Ab initio calculations of structures and stabilities of (NaI) nNa1 and ( CsI) nCs1 cluster ions
Texto completo
Figure
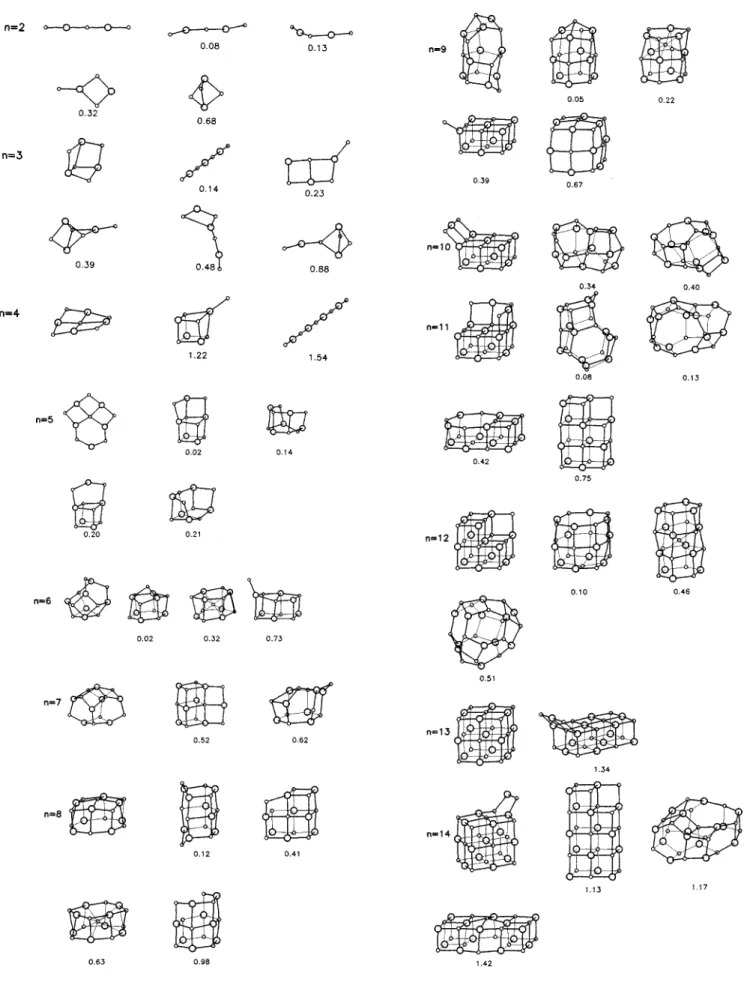

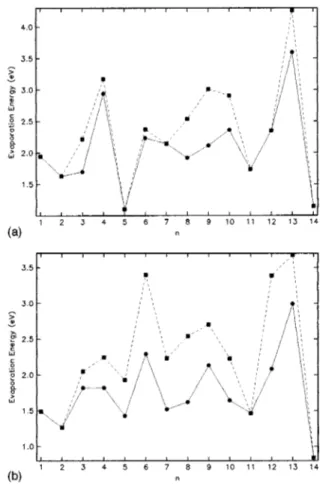
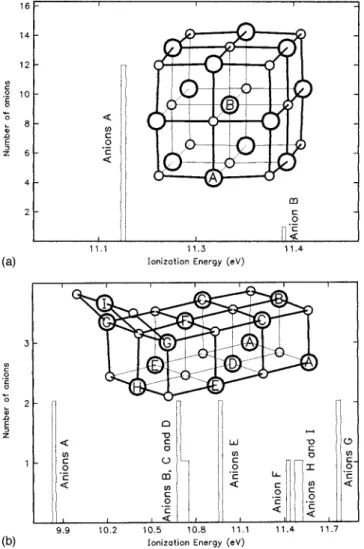
Documento similar
In the preparation of this report, the Venice Commission has relied on the comments of its rapporteurs; its recently adopted Report on Respect for Democracy, Human Rights and the Rule
a) The forensic-clinical interview is a reliable and valid instrument for measuring psychological injury in cases of IPV. Moreover, it is also valid in fulfilling
The faculty may have uploaded some file with the complete timetable by subjects.. 7) Scroll down and click on the academic course. You will find all the classes grouped by
It could maybe seem somehow insubstantial to consider an actual increase in computing power produced merely by a change in representation, consid- ering that computable functions
I also hope the book prepares the path for the new generation of telescopes, and most of all, sets the mood for cooperation amongst the 8-10 m facilities, as well as between these
In the previous sections we have shown how astronomical alignments and solar hierophanies – with a common interest in the solstices − were substantiated in the
well as powerful and versatile method for the modification of surfaces at the molecular level. SAMs of electroactive molecules have been exploited as electrochemical switches in
In this work we have been used classic and ab initio molecular dynamics simulations combined with electronic structure calculations to reproduce en- tire sequences of collisions and
