.
3.3. PREPARACIÓN DE LAS MUESTRAS
Las muestras fueron preparadas en u n amortiguador de fosfatos/boratos
conteniendo KCI 0.1 M., a u n p H inicial de 7.
Una vez disuelta la cantidad de proteína requerida en cada análisis, se
titularon c o n HCI y NaOH de concentración 1 M. El volumen de ácido o base que
se adiciona puede considerarse despreciable y así la concentración de las muestras
prácticamente no varió durante el experimento.
3.4. ESPECTROS DE FLUORESCENCIA
La variación de la intensidad de fluorescencia con el pH a temperatura
ambiente (25°C) fue seguida c o n u n fluorómetro modelo LS-5 de Perkin Elmer. El
intervalo de p H fue de 4 a 9 c o n Concentración de proteína de 0 . 1 mg/ml en un
amortiguador de fosfatos/boratos a p H = 7 conteniendo KCI 0.1 M. Los iones del
amortiguador n o interfieren c o n la emisión observada. La longitud de onda de
excitación fue de 282 nm haciendo un barrido de emisión de 2 8 5 a 5 0 0 n m se
obtienen dos bandas c o n máximos centrados alrededor de 293nm y 352nm. La
velocidad de barrido fue de 120 nm/min. En la Figura 3 se presentan las partes
principales del equipo, las cuales se describen bre~emente’~’’.
El equipo consta de una fuente de excitación (1) consistente de una lámpara de xenón pulsada de 1 5 W. La luz de esta lámpara es colimada por u n espejo
elipsoidal (2) y reflejada por otro espejo hacia una rejilla de difracción (3) , que se
emplea para seleccionar la longitud de onda de la luz que incide sobre la muestra.
La luz de excitación seleccionada por la rejilla de difracción incide sobre u n “divisor
de haz” (4) de tal manera que, aun cuando la mayor parte de la luz pasa através
de él, una pequeña fracción es reflejada para ser empleada como referencia por el
sistema de detección. La luz que atraviesa la rejilla es reflejada por dos espejos
antes de incidir sobre la muestra (7) colocada en una celda de cuarzo. Como
consecuencia de s u desexcitación, la muestra emite radiación electromagnética
(fluorescencia) la cuál es enfocada por dos espejos a la entrada de una segunda
rejilla de difracción (8) que permite determinar la longitud de onda de esta radiación
2
M
8
9 9
Fig. 3 Componentes principales del fiuorómetro LS-5
M = Espejos cóncavos E = Espejos planos
1 . -Fuente de excitación
2. -Espejo elipcoidal (colimador)
3. -Rejilla de difracción
4.- Divisor de haz
5.- Celda de rodarnina 6.- Obturador de haz
7.- Muestra
la cual es detectada por medio de un tubo fotomultiplicador (9). De aquí la señal pasa através de la electrónica de amplificación para finalmente presentar el espectro en un graficador.
3.5. ESPECTROS DE DICROíSMO CIRCULAR
Los espectros de dicroísmo circular (DC) se obtuvieron a 25°C en un espectropolarímetro JASCO J-500A en la región donde absorben los residuos aromáticos (250-320 nm); se empleó una celda de 1 .O cm. de recorrido Óptico con concentraciones de proteína de 0.276 mg/ml para papaína, 0.45 mglml para quirnopapaína y 0.451 rng/ml para proteinasa
Q,
en un amortiguador de fosfatos/boratos a pH 7.0. En la Figura 4 se se presenta un esquema de las partes principales del equipo las cuales serán descritas b r e ~ e m e n t e ' ~ " .a) Fuente radiación. En este equipo la fuente de radiación es una lampara de arco de xenón de alta intensidad.
b) Monocromador. Consta de dos prismas; la abertura de las rendijas puede controlarse manual o automáticamente con el "Sistema para control de rendija". Con esta última opción puede "barrerse" un espectro manteniendo constante el "ancho de banda espectral".
c) Polarizador. Su función es transmitir un haz de radiación polarizada en un plano.
d) Modulador electro-óptico. Esta es una de las partes Ópticas más importantes del equipo. Consiste de una placa de fosfato dideuterado de potasio cubierta con un material conductor. Cuando una diferencia de potencial (alternante en signo) se aplica entre las caras del modulador, la radiación que emerge del mismo se encuentra constituída de pulsos alternantes de luz circularmente polarizada "izquierda" y "derecha".
e) Compartimiento de la muestra. Provee el espacio necesario para colocar celdas cilíndricas de hasta 1 O cm de longitud.
f) Detector. En este componente la luz que emerge de la muestra es enfocada sobre el cátodo de un fotomultiplicador. Si la muestra es "ópticamente activa", la intensidad de la luz incidente en el detector variará con la misma
frecuencia del voltaje aplicado al modulador. Como resultado de esta fluctuación
en la intensidad, la fotocorriente generada tendrá u n comportamiento similar.
Finalmente la fotocorriente pasa a u n preamplificador que la convierte en una señal
de voltaje.
g) Sistema de medición. Este sistema recibe la señal fluctuante del detector,
la amplifica y la transforma en diferencia de absorción entre luz circularmente
polarizada "izquierda" y "derecha". La diferencia de absorción es convertida a
grados de elipticidad e indicada en la escala horizontal del sistema gráfico.
4.
RESULTADOS
4.1 DlCROlSMO CIRCULAR
Bandas de Absorción
Las señales de DC debidas a residuos aromáticos involucran transiciones de
un electrón en u n orbital IT del estado base a u n orbital n" vacío de mayor energía (estado excitado). Cada anillo aromático tiene una grán cantidad de estas
transiciónes electrónicas (
n-n"),
algunas de las cuales ocurren en el UV-cercanoy otras en el UV-lejano.
Transiciónes Vibrónicas
Los movimientos electrónicos durante la excitación son usualmente también
acompañados por cambios en la energía vibracional del nucleo. La combinación de
la excitación vibraciónal y electrónica ha sido designada bajo el término de
"transición vibrónica." Para cada estado electrónico excitado de una cadena lateral
de residuo aromático, exsisten muchas transiciónes vibrónicas; es decir, un
electrón del nivel electrónico basal puede ser excitado a cualquiera de los muchos
estados excitados vibrónicos diferiendo solamente en s u energía vibracional.
En la región de U V cercano, el espectro de DC de una proteína dada es
generalmente diferente de los espectros de otras proteínas no relacionadas
estructural o funcionalmente. Como podemos ver (Fig. 5). los espectros de las
proteasas estudiadas tienen una forma similar, lo cual es indicativo de que existen
ciertas semejanzas en la estructura tridimensional de estas enzimas.
Las asignaciones de las bandas, es decir, el tipo de residuo aromático
responsable de la presencia de cada banda, está basada en la información obtenida
por otros autores que utilizaron compuestos modelo de bajo peso molecular
principalmente amidas y ésteres de triptofano, tirosina y fenilalanina'58.59,60'. Cada
uno de los cuatro tipos de cadenas laterales que tienen bandas de DC antes de
250nm tienen ciertas características distintivas que pueden permitir su identificación; estas características se resumen de la siguiente forma'58':
a) Las bandas de DC de disulfuro son anchas y se extienden através del UV- cercano; algunas veces, estas bandas son más facilmente detectadas por su longitud de onda que se extiende hasta alrededor de los 310 nm.
b) Las transiciones vibrónicas del triptofano son de dos clases. En la primera clase, la transición de menor energía presenta una banda alrededor de 300 nm, acompañada de una segunda banda situada unos 4 nm de la primera. En la segunda clase, la primera banda se encuentra entre 289 y 293 nm y es acompañada por otra situada 7 nm hacia menor longitud de onda. En ambos casos se presentan otras bandas que son difíciles de resolver cuando hay más de un tipo particular de residuo aromático (como en el caso de las proteínas).
c) En el caso de los residuos de tirosina la transición de mayor energía tiene una posición entre 275 y 282 nm, mientras que la de menor energía tiene una posición entre 282 y 289 nm, dependiendo del medio en el que se encuentre la molécula. En un medio hidrofóbico la banda está situada cerca de 289 nm, mientras que en un medio hidrofílico la posición se mueve hacia 282 nm. Se observan otras bandas en longitudes de onda menores, aproximadamente a 7 y
12nm de la primera banda.
d) Los residuos de fenilalanina muestran señal de DC sólo en longitudes de onda inferiores a 270 nm. La progresión de bandas que ha sido observada tiene un espaciamiento muy variable dependiendo del compuesto. Más aún, es frecuente que tales bandas alternen en signo, lo cual hace más difícil la asignación.
e)
Consideraciones teóricas y experimentales indican que algunas interacciones tienen mayor probabilidad de producir señal de DC con más intensidad que otras. Cuando una cadena de tirosina o triptofano se encuentra a menos de 1OA
de otra cadena lateral aromática (histidina, tirosina, triptofano,Fig. 5 Espectros de dicroísrno circular (250-320
nrn)
a pH 7.0 de:( a ) papaína (conc. 0.276 mgiml)
( b ) qiiirriopapaína (conc. 0 . 4 5 mgiml)
( c ) proteinasa
Q
!conc. 0.45 rnghil)fenilalanina), la señal de DC en el UV-cercano es especialmente intensa debido al
acoplamiento p-p. El acoplamiento entre la transición del UV-cercano de una
cadena lateral de tirosina o triptofano y las transiciones
n-n*
del enlace peptídicocerca de 8A pueden tambien dar bandas de DC apre~iable'~''.
Las principales diferencias observadas entre los espectros de las tres enzimas
estudiadas a p H 7 fueron (Fig.5):
ai La banda negativa entre 290 y 320, asignada a un residuo de triptofano, es
notablemente mayor en quimopapaína que en las otras dos proteasas.
b)
Los espectros de papaína y proteinasa 0 tienen estructura fina en el intervalo de2 6 0 a 290 nm, mientras que en quimopapaína ésta no se observa.
c i El espectro de papaína es notablemente más grande que el de las otras proteínas
Haciendo uso de los razgos anteriores realizamos la asignación de las bandas en los
espectros obtenidos y se comparan las curvas obtenidas a partir de la titulación de
cada una de de las tres proteínas estudiadas seguidas por dicroísmo circular a
diferentes longitudes de onda (A). En seguida se presentan los resultados
obtenidos,
A = 298 nm
En el intervalo de pH de 7.5 a 9 la tendencia de las curvas es m u y similar
(Fig. 6 ) , y dado que a esta longitud de onda los residuos de tirosina y fenilalanina
no contribuyen a los espectros, los cambios observados pueden atribuirse a
cambios en el microambiente de los residuos de triptofano; además en este
intervalo de p H la cadena lateral de los residuos de cisteína se ioniza. AI analizar
la estructura tridimensional de la papaína y la proteinasa
n
puede verse que elresiduo triptofano 26 es el único que tiene una cisteína cercana (la del sitio activo),
m
por lo que podemos asumir que la posición de este triptofano c o n respecto a la
cisteína para las tres proteasas.
En el intervalo de p H de 9 a 11 la tendencia de la curva de papaína es m u y
diferente de los obtenidos para quimopapaína y proteinasa
C
2
(que son m u ysimilares entre sí). Esto puede deberse a que el triptofano que provoca la señal de
DC se encuentre rodeado de grupos que interaccionan fuertemente c o n él,
provocando menor movilidad de la molécula, o bien debido a que en este intervalo
de p H los residuos que se ionizan pueden ser lisinas y tirosinas, y la desprotonación
de estos grupos pueda inducir la formación de u n entorno más asimétrico,
favoreciendo la señal.
Dentro de una proteína, pueden existir interacciones electrostáticas entre las
cadenas laterales cargadas de algunos residuos. Dado que a p H neutro u n grupo
podrá estar cargado positivamente y otro negativamente, hay una fuerza
electrostática entre ellos. Tales enlaces se romperán si la proteína es llevada a
valores de p H muy altos o m u y bajos ya que alguno de los dos grupos perderá su
carga. La mutua repulsión entre los numerosos grupos cargados similarmente
cuando la solución es m u y ácida o m u y básica contribuirá a la inestabilidad de la
estructura plegada bajo estas condiciones, lo cual se observa por el cambio de la
tendencia de las curvas a valores de pH mayores que 11.
A = 287 nm
En los residuos de tirosina la transición de menor energía tiene una posición
entre 282 y 289 nm, dependiendo del medio en el que se encuentre la molécula.
En un medio hidrofóbico la banda está situada cerca de 289 nm, mientras que en
un medio hidrofílico la posición se corre hacia 282 nm, por lo que podemos pensar
que esta señal sea debida a uno (o más) residuos de tirosina ocultos (entorno más
asimétrico). La magnitud de esta banda (Fig. 7) es más intensa para la papaína que
para las otras dos proteínas en todo el intervalo de p H estudiado. Esto es debido,
probablemente, a que la papaína tiene más residuos de tirosina ocultos (1 2) que las otras dos
o
también debido quzá a que la papaína tiene cadenas laterales deotros residuos aromáticos más cercanos que en quimopapaína y proteinasa
n
.
o
ff'
"d
c",' ,pL; /"c
,, ,/'id
q'"
Oi i
Sin embargo, a esta longitud de onda la banda de triptofano también tiene señal
de DC. AI comparar el porcentaje de exposición de los residuos de triptofano
conservados en papaína y proteinasa
f2
se encuentra que son muy similares por loque el incremento en la señal de papaína lo podríamos atribuir al único residuo de
triptofano no contenido en quimopapaína y proteinasa
f2,
al triptofano 69. Esprobable que este residuo tenga cadenas laterales de residuos aromáticos a una
distancia de 1
OA
o menos que producen el incremento en la señal de DC.A = 278 nm
La transición de DC c o n mayor energía proveniente de tirosina está localizada
entre 2 7 5 y 282 nm por lo que la señal a 2 7 8 nm puede ser causada por dicha
transición y es la acompañante de la señal a 287nm. A ú n cuando la forma de las
curvas obtenidas como función del cambio del p H en las tres proteínas estudiadas
es m u y similar (Fig. 8), la correspondiente a papaína es mayor. Esto debido, quizá,
a que este residuo se encuentra más estabilizado por fuertes interacciones
electrostáticas c o n porciones polares de otros aminoácidos o cadenas laterales de
residuos aromáticos que en las otras dos proteínas. Dado que al aumentar el p H
llega u n momento en que las curvas tienden a bajar bruscamente alrededor de pH
11, esto viene a reforzar la afirmación de que esta banda es producida por
tirosinas ocultas; al aumentar el pH éstas quedan expuestas al solvente y su
actividad óptica se v e disminuída.
A = 255 nm
En la Fig. 9 se presentan las curvas obtenidas a partir del efecto del cambio
en el pH en papaína, quimopapaína y proteinasa
f2
seguido por DC a una longitudde onda de 2 5 5 nm. Para valores de p H menores de 9.5 las curvas son m u y
similares, mientras que para valores de pH mayores la curva de papaína se hace
aún más negativa que las curvas de quimopapaína y proteinasa
f2
en donde seobserva un cruce hacia valores positivos de la elipticidad molar. A p H mayor de
12.5 las tres curvas nuevamente tienen la misma tendencia, esto lo podemos
interpretar si suponemos que la señal es debida a la titulación de una tirosina
oculta, ya que a p H mayor de 12.5 las proteínas se han desnaturalizado dejando
expuestas las tirosinas al solvente, lo que da lugar al aumento en la señal. Por lo
tanto la elipticidad molar a 2 5 5 nm puede deberse a la ionización de residuos de
tirosina ocultos (pKaiti,..i..i = 9 . 5 - I 0 . 5 ) . Por otro lado, el microambiente de estos
residuos en papaína es diferente a los de quimopapaína y proteinasa
n,
es decir,probablemente en papaína dichos residuos se encuentren cerca de algunos anillos
aromáticos, ya que en compuestos modelo, aparentemente, la presencia de u n
segundo anillo aromático cercano al anillo fenolato causa u n aumento considerable
en la intensidad de DC'58i.
A esta longitud de onda la cadena lateral de los residuos de fenilalanina producen estructura fina en DC, sin embargo, de acuerdo a la literatura la señal del
tirosilo es hasta ocho veces más intensa que la de fenilalanina'58', por lo que esta
banda n o es atribuible a este residuo.
LL
w
O (D,..e-
a---
/&I
I N
i -
m
Lo
o
II
O O O
O
O 0 Od N N d CD a3
m
a7-
m
v>
m
Co
r- .-a)
n
b
+a I- T T In7 (d
C (d Q ([I Q
o
I
.-.
4.2 FLUORESCENCIA
La fluorescencia de las proteínas es originada por los residuos de
fenilalanina, tirosina y triptofano; estos residuos tienen las siguientes
características espectrales en agua a p H neutro'B61:
Compuesto Absorción Fluorescencia
. .
.
,.
.
,.
.
.
. . .
,. . . .
,.
..
.Amax( nm)..
..q,,JM-'cm.')..
. . .
.A,,,(nm).. . . .e..
. .
.sensitividad(&,,@)Triptofano 280 5600 348 0.20 1100
Tirosina 274 1400 303 0.14 200
Fenilalanina 257 200 282 0.04
a
Donde E,,, es el coeficiente de absorción molar y @ es la eficiencia cuántica,
definida como la razón de fotones emitidos a los fotones absorbidos.
En las proteínas que contienen los tres aminoácidos aromáticos la
fluorescencia es dominada usualmente por los residuos de triptofano, debido a que
s u absorción y s u eficiencia cuántica son mayores que los valores respectivos para
tirosina y fenilalanina, como se aprecia en el cuadro anterior. La emisión de
fenilalanina no se observa, debido a que su sensitividad es m u y baja. Otro factor
en la fluorescencia es la transferencia de energía entre los residuos. Por ejemplo,
la fluorescencia de fenilalanina también es m u y débil, debido a que s u emisión es
fuertemente disminuida por la transferencia de energía hacia los otros dos
aminoacidos aromaticos; esto debido al traslape entre la banda de emisión de
fenilalanina y las bandas de absorción de tirosina y triptofano (absorben
fuertemente alrededor de 280 nm, donde emite la fenilalanina). En proteinas que
contienen tanto tirosinac como triptofanos, la fluorescencia de tirosina es
disminuida debido escencialmente a tres causas:
1
.-
La emisión del triptofano es m u y fuerte.2.- La emisión de triptofano en proteinas plegadas se desplaza hacia longitudes de
onda más cortas, hacia la emisión de tirosina; y
3.- Debido a que puede ocurrir transferencia de energía no radiativa desde residuos de tirosina hacia residuos de triptofano en la proteina nativa.
Los cambios en la conformación de la proteina, como el desdoblamiento, producen grandes cambios en la emisión fluorescente. En el caso de las proteinas que no contienen triptofano, la emisión de tirosina cambia escencialmente en su intensidad.
En proteinas que contienen triptofano, se producen cambios tanto en la longitud de onda, como en la intensidad de emisión, que se observan durante el desdoblamiento. La emisión del triptofano de una proteina nativa puede ser más grande o más pequeña que la emisión del triptofano libre en solución acuosa. Consecuentemente, se pueden producir incrementos o decrecimientos en la intensidad fluorescente durante el desdoblamiento de la proteina. El máximo de la emisión se desplaza desde longitudes de onda cortas hacia los 350 nm, lo cual corresponde al máximo de la emisión de triptofano cuando éste se encuentra en solución acuosa. La localización exacta de este máximo depende en parte de la naturaleza y concentración del amortiguador. En un ambiente hidrofóbico, como lo es el interior de una proteína, la emisión del triptofano ocurre a longitudes de onda cortas'6E'.
La fluorescencia de grupos aromáticos es instantaneamente disminuída por choques de la molécula excitada con algunas moléculas pequeñas, como O,, I-, y acrilamida. Como es de esperar, en las cadenas aromáticas de la superficie de una proteína la emisión fluorescente en disminuída por choques con las pequeñas moléculas del solvente. Algo más sorprendente es que la emisión de muchos residuos internos puede ser ligeramente destruída por moléculas neutras como O, y acrilamida, lo cual sugiere que estas moléculas pueden difundirse hacia el interior de la proteína dentro del tiempo de vida del estado excitado. Inhibidores cargados como I-, son un orden de magnitud menos eficientes; esta notable diferencia refleja, presumiblemente, la inhóspita naturaleza no polar del interior de la proteína a moléculas cargadas. Sin embargo interpretaciónes detalladas de dichos estudios no son posibles debido a que estas se complican por fenómenos de transferencia de energía entre diferentes grupos dentro de la proteína, por su variación en el
rendimiento cuántico, por posibles perturbaciones a la estructura de la proteína
debida a la excitación de grupos aromáticos y por posibles enlaces de los
inhibidores c o n sitios dentro de la p r ~ t e í n a ' ' ~ ' .
En la Figura 10 se presentan los espectros de fluorescencia de las tres proteasas c o n una concentración de 0.1 mg/ml, en condiciónes de p H similares,
y excitando a 282 nm en todos los casos. Podemos observar que las tres proteasas
presentan espectros similares entre sí, es decir, estan compuestos por dos bandas
centradas alrededor de 293 nm y 3 5 2 n m que corresponden a la emisión de
tirosinas y triptofanos respectivamente"*'. Sin embargo, la intensidad de la banda
correspondiente a tirosinas en proteinasa
n,
es mucho menor c o n respecto a laque se observa en las otras dos proteinas; en contraste, la banda debida a
triptofanos es mucho mayor.
En la Figura 1 1 se presentan las curvas de intensidad fluorescente obtenidas
a partir del efecto del pH sobre la emisión de tirosinas (293 nm) para papaína,
quimopapaína y proteinasa
n.
De acuerdo a lo dicho al principio de esta sección,el efecto del solvente sobre los emisores provoca la destrucción de la luminiscencia
(disminución en la eficiencia cuántica); en la figura podemos observar que hacia
valores de p H m u y altos o m u y bajos la intensidad fluorescente es pequeña y del
mismo valor aproximadamente, sabemos que a estos valores de p H las proteínas
se encuentran desnaturalizadas y por lo tanto, podemos pensar que la mayoría de
los residuos emisores se encuentran expuestos al solvente; entonces para valores
de pH intermedios la fluorescencia es debida tanto a residuos internos como
externos.
La curva obtenida para papaína presenta dos máximos de emisión alrededor
de pH 4 y 9 aproximadamente, esto no se observa para las otras dos proteínas.
Como se dijo anteriormente, la luminiscencia de residuos internos se puede ver
disminuída debido a la difusión de pequeñas moléculas del solvente que inhiben la
fluorescencia de estos residuos'64'. Una intrpretación de los máximos observados
en papaína se puede dar si suponemos que la constante de difusión de esta
proteína es m u y sensible a los cambios en el pH, tomando u n valor mínimo para pH
alrededor de 4 y 9, después de estos valores empieza a ser importante la inhibición de la luminiscencia por efecto de la desnaturalización.
En la Figura 12 se presentan las curvas de intensidad fluorescente a 3 5 2 n m
como función del p H para las tres proteínas. Las curvas crecen conforme se
aumenta el pH, hasta alcanzar u n máximo alrededor de 7 . 5 . D e acuerdo a los
resultados obtenidos por Barel y Glazer (1 969) la curva de fluorescencia contra pH
para papaína corresponde a una curva de titulación de u n grupo de pKa de 6.6 que
ha sido interpretado como el reflejo de la interacción entre triptofano y una cadena
lateral de histidina (imidazol) protonada que forma parte del sitio activo'"'. D e
acuerdo a la estructura tridimensional de la papaína, los residuos 1 7 7 y 1 8 1 de triptofano están m u y cercanos a la histidina del sitio activo (159); c o n estos
resultados n o podemos atribuir la emisión fluorescente a alguno de estos dos
residuos; sin embargo al analizar las curvas a valores de p H arriba de 1 2
encontramos que estas decrecen rápidamente lo que nos indica que el residuo
emisor debe ser c o n mayor probabilidad un triptofano interno. Analizando la
estructura tridimensional de la papaína y la proteinasa
f2
encontramos que elresiduo de triptofano 177 en papaína (1 81 en quimopapaína y proteinasa
ni
estaexpuesto mientras que el triptofano 1 8 1 (1 8 5 en quimopapaína y proteinasa 0 )
esta oculto dentro de la proteína, por lo que podemos pensar que este último
residuo junto c o n la histidina son los responsables de la fluorescencia. Dado que
el mismo comportamiento se observa para las otras dos proteínas podemos decir
que la quimopapaína y la proteinasa
n
no sólo contienen u n residuo en un ambienteanálogo al triptofano 1 8 1 de la papaína sino que además se encuentra cercano a
él un residuo de histidina que al protonarse provoca el incremento en la emisión.
Analizando la estructura tridimensional de papaína y proteinasa
f2
observamos queel residuo de histidina del sitio activo y el triptofano responzables de la emisión
estan a una distancia menor a 1 O
A,
y de acuerdo c o n C r e i g h t ~ n ' ~ ~ ' estas condiciónes propicia para que se presente un proceso de transferencia de energía; como se
sabe'65', este proceso decrece fuertemente c o n la separación entre el donador y el
activador; así, el "reflejo" mencionado en la explicación dada por Barel y Glazer
(1969) para la fluorescencia en papaína bién puede interpretarse como u n
mecanismo de transferencia de energía entre la histidina (donador) y el triptofano
(activador). Por otro lado, se observa que la intensidad de la banda de
quimopapaína es más pequeña que la de las otras dos proteasas; de acuerdo con
la interpretación que hemos dado para la fluorescencia en papaína y proteinasa 0,
podemos suponer que en el caso de quimopapaina (en la que n o se conoce s u
estructura tridimensional) el residuo de histidina se encuentra más alejado del
triptofano que en las otras dos.
Cuando el p H esta entre 8 y 1 1 . 5 las curvas cambian nuevamente, esto
debido probablemente, al cambio en el microambiente del emisor debido a la
ionización de tirosinas (pKa= 9.5-10.5) y /o lisinas (pKa= 9.8-10.4) cercanas.
AI llevar el pH mas allá de 11.5, la intensidad de fluorescencia disminuye en
las tres proteínas; esto
se
debe, probablemente, a la aparición de nuevos canalesde des-excitación n o radiativos, debido a la interacción del emisor c o n el medio.
Intensidad de emisión íu.a.1
285 315 345 375 405 435 485 495 525
Long. da onda (nrn) Long. de onda (nm)
Intenaided da misión 1U.a )
25 ~
285 31i 3zl5 375 405 435 465 495 5 2 5
~ c n g ae onda ( n m )
Fig. 10 Espectros de fluorescencia, excitando a 282 nm, con concentración de
proteína. 0.lmgiml (ai papaína ípH = 6.86)
(b) quimopapaína (pH =7.361
W
u.
W
i
I
0, 03 L b C 1co
m
dm
m
Cm
Q
m
.-
n
m
Cm
Q
m
Q
.-
--
a
Ca
a
a
a
.-
*
a
Ca
a
a
Q
O
3.-
E
a
$-
.-
a
cn
a
C a,O
.-
-
a
@
E
c
N a3 N m O U Cm
O XI
o, c -n i w
m u
m.0
e,.-
E Q
" Cc
ü
CONCLUSIONES
1
.-
Estas proteínas son sistemas muy complejos, debido al gran número de residuos aromáticos que poseen.2.- A pesar de esto, los espectros de DC a pH 7.0 son cualitativamente similares; las diferencias cuantitativas podrían explicarse, como una primera aproximación, en base a los diferentes contenidos de aminoácidos aromáticos.
3.- AI variar el pH, el comportamiento de algunas bandas de DC atribuibles a tirosinas y triptofanos es diferente en las tres proteinasas. Esto sugiere que el microambiente, y las interacciones con grupos vecinos, de algunos de estos residuos varían de proteína a proteína.
Estas características espectroscópicas observadas en el UV-cercano pueden también tener contribución en los espectros de DC en la zona de peptídicos (UV- lejano) intensificando las diferencias existentes entre estas tres
proteína^'^''.
4.- Las tirosinas tienen posiciones menos conservadas en la secuencia de aminoácidos (tabla ii), por lo que sus propiedades fluorescentes muestran variaciones que contrastan notablemente de una proteína a otra.
5.- Los procesos difusivos de moléculas de solvente hacia el interior de las proteínas afectan notablemente la emisión fluorescente, siendo más dramático el efecto en papaína, en donde se observa una fuerte dependencia de la emisión con el cambio en el pH.
6.- La dependencia de la fluorescencia de triptofano con el pH es muy parecida (en su forma) en los tres casos. La razón de esta observación, de acuerdo a nuestra interpretación, es que solamente uno de estos residuos es el responsable de la mayor parte de la intensidad observada. Este residuo es el 181, el cual es excitado, probablemente, por un proceso de transferencia de energía desde la histidina del sitio activo. Además, los otros residuos de triptofano tienen posiciones