METROPOLITANA"
DIVISION DE CIENCIAS BASICA E INGENIERIA
DEPARTAMENTO DE QUIMICA
'ESTUDIO SOBRE LA
CARACTERIZACION DE
L A S
PROPIEDADES DE MOJADO DE ROCAS
FRACTURADAS'
Tesis que Presenta el
Alumno
José Antonio Moreno Razo
Matrícula 92321769
Para la Obtención del Grado:
Licenciado en Quimica
Asesor
METROPOLITANA”
DIVISION DE CIENCIAS BASICA E INGENIERIA
DEPARTAMENTO DE QUIMICA
’ESTUDIO SOBRE LA
CARACTERIZACION DE L A S
PROPIEDADES DE MOJADO DE ROCAS
FRACTURADAS’
Tesis que Presenta el Alumno
José Antonio Moreno Raza
Matrícula 92321769
Para
la
Obtención del Grado:
Licenciado en Quámica
Asesor
Dr. Armando Dominguez Ortiz
L A S PROPIEDADES DE MOJADO DE ROCAS
FRACTURADAS"
José
Antonio
Moreno
Razo
0.1 INTRODUCCIóN
. . .
60.2 DEFINICION DE MOJABILIDAD
. . .
70.3 MOJADO DE UNA SUPERFICIE IDEAL
. . .
100.3.1 Las Tensiones Interfaciales y la Superficie Sólida
. . .
100.4 HISTERESIS EN EL ANGULO DE CONTACTO
. . .
160.4.1 Definición de Histéresis
. . .
160.5 MEDICIONES DEL ANGULO DE CONTACTO
. . .
180.5.1 Método de la Elevación Capilar
. . .
190.5.2 Método de la Placa de Wilhelmy
. . .
190.5.3 Método del Anillo
. . .
200.5.4 Medidas del Volumen y Peso de la Gota
. . .
220.5.5 Método de la Gota Pendiente
. . .
230.6 MOJADO EN SUPERFICIES DE BAJA ENERGIA
. . .
230.7 MODELOS MOLECULARES Y TEORIA DE DISPERSION
. . .
260.7.1 Teoría de Lifshitz
. . .
280.7.2 Modelo de Hough y White
. . .
280.7.3 Modelo de Israelachvili
. . .
300.7.4 Mojabilidad Interfacial
. . .
320.8 INTRODUCCION
. . .
400.9 POROSIDAD
. . .
530.9.1 Defición de Porosidad
. . .
530.9.2 Determinación de la Porosidad
. . .
57O
.
11.1 Medio isotrópico. . .
680.11.2 Unidades de la Permeabilidad
. . .
71O
.
11.3 Validez de la ley de Darcy. . .
720.11.4 Tipos de permeabilidad
. . .
78O
.
11.5 Término de Forchheimer. . .
790.11.6 Permeabilidad de Klinkenberg
. . .
811 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Mojado de una gota en una superficie ideal
. . .
8Medición del ángulo de contacto
. . .
9Trabajo de Adhesión
. . .
13Trabajo de Cohesión
. . .
14Angulo de Contacto como Función del Cubrimiento de la Superficie
. . .
17Método de elevación capilar
. . .
20Métodos de la Placa de Wilhelmy: a) Separación; b) Estático
. . .
21Medida de la tensión interfacial por el método del anillo
. . .
22Separación de una gota desde la punta de un tubo estrecho
. . .
23Modelo de Hamaker
. . .
27Modelo de Israelachvili
. . .
31Triángulo de Neumann
. . .
32Cronología de importantes escritos
. . .
49Cronología de importantes desarrollos
. . .
51Respresentación un Medio Poroso
. . .
54Porosidad de bolas de dos tamaños diferentes (dl =diámetro de las pequeñas, d2 =diámetro de las mayores), los valores reportados es el co- ciente de d l l d 2
. . .
55Representación esquemática de distintos tipos de rocas indicando su relación entre su textura y porosidad
. . .
5621 22
23
24
25
26
27
28 29
30
Curva Esquemática de la relación entre el flujo y el gradiente hidráulico
. .
73Representación Esquemática del flju através de Medios Porosos
. . .
74Modelo de Arena par Flujo Rectilíneo Horizontal
. . .
75Flujo Vertical. ( u ) Flujo Libre; (b) Flujo Descendente con Columna de Fluido; (e) Flujo ascendente con Columna de Fluido
. . .
77Modelo para el Flujo Radial de Fluidos hacia el pozo
. . .
78Volumen Elmental Representativo,VER
. . .
80Concepto de Deslizamiento un Gas
. . .
84Efecto de la Presión en el deslizamiento de un Gas
. . .
85Efecto del Peso Molecular en el deslizamiento de un Gas
. . .
851
2
3
4
5
6
7
8
9
10
1 1
Tensión Superficial crítica de varios grupos determinados por Zisman
. . . .
34Constantes de Hamaker ( x~O-~OJ) calculadas por Hough y White (C=alcano; A=aire; W=Agua)
. . .
34Constantes de Hamaker ( x 10-20J) para diferentes materiales calculadas por Hough y White (M=material; A=aire; W=Agua)
. . .
35Distancias de Corte (LC = Do.nm). Constantes de Hamaker ( x~O-~OJ). Tensiones Interfaciales (20C. y Densidades (20C. 10-3Kgm-3)
. . .
35Angulos de Contacto Teóricos y Experimentales en PTFE; Constantes de Hamaker ( x 1 0 - 2 0 4 . asumiendo distancias de corte iguales. (S=PTEF; L=Alcano; A=aire)
. . .
36Porosidades de Empaquetados Regulares . . . 90
Análisis granulométrico
. . .
90Varlores de la porosidad eficaz para diferentes materiales
. . .
91Técnicas de Medición de la Porosidad
. . .
92Relaciones Empíricas obtenidas por Teodorovich et . al
. . .
930.1
La mojabilidad se refiere a la respuesta cuando un líquido entra en contacto con una
superficie sólida inicialmente en contacto con otro fluido, en la cual varias posibilidades
pueden existir, primero, que el líquido pueda moverse sobre el sólido, desplazando al fluido
original, y finalmente llegar a detenerse cuando el ángulo entre la interfase sólido-fluido y
sólido-líquido alcanza un cierto valor, un úngulo de contacto. De otra manera, el líquido
puede esparcirse sin límite; desplazando al fluido original, en tal situación el ángulo de
contacto es de O O . La importancia de la mojabilidad en recubrimientos, adhesión, deter-
gencia, lubricación y otras operaciones en las cuales los líquidos se aplican directamente
a las superficies sólidas. Esto también controla la imbibición espontánea de líquidos en
medios porosos. Aunque el significado de la mojabilidad ha sido cuestionado, su cuantifi-
cación se basa en el ángulo de contacto, y éste tiene una larga historia.
Thomas Young, en 1805 [Young, 18051 relacionó el ángulo de contacto con la energía de
superficie de las interfases en la unión de las tres fases (línea de tensión), y J. Willard
Gibbs [Gibbs, 19281 en 1878 expresó su relación en forma matemática, dándole al ángulo
de contacto una propiedad termodinámica.
La primera aplicación convincente de la energía de una superficie fue realizada por W.
A. Zisman et al. [Fowkes, 19641 en un trabajo realizado durante y después de la Segunda
Guerra Mundial en un Laboratorio de Investigación Naval. Zisman evitó los riesgos de
la irreproducibilidad por sus cuidadosas restricciones en sus sistemas y procedimientos.
En muchos casos fijó su atención en superficies sólidas y limpias, suaves, de 'baju energia' (superficies que no se contaminan fácilmente) en contacto con líquidos puros, los cuales
fueron incapaces de penetrar ó químicamente inactivos con la superficie. Las mediciones
de los ángulos de contacto, fueron reportados en la situación en la cual el líquido no
avanza sobre la superficie sólida (Úngulos de contacto estúticos). Bajo tales condiciones,
no únicamente la reproducibilidad de las ángulos de contacto, para un sólido dado, sino
también el gráfico de los valores de los ángulos de contacto versus l a s tensiones superfi-
ciales de los líquidos usados. Tales 'grúficos de Zismun' permiten extrapolar linealmente
la tensión interfacial a la condición tal que cos(6) = 1, dando como resultado la 'tensión
La mayor desventaja en las mediciones de los ángulos de contacto, la cual Zisman evitó,
es la histéresis, generalmente la histéresis se observa entre los úngulos de avance (OA) 3 retroceso (OR) y su relación con la heterogeneidad química de la superficie y su morfología,
esto fue dado a conocer en un artículo de Johnson y Dettre en 1969 [Johnson, 19691,
'.
..
los a'ngulos de contacto en una medición común y exitosa de la mojabilidad. Esto da información acerca de la energia de superficie y la heterogeneidad de la superficie. Su medición es sensible a contaminación en la superficie y controla muchos procesos técnicos.
. .
'Muchos autores se centran en medir en superficies de baja energía, del tipo estudiado
por Zisman, en contacto con gases u otros líquidos. Ellos incorporan desarrollos re-
cientes en entender las fuerzas de dispersión y reexaminar las gráficas de Zisman, como
los son los conceptos de Girifalco y Goog [Girifalco, 19751, Fowkes [Fowkes, 19641 y Neu-
mann [Neumann, 19741 en esa dirección. También actualmente se han usado técnicas
experimentales para medir los ángulos de contacto y evaluar, en particular los efectos de
la solubilidad de la superficie.
Los desarrollos más importantes en cuestión de mojabilidad en los pasados cincuenta años,
se pueden dividir en tres categorías:
1. Creciente entendimiento de las interacciones moleculares, dentro del líquido y a
través de las interfases líquido-sólido y líquido-líquido, responsables de la mojabili-
dad.
2. Un mejor entendimiento de la dinámica del mojado.
3. Un reconocimiento de la importancia del fenómeno de la mojabilidad en procesos y productos para los cuales el entendimiento era mínimo o nulo.
0.2
DEFINICION DE MOJABILIDAD.
La mojabilidad, no necesariamente involucra la interacción de un sólido. La mojabilidad
en presencia de un fluido inmiscible [Anderson, 19861. Puede ocurrir que el líquido cubra
la superficie, penetre en el sólido poroso ó el desplazamiento de un líquido por otro. Esto
puede ayudar a caracterizar la superficie y determinar las interacciones sólido/líquido.
VAPOR
Figura 1: Mojado de una gota en una superficie ideal.
La mojabilidad muchas veces es descrita por la gota asentada. Un diagrama
esquemático se presenta en la figura 1. El ángulo de contacto es una mediada de la
mojabilidad, y se mide hacia la fase más densa en el punto-tangente a la coexistencia de
las tres fases, figura 2.
A
valores pequeños del ángulo de contacto, la mojabilidad es altay a valores del ángulo de contacto altos, la mojabilidad es pobre. Los ángulos de contacto son siempre menores que 180' (el ángulo de contacto más alto observado, mercurio, (Hg),
en una superficie de vidrio, se ha reportado y tiene un valor de 148' [Young, 19281. En
sistemas que tiene más de un ángulo de contacto estables se dice que muestran histéresis
en el ángulo de contacto.
La interacción de superficies de baja energía con líquidos es a través de
fuerzas de dispersión o de van der Wals. La teoría de Hamaker-Lzfshitz (H-
L) [Hamaker, 1953, Lifshitz, 19561 proporciona un entendimiento preciso en las fuerzas
VAPOR
4
Ysv
YQL
SOLIDOFigura 2: Medición del ángulo de contacto.
otro por Israelachvili [Israelachvili, 19731, Lifshitz los aplicó en la teoría del mojado. Su
trabajo se centra en evaluar los conceptos y teorías de Zisman, Girifalco y Goog, Fowkes
y otros. La teoría de
H-L
requiere modificaciones en los conceptos de Zisman, i.e., elefecto del empacado molecular, la estructura de la superficie y la longitud de la cadena
de las monocapas en el mojado. Por otro lado, la teoría H-L ignora la posibilidad de la
penetración de las moléculas del líquido mojante dentro de la superficie del sustrato.
Los gráficos de Zisman (cos(@ us. la tensión superficial del líquido mojante) se han
usado por mucho tiempo para caracterizar la mojabilidad en superficies de baja energía.
Estos gráficos dan un buen ajuste empírico a los datos experimentales. El intercepto de
éstas curvas con el eje del cos(8) = 1 se conoce como la tensión superficial critica, Sin
embargo, las pendientes en los gráficos de Zisman han sido virtualmente ignorados, estas
contienen mucha información, particularmente acerca de la solubilidad de la superficie.
Una controversia adicional en la literatura del mojado ha sido la ecuación de estado de
superficie de Neumann [Neumann, 19741. Muchos otros problemas que se notan en la
literatura son causados al aplicar ecuaciones de equilibrio a sistemas en no-equilibrio.
Otros problemas ocurren cuando el ángulo de contacto se aproxima a cero. Algunos
confusiones existen entre la fuerza de superficie y la energia libre de superficie.
del mojado y las interacciones entre las fases. Esto es porque la tensión superficial, la
tensión interfacial y la presión de esparcimiento se pueden evaluar. Li et al. [Li, 19911
sugiere que los sistemas líquido/líquido/vapor no siguen las mismas leyes que los sistemas
sólido/líquido/vapor, sin embargo, los sistemas sólido/líquido/vapor tienen un mayor
importancia en detergencia, recuperación secundaria del petróleo y en muchos procesos
biológicos. La ecuación de Bartell-Osterhof (B-O) es la que relaciona el contacto
interfacial con los ángulos de contacto medidos en aire.
La clave para entender la mojabilidad es el entendimiento que, ésta se determina entre . " ,
las fuerzas de adhesión entre el líquido y el sólido y las fuerzas de cohesión en el líquido.
:?
(/'' 6
Las primeras causan que el líquido cubra al sólido y las segundas obligan al líquido a
formar una gota. El ángulo de contacto se determina por la competencia entre estas dos
fuerzas.
p,
r::
(? i.
3 :
r,:
;:2
F;".h.?;,*+
p "
c-1 A
. r
0.3
MOJADO DE UNA SUPERFICIE IDEAL.
Casi todas las teorías del mojado usan el mismo modelo de superficie ideal. Esta
superficie no presenta histéresis, es suave y no deformable. Cuando una gota de un
líquido se coloca en tal superficie, se asume un ángulo característico, figura 2. El ángulo
de contacto es independiente de la gravedad [Johnson, 19591.
0.3.1
Las Tensiones Interfaciales
y
la
Superficie Sólida.
Tensión Superficial sólido/vacío.
-ys/o, es comúnmente llamada tensión superficial, es el trabajo reversible para formar una
donde, Gs/,, es la energía libre de Gibbs de exceso de la interfase sólido/vacío, T la
temperatura absoluta, p la presión, Aslo el área de la superficie sólida y Nj es el número
de moles de la superficie de exceso de la j - esirno componente.
En la ecuación ( 1) indica que la derivada se realiza a T, p y el número de componentes
de exceso, Nj, constantes.
Tensión Superficial sólido/vapor.
YSlv, la tensión superficial sólido/vapor, es el trabajo reversible asociado para formar una
superficie unitaria cuando la superficie esta en equilibrio con el vapor saturado de un
líquido, esto se define en la ecuación ( 3):
Ys/v = Ys/o - XTT, f.
donde:
donde l?s/v es la energía libre de Gibbs de exceso del vapor en le sólido, p es la presión y
po es la presión de vapor del líquido y R es la constante de los gases.
Tensión Interfacial sólido/líquido.
Ts/l es la tensión interfacial sólido/líquido, es el trabajo reversible para formar una unidad
donde, Gs/l es la energía libre de Gibbs de exceso de la interfase sólido/líquido y A,/1 el
área sólido/líquido.
Gibbs obtuvo la misma ecuación minimizando la energía libre del sistema en un
desplazamiento virtual de las superficies e interfases [Johnson, 19591. Gibbs enfatizó que
es muy difícil medir ys/l y yslv. El consideró que la ecuación ( 6) puede ser mas simple y
más general que la ecuación de Young:
donde:
7rs/l mide la tendencia de un líquido a esparcirse. Esta es una cantidad negativa que Gibbs llamó: "la tensión superficial del fluido en contacto con el sólido". "/l/v mide
la tendencia de la gota a contraerse bajo las fuerzas de tensión. Las interacciones de
éstas presiones superficiales y tensiones superficiales se pueden representar por vectores,
figura 2. Para superficies de baja energía, 7rsIv es cero ó cercano a cero cuando 8 es
mayor que cero. Esto es una observación experimental. Además se explica asumiendo
que el líquido no se absorbe en un sólido el cual tiene una tensión superficial más baja
que la del líquido. En este caso, la ecuación ( 6) se simplifica en la ecuación ( 9), La cual
muestra la competencia entre fuerzas adhesivas, ns/l y fuerzas cohesivas,
y+,.
Para que ocurra un mojado completo solo se requiere que la tensión superficial del sólido
sea más grande que la tensión interfacial del líquido. De la ecuación de Young, se observa
que esta es condición necesaria pero no suficiente.
Figura 3: Trabajo de Adhesión.
De la ecuación ( 9) se nota que cuando una área unitaria del líquido es separada
de un sólido, una área unitaria de la interfase sólido/vapor y una área unitaria de
la superficie líquido/vapor se crea y una área unitaria de la interfase sólido/líquido
desaparece. El procedimiento de separación deja una capa de vapor absorbida en la
superficie del líquido.
El trabajo de cohesión Wc, figura 4, es una medida de las interacciones moleculares para
moléculas simétricas, se define por:
La ecuación anterior, se deduce separando una columna de líquido en dos partes,
creando dos superficies líquido/vapor. La energía libre de éste proceso es 2../1/,. La orientación de las moléculas puede ocurrir durante este proceso.
De estas dos cantidades podemos definir la ecuación ( 11) la cual ilustra la competencia
entre las fuerzas de cohesión, W,, y las fuerzas de adhesión, Wa.
Figura 4: Trabajo de Cohesión.
este también se puede definir como:
Este coeficiente únicamente puede ser negativo o cero. Aunque algunos textos sugieren
la posibilidad de que pueda ser positivo, esto se confunde con W:, el coeficiente de
esparcimiento en no-equilibrio, definido por:
Cuando una gota de líquido inicialmente en contacto en una superficie sólida W:,
puede ser positivo. Harkins [Harkins, 19521 llama a W: como el coeficiente inicial de
esparcimiento y Ws el Coeficiente final de esparcimiento. Como ocurre adsorción en
la interfase sólido/vapor, W: decrece hasta alcanzar un valor de cero ó negativo en el
equilibrio.
adsorben en el sólido.
Cuando 6' = O, la ecuación de Young se reduce a:
% / u = Ys/u - Ys/l
y la ecuación de Gibbs-Young:
la ecuación ( 18) es conocida como la regla de Anton08 [Antonoff, 19071.
Han existido algunas objeciones en las ecuaciones ( 18) y ecuación ( 19) en base en que
la ecuación de Young no es aplicable cuando 6' es cero. De hecho, la ecuación de Young
es válida cuando 6' es cero. Tales objeciones se pueden resolver por la distinción entre las
tensiones de adhesión en equilibrio y no-equilibrio. Una excelente discusión se encuentra
en el libro de Landau y Lifshitz [Landau, 19581.
Un problema más serio es cuando la ecuación de Antonoff se aplica a sistemas que tienen
un ángulo de contacto finito. Donde el criterio para la validez de la regla de Antonoff es
un ángulo de contacto cero, y aplicada donde el ángulo de contacto no es cero produce
resultados absurdos.
La ecuación ( 4) muestra que xs/, se determina por la integración de
rSju
us. p de p apo, un criterio para un ángulo de contacto finito es que
rsru
sea finito en p = po. Como p se aproxima a po, la capa absorbida puede llegar a ser más ancha que la película de lasuperficie exterior que tienen las propiedades del seno de líquido; esto es, la superficie
más externa no se ve afectada por el sustrato, Harkins llama a ésta película duplex. El
trabajo de formación de una área unitaria de la superficie completa y s / u de una película
duplex es ~ l más el trabajo de formación de la interfase interna / ~ ys/l.
Y s / u = Y l / u - Ys/l (20)
7rSlv puede llegar a ser significativo y no puede ser ignorado.
0.4
HISTERESIS EN EL
ANGULO DE CONTAC-
TO.
La histéresis en el ángulo de contacto es causada por la existencia de muchos estados termodinúmicos metaestables, para sistemas que tienen tres fase (sólido/líquido/vapor). A diferentes ángulos de contacto se les asocia un estado metaestable. El máximo ángulo
estable se le llama úngulo de avance, t9A. El mínimo ángulo estable se refiere como el
Úngulo de retroceso, OR. La histéresis es la diferencia entre estos dos.
De acuerdo con Adamson [Adamson, 19901, existen tres tipos de causas que producen
histéresis en el ángulo de contacto. La primera causa es la contaminación del sólido ó
líquido. Una limpieza rigurosa puede dar buenos resultados en el ángulo de contacto. La segunda, rugosidades en la superficie, definitivamente causan histéresis en el ángulo de contacto y la tercera causa que aparece es la inmovilidad de la superficie en una
escala macromolecular. Por ejemplo, en el caso de un líquido y una superficie sólida, es
necesario que la película de vapor absorbida sea mobible.
0.4.1
Definición de Histéresis.
La Histéresis es la causa de estados metaestables en la interfase sólido/líquido/vapor.
Cada uno de éstos estados metaestables es caracterizado por un ángulo de contacto.
Estos estados pueden ser producto de heterogeneidades en la superficie, rugosidades en
la superficie o superficies deformables. Las barreras entre los estados metaestables tienen
estados que pueden ser más grandes que k T . Si hay una vibración en el sistema, las
barreras de energía necesariamente pueden ser más grandes que la energía de vibración.
La causa mayor de histéresis es la heterogeneidad en la superficie. Pequeñas zonas de
necesariamente deben ser macroscópicas
,
las regiones pueden ser pequeñas más pequeñas que 4 ó 5 nm de diámetro.Porcentaje del Area Cubierta
Figura 5: Angulo de Contacto como Función del Cubrimiento de la Superficie.
Johnson y Dettre [Johnson, 19641 analizaron un modelo de superficie heterogénea
con bandas circulares concéntricas. El resultado obtenido esta dado en la figura 5. El
eje X es la fracción de la superficie cubierta con un material que produce un ángulo
de contacto calculado. El eje Y es el ángulo de contacto calculado. La curva superior
corresponde al úngulo de contacto de avance,thetaA y la curva inferior al úngulo de contacto de retroceso,thetaR. De acuerdo con su modelo, a los úngulos de avance se les asocia con regiones de baja mojabilidad y a los úngulos de retroceso con regiones de alta
mojabilidad.
La figura 5 también incluye una curva para una superficie heterogénea, en la cual
las áreas individuales de los dos componentes de la superficie son también producto
de pequeños estados metaestables. Esta curva, etiquetada con 8, corresponde a un
mínimo de energía libre para el sistema [Gibbs, 19281. Esta se calcula por la ecuación de
Cassie [Girifalco, 1975, Good, 1958, Good, 19601, ecuación ( 21).
0 es ángulo de contacto del líquido en la superficie heterogénea,
O1
y 02 son los ángulosde contacto de cada uno de los componentes, f i y f2 son sus respectivas fracciones en la
superficie. Como el tamaño de las regiones heterogéneas puede ser pequeño, las curvas
exteriores envuelven a la curva de Cassie.
0.5
MEDICIONES DEL ANGULO DE CONTAC-
TO.
El fenómeno de la tensión interfacial es conocido desde la antigüedad, las primeras obser-
vaciones cuantitativas fueron hechas por el científico árabe Algacini, en el siglo XIII, y lo
describe en su libro: ’La balanza de la sabiduria’ [Pugacevich, 19751.
Más tarde éste fenómeno fue estudiado por otros científicos entre los cuales podemos
citar a Leonardo da Vinci, Newton, Thomas Young, Gauss, Poisson, Mendeleev, Van der
Waals, Bohr, Einstein, Schodinger y muchos más.
Las primeras mediciones que se efectuaron para calcular la tensión interfacial en sólidos
fue realizada por Mervo [Pugacevich, 19751 empleando mercurio. Realmente la expan-
sión del estudio de la tensión interfacial en el ámbito científico comenzó a principios del
siglo XIX en el que se desarrollaron varias teorías acerca de la tensión interfacial, una
de las más importantes fue realizada por Young y Laplace, que establece una relación
entre la diferencia de presiones sobre los lados interno y externo de una superficie curva
de un líquido. El fenómeno de la tensión superficial es puramente físico el cual se debe
básicamente a fuerzas intermoleculares que actúan entre las moléculas de la superficie
líquida o cercanas a ella y estas fuerzas difieren de las que se ejercen entre moléculas a
mucha profundidad del interior del líquido.
0.5.1
Método de la Elevación Capilar.
Es un método exacto para determinar las tensiones interfaciales, cuando se usa cor-
rectamente. Puesto que las mediciones no implican una perturbación en la superficie,
es posible seguir efectos del tiempo lentos. La elevación de un líquido por un capilar
estrecho viene dada por:
rhgnP
y = 2cos(B)
para un ángulo de contacto cero se reduce a:
1
y = -rhgAp
2 (23)
Para hacer medidas precisas es necesario una corrección debido al menisco. En un capilar
estrecho el menisco es aproximadamente esférico y, por lo tanto:
En el caso de capilares más anchos, hay que tener en cuenta que el menisco no tiene
exactamente la forma hemisférica [Adamson, 19901.
En la práctica, el método de elevación capilar solo se usa cuando el ángulo de contacto
es cero.
0.5.2
Método de la
Placa de Wilhelmy.
Del brazo de una balanza se cuelga una placa de mica que se sumerge parcialmente en
un líquido [Wilhelmy, 18631, como se indica en la figura 7.
o Cuando se usa como método de separación de la superficie, ver figura 7a, el depósito
Figura 6: Método de elevación capilar.
en el punto de despegue. Para una lámina de longitud x, anchura y y peso W,
suponiendo que el ángulo de contacto es cero:
e El método de la lámina también puede utilizarse como un método estático, figura
7b, para la medida de cambios de tensión superficial. Se mide la variación de la
fuerza necesaria para mantener la placa a un nivel de inmersión determinado al
cambiar la tensión superficial.
0.5.3
Método del
Anillo.
En este método se determina la fuerza necesaria para separar un anillo de la superficie,
bien suspendiendo el anillo del brazo de una balanza ó utilizando un hilo de torsión
(tensiómetro Noiiy [Lecomte, 19191). La fuerza para desprenderlo esta relacionada con la
a) SEPARACION b) ESTATICO
Figura 7: Métodos de la Placa de Wilhelmy: a) Separación; b) Estático.
Pf
y = -
47rr
donde f es el empuje aplicado al anillo, en dinas, r el radio medio del anillo y
p
un fac- tor de correlación. Para tener un ángulo de contacto cero y, por lo tanto, constante, seutilizan anillos de platino cuidadosamente limpios con ácidos fuertes o flemeándolos. Es
esencial que el anillo repose plano en una superficie tranquila. Para medidas en interfases,
el líquido inferior debe mojar con preferencia la anillo.
El factor de corrección
P
tiene en cuenta que las fuerzas de tensión no están dirigidas ver-ticalmente y también la complicada forma del líquido que cuelga del anillo en el momento
de despegarse; por lo tanto, depende de las dimensiones del anillo y de la naturaleza de
la interfase.
Harkins y Jordan [Harkins, 19301 han tabulado valores de ,O que también pueden calcularse
de la ecuación de Zudeima y Waters [Zuidema, 1941
3.
4b 1
7r2 R2 4R7r (pl - p2)
(P
- u)2 = “f
Donde p1 y p2 son las densidades de la fase interior y superior, u = 0.725 y
b
= 9.075 xI " _ _ . . . I ... I ....
I
"""""""""""- AGUA _ _ _ _ _ - _ _Figura 8: Medida de la tensi6n interfacial por el método del anillo.
0.5.4
Medidas del Volumen
y
Peso de la Gota.
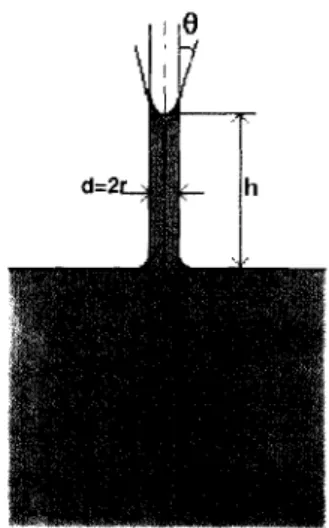
Consisten en medir el peso o el volumen de un agota de un líquido que se desprende
lentamente de la punta de un tubo estrecho montado verticalmente (figura 9j. En el
momento de desprenderse las gotas:
donde m es la masa de la gota, v el volumen, p la densidad del líquido, r el radio del
tubo y Q> un factor de corrección. Este factor es preciso porque:
o la gota formada no se desprende completamente de la punta del tubo y
o la fuerzas de tensión superficial muy raramente son son verticales, depende del
valores de
a.
Se observa que son preferibles valores de"&
comprendidos alrededor de 0.6 y 1.2.X
Figura 9: Separación de una gota desde la punta de un tubo estrecho.
0.5.5
Método de
laGota
Se basa en fotografiar una gota en
imétrico. A partir de la variación
Pendiente.
formación, o proyectando su imagen en papel mil-
de las dimensiones de la gota se puede calcular la
tensión superficial o interfacial [Adamson, 19901.
0.6
MOJADO EN SUPERFICIES DE BAJA ENER-
GIA.
Zisman et al. [Zimerman, 19641, iniciaron los estudios de mojabilidad. Bertell et al., y
algunos otros introdujeron algunos conceptos importantes. En el tiempo de Zisman,
cuando hicieron sus primeras investigaciones, la mayor característica de la mojabilidad
su pobre reproducibilidad. Muchos artículos fueron escritos en ese tiempo con títulos
como: 'Es el AnguZo de Contacto es una Propiedad Termodinámica?'
La mayor parte de su trabajo sobre superficies fluorinadas; algunas de sus conclusiones
o Cuando superficies de baja energía fueron mojadas por alcanos, el coseno del ángulo
de contacto us. la tensión superficial del líquido mojante es una línea recta (gráfico
de Zisman)
0 La intersección de ésta línea recta con el cos(0) = 1 se le llama tensión superficial
crítica ó T~ [Kobza, 20001, Figura 13
o Cuando se miden ángulos de contacto en superficies de baja energía por líquidos que
pueden formar puentes de hidrógeno internos o en otro caso asociarse, los gráficos
de Zisman muestran una curvatura.
o Los grupos químicos más alejados de la cadena determinan la mojabilidad.
La repulsión de los grupos químicos fluorocarbonados e hidrocarburos en la interfase
decrece en el orden:
La tabla 1 lista las tensiones superficiales críticas de varios grupos determinados
por Zisman.
En tiempos posteriores, los datos fueron reproducibles e intentos por explicar los
resultados que Zisman había obtenido. Un resultado intrigante fue que para hidro-
carburos en superficies de baja energía de fluorocarbonados, el ángulo de contacto
era una función única de la tensión interfacial crítica del líquido. Esto fue el foco de
muchos trabajos teóricos de Girifalco y Good [Girifalco, 1975, Good, 1958, Good, 19601, argumentaron que si las tensiones interfaciales (energías libres de superficie) son el
resultado únicamente de las fuerzas de dispersión, entonces las reglas de combinación
La relación de estados de Barthelot es que la constante de interacción de
dispersión entre dos moléculas diferentes del gas es la media geométrica de
l a s
con-stantes de interacción de esas moléculas. La ecuación ( 30) solo puede ser aplicada cuando:
o la superficie tiene la misma estructura que en su seno,
o las interacciones entre las moléculas son de wan der Waals.
Combinando la ecuación ( 30) con la ecuación de Young y haciendo T , / , = O, implica:
cos(0) = 2
J
- y40 - 1 yil,la ecuación ( 31) no predice una relación lineal entre 0 y sin embargo:
Girifalco y Good reconocieron las limitaciones e introdujeron
@.Una interpretación mejor fue realizada por Fowkes [Fowkes,
un parámetro ajustable
19671. El reconoció que
los factores críticos que controlan la mojabilidad son las interacciones entre las fases es
a través de las interfases. Sugirió que estas interacciones fueran relacionadas con las
clases de fuerzas que actúan entre las moléculas de cada fase. Estas fuerzas pueden ser
independientes una de la otra. Por lo tanto, si ambas, puentes de hidrógeno y fuerzas de dispersión actúan a través de la interfase, el trabajo total de adhesi-”on puede ser el trabajo dispersivo de adhesión más el trabajo de adhesión debido a los puentes de
hidrógeno. La ecuación ( 31) se puede reescribir en términos de interacciones. Estas
interacciones están definidas por las componentes (y,“ y y+) de las tensiones superficiales
Para sistemas donde solo existe interacción únicamente por puentes de hidrógeno y
fuerzas de dispersión.
w,
=w,“
w,“+
(34)donde
W,d
es el trabajo de adhesión por fuerzas de dispersión yW,h
es el trabajo deadhesión por puentes de hidrógeno.
Owens y Wendt [Owens, 19691, Kaelble [Kaelble, 19701 y otros también aplicaron los
conceptos de Fowkes:
w,
=w,”+w,“+wap
(35)donde W,P es el ’trabajo de adhesión polar’, Fowkes [Fowkes, 19671, mostró que todas la
interacciones a través de la interfase se ajustan a dos tipos: dispersión y úcido-base. Esto
requiere dos términos en la ecuación del trabajo de adhesión porque para cada grupo
ácido puede haber un correspondiente grupo básico. Esto implica:
donde A/B representa una interacción ácido-base y B/A una interacción base-ácido.
0.7
MODELOS MOLECULARES
Y
TEORIA DE
DISPERSION.
Las fuerzas de dispersión son causadas por las fluctuaciones de las nube electrónica que
rodea a los átomos. El entendimiento entre las fuerzas de van der Waals únicamente
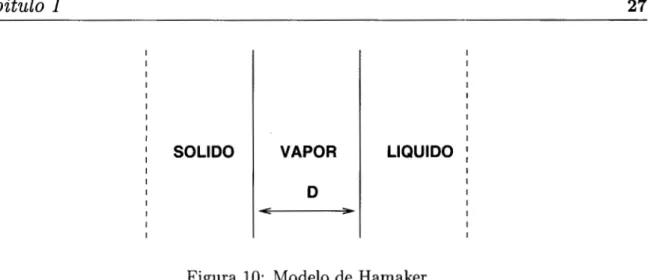
puede ser posible con el desarrollo de la mecánica cuántica. Hamaker [Hamaker, 19531
fue el primero en mostrar que las fuerzas intermoleculares de dispersión pueden contar
I I I I I I I I I I I I I I I I I I
en la figura 10.
SOLIDO VAPOR
I I I I I I I I I LIQUIDO I I I I I I I
Figura 10: Modelo de Hamaker
Las fases sólida y líquida tienen una forma infinita separadas por un vacío ó vapor,
La energía de interacción entre los dos planos separados por una distancia D, esta dada
por la ecuación de Hamaker.
donde A,/1 es la contante sólido/líquido de Hamaker y D la distancia de separación entre
los planos en el equilibrio. En la aproximación más cercana
D
=Do.
Esta separación sedetermina por un balance entre las fuerzas atractivas de dispersión y fuerzas de repulsión
de Born. Hamaker dedujo A,/1 por la integración de las interacciones de dispersión entre
todos los elementos en las dos fases.
donde las v’s son los elementos de volumen de las dos fases, las q’s son las correspondientes
densidades de átomos por unidad de volumen, X es la contante de interacción molecular
Padday y Unffindell [Padday, 19681 calcularon A,/l de la teoría molecular de Moelwyn-
Hughes y ajustando Do, ellos calcularon la tensión superficial de alcanos con mucha
exactitud. Ajustando los datos pudieron calcular las interacciones entre grupos más que
entre átomos. Sus valores calculados de A,/1 fueron diferentes a los valores calculados
por la teoría de Lifshitz.
0.7.1
Teoría de Lifshitz.
La teoría de Lifshitz [Lifshitz, 19561 de las fuerzas de dispersión es una de la teorías
más exactas en la física de superficies. Su desarrollo elimina muchos de los problemas
asociados con la con la aproximación de Hamaker.
El modelo de Lifshitz es el mismo como Hamaker. La diferencia entre las dos teorías es
el camino para determinar la constante de Hamaker. El método de Hamaker determina
la constante integrando ó sumando las interacciones moleculares. El método de Lifshitz
determina la constante del espectro de absorción electromagnética. La ventaja en los
cálculos de Lifshitz hoy en día es que las mediciones ópticas se pueden realizar en objetos
macroscópicos homogéneos.
0.7.2 Modelo de Hough
y
White.
El trabajo de Hough y White [Hough, 19801 tiene un mayor interés. El punto de partida
donde h es la constante de Planck, k' la constante de Boltzmann y E la energía entre la
mitad del espacio entre los planos 1 y 3 separados por 2. La prima en la sumatoria indica que el término n = O. La prima en la contante de Boltzmann se usa para distiguirla del
índice k empleado por Hough y White.
~(25)
está relacionado con la respuesta dieléctrica€ ( u ) , donde w es la frecuencia de radiación electromagnética.
Hough y White hicieron un cambio de variable para convertir las ecuaciones anteriores
en una forma más resumida:
A 1 2 3 - E 1 2 3 =
12TL
donde A123 es la constante
es la distancia media entre
3k'T O3
A123 "
c
2 0
(43)
sólido/líquido de Hamaker definida por la ecuación ( 44) y
L
los espacios 1 y 3:
donde
x
= 2kL. A123 es dependiente de las propiedades del material del sistema atravésde la fución q
(ZC) .
Los ángulos de contacto se pueden calcular de la ecuación ( 46)
Para un líquido simple donde las moléculas no se orientan en la superficie:
- Al/, -
2 4 ~ L2
donde L es la separación del líquido. Sustiyendo Yllv de la ecuación ( 3):
2As/1 - 2 4 r L 2 ~ , / ,
cos(8) = - 1
Al/,
Hough y White asumen que 7rslv es cero, lo cual produce la ecuación aproximada:
2As/1 Al/, cos(e) = _ _ -
La tabla 2 muestra las constantes de Hamaker
alcanos y agua. La tabla 3 muestra las constantes
calculadas por Hough
de Hamaker calculadas
y White para
para diferentes
materiales. La tabla 4 da las constantes de Hamaker, tensiones superficiales, densidades
y distancias de corte para alcanos. La tabla 5 d a cálculos teóricos realizados para
el politretafluoroetileno (PTFE) por Hough y White, usando la ecuación ( 43). Los
resultados experimentales se tomaron de Fox y Zisman [Fox, 19501.
0.7.3
Modelo de Israelachvili.
La ecuación de Hamaker y Lifshitz únicamente se aplica a sólidos homogéneos. Is-
raelachvili [Israelachvili, 19731 extendió el análisis de Lifshitz a sistemas donde el sólido
es dopado por una película ó monocapa. La figura 11 muestra su moldelo.
Esto es similar al modelo de Hamaker-Lifshitz, excepto que una película se adhiere
I I I I I I
i
SOLIDO I I I I I I I FILM T-
-
VAPOR D I I I I I ILIQUIDO
I
I I I I I I IFigura 11: Modelo de Israelachvili.
al 20%.
Do
es un parámetro ajustable, esta relacionado con la distancia entre los planos.All, es la constante de Hamaker líquido/substrato y Allf es la constante de Hamaker líquido/película.
Ordinariamente, uno asocia la mojabilidad con un sustrato sólido. Sin embargo, el moja-
do de un líquido por otro puede ser extremadamente útil para entender la termodinámica
y probar las tareas de mojado. Los estudios líquido/líquido tiene papel dominante en
el desarrollo de todas las teorías del mojado. Los conceptos anteriores pueden aplicarse
sistemas líquido/líquido/sólido definiendo, 8, como el ángulo de contacto equivalente:
cos(8,) = YLl/V - YLI/L2
Y L d V
(53)
Los subindices L1 y L2 se refieren a fluidos inmiscibles. 8, no puede medirse directamente
pero puede calcularse de la ecuación ( 46) de las mediciones independientes de las
tensiones interfaciales y los ángulos interfaciales.
Las relaciones geométricas en la línea de las tres fases líquido/líquido/vapor se definan por el triángulo de Neumann [Neumann, 18941 y se muestra la figura 12. Los vectores
triángulo, pero ,B no es Be.
Figura 12: Triángulo de Neumann.
0.7.4
Mojabilidad Interfacial
La Ecuación de Bartell-Osterhof.
La mojabilidad interfacial es el mojado de un sólido por un líquido que está rodeado
por un segundo. Un punto de partida para el estudio de la mojabilidad interfacial es
usualmente la ecuación de Bartell-Osterhof.
El subídice w, designa la fase acuosa (líquido 1) y b la fase aceitosa (líquido 2), f9L es el ángulo ángulo de contacto del aceite en el sólido medido a través de la fase acuosa. La
ecuación de Young para una gota de agua en una superficie con agua rodeada por aceite,
en equilibrio:
donde el ángulo de contacto f9L se mide a través del agua.
La ecuación de Young para el sistema sólido/agua/vapor:
Yw/w cos(Qw) = Ys/o - Ys/w
La ecuación de Young, sino se considera adsorción es:
Melrose [Melsrose, 19671 extendió la derivación incluyendo la adsorción. El trabajo de
Superficie ,-yc (mJ m-2, 20C)
-CF3 6
-CF2H 15
-CF2- 18
-CH3 cristal 22
- C H3 monocapa 24
-CH2- 31
Polimeros ”
Politetrafluroetano ”
Polietileno 18.5
Tabla 1: Tensión Superficial crítica de varios grupos determinados por Zisman.
Alcano C-A- C C- W- C C- W-A C-A- W C-A-
Weal
W- C-Apentano 3.75 0.336 0.153 3.63 3.72 0.108
Hexano 4.07 0.360 -0.004 3.78 3.88 0.285
Heptano 4.32 0.386 -0.118 3.89 4.00 0.423
Octano 4.50 0.410 -0.200 3.97 4.08 0.527
Nonano 4.66 0.435 -0.275 4.05 4.15 0.624
Decano 4.82 0.462 -0.344 4.11 4.22 0.719 Pentadecano 5.23 0.540 -0.519 4.28 4.40 0.964
Agua 3.70
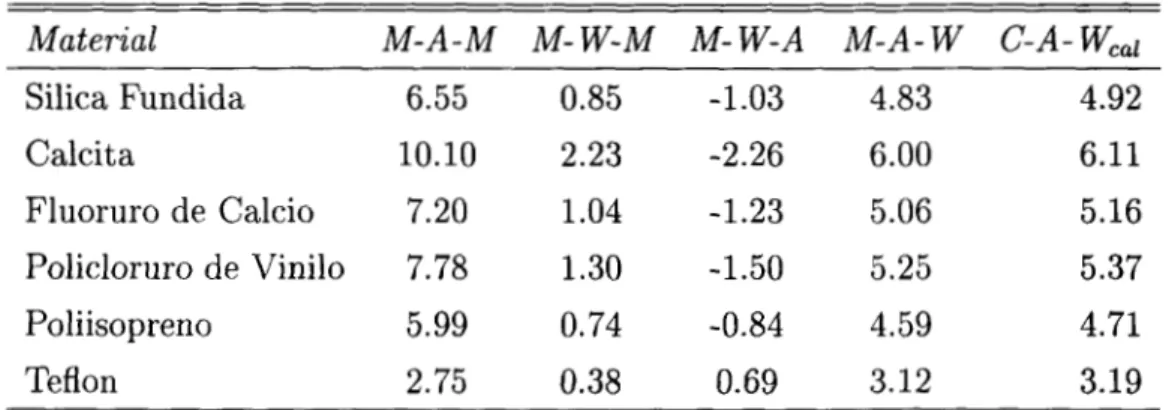
Material M-A-M M- W-M M- W - A M - A - W C-A-
Weal
Silica Fundida 6.55 0.85 -1.03 4.83 4.92
Calcita 10.10 2.23 -2.26 6.00 6.11
Fluoruro de Calcio 7.20 1 .O4 -1.23 5.06 5.16
Policloruro de Vinilo 7.78 1.30 -1.50 5.25 5.37
Poliisopreno 5.99 O. 74 -0.84 4.59 4.71
Teflon 2.75 0.38 0.69 3.12 3.19
Tabla 3: Constantes de Hamaker ( x 10-20J) para diferentes materiales calculadas por
Hough y White (M=material; A=aire; W=Agua).
6 4.07 18.4 0.6603 7 4.32 20.1 0.6838
8 4.50 21.6 0.7025
9 4.66 22.9 0.7176 10 4.82 23.8 0.7300
11 4.88 24.7 0.7402
12 5.03 25.4 0.7487
13 5.04 26.0 0.7564
14 5.05 26.6 0.7628
15 5.10 27.1 0.7685
16 5.23 27.4 0.7733
Agua 3.70 73.0 0.9970
L:
(DO
1
0.1785 0.1712 0.1685 0.1660 O. 1644 0.1637 0.1618 0.1622 0.1604 0.1594 0.1588 0.1507 0.1420
Tabla 4: Distancias de Corte
(LC
= Do, nm), Constantes de Hamaker ( x ~ O - ~ O J ) , Ten-Pentano Hexano Heptano Octano Nonano Decano Undecano Dodecano Tridecano Tetradecano
Pent adecano
Hexadecano
3.75 3.77 3.74
3.91 3.93 4.06
4.03 4.05 4.31
4.11 4.13 4.49
4.18 4.21 4.66
4.25 4.28 4.81
4.28 4.30 4.87
4.35 4.37 5.03
4.35 4.38 5.04
4.38 4.40 5.09
4.40 4.42 5.15
4.43 4.45 5.22
1 .O06
0.924 O. 869 0.831 0.798 0.768 0.758 0.730 0.728 0.719 0.709 0.698
moja moja
22 12
29 21
34 26
37 32
40 35
41 39
43 42
43
44 44
45
46 46
Tabla 5: Angulos de Contacto Teóricos y Experimentales en PTFE; Constantes de Hamak-
[Adamson, 19901 Physical Chemistry of Surface, Adamson, A. W.; Edt. John Wiley &
Sons. Inc. 1990.
[Anderson, 19861 Anderson W. G.,Jour. Pet. Tech., 1125, Oct. (1986)
[Antonoff, 19071 Antonoff, G., (a) J. Chem. Phys.,5, 364 (1907), (b) J. Chem. Phys.,5,
372 (1907)
[Bertholet, 18981 Bertholet,
D.,
Compt. Rend., 126, 1703 (1898)[Fowkes, 19641 Fowkes, F.
M.,
Advances in Chemistry Series, 43, American ChemicalSociety, Whashington, DC., 1964
[Fowkes, 19671 Fowkes, F.
M.,
Wetting, SCI Monograph NO. 25, Soc. Chem. Ind., London,1967, p 3.
[Fowkes, 19641 Fowkes,
M.
F., Acs Adw. In Chem. Series 43, 99 (1964)[FOX, 19501 Fox, W. H. y Zisman, A. W., J. Coll. Sci.,5, 514 (1950)
[Gibbs, 19281 Gibbs, W.
J.,
The collected Works of Thermodinamics, Vol. 1, Yale Uni- versity Press. New Haven, 1928.[Girifalco, 19571 Girifalco, A. L. y Good, J.
R.,
J.
Phys. Chem., 61, 904 (1957)[Girifalco, 19751 Girifalco, A. L. y Good, J.
R.,
J . Phys. Chem., 61, 904 (1957);[Good, 19581 Good,
J.
R.,
Girifalco, y Kraus, G.,J.
Phys. Chem.,62, 1418 (1958)[Hamaker, 19531 Hamaker, C. H., Physica,4, 1058 (1953)
[Hamaker, 19531 Hamaker, C. H., Physica,4, 1058 (1953)
[Harkins, 19521 Harkins, D. W., The Physical Chemistry of Surface films, Reinhold, New
York, 1952, Cap. 2.
[Harkins, 19191 Harkins, W. D. y Brown,
F.
E.,J.
Amer Chem. Soc., 41, 499 (1919)[Harkins, 19301 Harkins, W. D. y Jordan,
H.
F.,
J.
Amer. Chem. Soc., 52, 1751 (1930)[Hough, 19801 Hough, B. D. y White, E. L., Adv. Coll. Inter Sci., 14, 1 (1980)
[Israelachvili, 19891 Israelachvili, N. J. y Gee,
L.
M., Langmuir,5, 288 (1989)[Israelachvili, 19731 Israelachvili,
N.
J.,J.
Chem. Soc., Faraday II, 69, 1729 (1973)[Johnson, 19591 Johnson,
E.
R.
Jr.,J.
Phys. Chem, 63, 1655 (1959)[Johnson, 19691 Johnson,
E.
R.
y Dettre, H.R.,
in Surface and Colloid Science, Vol. 2(E.
Matijevic, ed.), Wiley-Interscience, New York, 1969[Johnson, 19641 Johnson,
E.
R.,
y Dettre, H.R.,
J.
Phys. Chem., 6 8 , 1744 (1964)[Kaelble, 19701 Kaelble, H. D.,
J.
Adhesion, 2, 66 (1970)[Kobza, 20001 Kobza, K.; Gestwicki, J . E. y McGrath, J. L.,
J.
Chem. Educ, 77, 63 (2000)[Landau, 19581 Landau, D. L. Y lisfshitz,
M. E.,
Statistical Physics, Pergamon, London,1958, p. 471-473.
[Lecomte, 19191 Lecomte du Noüy,
P.,
J.
Gen. Physiol., 1, 521-524 (1919)[Li, 19911 Li,
D.,
Moy, E. y Neumann, W.A.,
Langmuir, 7 , 1833 (1991)[Melsrose, 19671 Melrose,
J.
C., Wetting SCI Monograph No. 25, Soc. Chem. Ind., Lon- don, 1967, p123-143.[Neumann, 18941 Neumann, F. Vorlesungen uber die Theorie der Capillaritat, B. G. Teub-
ner, Leipzing, 1894
[Neumann, 19741 Neumann, W.
A.,
Adv. Coll. Inter. Sci., 4, 1 (1974)[Owens, 19691 Owens, K. D. Y Wendt, C.
R.,
J.
Appl. Polym. Sci., 13, 1741 (1969)[Padday, 19681 Padday, F.
J.
y Uffindell, N. D.,J.
Phys. Chern., 72, 1407 (1968)[Pugacevich, 19751 Pugacevich, P. P., Experimental Thermodynamics, Vol. 2, cap. 20,
Butterworths, London, 1975.
[Schhultz, 19271 Schhultz,
S.;
Tsutsumi, K. y Donnet, B.J.,
Ind. Eng. Chem., 19, 1277 (1 927)[Wilhelmy, 18631 Wilhelmy, L., Ann Physik, 119, 177-217 (1863)
[Young, 19281 Young, F.
T.
y Harkins,H.
D., Internat. Critical Tables, 4, 434 (1928)[Young, 18051 Young, T., Phil. Trans., 95,65 y 82 (1805)
[Zimerman, 19641 Zimerman, W.
A.,
Adv. Chem. Ser. Vol. 43(R.
F. Gould, ed) Am. Chem. Soc. Washington,D.C.,
1964, p l .0.8 INTRODUCCION
Para resolver problemas prácticos de interés en los campos de la Hidrogeología, Ingeniería
de la Agricultura, Ingeniería Petrolera, Ingeniería Ambiental, Física de Suelos y Geofísica es necesario tener estimaciones reales de parámetros hidráulicos como: permeabilidad,
conductividad hidrúulica y porosidad. Desde los trabajos pioneros de Darcy, Dupuit,
Forchheimer y otros en Europa en la segunda mitad del siglo XIX, un cuerpo sustancial
de literatura se ha acumulado en diversos campos de las ciencias de la tierra y métodos
pertinentes en ingeniería, para estimar las características hidráulicas. Para estudiosos
de ciencias de la tierra es de gran importancia no únicamente de cómo las ideas están
relacionadas en la caracterización hidráulica que históricamente se les ha involucrado
sino también descifrar las nociones fundamentales en los métodos involucrados. Los
métodos usados hasta hoy en día tienen algo en común: una ecuación empirica de
movimiento, familiarmente conocida como la ley Darcy, la cual da una noción formal
de la permeabilidad y la ecuación de conducción de calor originalmente propuesta por
Fourier en 1807, la cual establece un modelo de trabajo para los procesos de difusión en
las ciencias físicas. En la ecuación de movimiento está inmersa en la ecuación de difusión.
Intrínsecamente a la ecuación de transporte por difusión están los parámetros de con-
ductividad hidráulica y capacidad hidráulica. En cambio, la capacidad hidráulica incluye
entre otras propiedades: la porosidad del medio. La capacidad hidráulica representa la
cantidad de agua liberada del almacenaje por un cambio unitario en la presión debido a
la combinación de tres procesos independientes: cambio en el volumen del poro, cambio
en la saturación de agua y una expansión del agua. La ecuación de difusión proporciona
los fundamentos para la caracterización hidráulica. Ultimamente, todos los métodos
para la caracterización hidráulica consisten en ajustar los datos del campo a la ecuación
de difusión y el mejor ajuste en la combinación de los parámetros que concuerdan
con los datos del campo. Por lo tanto, los métodos de caracterización hidráulica son
métodos inversos concordantes con estimación de parámetros compatibles con el modelo
de difusión. La ley Darcy juega un papel central en el estudio del flujo de fluidos en
medios porosos. El movimiento de fluidos en materiales geológicos está basado en el
de calor en sólidos. Como una consecuencia, el modelo de trabajo matemático para el
transporte de fluidos en materiales geológicos es una ecuación diferencial parcial de la
conducción del calor, originalmente por propuesta por Fourier (Theorie de la Propagation de la Chaleur duns les solides, manuscrito sometido al Instituto de Francia en 1807). El trabajo de Fourier, nunca fue formalmente publicado [Gratton-Guinness, 19721. Después
de mucho trabajo adicional, (Laplace, Lagrange y Lacroix) el trabajo clásico de Fourier
fue publicado en 1822.
La construcción de termómetros de confianza para las mediciones precisas del calor fue
crítico en el desarrollo de la ciencia del calor y durante la mitad del siglo XVIII. Aunque
el termómetro de mercurio (Hg) fue construido en Francia por Boulliau en 1659, los
termómetros exactos con escalas bien definidas fueron posibles hasta la mitad del siglo
XVIII por Fahrenheit en 1724 y Celsius en 1742 [Cajori, 18981. Con la capacidad de estos
instrumentos, Joshep Black, un pionero en la química cuantitativa moderna, descubrió el calor latente y la capacidad calor@ca en un calorímetro y determinó estas cantidades en 1760, sin embargo, é1 nunca publicó sus resultados. Las primeras mediciones publicadas
de estas cantidades se le atribuyen a Lavoisier y Laplace (’Mernoire sur la Chaleur’)
presentado en la Real academia de Francia en 1783. Fourier en su manuscrito de 1807
de la propagación del calor, introdujo el parámetro de conductiwidad (que éI llamó ’conductiwidad espec$ca interna
’)
en términos matemáticos precisos.En su trabajo más importante, ’Hydrodynamica’ publicado en 1738, Daniel Bernoulli,
identificó tres componentes que provienen de la energía mecánica, del movimiento de
un fluido: energia potencial debida a la gravedad, energia elcistica debida a la presión y energia cinética. Trabajando en esta dirección, Ohm en 1827 [Ohm, 18271, determinó experimentalmente la relación inversa entre la corriente eléctrica y el voltaje a través
de un conductor, la constante de proporcionalidad es la resistencia eléctrica debido al
cuerpo conductor. Por lo tanto, la resistencia es una función del material de conducción.
Poiseuille [Poiseulle, 18421, un médico, interesado en el mecanismo del flujo de sangre
a través de las venas de animales y humanos, estudió los mecanismos que gobiernan
el flujo de fluidos a través de tubos capilares. Estos experimentos meticulosos fueron
es directamente proporcional a la presión sobre el tubo y al área de la sección transversal
e inversamente proporcional a la longitud del tubo, la constante de proporcionalidad se
representa por K . De acuerdo con Herschel [Herschel, 19401, la expresión comunmente
usada involucra la ley a la cuarta potencia en el radio del capilar que no fue propuesta
por el mismo Poiseuille. Después la función la derivó James Clerk Maxwell, integrando
las ecuaciones de Newton de la viscosidad aplicadas a tubos cilíndricos. En Alemania,
Hagen [Hagen, 18391, obtuvo resultados experimentalmente similares.
Es fácil notar de los precedentes al tiempo de Darcy se aventuraron en estudios experi-
mentales de conducción en filtros de arenas para suministrar agua a Dijon [Darcy, 18561, un marco bien definido fue preparado para el diseño de experimentos y la interpretación
de los resultados, gracias a las contribuciones de Fourier, Ohm y Poiseuille. Podemos
asumir que Darcy fue consciente de estos desarrollos y lo usó para su trabajo. Es
pertinente hacer notar las diferencias entre las formas matemática de la ley de Darcy y
la ley Ohm. En la ley de Ohm se considera la resistencia del cuerpo conductor como un
todo. La resistencia, como aparece en la ley de Ohm, es una integral, evaluada sobre el
cuerpo como un todo. En la ley de Darcy que tiene una forma exactamente la ley de
Fourier para la conducción del calor e involucra derivadas parciales del potencial. La ley
de Darcy en observaciones experimentales tiene una forma compatible con la ecuación
diferencial, la ley de Ohm, es inherente a ecuaciones integrales y flujos netos.
Inmediatamente en la contribución de Darcy, analogías a la conducción del calor fueron
inmediatamente propuestas por ingenieros en Austria, Francia y Alemania para resolver
problemas prácticos en el flujo de acuíferos y la segunda mitad del siglo X I X . Aunque la ecuación de Fourier, esta direccionada al proceso de conducción de calor, estos ingenieros
civiles restringieron por asimismo a sistemas en flujo estacionario. Mientras que el
proceso de transporte involucra dos parámetros (conductancia y capacitancia), el estado
estacionario involucra únicamente el parámetro de conductividad. Jules-Juvenal Dupuit,
un contemporáneo de Darcy, fue un ingeniero teórico orientado a los problemas de flujo
en canales abiertos. El capítulo, que se refiere a los filtros en su libro de flujos en canales
abiertos [Dupuit, 18631 probó que puede ser una referencia en el tema. Es interesante
que Dupuit, inició de los principios hidráulicos del flujo en canales abiertos, derivó una
empírica de Darcy. Integrando las ecuaciones del movimiento sobre un dominio radial,
Dupuit obtuvo la solución para el flujo estacionario en acuíferos confinados y en acuíferos
no confinados. Otra importante figura en este periodo fue Joshep Boussinesq, quien, en
la investigación del papel de la fricción en flujo laminar de fluidos y (líquidos y gases),
obtuvo la expresión por el flujo de agua en una fractura idealizada de planos paralelos,
ahora referida como la ley cúbica [Boussinesq, 18681. En Alemania, Adolf Theim, y,
después, su hijo, Gunther Thiem, iniciaron el trabajo pionero del flujo de agua y en
filtros, especialmente en el estudio del flujo de agua pozos. A ellos también se les acredita
una extensa colección de información en este tema. Aunque el después llegó a estar
consciente de las contribuciones de Dupuit y Darcy, Adolph Thiem independientemente
obtuvo la expresión para el flujo radial estacionario del agua en acuíferos confinados y
no-confinados. En ese campo de la Hidrología, Gunther Thiem [Thiem, 19061, que es
ampliamente conocido por la ecuación que describe el flujo radial estacionario del agua
en acuíferos confinados, aunque la solución fue obtenida posteriormente por Dupuit.
La palabra 'groundwater', (grundwasser en alernún) aparece la literatura por la época
de 1880 en el trabajo de Adolph Theim. Quizás el investigador más conocido de esa
época fue Phillipp Forchheimer de Australia. El tempranamente reconoció los conceptos
de líneas isopotenciales y líneas de pujo con aplicación a filtros de agua subterránea, y
extendió estos conceptos sistemáticamente a la generación de los flujos netos como una
medida cuantitativa analizando el flujo estacionario en campos, incluyendo el flujo de
agua a pozos variando las condiciones geométricas. Forchheimer formalmente escribió
sobre la ecuación de Laplace [Forchheimer, 18981, que describe el flujo estacionario de
agua subterráneas.
En Estados Unidos, Slichter [Slichter, 18991 en 1899, pionero en el estudio de sistemas de
flujos subterráneos, analizando matemáticamente el flujo estacionario de agua a través de
medios geológicos. Slichter desconocía los trabajos de Forchheimer y formuló la ecuación
de Laplace independientemente. Estudiando
l a s
propiedades geométricas de variosempaquetados esféricos, Slichter identificó el componente geométrico y los componentes
del arrastre viscoso de la conductividad hidráulica.