Capítulo 8
229
8.1 Consideraciones termodinámicas
Las aleaciones amorfas estudiadas tienen un contenido en GeSe2 comprendido entre el 50 y el 67 mol %. Este hecho es de suma importancia para el desarrollo de la modelización. Desde el punto de vista termodinámico, cabe destacar en primer lugar que el sistema GeSe2 -Sb2Se3 constituye un sistema de tipo eutéctico en el que los equilibrios sólido-líquido pueden modelizarse despreciando la solubilidad en fase sólida y considerando que la disolución líquida es fuertemente asociada con especies del tipo Sb2Se3 y GeSe2 (Clavaguera et al. 1985). En la Figura 8.1.1 se muestra el diagrama de fases y sus proyecciones para los equilibrios metastables de las composiciones límite estudiadas, con indicación explícita de la temperatura de liquidus del equilibrio metastable.
0.2 0.4 0.6 0.8
mol% GeSe2 400 600 800 1000 T e m p e ra tu ra ( K ) GeSe2 Sb2Se3
TliquidusSb2Se3= 790 K
TliquidusGeSe2= 692 K
(metastable)
0.2 0.4 0.6 0.8
mol% GeSe2
400 600 800 1000 T e m p e ra tu ra ( K ) GeSe2 Sb2Se3
TliquidusSb2Se3= 724 K TliquidusGeSe2 = 812 K
(metastable)
a) Proyección de los equilibrios metastables mostrando las temperaturas de liquidus estable y metastable para las aleaciones de composición
(GeSe2)0.5(Sb2Se3)0.5
b) Proyección de los equilibrios metastables mostrando las temperaturas de liquidus estable y metastable para las aleaciones de composición
(GeSe2)0.67(Sb2Se3)0.33 Fig. 8.1.1: Diagrama de fases del
Capítulo 8
Así, para la discusión subsiguiente hay que tener en cuenta que a una determinada temperatura T, el sub-enfriamiento de una aleación de (GeSe2)α(Sb2Se3)1-α para la formación de GeSe2 debe medirse a partir de una temperatura de liquidus comprendida entre 692 K (α=0.5) y 812 K (α=0.67). De manera equivalente, el sub-enfriamiento para la formación de Sb2Se3 debe medirse a partir de una temperatura de liquidus comprendida entre 790 K (α=0.5) y 724 K (α=0.67).
En segundo lugar, también pueden deducirse las entalpías de las distintas fases metastables (vidrio y líquido sub-enfriado) a partir de la entalpía de fusión y las capacidades caloríficas medidas experimentalmente. En la figura 8.1.2 se muestra tal representación para la composición eutéctica.
400 500 600 700 800
Temperatura (K) -150
-100 -50 0 50
HT
H7
5
4
(
J/
g
)
∆H fusión vidrio
liquido sub-enfriado
estado cristalino
liquido estable
parcialmente cristalino
∆H
1r ∆H2r
Fig. 8.1.2: Entalpía del líquido estable, líquido sub-enfriado y vidrio obtenido por temple
231 SEM-EDAX muestran que el primer pico de cristalización corresponde a la cristalización eutéctica, i.e. precipitación simultánea de los dos compuestos GeSe2 y Sb2Se3 cabe deducir que el segundo pico se ve retardado por razón de la difusión a través de la intercara dificultada por la acumulación de soluto en la misma.
En tercer lugar, es muy ilustrativo analizar el comportamiento de la temperatura de transición vítrea. En la Fig. 1.1 se muestra cómo Tg aumenta con el contenido en GeSe2 de la aleación vítrea. Por otra parte en el Cap. 3 se indican varios resultados de interés respecto a la evolución de Tg. Con referencia a las muestras con el 50% molar en GeSe2, Tg aumenta al efectuar un segundo calentamiento (Fig. 3.2) lo que estaría de acuerdo con un enriquecimiento de la matriz amorfa en GeSe2 y, por consiguiente, o bien con una cristalización parcial de Sb2Se3 durante el primer calentamiento o bien a la cristalización eutéctica de ambos compuestos seguida de la fusión y posterior solidificación parcial prioritaria de Sb2Se3. Puesto que el primer calentamiento a 10 K/min hasta 773 K da lugar, para esta muestra, a una cristalización casi total (ver difractograma, Fig. 6.6) en dos picos (ver fig. 4.30a) hay que concluir que la fracción en volumen de muestra que no cristaliza durante el enfriamiento es más rica en GeSe2 que la inicial. Además, este amorfo residual es muy estable ya que no recristaliza durante un segundo calentamiento (ver Fig. 4.30a).
Si aplicamos este mismo análisis a las aleaciones con el 58% molar en GeSe2 resulta que Tg disminuye ligeramente al efectuar un segundo calentamiento (Fig. 3.3) lo que estaría de acuerdo con un enriquecimiento de la matriz amorfa en Sb2Se3 y, por consiguiente, o bien con una cristalización parcial de GeSe2 durante el primer calentamiento o bien a la cristalización eutéctica de ambos compuestos seguida de la fusión y posterior solidificación parcial prioritaria de GeSe2. Puesto que el primer calentamiento a 10 K/min hasta 773 K da lugar, para esta muestra, también a una cristalización casi total hay que concluir que la fracción en volumen de amorfo remanente después de la solidificación es ligeramente más rica en Sb2Se3 que la inicial. Además, este amorfo residual no es muy estable ya que recristaliza durante un segundo calentamiento (ver Fig. 4.30b).
Capítulo 8
comparar las diferentes curvas DSC de calentamiento. Durante el primer calentamiento, además, casi no se produce ninguna cristalización y, por lo tanto, apenas hay fusión. En los calentamientos sucesivos se van destacando dos procesos sucesivos de cristalización que acaban aparentándose a los observados para las aleaciones con un 50 y un 58% molar en GeSe2. Si se detiene el calentamiento y se hace un recocido a 703 K, se constata la aparición de estructuras lamelares rodeadas por estructuras fibrosas ( ver Fig. 7.20 y 7.21) equivalentes a las obtenidas para las aleaciones con un menor contenido en GeSe2. Este hecho está de acuerdo con la ligera disminución de Tg en los sucesivos ciclos puesto que la matriz amorfa va empobreciéndose en este compuesto.
Todas estas consideraciones constituyen la base del desarrollo de este capítulo.
8.2. Evaluación de magnitudes cinéticas
8.2.1. Viscosidad
Conocidos los valores de la temperatura de transición vítrea en función de la composición, un primer intento de modelizar el comportamiento de la viscosidad del líquido sub-enfriado es admitir que su viscosidad está comprendida entre el valor de 10-2 Poise para la temperatura eutéctica y el valor de 1013 Poise a la temperatura de la transición vítrea. Además, vamos a considerar que la viscosidad sigue una ley de Vogel-Fulcher (ver ecuación 2.2.17). Dados los valores de Tg de las aleaciones consideradas, podemos representar el comportamiento de la viscosidad en función de la temperatura reducida, Tr, definida por
Tr = T/Teut (8.2.1)
233 Fig. 8.2.1: Variación de la viscosidad, en escala logarítmica, en función de la temperatura para dos valores de
la temperatura reducida de transición vítrea.
8.2.2. Nucleación
La evaluación de la frecuencia de nucleación se basa en la ecuación (2.2.41) con un
coeficiente de difusión cuyo valor depende del tipo de transformación. En las primeras etapas
de la cristalización tomaremos un coeficiente de difusión dado por la ecuación (2.2.42). Es
decir, se admite que la composición del líquido sub-enfriado no cambia puesto que la
cristalización es “globalmente” eutéctica. Aquí el término globalmente hay que entenderlo en
el sentido de que vamos a distinguir la nucleación de cada uno de los compuestos, GeSe2 y
Sb2Se3, utilizando siempre la misma expresión para el coeficiente de difusión.
Las diferencias entre los valores de la frecuencia de nucleación del compuesto que
germina se establecen claramente al considerar el valor de la barrera de formación de un núcleo
de tamaño crítico, W*, ecuación (2.2.26). Admitiremos que el factor de Turnbull, a, definido
por la ecuación (2.2.33) tiene un valor fijo, pero admitiremos valores diferentes para cada
compuesto. Por otra parte, la modelización del diagrama de fases permite considerar valores
de Dgv precisos en cuanto al grado de sub-enfriamiento relativo a cada compuesto a una
Capítulo 8
completar dicha evaluación. Efectivamente, en la Fig. 8.2.2 se muestra el valor de ∆gvr definido por
∆gvr =Vm·∆gv/∆Hm (8.2.2)
Se observa en esta figura que, para un valor fijo de la temperatura, a medida que aumenta el contenido enGeSe2 el valor de ∆gvr para la formación de Sb2Se3 aumenta, mientras que disminuye el correspondiente valor para la formación de GeSe2. En la Tabla 8.1 se presentan los valores utilizados para el cálculo de las curvas mostradas en la figura.
Pasamos a describir los posibles comportamientos de la frecuencia de nucleación cuando el factor de Turnbull oscile entre 1/4<α<1/2. Para ello haremos referencia a las
representaciones gráficas de las figuras 8.2.3 a 8.2.6 que corresponden, respectivamente, a modificar α cuando se pasa desde una composición con un 50 % molar de GeSe2 hasta un 67 % molar y la formación de cristales corresponde a GeSe2 o a Sb2Se3.
Tabla 8.1: Valores del salto de capacidad calorífica a la transición vítrea, ∆Cp, entropía de fusión de cada compuesto, ∆Sm, máxima temperatura de nucleación de cada compuesto, Tmax.
% mol GeSe2 ∆Cp (J/g) ∆Sm(GeSe2 )
(J/g·K)
∆Sm(Sb2Se) (J/g·K)
Tmax(GeSe2 )
(K)
Tmax(Sb2Se) (K)
50 0,25 0,11 0,142 535 (α=0,4)
545 ( ,, 0,3) 555 (,,0,25)
542 (α=0,4)
553 ( ,, 0,3) 564 (,,0,25)
67 0,25 0,094 0,13 601 (α=0,4)
615 ( ,, 0,3) 627 (,,0,25)
553 (α=0,4)
235
500 600 700 800
Temperatura (K) 0 0.1 0.2 0.3 gvr p a ra S b2 Se 3 ∆
%molar de GeSe2
50
67
500 600 700 800
Temperatura (K) 0 0.1 0.2 0.3 gvr p a ra G e S e2 ∆
%molar de GeSe2
50
67
a) Valor de ∆gvr para la formación
de Sb2Se3 en las aleaciones de
composición 50 y 67 %molar en GeSe2.
b) Valor de ∆gvr para la formación
de GeSe2 en las aleaciones de
composición 50 y 67 %molar en GeSe2.
Fig. 8.2.2: Diferencia de energía libre entre el líquido sub-enfriado y
el cristal.
Es las figuras 8.2.3 a 8.2.6 se ha representado el término exponencial de la frecuencia de nucleación en función de la temperatura. En todos los casos el eje vertical tiene un origen arbitrario, lo que facilita ver como varia la temperatura del máximo de este término al variar el valor de α. Haciendo referencia a la Fig. 2.2.5, el intervalo de temperaturas en que la
frecuencia de nucleación es cercana al valor máximo se amplia hacia valores más elevados al reducir el valor de α y, también, considerando el mismo valor de α la formación de Sb2Se3
requiere temperaturas más elevadas que la formación de GeSe2, cuando la composición
global del vidrio es (GeSe2)0.5(Sb2Se3)0.5, mientras que esta tendencia se invierte para la
composición (GeSe2)0.67(Sb2Se3)0.33. Este resultado no puede sorprender ya que el vidrio de
composición (GeSe2)0.67(Sb2Se3)0.33 está más sub-enfriado respecto a la formación de GeSe2
Capítulo 8
520 540 560 580
Temperatura (K) -( N W *+ G ') /R T ( c e ro a rb itr a ri o ) ∆ α 0.25 0.3 0.4
Fig. 8.2.3: Término exponencial de la frecuencia de nucleación de GeSe2 cuando se parte de una
aleación de composición (GeSe2)0.5(Sb2Se3)0.5. Las divisiones en el eje vertical corresponden a dos unidades.
540 560 580 600
Temperatura (K) -( N W *+ G ') /R T ( ce ro a rb itr a ri o ) ∆ α 0.25 0.3 0.4
Fig. 8.2.4: Término exponencial de la frecuencia de nucleación de Sb2Se3 cuando se parte de una
aleación de composición (GeSe2)0.5(Sb2Se3)0.5. Las divisiones en el eje vertical corresponden a la unidad.
8.2.3. Crecimiento cristalino
237 los valores de U·3πao2ηoN/RT con ηo el factor pre-exponencial de la viscosidad.
En esta figura se ve claramente que la velocidad de crecimiento de ambas fases para una composición fija del vidrio tiende al mismo valor a baja temperatura; es decir, dicha
velocidad sólo depende de la viscosidad cuando el sub-enfriamiento es muy elevado.
580 600 620 640 660
[image:10.595.226.419.202.403.2]Temperatura (K) -( N W *+ G ') /R T ( c e ro a rb it ra ri o ) ∆ α 0.25 0.3 0.4
Fig. 8.2.5: Término exponencial de la frecuencia de nucleación de GeSe2 cuando se parte de una
aleación de composición (GeSe2)0.67(Sb2Se3)0.33. Las divisiones en el eje vertical corresponden a la unidad.
540 560 580 600
Temperatura (K) -( N W *+ G ') /R T ( ce ro a rb itr a ri o ) ∆ α 0.25 0.3 0.4
Fig. 8.2.6: Término exponencial de la frecuencia de nucleación de Sb2Se3 cuando se parte de una
[image:10.595.226.419.475.672.2]Capítulo 8
550 600 650 700 750 800 850
Temperatura (K) -14
-12 -10 -8 -6 -4
L
n
[U
?3
N
/R
T
]
50% mol. GeSe2
GeSe2 Sb2Se3
67% mol. GeSe2
GeSe2 Sb2Se3
π
η o o
a
[image:11.595.177.407.89.306.2]2
Fig. 8.2.7: Dependencia con la temperatura de la velocidad de crecimiento ( en escala logarítmica) de cada fase para las composiciones límite estudiadas.
Por el contrario, es la temperatura de liquidus, estable o metastable, la que fija prioritariamente el comportamiento de la velocidad de crecimiento cuando el régimen de cristalización corresponde a bajo sub-enfriamiento. Por otra parte el valor máximo que alcanza la velocidad de crecimiento depende de la viscosidad. Así, el valor relativamente inferior de la velocidad de crecimiento para una aleación con un 67 % molar en GeSe2 hay que atribuirlo principalmente al hecho de que su viscosidad es más elevada, tal como se indica en la Fig. 8.2.1.
8.3. Cinética de cristalización
239
8.3.1. Cinética de cristalización de las muestras (GeSe
2)
α(Sb
2Se
3)
1-αdonde
α∈
{.50, .58}
Aparte de las consideraciones termodinámicas y cinéticas, el análisis morfológico de la evolución de la cristalización realizado por microscopía óptica y electrónica de barrido revela que en las mismas etapas de la cristalización se desarrollan estructuras fibrosas con crecimiento piramidal con gérmenes cristalinos distribuidos en el seno del volumen y, en algunos casos, acompañados de estructuras lamelares. Las estructuras piramidales se desarrollan aumentando de longitud y de grosor durante el primer pico. A su vez las estructuras lamelares se desarrollan en formaciones alineadas de manera más acentuada en muestras tratadas hasta desarrollarse el segundo pico de cristalización.
El análisis de composición con la técnica de la distribución espectral de la energía de los rayos-X dispersados (EDX) permite identificar las variaciones de composición asociadas a las estructuras fibrosas y lamelares. Así, las estructuras fibrosas se identifican con gérmenes cristalinos de Sb2Se3 mientras que las estructuras lamelares con gérmenes cristalinos de GeSe2.
Así, los resultados experimentales indican que los primeros gérmenes que se desarrollan corresponden a Sb2Se3: Ahora bien, por el hecho de que esta fase tiene una composición más rica
en Sb que la inicial, el entorno inmediato a dichos gérmenes se enriquece en Ge. Tal como resulta del estudio de vidrios más ricos en GeSe2, una composición enriquecida en GeSe2 puede ofrecer
mayor resistencia a la cristalización, ya que es mejor formador de vidrio. Sin embargo, durante el posterior crecimiento de los gérmenes de Sb2Se3 se desprende localmente el calor de
cristalización y este aporte de calor puede ser suficiente para inducir una incipiente cristalización de la fase GeSe2, tal como efectivamente se observa experimentalmente. Además, en zonas en las
que no se ha iniciado la formación de Sb2Se3, existe también la posibilidad de nucleación directa
de GeSe2.
Para determinar el comportamiento cinético de las muestras, vamos a considerar únicamente las primeras etapas de la primera cristalización. Concretamente, tomaremos los datos correspondientes a una fracción transformada del 10 % del primer pico de cristalización tanto en régimen isotermo como en régimen no-isotermo. Los únicos parámetros sobre los que se efectúa el ajuste entre la modelización y los datos experimentales corresponden al valor de la energía interfacial entre núcleo y líquido sub-enfriado, o bien factor de Turnbull α, y a la
Capítulo 8
solo consideramos las etapas iniciales de crecimiento cristalino, vamos a considerar que éste está limitado por la interface (ecuación 2.2.55) y que la frecuencia de los sitios sobre la superficie del núcleo disponibles para el crecimiento que aparece en dicha ecuación vale f = 1.
En las figuras 8.3.1 y 8.3.2 se presentan los resultados del ajuste entre modelización y datos experimentales en forma de curvas T-T-T y T-HR-T, respectivamente para la aleación con un 50 % molar en GeSe2. La forma de presentación sigue la misma pauta en ambas gráficas. Se muestran los valores calculados para la cristalización eutéctica del líquido sub-entriado correspondientes a tres valores de la fracción transformada: 10-6 (considerado como el inicio de la transformación, aunque apenas detectable por DSC), 10-3 y 10-1. Esta última curva es la que se compara con los datos experimentales. Para visualizar la influencia de la competición entre las dos fases cristalinas, también se representan las curvas T-T-T correspondientes a aquella fase que ofrece mayor resistencia a la cristalización, que en estas figuras es la de GeSe2.
10
Tiempo (s)
580 600 620 640 660 680
T
e
m
p
e
ra
tu
ra
(
K
)
50 % molar de GeSe2
exp. 10-1
10-3
10-6
Cristaliz. eutéctica
Cristaliz. de GeSe2
103
102 105
10-1 10-3 10-6
[image:13.595.160.421.354.599.2]104
Fig. 8.3.1: Curvas T-T-T calculadas a partir de los valores indicados en el texto y en la Tabla 8.1.
241
1 10
Velocidad de calentamiento (K/s)
580 600 620 640 660 680
T
e
m
p
e
ra
tu
ra
(
K
)
50 % molar
de GeSe
2exp. 10-1 10-3 10-6 10-1 10-3 10-6 Cristaliz. eutéctica Cristaliz. de GeSe2
[image:14.595.173.436.90.341.2]10 -2 10 -1 102
Fig. 8.3.2: Curvas T-HR-T calculadas a partir de los valores indicados en el texto y en la Tabla 8.1.
560 600 640 680
Temperature (K)
Sb2Se3
GeSe2
I
u
I (
cm
-3s
-1)
2·109 1·109 2·10-5 1·10-5u
(
cm
·s
-1)
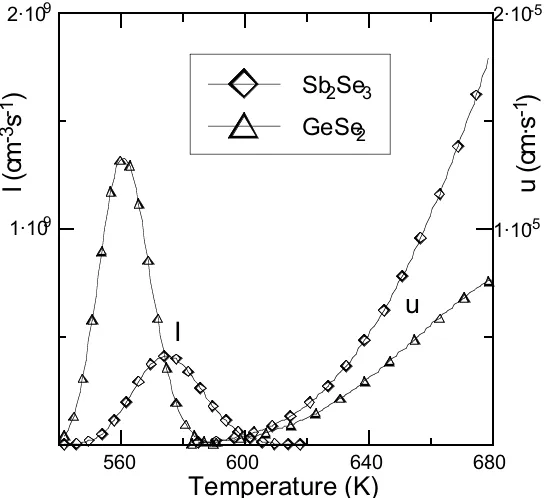
Fig. 8.3.3: Frecuencia de nucleación, I, y velocidad de crecimiento cristalino, u, de las fases GeSe2 (α=0.25) y
Sb2Se3 (α=0.26).
[image:14.595.170.442.388.637.2]Capítulo 8
a los valores calculados. Ello se debe a que en el cálculo se ha omitido la influencia de la velocidad de calentamiento previa a la isoterma. Sin embargo, si consideramos la Fig. 8.3.2 se observa que los valores calculados para un calentamiento a 40 K/min (o 0.67 K/s) hasta 600 K suponen una incipiente cristalización de la muestra (fracción transformada entre 10-6 y 10-3). Esta transformación no se aprecia calorimétricamente pero es suficiente para producir núcleos que pueden avanzar la posterior cristalización en régimen isotermo. La inducción de núcleos durante el calentamiento previo de las muestras es probablemente la causa de que no se obtenga el mismo valor de la energía de activación en régimen isotermo que en régimen no-isotermo (ver Cap.4)
El acuerdo entre los datos experimentales y los calculados es bastante bueno cuando se analiza el comportamiento en régimen de calentamiento continuo, tal como se muestra en la Fig. 8.3.2. En este caso el aumento progresivo de la velocidad de calentamiento retarda, en temperatura, la cristalización. Ello se debe a que la densidad de núcleos formados a una determinada temperatura depende de la velocidad a la que se ha atravesado el intervalo de temperaturas para el que la frecuencia de nucleación es apreciable.
8.3.2. Cinética de cristalización de las muestras (GeSe
2)
α(Sb
2Se
3)
1-αdonde
α
∈
{.63, .67}
243 la vez GeSe2 y Sb2Se3, probablemente debido a la presencia de núcleos pre-existentes formados durante la solidificación en el calorímetro. Todos estos resultados se han corroborado por difracción de Rayos X. Así, el análisis de la evolución de los difractogramas de calentamiento a 0.57 K/min en función de la temperatura de una muestra de (GeSe2)0.67(Sb2Se3)0.33 recién preparada por temple con la de una muestra previamente enfriada a 200 K/min en el calorímetro (ver Fig. 6.16b y 6.17) indican claramente como hay núcleos de GeSe2 en la muestra enfriada en el DSC. Además, el proceso de cristalización de esta muestra se avanza en temperatura pero conduce, igual que en la otra muestra, a una cristalización eutéctica. El desarrollo de estructuras esferulíticas en las muestras enfriadas por DSC (ver Fig. 7.23 y 7.26) también apoya la observación de núcleos pre-existentes. Estos serían centros preferenciales de desarrollo de la cristalización eutéctica en forma de agregaciones esferulíticas de lamelas y fibras.
Según se desprende del análisis de las fig. 8.2.5 y 8.2.6, la temperatura a la cual la frecuencia de nucleación es máxima es más elevada para la fase GeSe2 que para la fase Sb2Se3. Ahora bien, la velocidad de crecimiento (fig. 8.2.7) de la fase GeSe2 tiene valores apreciables en un mayor intervalo de temperaturas que la de Sb2Se3, lo que facilita que aparezca en el primer calentamiento, aunque en pequeña proporción, como fase cristalina. Este análisis justificaría la variación de las curvas de DSC, en número y forma de los picos, con la velocidad del primer calentamiento (ver Fig. 4.44 a 4.46).
Si nos fijamos en los resultados obtenidos durante el tercer calentamiento, la forma de los picos de cristalización en función de la velocidad de barrido si permite un análisis en términos de energía de activación del proceso. Así se ha hecho en el cap. 4. Ahora bién, los valores obtenidos de la energía de activación, 840 J/mol para el primer pico, son muy inferiores a los obtenidos para las aleaciones con un 50% molar en GeSe2, 274 kJ/mol en el primer pico. Una vez más este resultado tiende a confirmar la necesidad de una menor activación del proceso debido a la presencia de gérmenes precursores formados durante las etapas previas de tratamiento térmico.
Capítulo 8
de calentamiento supera los 2,5 K/min. Sólo después de la fusión y enfriamiento a velocidades más lentas que las del temple se puede observar una transformación completa a fases cristalinas.
Conclusiones
En esta memoria, se ha estudiado el comportamiento térmico, la termodinámica, la
evolución morfológica así como los mecanismos que rigen el proceso de la cristalización de
los materiales calcogenuros del sistema binario Sb2Se3-GeSe2, especialmente las aleaciones
(GeSe2)α(Sb2Se3)1-α (α= 0, 0.18, 0.5, 0.58, 0.63, 0.67). Las aleaciones se han preparado por
temple en agua (o enfriamiento lento) de una disolución líquida previamente homogeneizada
en fase líquida estable durante unas 12 horas.
Las curvas calorimetricas obtenidas por calentamiento de la aleación amorfa
muestran varios accidentes térmicos. La primera transformación observada es la transición
vítrea; y se corresponde con un aumento de la capacidad calorífica de la muestra al pasar del
estado amorfo al líquido sobre enfriado. La cristalización se visualiza por calorimetria por la
aparición de dos picos exotérmicos en las curvas DSC de calentamiento.
Se realizó un estudio cinético a nivel de la transición vítrea sobre todas las muestras
amorfas (GeSe2)α(Sb2Se3)1-α (α= 0.5, 0.58, 0.63, 0.67):
- Determinando por medidas calorimétricas la relajación estructural de las muestras.
- Estudiando la influencia de la velocidad de calentamiento, de enfriamiento, en la
transición vítrea.
- Estudiando la influencia del recocido sobre la temperatura de la transición vítrea.
- Determinando por medidas calorimétricas la capacidad calorífica y la entalpía tanto de
las aleaciones recién preparadas como aquellas sometidas a relajación.
Se realizó un estudio cinético de cristalización y de recristalización de todas las
aleaciones estudiadas del sistema binario Sb2Se3-GeSe2.
- Se ha efectuado un análisis, mediante el método de Kissinger y el método de los
múltiples barridos que permiten determinar el valor de la energía de activación.
- Se ha efectuado la determinación de las curvas T-HR-T y T-T-T tanto para un
tratamiento no isotérmico como por un tratamiento isotérmico a nivel de cristalización
como de recristalización de algunas de las aleaciones estudiadas.
- Se ha realizado un estudio de la influencia de la relajación estructural sobre el proceso de
cristalización de la aleación eutectica y como evoluciona este último proceso tanto en
función de la temperatura como en función del tiempo del recocido.
Se realizó un estudio analítico de cristalización mediante difracción de rayos X llevando
a cabo:
- La identificación de los fases presentes después la cristalización de las aleaciones vítreas,
248
- Determinación de los espectros de rayos X de la evolución desde un estado amorfo a un
estado cristalino tanto en función de la temperatura como del tiempo.
Se realizó un estudio morfológico de la evolución de los productos de la cristalización y
de la recristalización de todas las aleaciones estudiadas:
- La cristalización de las aleaciones vítreas (GeSe2)α(Sb2Se3)1-α (α= 0.5, 0.58, 0.63, 0.67)
nos permite revelar la existencia de dos tipos de estructuras cristalinas de morfologías diferentes:
• Estructuras fibrosas piramidales
• Estructuras lamelares
El tratamiento térmico cíclico de las aleaciones cuya fracción molar es 0.63 y 0.67 remite
revelar la existencia de unas estructuras esferuliticas. Es decir, una crecimiento cristalino
primario de las lamelas en forma de una cruz y alrededor de estas lamelas se agregan las
fibras.
- La recristalización de las aleaciones cristalinas Sb2Se3 y (GeSe2)0.19(Sb2Se3)0.81 permite
revelar la existencia de dos tipos de estructuras cristalinas sucesivamente de morfológicas diferentes:
• Estructuras granulosas ordenadas en forma lineal encima de las
lamelas.
• Estructuras en forma de “V”
El análisis de composición con la técnica de la distribución espectral de la energía de
los rayos X dispersados (EDX) permite identificar las composiciones asociadas a las
estructuras fibrosas y lamelares en el caso de la cristalización de las aleaciones amorfas y de
otra parte a las estructuras granulosas y aquellas en forma de “V” en el caso de la
recristalización de las aleaciones cristalinas.
Las estructuras fibrosas se identifican como Sb2Se3 mientras que aquellas estructuras
lamelares como GeSe2. De otro lado, las estructuras morfológicas correspondientes a la
recristalización tanto la morfología granulosa como aquella en forma de “V” corresponden a
Conclusiones
Se ha determinado el modelo cinético de cristalización de la muestra estudiada, que
en general explica los procesos de nucleación y crecimiento cristalino y recibe el nombre de
modelo de Johnson-Mehl-Avrami-Erofeev.
Se ha hecho una modelización de la cinética de cristalización con la cual se ha
llegado a la determinación de la frecuencia de nucleación I y la velocidad de crecimiento
cristalino U de ambos gérmenes tanto para Sb2Se3 como para GeSe2, en función de la
Perspectivas
Perspectivas
De los resultados obtenidos en este estudio, se pueden destacar varias líneas que
permitirán profundizar en la investigación de la formación de vidrio y cristalización de
aleaciones del sistema binario Sb2Se3-GeSe2
Ampliar el estudio cinético realizado de los mecanismos de cristalización cíclica de las
aleaciones (GeSe2)α(Sb2Se3)1-α para valores de α superiores concretamente en lo referente a
los siguientes puntos:
• Ver hasta que punto la cristalización cíclica de estas aleaciones puede controlarse
mediante la velocidad de calentamiento y enfriamientoutilizados.
• Modelización de la cinética de cristalización en función de la densidad y naturaleza de
los núcleos pre-existentes.
Como segunda línea, será interesante hacer un estudio de relajación estructural de las aleaciones (GeSe2)α(Sb2Se3)1-α cuya α es superior a 0.5 debido a su importancia en el
dominio de la formación de los vidrios.
• Estudio detallado de la influencia de la relajación en el proceso de cristalización de estas
aleaciones.
• Estudio por Mössbauer (usando una fuente de Sb121) en el rango del líquido sub-enfriado
durante la relajación para ver las transiciones energéticas y el comportamiento del
Referencias
Aballe.M, J.López Ruiz, J.M.Badía y P.Adeva, 1996, Microscopia Electronica de Barrido y
Micoanálisis por rayos X
Abdel-Rahim M.A., Moharram A.H., Dongol M. y Hafiz M.M, 1990, J. Phys. Chem. Solids 51,355.
Abdl El-Salam F. and E. Abd El-Wahabb, 1992, Vacuum 43-8, 849-853
Adalbert Felz, 1993, Amourphous Inorganic Materials and Glasses,
Adam G. et Gibbs J.H, 1965, J.Chem. Phys., 43, 193
Afify. N, M. A. Abdel-Rahim, A. S. Abd El-Halim and M. Hafiz, 1991, J. Non-Cryst.
Solids 128, 269-278.
Angell C.A, 1966, J. Phys. Chem., 70, 2793.
Angell C.A, 1988, J. Non Cryst. Solids, 102, 205
Angell.C.A, 1968, J.Amer. Ceram.Soc. 51, 117.
Avrami .M, 1941, J. Chem. Phys.9, 177.
Avrami. A, 1939, J. Chem. Phys.7, 1103. Avrami. M, 1940, J. Chem. Phys.8, 212
Bailey.A. R., 1984, M. Sc., Ph. D, D. I. C, F. I. M. Introductory practical Metallography.
Baró.M.D, N. Clavaguera, S.Bordas, M. T. Clavaguera-Mora et J. Cesas-Vázquez, 1976,
JCAT.
Barralis.J, G. Maeder, 1986, Précis de métalurgie 3éme édition, ISSN 0765-5142.
Becker R. et Döring W, 1935, Ann. Phys., 24, 719. Bergman.C et al, 1984, J. Non Cryst. Solids, 70, 367
Bestul.A.B. et Chang S.S, 1964, J. Chem Phys., 40, 731.
Boolchand P, 1986, Hyperfine Interactions 27, 3-14
Bordas. S, B Legendre and M.T. Clavaguera-Mora, 1982, Thermochimica Acta 56, 161
Bordas. S, N. Clavaguera, M.D.Baró, M.T.Clavaguera Mora et J.Casas-Vazquez, 1976,
Proceding journées de calorimetrie et d’Analyse thérmique, Besancon, AFCAT, Vol VII, p.
3-12- 1.
Bordas.S, 1977, Tesis Doctoral, U.P.B.
Bordas.S, M. T. Clavaguera-Mora and B. Legendre, 1982, Thermochimica Acta, 56, 161
182.
Bordas.S, M. T. Clavaguera-Mora and N. Clavaguera, 1990, J. Non-Cryst. Solids 119, 232.
Borisova Z. U., A.V. Pazin, Solid State Chemistry, Myuller R. L. and Borizova Z. U., 1966,
Consult, Bureau, New York, p. 63.
Cernosková. E, Z.G. Ivanova and V. Pamukchieva,1998, Thermochimica acta, 316, 97-100
Clavaguera N. and Clavaguera-Mora M.T, 1996, Mat. Res. Soc. Symp. Proc.Vol. 398.
Clavaguera, M. T. Clavaguera-Mora, J. Onburia, 1985, Journal of materials science 20,
3917-3925.
258
Clavaguera. N, S. Bordas, M. Geli et M. T. Clavaguera-Mora. 1980, J. Calor. 8. Anal.
Therm, Vol XI.
Clavaguera-Mora .M:T, S. Suriñach, M. D. Baró, A. Otero and N. Clavaguera, 1992, Thermochica Acta, 203, 379-389.
Clavaguera-Mora M. T., 1989, Thermochimica Acta, 148, 261
Clavaguera-Mora M. T., 1996, Amorphous Insulators and Semiconductors, Nato Asi,
Vol.23, P.45-69
Clavaguera-Mora M.T and N.Clavaguera, 1997, J. of Alloys and compounds 247, 93-97
Clavaguera-Mora M.T, 1989, J. Thermal Analysis, 35, 1787
Clavaguera-Mora M.T, 1989, Thermochimica Acta, 148, 261
Clavaguera-Mora M.T, 1989, Thermochimica Acta, 148, 261
Clavaguera-Mora M.T, 1998, Thermochimica Acta 314, 281-289
Clavaguera-Mora M.T., C.Comas, R. Ferro, G. Borzone, J.Fontan, N.Clavaguera, 1997,
J.Chem Phys, 94, 1116-1120
Clavaguera-Mora M:T, S. Suriñach, M. D. Baró and N. Clavaguera, 1993, Solid State
Ionics, 63-65, 268-273.
Clavaguera-Mora. M.T, Baró M.D, Suriñach.S, Saurina.J, Clavaguera. N, 1991, J. Non Crys
Solids, 131-133, 479
Cohen M. H., R. G. Neale and A. Paskin, 1972, J. Non Cryst. Solids, 8-10, 885.
Cohen M.H. et Turnbull D, 1959, J.Chem.Phys., 31, 1164.
Cornet.J, 1975, Rev. de Phys. Appl, 10, 409.
Cortes.P, S. Montserrat, J. Ledru, J.M. Saiter, 1998, J.Non Crys Solids 235-237, 522-526
Davies,.R.O, Jones, G.O, 1953, Adv. Phys, 2, 370
Debye.D, 1915, Ann. Phys., 46, 809
Decottignies M., Phalippou J. et Zarzycki J. C. R, 1977, Acad. Sci. , 285 C, 265.
Di Marzio E.A.et Gibbs J.H. 1958, J. Chem Phys., 28, 807. Donald W. Henderson, 1979, J. Non Cryst. solids 30, 301-315
Doolittle A.K. 1951, J Appl. Phys., 22, 1471.
El Souaidi M., Clavaguera-Mora M.T and N. Clavaguera, 1996, designed Poster. Sozopol,
Bulgaria. May 26 – June 8, 1996
El Souaidi M., M.T.Mora, T. Pradell, D. Crepo, and N. Clavaguera, 1997, Proceeding of the
5th International Workshop on Non-Crystallization Solids “Non-Crystalline and Nanoscale Materials, pgs. 49-54
Flower R. et Guggenhem E.A, 1952, Statistical Thermodynamics. Cambridge Univ. Press (N.Y.), P.340.
Fox T.G. et Flory P.J, 1950, J.Appl. Phys., 21, 581. 1951, J. Phys.Chem., 55, 221., 1954,
J.Polymer Sc., 14, 315.
Referencias
Frumar.M y al, 1972, J. Z. Chem, 16, 25
Geli. M, S. Bordas, N. Clavaguera and M.T. Clavaguera-Mora, 1980, “Jornadas cientificas
sobre ceramica y Vidrio”, Edited by C.de la fuente (Sociedad Española de Ceramica y Vidrio, Madrid) p. 196
Geli.M, S. Bordas, N. Clavaguera and M. T. Mora, 1980, Jornadas Cientificas sobre
Ceramica y Vidreo, 197-211.
Gibbs J.H. et Di Marzio E.A, 1958, J. Chem Phys., 28, 373.
Giridhar.A, P. S. L. Narasimham and S. Mahadevan, 1980, J. Non cryst.solids 37, 165.
Giridhar.A., P. S. L. Narasimham and S. Mahadevan, 1981, J. Non cryst.solids 43, 29.
Guinier.A, 1964, Théorie et téchnique de la radiocristallographie Dunod, Paris.
Gutzow.I and A.Dobreva, 1996, Amorphous Insulators and Semiconductors, Nato Asi,
Vol.23, P.21-43
Haggerty.J.S, Cooper.A.R and Heasley.J.H, 1968, Phys.Chem. Glasses 9, 47, (1968); 9, 132.
Haistly R. W. and Kerbs H., 1969, J. Non Cryst. Solids, 1, 399. Hatakeyama.T and F.X.Quinn, 1994, Thermal Analysis.
Herbert Ipser, Michele Gambino, and Wilfried Schuster. 1982, Monatshefte fur chemie 113,
389-398.
Hilling W.B. dans 1962, (Symposium on nucleation and crystallization in glasses and melts)
Amer. Cer.Soc.éd.
Hilton A.R., Hayes D.J. y Rechlinnn M.D, 1975, J. Non-Cryst. Solids 17, 319.
Hilton.A.R and D. J. Hayes, 1975, J. Non-cryst. Solids 17, 319, 339.
Hodge. I.M, 1994, J. Non Cryst. Solids, 169, 211
Hopper.R.W, G.Scherer and Uhlmann, 1974, J.Non Cryst Solids, 15, 45-62
Kauzmann W. 1948, Chem, Rev., 43, 219.
Kissinger H. E., 1957, Anal.Chem. 29, 1702.
Klocek P., Roth M. y Rock R.D, 1987, Opt. Eng. 26, 88.
Klug.H.P and L.E.Alexander, 1974, X-ray Diffraction Procedures. Wiley, New York.
Kraus W.y Nolze G., 1996, J. Appl. Cryst. 29, 301.
Kudlik, C. Tschirwitz, T. Blochowicz, S.Benkhof and E. Rössler, 1998, J. Non crys solids,
235-237, 496-411.
Larmagnac.J.P, J. Grenet and P.Michon, 1981, J. Non Cryst Solids, 45, 157.
Lasocka.M, 1978, Mater. Sci. Eng., 23, 5655
Lombardi G., 1977, For better thermal analysis, Instituto di mineralogia e petrografia
dell´università de Roma. Roma.
Mackenzie R.C, 1972, Procc. ICTAIII, Birkhäuser Verlag, Basel, Stuttgart, 1, 609
Mahadevan.S, A. Giridhar and A. K. Singh, 1983, J. Non-Cryst. Solids 57, 423.
Mills.K.C and Richardson.M.J, 1973, Thermochim.Acta, 6, 427.
260
Moynihan. C.T, A.J. Easteal, J.Wilder and J.Tucker, 1974, J.Phys.Chem., 78, 2673.
Mraw.S.C and Naas.D.F, 1979, J.Chem. Therm. 11, 567.
Narasimham P. S. L, A. Giridhar and S. Mahadevan, 1981, J. Non-Cryst. Solids 43, 301. Nasser Afify, 1990, J. Non-Cryst. Solids 126, 130-140
Norban.B, D. Perching, R.N. Enzweiler, P Boolchand, J.E.Griffiths and J.C. Phillips, 1987,
Phys Review B,36, 15, 8109-8114
Onorato.P.I.K, Uhlman.D.R and Hopper.R:W, 1980, J.Non Crys. Solids 41, 189-200
Rock D, 1985, Photonic Spectra (USA) 19, 77.
Ruffolo.D and P.Boolchand, 1985, Physical review letters, 55, 2, 242-245
Saurina J., M.T. Clavaguera- Mora and Narcis Clavaguera, 1997, Proceeding of the 5th International Workshop on Non-Crystallization Solids “Non-Crystalline and
Nanoscale Materials , pgs. 581-587.
Savage J.A., Webber P.J. y Pitt A.M, 1987, J. Mater. Sci. 13, 859.
Simon.R: Resolving power on the SEM, 1969, J.Appl. Phys.40, 1851
Sreeram.A.N, D.R.Swiler and A.K.Varshneya, 1991, J. Non Crys Solids 127, 287-297.
Stephens. R.B, 1976, J. Non Crys Solids, 20.
Temple. J.Back, 1974, Principes and Thechniques of SEM, vol1 M. A. Hayat ed., Van
Nostrand Reinhold, New York.
Thornburg.D.D and Johnson.R.J, 1975, J.Non Cryst Solids, 17, 2.
Turnbull .D and J. C. Fisher, 1949, The Journal of chimical Physics , volume 17, Num 1.
Turnbull D, 1969, Contemp.Phys., 10, 473.
Turnbull D. et Cohen M.H. 1961, J:Chem Phys., 34., 34, 120.
Turnbull D. et Fischer J.C, 1949, J.Chem Phys., 17, 71.
Uhlmann D. R., 1969, Mat. Sc. Res. V4 Ch. 9.
Uhlmann D.R, 1972, J.Non Cryst Solids, 7, 337.
Uhlmann D.R, 1977, J.Non Cryst Solids, 25, 34. Uhlmann.D. R., 1972, J. Non-cryst. solids 7, 337-348.
Villars. P and L.D. Calvert, 1985, Pearson´s Handbook of Crystallographic data for
intermetallic Phases, American society for metals.
Volmer. M, et Weber, 1925, A. Z. Phys.Chem., 119, 277.
Williams M.F., Landel R.F et Ferry J.D, 1955, J.Am Chem. Soc., 77, 3701.
Xu.Q, Ichikawa.K, 1986, J.Phys. Chem, 19, 7145
Zarzycki. J, 1982, les verres et l´état vitreux, Masson, Paris.
Estudio estructural y morfológico mediante microscopia electrónica de barrido (SEM)
El estudio morfológico por SEM de la muestra recristalizada hasta la zona “b” de la
figura 7.33 nos permite revelar la existencia de unas inclusiones de tipo granuloso distribuidas sobre las estructuras lamelares. Estas estructuras granulosas están alineadas en la
[image:29.595.94.509.201.434.2]misma dirección que las lamelas. Estas estructuras tienen un tamaño del orden de 0.5 ± 0.1 µm como se muestra en las fotografías “a” y “b” de la figura 7.35.
Fig.7.35: Fotografías (a) y (b) de la muestra recristalizada (zona “b” de la Fig.7.33).
7.4. Análisis de composición con la microsonda electrónica (EDX)
A continuación, vamos a analizar todas estas estructuras morfológicas encontradas
tanto a nivel de la cristalización de las muestras como por la recristalización.
Para la identificación de estas estructuras usamos el método de distribución espectral
de la energía de los rayos X dispersados. Este método de análisis nos va a permitir identificar
todas las fases cristalinas correspondientes a cada estructura morfológica encontrada
previamente.
Gracias al método de análisis de la distribución espectral de la energía de los rayos X
dispersados (EDX), hemos llegado a la identificación de la composición de las muestras.
El EDX es una técnica reciente y su utilización es muy importante para el análisis de
la homogeneidad en composición de la muestra.
222
SEM
Jeol, modelo JSM840
Microanlisis: Link An 10000.EDS
Antes de presentar los resultados del análisis de composición reproducimos, en la
tabla siguiente, las energías correspondientes a las líneas de los elementos de interés en este
estudio, según la recopilación de ASTM Data Series DS 46.
Tabla 7.1: Las energías de los tres elementos (Ge, Se y Sb) según ASTM, DS 46.
Elemento Línea
Rayos-X
Energía (keV) Intensidad
relativa
L1 1.036 1
Ln 1.068 1
Lα1,2 1.188 100
Lβ1, 1.218 35
Lβ4 1.286 1
Lβ3 1.294 1
Ge Kα2 9.854 50
Kα1,2 9.874 150
Kα1 9.885 100
Kβ3 10.976 7
Kβ1 10.980 14
Kβ5 11.073 0.05
Kβ2 11.099 0.5
Ll 1.204 1
Ln 1.244 1
Lα1,2 1.379 100
Lβ1, 1.419 35
Lβ3,4 1.490 2
Se Kα2 11.179 50
Kα1,2 11.207 150
Kα1 11.220 100
Kβ3 12.437 8
Estudio estructural y morfológico mediante microscopia electrónica de barrido (SEM)
Kβ5 12.594 0.05
Kβ2 12.650 1
Ll 3.188 7
Ln 3.436 7
Lα2 3.595 10
Lα1 3.604 100
Lβ1 3.843 75
Lβ4 3.886 4
Lβ3 3.932 6
Sb Lβ6 3.979 1
Lβ2,15 4.100 17
Lβ7 4.125 0.1
Lβ10 4.161 0.01
Lβ9 4.170 0.01
Lγ5 4.228 0.1
Lγ1 4.347 8
Lγ2,3 4.599 2
Lγ4 4.696 0.1
No se indican ni las líneas M del “Sb” (por tener energías inferiores a 0.8keV) ni las
líneas K del “Sb” (por tener energías superiores a 26 keV).
Lo más importante del análisis consiste en diferenciar las estructuras fibrosas
piramidales y las estructuras lamelares que se forman al cristalizar la muestra amorfa.
También analizar las estructuras cristalinas granulares y aquellas en forma de “V” en el caso
de la recristalización de las aleaciones (Sb2Se3)0.81(GeSe2)0.19 y Sb2Se3.
En la figura 7.36, se muestra un espectro de la energía de los rayos X dispersados
correspondiente a un área que contiene las dos estructuras (fibrosas piramidales y lamelares)
de una muestra cristalizada de (Sb2Se3)0.5(GeSe2)0.5. El análisis por EDX de un área donde se
encuentran los dos tipos de estructuras tanto fibrosa piramidal como lamelar de las muestras
cristalinas (Sb2Se3)1-α(GeSe2)α donde α∈0.58, 0.62, 0.67 da el mismo espectro de la
energía de los rayos X que el señalado en la figura 7.36.
En este espectro figuran tanto los picos de la energía del “Se” y “Ge” como del “Sb”.
224
cristalina sobre todo en el área donde se realizó el análisis de la distribución espectral de la
energía de los rayos X dispersados.
Un estudio similar realizado sobre una zona puntual de una estructura fibrosa piramidal de una muestra (Sb2Se3)0.5(GeSe2)0.5 metalizada con oro, permite la obtención del
espectro representado en la figura 7.37. En este ultimo espectro figuran los picos de la
energía correspondientes a los elementos “Sb”, “Se” y “Au”. Eso confirma que las
estructuras fibrosas piramidales corresponden al compuesto Sb2Se3. La presencia del pico
[image:32.595.92.511.241.495.2]correspondiente al “Au” es debido a la metalización de la muestra.
Fig.7.36: Espectro de la energía de los rayos X dispersados sobre una área constituida de unas estructuras
Estudio estructural y morfológico mediante microscopia electrónica de barrido (SEM)
Fig.7.37: Espectro de la energía de los rayos X dispersados sobre una zona puntual en el interior de unas
estructuras fibrosas piramidales.
Fig.7.38: Espectro de la energía de los rayos X dispersados sobre una área constituida de unas estructuras
[image:33.595.134.493.481.730.2]226
En la figura 7.38 se muestra un espectro puntual realizado sobre una estructura
lamelar de una muestra semi-cristalizada de (Sb2Se3)0.5(GeSe2)0.5. Los picos presentes en este
espectro corresponden a los de los elementos “Ge”, “Se” y “C”. Lo que confirma entonces que las estructuras lamelares corresponden al compuesto GeSe2. La presencia del pico de “C”
en el espectro de la energía es debido a la metalización de la muestra con el carbón.
En el caso de la recristalización de las aleaciones (Sb2Se3)0.81(GeSe2)0.19 y Sb2Se3, la
recristalización da como lugar a la existencia de unas estructuras sucesivamente en forma de
“V” y otras granulosas sobre las lamelas.
El análisis de la distribución espectral de la energía de los rayos X dispersados en
estas estructuras permite obtener un espectro muy similar a aquel obtenido en el caso de las
estructuras fibrosas piramidales (Fig.7.37). Lo que permite constatar la existencia del
compuesto Sb2Se3 como resultado de la recristalización de las aleaciones
(Sb2Se3)0.81(GeSe2)0.19 como Sb2Se3 sucesivamente en forma de “V” y de granulos encima de
Capítulo 7
Fig.7.23: Estructuras lamelares en forma en forma de "cruz".
Fig 7.24: Estructuras piramidales creciendo para dar lugar a la existencia de fibras.
Fig 7.25: Estructuras lamelares y fibrosas agregándose para formar una estructura
esferulítica.
Fig 7.26: Estructuras esferulíticas formadas por las lamelas y las fibras agregándose alrededor
215
ii. Proceso de Solidificación:
El estudio de la evolución térmica de la solidificación en régimen de
enfriamiento continuo de la muestra ha sido realizado desde la temperatura 500ºC
hasta 200ºC usando una velocidad de enfriamiento de 5K/min. (la velocidad de
calentamiento que se usó hasta la fusión de la muestra fue 5K/min).
La curva siguiente presenta el resultado térmico de la solidificación del líquido
fundido dentro del DSC. Se trata de un pico exotérmico de una cristalización de la
muestra por enfriamiento lento.
La serie de fotografías de la figura 7.28 ha sido realizada en zona a de la curva
señalada en la figura 7.27.
600 625 650 675 700 725
-0.10 -0.05 0.00 0.05 0.10
Zona a β=5K/min
β' =5K/min
Solidificación de la muestra
F
luj
o de
Ca
lor (---> E
ndo)
Temperatura (K)
Fig.7.27: Representación de la curva de la solidificación de la muestra indicando la zona a donde se efectuó el estudio morfológico por SEM.
La fotografía “a” de la figura 7.28 muestra la aparición de unos gérmenes
cristalinos de dos tipos distintos. Unos que tienen unas formas lamelares y otros de
tipo fibroso.
La fotografía “b”, muestra el desarrollo de las estructuras lamelares
agregándose para formar una estructura en forma de "cruz" en el volumen amorfo de
la muestra.
A medida que la temperatura decrece, una activación importante de la
[image:36.595.137.469.297.531.2]Capítulo 7
En la fotografía “c”, se muestra una ramificación de unas estructuras fibrosas
alrededor de la estructura en "cruz" formando unas esferulitas distribuidas en distintas
zonas del volumen amorfo de la aleación.
Fotografía “a”: Formación de estructuras lamelares y fibrosas
Fotografía “b”: Estructuras lamelares formando una "cruz"
[image:37.595.196.399.431.685.2]Fotografía “c”: Agregación de las fibras alrededor de las lamelas formando una estructura esferulítica
217
7.3. Estudio morfológico de la recristalización de las muestras
(Sb
2Se
3)
0.81(GeSe
2)
0.19; Sb
2Se
3Los resultados de rayos X muestran que las aleaciones
(Sb
2Se
3)
81.2(GeSe
2)
18.8y Sb
2Se
3 obtenidas por enfriamiento lento o bien por temple en agua forman una fasecristalina.
El estudio morfológico de las muestras por microscopio electrónico de barrido
(SEM) permite de revelar la existencia de unas estructuras lamelares; en ambos casos
tanto para la muestra obtenida por enfriamiento lento como por aquella obtenida por
temple en agua.
7.3.1. Estudio morfológico de la muestra recristalizada
(Sb
2Se
3)
0.81(GeSe
2)
0.19En las fotografías “a” y “b” de la figura 7.29 se presentan dos micrografías de
muestras de la aleación
(Sb
2Se
3)
81.2(GeSe
2)
18.8.
Una fue obtenida por enfriamientolento y la otra por temple en agua. Se constata que ambas muestras de esta aleación
[image:38.595.94.503.465.712.2]tienen una morfología lamelar.
Fig.7.29:
Fotografía “a”: Muestra obtenida por
enfriamiento lento
Fotografía “b”: Muestra obtenida por
Calorimétricamente, la muestra obtenida por enfriamiento lento no da ningún pico
exotérmico, justificando que no hay ningún proceso de recristalización, mientras que la muestra obtenida por temple en agua recristaliza y da como resultado un pico exotérmico
cuya área es pequeña, tal como se muestra en la Fig.7.30.
450 500 550 600 650 700
-0.025 -0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010
α = 0.5 β = 40K/min
F
luj
o de
Ca
lor ( ---> E
ndo)
[image:39.595.136.470.188.432.2]Temperaturas (K)
Fig.7.30: Caracterización térmica de la muestra por el DSC7
Haciendo un calentamiento continuo por DSC7 de la muestra templada en agua hasta
una fracción transformada de 0.5 se revela la existencia de unas estructuras distribuidas en
forma bien ordenada en el seno del volumen cristalino de la muestra. Se trata de una morfología de capas finas con inclusiones en forma de “V” tal como se ven en las fotografías
219
[image:40.595.97.504.72.344.2]Fotografía “a” Fotografía “b”
Fig.7.31: Microfotografías mostrando la existencia unas estructuras en forma de “V” encima de las lamelas.
Fig 7.32: Ampliación de una microfotografia de una estructura en “V”
La fotografía 7.32 representa una ampliación de la estructura en forma de “V”
distribuida en el seno cristalino del volumen de la muestra, especialmente encima de las
[image:40.595.175.417.376.630.2]Capítulo 7
7.3.2. Estudio morfológico de la muestra recristalizada (Sb
2Se
3)
El estudio calorimétrico de la muestra Sb2Se3 obtenida por temple en agua da como
resultado un pico exotérmico de recristalización cuya área es pequeña.
El estudio morfológico por SEM de las muestras recién preparadas indica la misma estructura
tanto para la obtenida por como enfriada lentamente. En la figura 7.34 se muestra como
ejemplo la morfología de una muestra obtenida por temple en agua y calentada hasta la zona
“a” de la figura 7.33. Esta microfotografía muestra estructuras lamelares de Sb2Se3 y
fracturas de dichas lamelas en forma fibrosa.
450 500 550 600 650 700
-0.02 -0.01 0.00 0.01 0.02
"b" "a"
Zonas de la realización del estudio morfológico por "SEM"
β = 20K/min Muestra obtenida por temple en agua
F
luj
o de
c
al
or ( --->E
ndo)
[image:41.595.158.451.263.487.2]Temperatura (K)
Fig.7.33: Caracterización térmica de la muestra por DSC
[image:41.595.192.407.520.749.2]207 aparecen aleatoriamente agujeros o poros dentro la muestra. Así se muestra en las
figuras 7.10 y 7.13.
Fig.7.8.1 Fig.7.9.1
Microfotografías (MO, X 400) visualizándose el crecimiento fibroso-piramidal y lamelar.
(Punto c de la curva de DSC )
[image:42.595.95.503.367.636.2]
Fig.7.10 Fig.7.11
Microfotografía mostrando los agujeros
(punto d de la curva de DSC )
Microfotografía indicando la forma paralela
de la propagación uniforme de los cristales
lamelares (punto d de la curva de DSC)”
En las zonas porosas la cristalización no se produce en volumen sino en
superficie lo que modifica ligeramente la morfología de los mismos. Sin embargo, el
Capítulo 7
Fig.7.12: Microfotografía mostrando las estructuras fibrosas piramidales
(punto d de la curva de DSC)
Al llegar al punto d pueden observarse zonas en que se han desarrollado ambas
estructuras (fig.7.11) y fig.7.12).
En las figuras 7.13 y 7.14 se muestran observaciones cuando el material se ha
tratado hasta el punto e. Alrededor de las estructuras fibrosas piramidales la fracción
del material amorfo ha disminuido a la vez que las estructuras lamelares se han
desarrollado.
[image:43.595.100.499.470.717.2]
Fig.7.13 Fig.7.14
Microfotografía mostrando los cristales
fibrosos dentro un agujero
(punto e de la curva de DSC )
Microfotografía correspondiente al punto e de
la curva de DSC mostrando la distribución
209 El desarrollo de las estructuras lamelares se acentúa cuando la muestra esta
tratada hasta el punto f como puede verse en las micrografías de las figuras 7.15 y
7.16. Particularmente, en la figura 7.16 se constata la presencia de la alternancia entre
estructuras lamelares y piramidales. En esta zona las estructuras fibrosas y lamelares
tienen una dirección común de crecimiento.
[image:44.595.90.508.180.432.2]
Fig.7.15 Fig.7.16
[image:44.595.194.404.474.719.2]Microfotografías SEM correspondiente al punto f de la curva de DSC
Fig.7.17 Microfotografía mostrando las estructuras fibrosas piramidales y lamelares
Capítulo 7
Una vez se llega al punto g de la curva de DSC no hay ningún cambio
significativo en la morfología estructural de las muestras (fotografía de la figura 7.17).
En esta figura se presenta una visión global de una zona mostrando fractura cristalina
donde se presenta en detalle la presencia de lamelas y estructuras fibrosas. Estos
resultados se confirman exactamente mediante los estudios estructurales por análisis
de difracción de rayos-X.
7.2. Estudio morfológico de las muestras (Sb
2Se
3)
1-α(GeSe
2)
α(con α∈0.63, 0.67)
La muestra obtenida por temple en agua sin ningún tratamiento térmico tiene
una estructura totalmente amorfa.
En la fotografía 7.18, se muestra un volumen amorfo en el cual el comienzo de
[image:45.595.201.398.351.606.2]la formación de los núcleos no ha empezado tener lugar todavía.
Fig 7.18: Microfotografia de una muestra amorfa obtenida por SEM
7.2.1. Estudio isotérmico de la muestra (GeSe
2)
0.67(Sb
2Se
3)
0.33El estudio de la evolución térmica de la cristalización en régimen isotérmico se
efectuó por calentamiento de la muestra (a la velocidad de β=10K/min) hasta una
temperatura previamente seleccionada, sometiéndola a continuación a un recocido
211
La figura 7.19 presenta el resultado calorimétrico de una muestra tratada
isotérmicamente a 703 K durante una hora.
0 500 1000 1500 2000 2500 3000
-0,10 -0,05 0,00 0,05 0,10
b a
La subida fue a 10K.min-1 Isoterma a 703 K
F
luj
o de
Ca
lor (W
/g) (----> E
ndo)
Tiempo ( s)
Fig.7.19: Representación de una curva isotérmica de cristalización indicando los puntos donde se efectuó el estudio morfológico.
El tratamiento isotérmico de la cristalización de la muestra hasta el punto a de
la curva señalada anteriormente revela la aparición de unos gérmenes cristalinos de
carácter lamelar (fig.7.20 ) .
A medida que aumenta el tiempo, estos últimos se desarrollan creciendo en el
seno del volumen amorfo, formando una estructura lamelar con una dirección
preferencial de crecimiento al como se ve en la fotografía señalada.
A medida que aumenta el tiempo, unos gérmenes de tipo fibroso empiezan a
aparecer justamente en el punto b señalado en la curva de DSC representada
anteriormente. Allí es donde se ha podido revelar la formación de gérmenes de tipo
fibroso creciendo paralelamente y perpendicularmente a las lamelas en el seno del
Capítulo 7
[image:47.595.91.509.69.328.2]
Fig.7.20: Estructuras lamelares. Fig 7.21: Estructuras fibrosas distribuidas (Punto a de la curva de DSC) encima de las lamelas. (Punto b de la curva de DSC)
7.2.2. Estudio no isotérmico de la muestra
i. Proceso de cristalización:
Si calentamos calorimétricamente la aleación (Sb2Se3)1-α(GeSe2)α (con α∈0.63, 0.67) a 10 K/min hasta llegar al pico de fusión y después enfriamos a 200K/min entonces obtendremos como resultado de un nuevo calentamiento un pico
exotérmico de cristalización de esta aleación. El segundo ciclo de calentamiento de
esta aleación da como resultado un pico exotérmico también. El estudio morfológico
de esta aleación durante el proceso de cristalización da las mismas estructuras
morfológicas obtenidas en el caso anterior (fibrosas piramidales y lamelares).
Si calentamos la aleación (Sb2Se3)0.33(GeSe2)0.67 por tercera vez (tercer
213
550 600 650 700 750
-0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6
Zona a β = 5K/min
β´=200K/min
F
luj
o de
c
al
or (W
/g) (--->E
ndo)
[image:48.595.154.456.90.301.2]Temperatura (K)
Fig.7.22: Representación de la curva DSC del tercer calentamiento continuo indicando la zona donde se efectuó el estudio morfológico por SEM.
El estudio de la evolución térmica de la cristalización en régimen no isotérmico
se efectuó para el tercer calentamiento. Las velocidades de calentamiento β y de
enfriamiento β’ de los dos ciclos precendentes fueron β = 5K/min y β´ = 200K/min.
En el tercer calentamiento, la muestra fue tratada hasta la zona a previamente
seleccionada para realizar los estudios morfológicos por SEM. En esta zona, se revela
la existencia de lo siguiente:
• Estructuras lamelares formando una agregación en forma de "cruz" en distintas zonas del volumen amorfo de la muestra (Fig.7.23).
• Estructuras fibrosas piramidales agregando alrededor de las lamelas (Fig.7.24 y 7.25).
• Las estructuras fibrosas que se agregan alrededor de la “cruz” para formar unas estructuras de tipo esferulítico radiales distribuidas en todo el volumen amorfo de la
aleación. Estas esferulitas van creciendo en todas las direcciones hasta la obtención
de una estructura totalmente cristalina (Fig.7.26).
Justamente después de la transición vítrea el liquido sub-enfriado empieza su
cristalización con la formación de estructuras lamelares. Al mismo tiempo unas
estructuras fibrosas empiezan a nacer agregandose alrededor de la lamelas, formando
una ramificación estructural (zona a de la curva de DSC de la fig. 7.22).
Al observarlas en el microscopio electrónico de barrido se revelan unas
estructuras con simetría esférica y radial. La distribución de las estructuras
Estudio analítico de cristalización mediante difracción de rayos X
10 20 30
0 200 400 600
254ºC (1H) 255ºC (2H)
25ºC (2H)
25ºC (1H)
Int
ens
ida
d (u.a
)
2θ(degree)
Fig.6.17a: Representación de los difractogramas a 25ºC y a 254±1ºC durante el primer (1H) y el
segundo calentamiento (2H) de la muestra (Sb2Se3)0.33(GeSe2)0.67 calentada a 0,75K/min
Los dos difractogramas registrados a 25ºC y a 254ºC de la muestra recién
preparada (Sb2Se3)0.33(GeSe2)0.67 calentada a 0.57K/min muestran la existencia de una
fase amorfa tal como se ve en la figura 6.17a. En cambio, los dos difractogramas
registrados a las mismas temperaturas de la misma muestra durante el segundo ciclo
de calentamiento muestran la existencia de unos núcleos de la fase cristalina GeSe2 a
25ºC. A 255ºC, la muestra es cristalina y los picos identificados en el difractograma
255(2H) de la Fig.6.16a corresponden a las dos fases GeSe2 y Sb2Se3.
La intensidad relativa en el caso de los difractogramas correspondientes a la
figura 6.17 es mucho menor que aquella correspondiente a la figura 6.16b. Esto es debido a la poca cantidad de la muestra utilizada en el caso del segundo ciclo de
[image:49.595.160.488.95.340.2]196
6.4 Recristalización de las muestras cristalinas Sb
2Se
3,
(Sb
2Se
3)
0.81(GeSe
2)
0.196.4.1 Estudio de la recristalización de la muestra
(Sb2Se3)
0.81(GeSe2)
0.19En las figuras 6.18 y 6.19, se presentan dos difractogramas de dos muestras,
sucesivamente, una obtenida por enfriamiento lento y la otra obtenida por temple en
agua de la muestra (Sb2Se3)0.81(GeSe2)0.19 .
Se constata que el difractograma correspondiendo a la muestra obtenida por
temple en agua contiene unos picos cuya anchura a media altura es mayor que aquella
de los picos del difractograma correspondiente a la muestra obtenida por enfriamiento lento.
De otra parte, se constata también que todos los picos del difractograma de la
muestra obtenida por enfriamiento lento son más agudos y más intensos
comparándolos con aquellos obtenidos por temple. Esto se justifica por el hecho de
que en la muestra obtenida por enfriamiento lento el tamaño cristalino es superior que
en aquella obtenida directamente por temple en agua
25.0 27.5 30.0 32.5 35.0 37.5 40.0
0 1000 2000 3000 4000 5000 6000 7000
Muestra enfriada lentamente y tratada calorimetricamente Muestra enfriada lentamente
Int
ens
ida
d ( u.a
)
[image:50.595.160.501.434.697.2]2θ ( degree)
Estudio analítico de cristalización mediante difracción de rayos X
Se constata que la muestra enfriada lentamente y calentada por DSC es un poco
mas cristalina que la muestra obtenida directamente por enfriamiento lento (Fig.6.18).
En la figura 6.19 se nota la diferencia entre el difractograma de la muestra
obtenida por temple en agua y recristalizada en el DSC y el difractograma de la
muestra obtenida directamente por temple en agua. La anchura a media altura de los
picos en el caso de la muestra enfriada rápidamente y calentada por el DSC es mucho menor que aquella obtenida en el caso de los picos del difractograma correspondiente
a la muestra obtenida directamente por temple en agua.
30.0 32.5 35.0 37.5 40.0
0 1000 2000 3000
Muestra templada en agua
Muestra templada en agua y tratada calorimetricamente
Int
ens
ida
d ( u.a
)
[image:51.595.159.499.234.500.2]2θ ( degree)
Fig.6.19: Difractogramas de una muestra templada en agua
Se realizó un estudio de difracción de rayos X en función de la temperatura
para ver como evoluciona la anchura a media altura de los picos en función de la
temperatura.
Para realizar este estudio, se eligió un rango angular donde figuran los picos
mas intensos y se hizo un estudio en función de la temperatura para ver el
comportamiento de estos picos.
Según lo que muestra la figura 6.20, aumentando la temperatura, la muestra
obtenida por enfriamiento lento tiene picos que devienen más finos que los de la
muestra obtenida por temple en agua.
Se constata que la anchura a media altura del pico de Sb2Se3 va decreciendo a
medida que aumenta la temperatura, y es menor en el caso de la muestra obtenida por
198
300 400 500 600 700
0.15 0.16 0.17 0.18 0.19 0.20 0.21 0.22 0.23 0.24
Sb2Se3 (212)
Muestra obtenida por enfriamiento lento Muestra obtenida por temple en agua
F
W
H
M
[image:52.595.137.471.90.363.2]Temperatura (K)
Fig.6.20: Representación de la anchura a media altura del pico (212) de la fase Sb2Se3 de las dos
muestras de la aleación (Sb2Se3)81.2(GeSe2)18.8.
6.4.2 Estudio de la recristalización de la muestra (Sb
2Se
3)
El compuesto Sb2Se3 obtenido por temple en agua y analizado
calorimétricamente por el DSC7 presenta un difractograma muy semejante al de la
muestra obtenida directamente por temple en agua. La única diferencia que se ve
comparando estos dos difractogramas es que en el caso de la muestra obtenida por
temple en agua y recristalizada por DSC los picos son más finos que los de la muestra
Estudio analítico de cristalización mediante difracción de rayos X
10 15 20 25 30 35 40 45 50
0 200 400 600 800 1000 1200 1400 1600
Sb2Se3 Obtenida por temple en agua
Sb2Se3 Obtenida por temple en agua y tratada por el DSC
Int
ens
ida
d (u.a
)
2θ (Degree)
Fig.6.21: Difractogramas de una muestra obtenida por temple en agua
Los dos difractogramas representados en la figura 6.21 son muy semejantes y no
presentan una gran diferencia. Este hecho indica que la ausencia de GeSe2 en esta
muestra da lugar a un tamaño medio de grano ya relativamente importante incluso en las