Neutral and Cationic [Bis(η1 amidosilyl) η5 cyclopentadienyl]titanium and zirconium complexes: synthesis, X ray molecular structures and DFT calculations
12
0
0
Texto completo
(2) FULL PAPER of a 14-electron metallocene species I. Structures of type III with two amido ligands would indeed have the formal electron count of I. Despite this, complexes III are special, since they do not bear an alkyl group ready for Ziegler⫺Nattatype insertion polymerizations. Nevertheless, we have reported[7] that they still have potential for polarized metal⫺olefin binding similar to, for instance, the 18-electron Cp3M⫹ series (M ⫽ Ti, Zr).[8] For this reason we decided to study the synthesis and chemical behaviour of [bis(amidosilyl)cyclopentadienyl]titanium and -zirconium complexes of type III. Here we report the syntheses of monocyclopentadienyl [M{η5-C5H3[SiMe2(NHtBu)]2}X3], (amidosilyl)cyclopentadienyl [M{η5-C5H3[SiMe2(NHtBu)][SiMe2(η1-NtBu)]}X2] and bis(amidosilyl)cyclopentadienyl [M{η5-C5H3(SiMe2η1-NtBu)2}X] group-4 metal compounds and their cationic [M{η5-C5H3(SiMe2-η1-NtBu)2}]⫹ derivatives. The X-ray molecular structures of the neutral [Ti{η5-C5H3[SiMe2(η1NtBu)]2}(CH2Ph)] and the cationic [Zr{η5-C5H3[SiMe2(η1NtBu)]2}]⫹[(CH2C6H5)B(C6F5)3]⫺ complexes were determined by diffraction methods and DFT calculations were carried out for the neutral and cationic species.. Results and Discussion The cyclopentadiene C5H4(SiMe2NHtBu)2 (1) was isolated as a yellow liquid by treatment of the reported bis(chlorosilyl)cyclopentadiene[9] with 2 equiv. of LiNHtBu in THF and was characterized by 1H and 13C NMR spectroscopy as a mixture of isomers (Scheme 2). The 1H NMR spectrum of 1 recorded at 20 °C indicated a mixture of isomers a, b, c and d in a molar ratio of 8:4:1.3:1.. Scheme 2. Isomers of C5H4(SiMe2NHtBu)2 observed by NMR spectroscopy. In comparison with other previously reported disilyl-substituted compounds C5H4(SiR3)2,[10] in which the 1,1-isomer (b; R ⫽ Me, Cl) is by far the major component, the steric hindrance caused by the bulkier substituents (R ⫽ NHtBu) means that the 1,3-isomer (a; two broad SiMe2 singlets) is the major component in the isomeric mixture of 1, being more favourable than the 1,1-isomer (b; one broad SiMe2 singlet), as shown in Scheme 2. The other two isomers (2,4- and 2,5-), with both silyl groups bound to sp2 carbon atoms, were less abundant components. All of these 2464. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck. isomers interconvert, probably through successive 1,2-shifts of the silyl groups,[10] and increasing temperatures increase the a/b ratio to 4:1 at 50 °C, 8:1 at 70 °C and more than 10:1 at temperatures higher than 80 °C. The broad signals observed for 1a at 20 °C appeared as four singlets when the 1 H NMR spectrum was recorded at ⫺40 °C. The tetraamide M(NMe2)4[11] and tetrabenzyl M(CH2Ph)4[12] compounds (M ⫽ Ti, Zr) were used to deprotonate the cyclopentadiene compound 1, which possesses three acidic protons, to generate the metal-coordinated cyclopentadienyl ligand. Monocyclopentadienyl Compounds Immediate deprotonation of the most acidic cyclopentadiene proton took place when a toluene solution of 1 was treated at room temperature with 1 equiv. of Zr(NMe2)4 to give the monocyclopentadienyl compound 3 (Scheme 3) as the unique reaction product after 1 h (100% NMR yield). When Ti(NMe2)4 was used, heating at 50 °C was required to complete the reaction, the analogous titanium derivative 2 being isolated. Compound 2 was isolated as a dark red oil in quantitative yield after removal of the toluene and extraction into hexanes. The related tribenzyltitanium and zirconium derivatives could not be isolated when M(CH2Ph)4 was used[13] because further deprotonation to give the mono- and bis(silylamido) benzyl derivatives took place very easily at room temperature. The different thermal conditions required to deprotonate the tribasic diaminocyclopentadiene 1 show that the deprotonation with irreversible toluene elimination by the tetrabenzyl compounds M(CH2Ph)4 is much easier than the reversible elimination of amine by the amido derivatives M(NMe2)4, and that the zirconium compounds are more easily deprotonated than the more crowded titanium analogues, in spite of their weaker Ti⫺C and Ti⫺N bonds.[14] In all reactions the first deprotonation involves the most acidic cyclopentadiene proton. The 1H NMR spectra of 2 and 3 in C6D6 each show equivalent silylamido groups with NH (δ ⫽ 1.04, 1.08 ppm), tBu (δ ⫽ 1.16, 1.15 ppm), and diastereotopic SiMe2 (δ ⫽ 0.41, 0.43 ppm; δ ⫽ 0.42, 0.44 ppm) resonances close to those observed for 1. Additionally, they each show an A2B spin system for the ring protons and one singlet for all three equivalent NMe2 groups. The expected spectral features were also observed in the 13C NMR spectra, which each show one downfield shifted ring Cipso resonance (δ ⫽ 127.8, 127.2 ppm) and ∆δ (δ Ctert ⫺ δ CMe) values[15] (15.3 ppm and 15.4 ppm) consistent with the presence of uncoordinated SiMe2⫺NHtBu groups. Single (Amidosilyl)cyclopentadienyl Compounds When toluene or THF solutions of complex 3, prepared at room temperature, were heated at 65 °C, complete transformation of 3 into 5 was observed after 3 h. Compound 5 was the only NMR-detectable product under these conditions, and it could be isolated as a brown solid and characterized by elemental analysis and NMR specwww.eurjic.org. Eur. J. Inorg. Chem. 2003, 2463⫺2474.
(3) Neutral and Cationic [Bis(η1-amidosilyl)-η5-cyclopentadienyl]titanium and -zirconium Complexes. FULL PAPER. Scheme 3. Synthesis of cyclopentadienyl- and [(amidosilyl)- and bis(amidosilyl)cyclopentadienyl]titanium and -zirconium amide and benzyl complexes. troscopy. A similar transformation of 2 was achieved when a toluene solution of previously isolated 2 was heated at reflux for 5 h. The deprotonation of only one of the SiMe2NHtBu substituents with elimination of amine NHMe2 occurred in a selective way to give pure 4, which was isolated as a dark red oil and characterized by elemental analysis and NMR spectroscopy. The related dibenzyl[ansa-(cyclopentadienylsilyl)amido]titanium compound 6 was produced when a toluene solution of 1 and Ti(CH2Ph)4 was heated (below 70 °C) for 5 h. Higher temperatures could not be used, since further deprotonation would then occur, yielding small amounts of 8. Under these conditions, 6 was the only reaction product that could be isolated, as a red-brown solid, and identified by elemental analysis and NMR spectroscopy. The third deprotonation could not be avoided when Zr(CH2Ph)4 was used, because deprotonation then occurs even at low temperature to give a mixture of 7 and other minor components. Because the products could not be separated, 7 was only characterized by NMR spectroscopy. Coordination of the η5-cyclopentadienyl ligand increases the acidity of the NHtBu amino protons [δ ⫽ 1.04 (2), 1.08 (3) ppm], and this is further increased after abstraction of the first amino hydrogen atom [δ ⫽ 0.86 (4), 0.85 (5) ppm]. For these reasons the reactions with the titanium reagents could be carried out selectively to give pure amido and benzyl compounds, whereas the tribenzyl compounds could not be isolated at room temperature. The 1H NMR spectra of the silylamido compounds 4⫺7 in C6D6 each show an ABC spin system for the three ring protons while the 13C NMR spectra exhibit five resonances for the inequivalent ring carbon atoms, as expected for asymmetric molecules. Four resonances (1H and 13C) for Eur. J. Inorg. Chem. 2003, 2463⫺2474. www.eurjic.org. the SiMe2 moieties and two for the tBu groups (1H and 13 C), along with two 13C resonances for their quaternary carbon atoms, are also observed for the two non-equivalent SiMe2NtBu and SiMe2NHtBu groups. Both can be easily distinguished by the different ∆δ values[15] observed in the 13 C NMR spectra, which are smaller [15.5 (4), 15.7 (5), 16.0 (6, 7) ppm] for uncoordinated NHtBu groups than for bridging NtBu groups [26.4 (4), 21.7 (5), 27.3 (6), 23.7 (7) ppm]. The amido complexes 4 and 5 also show two signals (1H and 13C) for non-equivalent NMe2 ligands, whereas the benzyl derivatives 6 and 7 show four 1H doublets and two 13 C resonances for diastereotopic methylene protons and carbon atoms of two non-equivalent benzyl ligands. The chemical shifts observed in the 1H and 13C NMR spectra for all of the ring substituents of the analogous titanium and zirconium complexes show very slight differences, whereas resonances for the additional NMe2 and CH2Ph ligands appear at a lower field for the titanium than for the zirconium analogues. The tert-butyl 1H and 13C resonances of the coordinated amido ligand are shifted downfield with respect to a free NHtBu group as a consequence of the π-donation to the metal centre, which is also consistent with a higher value of ∆δ. Bis(amidosilyl)cyclopentadienyl Compounds Complete deprotonation of the uncoordinated SiMe2⫺NHtBu groups in the dibenzyl complexes 6 and 7 required heating of their toluene solutions under reflux or at 70 °C, respectively, to give the bis(silylamido) compounds 10, isolated as orange crystals and 11, isolated as a brown solid. Both complexes were identified by elemental analyses and NMR spectroscopy. Temperatures higher than reflux in toluene were required to achieve similar deprotonation 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2465.
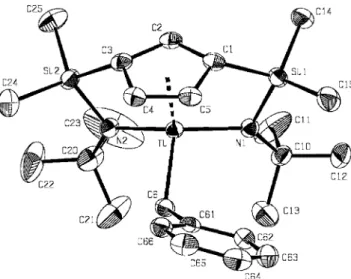
(4) FULL PAPER of the amido complexes 4 and 5. The reaction was monitored by 1H NMR spectroscopy with a C6D6 solution of 5 in a Teflon-valved NMR tube. Complete deprotonation was observed after 12 h of heating at 120 °C, giving the bis(silylamido) compound 9 as the only reaction product. This reaction was reversible, however, and when the solution was cooled to room temperature, 9 reacted in solution with the eliminated amine NHMe2 to give the starting product 5 quantitatively, and so could not be isolated in this way. Nevertheless, pure 9 could be obtained at a preparative level when the same thermal treatment of 5 was carried out in a Teflon-valved Schlenk tube after addition of 1 equiv. of the monobenzylzirconium compound 11, which reacts with amine in an irreversible reaction to give 9 with elimination of toluene (see Scheme 3). Similar deprotonation of the titanium compound 4 required heating at temperatures higher than 160 °C to afford the bis(silylamido)titanium derivative 8. This transformation was not quantitative, although pure 8 could be easily recovered because the reaction was not reversible on cooling to room temperature. Compounds 8 and 9 were isolated as brown and yellow solids, respectively, and identified by NMR spectroscopy. Formation of the bis(silylamido) compounds is more selective, as it requires heating at temperatures above 100 °C, except in the case of the benzylzirconium compound 11 (70 °C). For the same reason the formation of the amidozirconium compound 9 is reversible in the presence of free amine. The 1H and 13C NMR spectra of these diamido compounds 8⫺11 in C6D6 are consistent with the presence of a plane of symmetry, making the two silylamido groups equivalent. They show one singlet (1H) and two resonances (13C) for the tBu groups and two signals (1H and 13C) for the non-equivalent methyl groups of the SiMe2 moiety. Additionally, they show an A2B spin system for the ring protons and three signals for the ring carbon atoms, along with one resonance for the methyl (8, 9) and methylene (10, 11) protons and carbon atoms of the NMe2 and CH2Ph ligands, respectively. The ∆δ[15] values are larger than those observed for non-coordinated SiMe2NHtBu groups in monocyclopentadienyl (2, 3) and single silylamido (4⫺7) compounds, but lower than those observed for the metalcoordinated SiMe2-η1-NtBu groups in silylamido compounds (4⫺7). Orange crystals of 10 suitable for X-ray diffraction studies were isolated after extraction into hexane and slow cooling. The molecular structure of 10 is shown in Figure 1, and selected bond lengths and angles are listed in Table 1. The titanium atom is in a pseudo-tetrahedral environment defined by the methylene carbon atom of the benzyl ligand, the centroid of the cyclopentadienyl ring, and the two coordinated nitrogen atoms of the silylamido groups. This is the first example of a neutral compound with a tridentate cyclopentadienyl ligand tethered by two silylamido groups. Complex 10 shows Ti⫺N distances slightly longer than those reported for related [Ti{η5-C5R4SiMe2(η1-NR⬘)}X2] compounds with only one silylamido ligand (1.907⫺1.919 2466. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck. Figure 1. ORTEP drawing of the molecular structure of the major part of 10 in the solid state; thermal ellipsoids are drawn at the 50% probability level; the hydrogen atoms are omitted for clarity. Table 1. Selected interatomic distances [Å] and angles [°] for complexes [M ⫽ Ti (10), Zr (13)]; Cg denotes the centroid of the Cp ligand 10 M⫺N1 M⫺N2 M⫺C1 M⫺C2 M⫺C3 M⫺C4 M⫺C5 M⫺C6 M···C32 M⫺C33 M···C34 M⫺Cg Si1⫺N1 Si1⫺C1 Si2⫺N2 Si2⫺C3 N1⫺M⫺N2 N1⫺M⫺C6 N1⫺M⫺C33 N1⫺M⫺Cg N2⫺M⫺C6 N2⫺M⫺C33 N2⫺M⫺Cg C6⫺M⫺Cg C33⫺M⫺Cg Cg⫺C1⫺Si1 Cg⫺C3⫺Si2 C1⫺Si1⫺N1 C3⫺Si2⫺N2 M⫺N1⫺Si1 M⫺N2⫺Si2 M⫺C6⫺C61 M⫺C33⫺C32 M⫺C33⫺C34. www.eurjic.org. 1.9785(15) 1.9808(15) 2.3323(19) 2.2915(18) 2.3432(18) 2.3885(18) 2.381(2) 2.1615(19). 2.014 1.7593(15) 1.859(2) 1.7491(15) 1.862(2) 128.05(6) 104.63(7) 106.9 99.22(7). 13 2.0858(17) 2.0812(17) 2.4430(19) 2.406(2) 2.437(2) 2.467(2) 2.466(2) 3.019(2) 2.571(3) 3.057(3) 2.127 1.7579(17) 1.865(2) 1.7610(18) 1.870(2) 126.89(7) 105.86(7) 101.0 102.94(7) 100.8. 106.4 111.0 147.7 146.9 93.70(8) 93.92(8) 103.19(7) 103.53(8) 124.25(13). 121.1 150.1 150.5 93.65(8) 92.94(9) 105.56(8) 105.68(8) 94.56(15) 96.60(17). Eur. J. Inorg. Chem. 2003, 2463⫺2474.
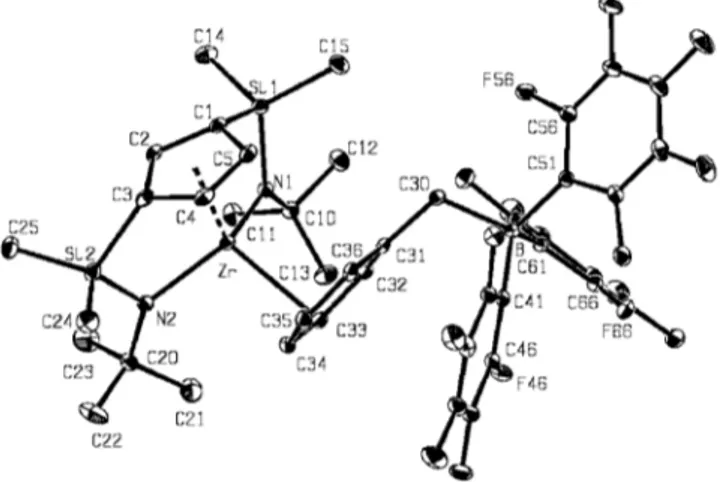
(5) Neutral and Cationic [Bis(η1-amidosilyl)-η5-cyclopentadienyl]titanium and -zirconium Complexes. Å)[2h,4g,16,17,18] but similar to the distance (1.971Å)[19] reported for R ⫽ Me, R⬘ ⫽ tBu, X2 ⫽ 2 NMe2 (see Table 2). This behaviour would be consistent with a weaker π-bonding contribution from the interaction between the nitrogen pπ orbital with the appropriate vacant metal dπ orbital due to the presence of a second amido group, which simultaneously increases the steric crowding and strain of the bicyclic structure. In contrast, the Si⫺N and Si⫺C distances observed for 10 are very close to those observed for all single silylamido compounds. The Ti⫺Cg distance is in the lower range found for singly bridged silylamido compounds. There is no deviation of the disilyl-substituted cyclopentadienyl ligand from η5-coordination, as deduced from the small differences in the Ti⫺C(ring) (2.347 Å average) and the C⫺C ring distances (1.419 Å average). The amido nitrogen atom exhibits the expected planar disposition with sp2 hybridization (the sum of bond angles around N is 360°) known for all structurally characterized silylamido complexes. The close Cg⫺Cring⫺Si angles (147.3° average) account for the strain in these bicyclic compounds. In addition, the Cg⫺Ti⫺N angles are similar, whereas the Ti⫺N⫺Si angles are significantly narrower and the C1⫺Si1⫺N angles more open than those found in single silylamido compounds. This is also consistent with the differences observed in the bond lengths for a system in which the Si⫺N distances do not change whereas the Ti⫺N distances are longer. The N1⫺Ti⫺N2 angle is very open. The Ti⫺C6 distance is in the known range for benzyltitanium compounds. The most significant structural feature of 10 is the orientation of the benzyl ligand, with its phenyl ring directed toward the cyclopentadienyl ring (see DFT calculations be-. FULL PAPER. low). A similar orientation has also recently been observed for [Ti{1,3-[CH2(2-C4H3N)]}2(η5-C5H3)(NMe2)].[20] Cationic Compounds Addition of C6D6 by vacuum transfer to a Teflon-valved NMR tube containing a mixture of B(C6F5)3 and the benzyl compounds 10 or 11 in a 1:1 molar ratio immediately gave the partially soluble cationic species [M(η5C5H3(SiMe2-η1-NtBu)2]⫹, isolated as [B(CH2Ph)(C6F5)3]⫺ salts 12 and 13 as orange solids, which were identified by NMR spectroscopy and X-ray diffraction studies.. Scheme 4. Formation of cationic bis(amidosilyl)cyclopentadienyl complexes. The structures of the cations in C6D6 solutions were established by NMR studies. Their 1H and 13C NMR spectra are consistent with the Cs symmetry also observed for the neutral benzyl precursor compounds 10 and 11. They each show one singlet (1H) and two resonances (13C) for the tBu groups, two signals (1H and 13C) for non-equivalent methyl groups of the SiMe2 groups, an A2B spin system for the ring protons and three signals for the ring carbon atoms.. Table 2. Selected bond lengths [Å] and bond angles [°] for related complexes Compound. M⫺N. M⫺Cg[a]. Cg⫺M⫺N. M⫺C. Si⫺N. Ref.. Ti[(C5Me4)SiMe2(η1-NBz)]Bz2. 1.919. 2.045. 107.4. 1.731. [16]. Ti[(C5Me4)(SiMe2{η1-NCHMePh})] Cl(CH2SiMe3) Ti[(C5H4)SiMe2(η1-NAr)]Cl2 Ti[(C5H4)SiMe2(η1-NAr){CH2B(C6F5)2} (C6F5)] Ti[(C5Me4)SiMe2(η1-NtBu)]Cl2 Ti[(C5Me4)SiMe2(η1-NtBu)](NMe2)2. 1.909(3) 1.914(3) 1.911(4) 1.907(4) 1.972(4) 1.924(5) 1.906(4) 1.919(9) 1.9785(15) 1.9808(15) 1.959(4) 1.946(5) 2.108(4)[b] 2.060(5) 2.064(4) 2.078 2.061(2). 2.030 2.016 2.033 2.030 2.083. 107.2 105.1 105.3 107.6 105.5. 2.131 2.158 2.106(5). 2.132 2.014. 2.233. 119.4 106.9 106.4 106.7 106.6 100.2. 2.198. 101.0. Ti(C5Me5)(NMe2)3 Ti[C5H3{SiMe2(η1-NtBu)}2](CH2Ph) [Ti{C5H3[SiMe2(η1-NtBu)]2}]⫹ [(CH2Ph)B(C6F5)3]⫺ Zr[(C5Me4)SiMe2(η1-NtBu)](NMe2)2 Zr[(C5H3){SiMe2(η1-NtBu)}{SiMe2(NMe2)}]Cl(CH3) Zr[(C13H8)SiMe2(η1-NtBu)](CH2SiMe3)2 [Zr{C5H3[SiMe2(η1-NtBu)]2}]⫹ [(CH2Ph)B(C6F5)3]⫺ [a]. Cg denotes the centroid of the cyclopentadienyl ring.. Eur. J. Inorg. Chem. 2003, 2463⫺2474. 2.0858(17) 2.0812(17) [b]. 1.997(5). 2.127. 101.0 100.8. 2.169(5). [17]. 1.755(3) 1.749(4). [2h] [4g] [19]. 1.722(4). [19]. [18]. 2.1615(19) 2.447(3). 2.311 2.232(3) 2.248(3) 2.571(3). 1.7593(15) 1.7491(15) 1.773(5) 1.788(5) 1.730(4) 1.742 1.738. [7]. [19]. [21] [22]. 1.7579(17) 1.7610(18). The M⫺N bond of the silylamido bridge.. www.eurjic.org. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2467.
(6) FULL PAPER The ∆δ[15] values for the tBu 13C signals are slightly larger than those observed for corresponding neutral compounds. A surprising effect is observed when the 1H and 13C NMR spectra of the neutral benzyl precursor compounds 10⫺11 are compared with those of the corresponding cationic titanium and zirconium derivatives 12⫺13: all of the NMR resonances for the cationic species are shifted upfield. It is also remarkable that this effect is particularly important for the ring CH proton located between the two silyl substituents. Furthermore, it is interesting to note that the ring Cipso resonance is the signal at highest field for the neutral compounds whereas for the cationic derivatives it appears between the other two ring C resonances. This unexpected behaviour seems to be associated with the anisotropy of the benzyl ring exerted in the particular structure of these cationic species. Orange crystals of 13, appropriate for a single-crystal Xray structure determination, were isolated from a C6D6 solution in the NMR tube. A perspective view of the molecular structure of 13 is shown in Figure 2, along with the non-hydrogen labelling scheme, and selected bond lengths and angles are listed in Table 1.. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck. tral complexes (see Table 2), indicating higher electron donation from the cyclopentadienyl ring. The amido nitrogen atom exhibits the expected planar disposition with sp2 hybridization (the sum of bond angles around N is 360°) known for all reported silylamido complexes. The constrained geometry of the Cg⫺Cring⫺Si⫺N⫺Zr cyclic systems is similar to that found for 12. The ring C1 and C3 atoms, bearing the amidosilyl arms, are pyramidally distorted almost to the same extent, as shown by the sums of their bond angles (349.4° average) and by the Cg⫺Cring⫺Si angles (150.3° average). The angles at Si and N are in the range found for 12, whereas the Cg⫺Zr⫺N angles (100.9° average) are significantly narrower than those observed in the related titanium cation 12 (106.65° average). This is consistent with the increased atomic radius of the Zr centre and is responsible for the slightly more constrained geometry shown by 13. The Zr⫺C33 bond [2.571(3) Å] is about 0.30 Å longer, whereas the distances to the neighbouring C32 and C34 atoms are about 0.8 Å longer than those observed for covalent Zr⫺C bonds, indicating an interaction of the empty hybrid metal orbital with the pπ orbital of the meta-Cphenyl atom of the borate anion.[23] The most significant feature of this structure is that the phenyl ring is oriented upward in the direction of the cyclopentadienyl ring, as also observed for the benzyl substituent in complex 10. Structure and Bonding of [Ti{η5-C5H3(SiMe2-η1-NtBu)2}]ⴙ Cations. Figure 2. ORTEP drawing of the molecular structure of 13 in the solid state; thermal ellipsoids are drawn at the 10% probability level and hydrogen atoms are omitted for clarity. The pseudo-tetrahedral geometry about the Zr atom of 13 is defined by the centroid of the cyclopentadienyl ring and the two N-donors of the appended (dimethylsilyl)amido arms, with the remaining coordination site occupied by C(33) of the phenyl ring of the benzylborate anion. This disposition is very close to that observed for complex 12[7] with the expected longer Zr⫺N and similar Si⫺N [1.773(5) Å; 1.788(5) Å for 12] bonds. In comparison with those in the related neutral [Zr{(η5-C5R4)SiMe2(η1-NR⬘)}X2] compounds containing only one silylamido ligand, the Zr⫺N distances in 13 are similar,[21,22] but slightly shorter than the distances reported[19] for R ⫽ Me, R⬘ ⫽ tBu, X2 ⫽ 2 NMe2. This behaviour is consistent with a similar π-bonding contribution from the two amido ligands to the more acidic zirconium cation. The Cg⫺Zr distance is significantly shorter than those observed in related single silylamido neu2468. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. In this section we present density functional calculations performed on the model compound [Ti{η5-C5H3(SiH2-η1NH)2}]⫹ as well as on the real molecule [Ti{η5C5H3(SiMe2-η1-NtBu)2}]⫹. The calculations for the model system serve to establish characteristic patterns in chemical bonding for bis(amidosilyl)cyclopentadienyl compounds of Ti and Zr. As mentioned above, these compounds can reasonably be regarded as 14-electron species, due to inclusion of the amido π-electrons. In the light of our previous analysis,[8c] we can expect that the bonding of [Ti{η5C5H3(SiR2-η1-NR)2}]⫹ cations should be governed by electrostatic interaction and ligand-to-metal donation, whereas back-bonding should play an only minor role. Bonding of the Model System [Ti{η5-C5H3(SiH2-η1NH)2}]ⴙ We first performed DFT calculations on the species with aryl ligands, [Ti{η5-C5H3(SiH2-η1-NH)2}]⫹[ηx-C6H5R] (R ⫽ H, CH3, CH2BF3⫺; x ⫽ 1, 2). For benzene, we considered different ‘‘up’’ and ‘‘down’’ coordination modes, as indicated in Scheme 5. For η1-coordination, ‘‘up’’ and ‘‘down’’ coordination modes are characterized by dihedral angles (C4⫺C1⫺M⫺Cg) close to 0 and 180°, respectively, C1 being the benzene carbon atom coordinating to the metal centre, and C4 being the benzene carbon atom in the position para to C1. Similarly, for η2-coordination, dihedral angles (X4⫺X1⫺M⫺Cg) close to 0 and 180° characterize ‘‘up’’ and ‘‘down’’ coordination modes, X1 being the midwww.eurjic.org. Eur. J. Inorg. Chem. 2003, 2463⫺2474.
(7) Neutral and Cationic [Bis(η1-amidosilyl)-η5-cyclopentadienyl]titanium and -zirconium Complexes. point between the two coordinating benzene carbon atoms, and X4 being the midpoint of the two corresponding benzene carbon atoms in the para position.. Scheme 5. The ‘‘up’’ and ‘‘down’’ coordination modes of the solvated model cation [Ti{C5H3(SiH2NH)2}]⫹. The optimized geometry of the naked cation [Ti{η5C5H3(SiH2-η1-NH)2}]⫹ and its main empty frontier molecular orbital are displayed in Figure 3. The HOMO of the aryl ligand, dominates the orbital interaction through donation to the empty dz2 orbital at the metal centre.. FULL PAPER. ring carbon atom in the position para to the methyl substituent. The relative energies for various investigated complex geometries, together with optimized Ti⫺L bond lengths, are collected in Table 3. Relative energies are determined for groups of isomeric compounds, in which the energy of the isomer with the largest absolute value in total bonding energy is set to zero, and energies of isomers are reported with reference to the energy of the isomer at zero energy. The energy differences are small, but significant. In particular, we note that η1-coordination is favoured for the cationic complexes by about 10 kJ/mol. The small energy differences are accompanied by a fairly large change in the Ti⫺C bond length, of about 0.20 Å, indicating the subtle nature of this bonding interaction. The bond energies (ΒΕs) of benzene and toluene amount to 89 kJ/mol and 100 kJ/ mol, respectively. Finally, complexes of the borate anion C6H5CH2BF3⫺ are characterized by the Ti⫺C bonds, which are about 0.20 Å shorter than those in the related benzene or toluene complexes. Electrostatic interactions between the anionic ligand and the cationic metal fragment have a pronounced influence on the Ti⫺L bond strength. Again, η1-coordination is favoured by 16 kJ/mol. Complexes with the Cation [Ti{η5-C5H3(SiMe2-η1NtBu)2}]ⴙ. Figure 3. Optimized geometry for the model [Ti{C5H3(SiH2NH)2}]⫹ and a main empty frontier MO. cation. We recall from simple Hückel considerations that the HOMO of benzene consists of a set of degenerate e1g orbitals, one of which is more likely to undergo η1-type donation, whereas the other would induce a preference for η2coordination. For toluene, the degeneracy of the e1g set is lifted, due to the antibonding hyperconjugation of the methyl group. In this case, the HOMO is most suited to η1coordination, having the largest orbital coefficient at the. We begin with the molecular geometry of [Ti{η5C5H3(SiMe2-η1-NtBu)2}{η1-C6H5CH2B(C6F5)3}], which has also been established by crystal structure determination.[7] By our analysis, we can expect the following features for this compound. Firstly, the [C6H5CH2B(C6F5)3]⫺ borate ligand should coordinate in η1-fashion, being oriented upwards toward the site of the cyclopentadienyl ring. This structural feature is observed in the crystal structure of this compound. Furthermore, from simple Hückel orbital considerations, we expect that the coordinating ring carbon atom should be located para to the boron substituent. However, the solid-state structure reveals coordination through a meta carbon atom rather than one in the para position. We therefore optimized two representative geometries for both meta and para coordination, as shown in Figure 4. For the meta coordination (Figure 4, a), inspection of the optimized geometries reveals the presence of short F···H distances (d 艐 2.40 Å), between the pentafluorophenyl rings and methyl groups of the metal fragment. For para coordi-. Table 3. Relative energies [kJ/mol] and metal-to-ligand bond lengths [Å] for coordination of the aryl double bond for various complexes of the type [{C5H3(SiH2NH)2}Ti]⫹[ηx-C6H5R] (R ⫽ H, CH3, CH2BF3⫺; x ⫽ 1, 2) R⫽H. η1 Erel[a] d(Ti⫺C)[b]. η2. ‘‘up’’[c]. ‘‘down’’. ‘‘up’’. ‘‘down’’. η1 ‘‘up’’. 0 2.46. 6 2.54. 9 2.69. 8 2.66. 0 2.43. η2 ‘‘up’’. R ⫽ CH2BF3⫺ η1 η2 ‘‘up’’ ‘‘up’’. 12 2.67, 2.69. 0 2.28. R ⫽ Me. 16 2.41, 2.66. [a]. The most stable isomer is set to zero. [b] Bond lengths between the C6H5R ligand and the metal centre. [c] See Scheme 5 for a definition of ‘‘up’’ and ‘‘down’’ coordination.. Eur. J. Inorg. Chem. 2003, 2463⫺2474. www.eurjic.org. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2469.
(8) FULL PAPER. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck. Figure 4. Relative energies and selected bond lengths for meta (a) and para coordination (b) of [Ti{η5-C5H3(SiMe2NtBu)2}{η1C6H5CH2B(C6F5)3}]; comparison with X-ray analysis. nation (Figure 4, b), in contrast, no such short contacts can be found. The presence of such intramolecular hydrogen bonds might be the key point that explains why the meta geometry is energetically favoured by 30 kJ/mol. To produce further evidence for our argument, we also optimized meta and para geometries for [Ti{η5-C5H3(SiMe2-η1NtBu)2}(η1-C6H5CH2BF3)]. As was to be expected, since no short F···H contacts can be achieved in this case, orbital interactions now favour the latter coordination mode by 13 kJ/mol. We then optimized the molecular geometry of [Ti{η5C5H3(SiMe2-η1-NtBu)2}(CH2C6H5)], which is displayed in Figure 5. The geometry of the neutral benzyl complex again exhibits ‘‘up’’ coordination, with a typical Ti⫺C bond length of 2.19 Å. The optimized geometry is in good agreement with the crystal structure of this compound. The reaction energy (∆E) for the formation of the benzyl complex from [Ti{η5-C5H3(SiMe2-η1-NtBu)2}{η1-C6H5CH2B(C6F5)3}], which involves the cleavage of a C⫺B bond, followed by a reorientation of the benzyl fragment, amounts to ⫺15 kJ/mol. The latter compound is therefore stable for kinetic, but not for thermodynamic, reasons. One factor. lending kinetic stability to the unusual η1-adduct is the energy of preparation of the B(C6F5)3 fragment, associated with the pyramidalization of the trigonal-planar-coordinated boron atom, which substantially increases the value for the C⫺B bond strength by the order of 80 kJ/mol.[8c]. Conclusion To conclude, we have synthesized a new type of [bis(η1amidosilyl)cyclopentadienyl]titanium and -zirconium complexes by direct deprotonation of the corresponding bis(aminosilyl)cyclopentadiene with metal tetraamide and tetrabenzyl derivatives under varying thermal conditions. The complexes obtained in this way may be classified into three categories: monocyclopentadienyl, ansa-(amidosilyl)cyclopentadienyl and bis[ansa-(amidosilyl)cyclopentadienyl] complexes. The first two categories show the structures and chemical behaviour expected for these well-known types of compounds. However, the bis[ansa-bis(amidosilyl)cyclopentadienyl] complexes are isolobal with previously reported tris(cyclopentadienyl) compounds and present reactivity pathways determined by the strong electrostatic character of the interactions, additionally facilitated by the unique accessible metal orbital. Treatment of the benzyl complexes with B(C6F5)3 gave the cationic species, which were characterized by X-ray diffraction methods, indicating that the counter-ion is coordinated to the metal atom through a single interaction with the phenyl m-carbon atom of the benzyl borate anion. Density functional calculations developed for the cationic complexes provide an explanation for their remarkable structural features, particularly the ‘‘up’’ (oriented toward the cyclopentadienyl ring) coordination of the benzyl group in the neutral benzyltitanium complex and of the ligand (solvent or counter-anion) bound in the cationic species.. Experimental Section. Figure 5. Optimized geometry of the benzyl complex [Ti{η5-C5H3 (SiMe2NtBu)2}(CH2C6H5)] 2470. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. General Remarks: All experiments were carried out under argon by use either of a Vacuum Atmospheres glove box or of standard Schlenk techniques. Hydrocarbon solvents and THF were distilled from Na/benzophenone and stored under argon prior to use.. www.eurjic.org. Eur. J. Inorg. Chem. 2003, 2463⫺2474.
(9) Neutral and Cationic [Bis(η1-amidosilyl)-η5-cyclopentadienyl]titanium and -zirconium Complexes 1,1-Bis(chlorodimethylsilyl)cyclopentadiene, Ti(NMe2)4,[11] Zr(NMe2)4,[11] Ti(CH2Ph)4,[12] Zr(CH2Ph)4[12] and B(C6F5)3,[24] were prepared by previously reported methods. NMR spectra were recorded with Varian Unity FT 300 and Varian FT 500 Unity Plus instruments in Teflon-valved tubes at probe temperature (20 °C). 1 H and 13C NMR chemical shifts were measured relative to the residual resonances of C6D6 used as solvent, but the chemical shifts are reported with respect to TMS. 19F NMR chemical shifts are referenced to external CFCl3. Coupling constants are reported in Hz. C, H and N analyses were carried out with a Perkin⫺Elmer 240 C analyzer. C5H4(SiMe2NHtBu)2 (1): A solution of LiNHtBu (1.61 g, 20.5 mmol) in THF (40 mL) was added at 0 °C to a solution of C5H4-1,1-(SiMe2Cl)2 (2.51 g, 10 mmol) in THF and the mixture was stirred for 12 h. The volatiles were removed under vacuum and the residue was extracted into hexane to give a mixture of isomers of 1 as a yellow liquid after removal of the solvent (2.92 g, 9.0 mmol, 90%). 1H NMR (C6D6, 20 °C) of the major component (1a): δ ⫽ ⫺0.05 (br. s, 6 H, SiMe2), 0.21 (br. s, 6 H, SiMe2), 1.12 (s, 9 H, NHtBu), 1.17 (s, 9 H, NHtBu), 3, 57 (m, 1 H, CpH), 6.50 (m, 1 H, C5H3), 6.71 (m, 2 H, C5H3) ppm. Component 1b: δ ⫽ 0.09 (s, 12 H, SiMe2), 1.15 (s, 18 H, NHtBu), 6.53 (m, 2 H, C5H4), 6.78 (m, 2 H, C5H4) ppm. [Ti{η5-C5H3-1,3-[SiMe2(NHtBu)]2}(NMe2)3] (2): A solution of Ti(NMe2)4 (0.65 g, 2.9 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (0.94 g, 2.9 mmol) was added by syringe. The resulting red solution was stirred at 50 °C for 5 h. When the evolution of gas had stopped, the solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, complex 2 was isolated as a deep red oil (1.45 g, 2.8 mmol, 97%). 1H NMR (C6D6, 20 °C): δ ⫽ 0.41 (s, 6 H, SiMe2), 0.43 (s, 6 H, SiMe2), 1.04 (s, 2 H, NHtBu), 1.16 (s, 18 H, NtBu), 3.13 (s, 18 H, NMe2), 6.31 (d, 2 H, C5H3), 6.60 (t, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.9 (SiMe2), 4.1 (SiMe2), 34.0 (NtBu), 49.3 (NtButert), 50.3 (NMe2), 119.9 (C5H3), 125.2 (C5H3), 127.8 (C5H3ipso) ppm. IR (Nujol): ν̃ ⫽ 3383 (N⫺H) cm⫺1. C23H53N5Si2Ti (503.76): calcd. C 54.84, H 10.60, N 13.09; found C 54.17, H 10.23, N 12.82. [Zr{η5-C5H3-1,3-[SiMe2(NHtBu)]2}(NMe2)3] (3): Compound 1 (0.05 g, 0.1 mmol) was added by syringe to a solution of Zr(NMe2)4 (0.041 g, 0.1 mmol) in C6D6 (0.6 mL). Complex 3 was immediately identified in the NMR spectrum as the unique product of the reaction. 1H NMR (C6D6, 20 °C): δ ⫽ 0.42 (s, 6 H, SiMe2), 0.44 (s, 6 H, SiMe2), 1.08 (s, 2 H, NHtBu), 1.15 (s, 18 H, NtBu), 2.98 (s, 18 H, NMe2), 6.50 (d, 2 H, C5H3), 6.78 (t, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.8 (SiMe2), 3.5 (SiMe2), 33.9 (NtBu), 45.6 (NMe2), 49.3 (NtButert), 120.5 (C5H3), 127.0 (C5H3), 127.2 (C5H3ipso) ppm. Pure 3 was only observed in NMR-tube experiments, while solid samples were always contaminated by small amounts of 5. [Ti{η5-C5H3-1-[SiMe2(NHtBu)]-3-[SiMe2(η1-NtBu)}(NMe2)2] (4): A solution of Ti(NMe2)4 (2.00 g, 8.9 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (2.91 g, 8.9 mmol) was added by syringe. The resulting red solution was heated at reflux for 5 h. When the evolution of gas had stopped, the solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, complex 4 was isolated as a deep red oil (4.10 g, 8.8 mmol, 98%). 1H NMR (C6D6, 20 °C): δ ⫽ 0.35 (s, 3 H, SiMe2NHtBu), 0.43 (s, 3 H, SiMe2NHtBu), 0.50 (s, 3 H, SiMe2NtBu), 0.54 (s, 3 H, SiMe2NtBu), 0.86 (s, 1 H, NHtBu), 1.12 (s, 9 H, NHtBu), 1.28 (s, 9 H, NtBu), 2.87 (s, 6 H, NMe2), 3.05 (s, Eur. J. Inorg. Chem. 2003, 2463⫺2474. www.eurjic.org. FULL PAPER. 6 H, NMe2), 6.12 (m, 1 H, C5H3), 6.31 (m, 1 H, C5H3), 6.50 (m, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.1 (SiMe2), 2.4 (SiMe2), 2.4 (SiMe2), 3.7 (SiMe2), 34.1 (NHtBu), 34.4 (NtBu), 49.1 (NMe2), 49.6 (NHtButert), 50.4 (NMe2), 60.8 (NtButert), 109.1 (C5H3ipso), 121.2 (C5H3), 122.5 (C5H3), 123.7 (C5H3), 130.7 (C5H3) ppm. IR (Nujol): ν̃ ⫽ 3385 (N⫺H) cm⫺1. C21H46N4Si2Ti (458.67): calcd. C 54.99, H 10.11, N 12.21; found C 54.67, H 9.97, N 12.04. [Zr{η5-C5H3-1-[SiMe2(NHtBu)]-3-[SiMe2(η1-NtBu)]}(NMe2)2] (5): A solution of Zr(NMe2)4 (3.37 g, 12.6 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (4.09 g, 12.6 mmol) was added by syringe. The resulting yellow solution was warmed to 65 °C for 5 h. When the evolution of gas had stopped, the solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, complex 5 was isolated as a light brown solid (6.19 g, 12.3 mmol, 98%). IR (Nujol): ν̃ ⫽ 3384 (N⫺H) cm⫺1. 1H NMR (C6D6, 20 °C): δ ⫽ 0.35 (s, 3 H, SiMe2NHtBu), 0.42 (s, 3 H, SiMe2NHtBu), 0.57 (s, 3 H, SiMe2NtBu), 0.59 (s, 3 H, SiMe2NtBu), 0.85 (s, 1 H, NHtBu), 1.11 (s, 9 H, NHtBu), 1.27 (s, 9 H, NtBu), 2.81 (s, 6 H, NMe2), 2.82 (s, 6 H, NMe2), 6.39 (m, 1 H, C5H3), 6.47 (m, 1 H, C5H3), 6.69 (m, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.3 (SiMe2), 2.7 (SiMe2), 2.8 (SiMe2), 3.0 (SiMe2), 33.9 (NHtBu), 34.9 (NtBu), 44.2 (NMe2), 44.5 (NMe2), 49.6 (NHtButert), 56.6 (NtButert), 111.4 (C5H3ipso), 122.6 (C5H3), 122.9 (C5H3); 124.7 (C5H3), 126.7 (C5H3) ppm. C21H46N4Si2Zr (502.01): calcd. C 50.24, H 9.24, N 11.16; found C 49.73, H 9.05, N 11.45. [Ti{η5-C5H3-1-[SiMe2(NHtBu)]-3-[SiMe2(η1-NtBu)]}(CH2Ph)2] (6): A solution of Ti(CH2Ph)4 (2.14 g, 5.2 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (1.69 gr, 5.2 mmol) was added by syringe. The resulting yellow solution was warmed to 70 °C for 5 h. When the evolution of gas had stopped, the solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, complex 6 was isolated as a red-brown solid (2.86 g, 5.1 mmol, 98%). 1H NMR (C6D6, 20 °C): δ ⫽ 0.21 (s, 3 H, SiMe2NHtBu), 0.27 (s, 3 H, SiMe2NHtBu), 0.37 (s, 3 H, SiMe2NtBu), 0.38 (s, 3 H, SiMe2NtBu), 0.72 (s, 1 H, NHtBu), 1.06 (s, 9 H, NHtBu), 1.44 (s, 9 H, NtBu), 2.46 (d, J ⫽ 10.5 Hz, 1 H, CH2Ph), 2.55 (d, J ⫽ 10.5 Hz, 1 H, CH2Ph), 2.81 (d, J ⫽ 10.5 Hz, 1 H, CH2Ph), 2.97 (d, J ⫽ 10.5 Hz, 1 H, CH2Ph), 5.83 (m, 1 H, C5H3), 6.14 (m, 1 H, C5H3), 6.83 (m, 1 H, C5H3), 6.87⫺7.20 (m, 10 H, CH2Ph) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 0.6 (SiMe2), 1.5 (SiMe2), 2.4 (SiMe2), 2.9 (SiMe2), 33.8 (NHtBu), 34.2 (NtBu), 49.8 (NHtButert), 61.5 (NtButert), 79.6 (CH2Ph), 83.7 (CH2Ph), 110.2 (C5H3ipso), 122.1 (C5H3), 122.5 (C5H3), 122.8 (C5H3), 123.0 (C5H3), 125.9 (C6H5), 126.8 (C6H5), 127.4 (C6H5), 128.5 (C6H5), 128.6 (C6H5), 128.7 (C6H5), 128.9 (C6H5), 129.8 (C6H5), 132.6 (C6H5), 149.6 (C6H5ipso), 150.1 (C6H5ipso) ppm. IR (nujol): ν̃ ⫽ 3349 (N⫺Η) cm⫺1. C31H48N2Si2Ti (552.78): calcd. C 67.36, H 8.75, N 5.07; found C 67.82, H 8.71, N 4.81. [Zr{η5-C5H3-1-[SiMe2(NHtBu)]-3-[SiMe2 (η 1-NtBu)]}(CH 2Ph) 2] (7): A solution of Zr(CH2Ph)4 (2.93 g, 6.43 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (2.09 g, 6.43 mmol) was added by syringe. The resulting yellow solution was warmed to 40 °C for 5 h. The solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, a mixture of complexes 7 and 11 was isolated. Formation of 7 was confirmed by IR and NMR spectroscopy, although pure 7 free of 11 could not be isolated. Data for 7: IR (Nujol): ν̃ ⫽ 3353 (N⫺Η) cm⫺1. 1H NMR (C6D6, 20 °C): δ ⫽ 0.33 (s, 3 H, SiMe2NHtBu), 0.34 (s, 3 H, SiMe2NHtBu), 0.35 (s, 3 H, SiMe2NtBu), 0.40 (s, 3 H, SiMe2NtBu), 0.69 (s, 1 H, NHtBu), 1.06 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2471.
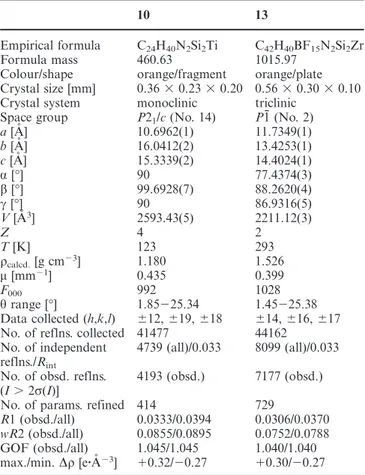
(10) FULL PAPER. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck. (s, 9 H, NHtBu), 1.13 (s, 9 H, NtBu), 1.66 (s, 1 H, CH2Ph), 1.72 (s, 1 H, CH2Ph), 2.12 (s, 1 H, CH2Ph), 2.17 (s, 1 H, CH2Ph), 6.21 (m, 1 H, C5H3), 6.35 (m, 1 H, C5H3), 6.60 (m, 1 H, C5H3), 6.90⫺7.30 (m, 10 H, C6H5) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 1.3 (SiMe2), 2.0 (SiMe2), 2.4 (SiMe2), 2.5 (SiMe2), 33.7 (NHtBu), 33.8 (NtBu), 49.7 (NHtButert), 54.9 (CH2Ph), 57.5 (NtButert), 57.9 (CH2Ph), 109.6 (C5H3ipso), 122.2 (C5H3), 122.5 (C5H3), 124.8 (C5H3), 125.6 (C5H3), 125.9 (C6H5), 126.4 (C6H5), 126.6 (C6H5), 127.8 (C6H5), 129.3 (C6H5), 129.6 (C6H5), 129.6 (C6H5), 129.8 (C6H5), 131.9 (C6H5), 145.8 (C6H5ipso), 146.2 (C6H5ipso) ppm.. (70 mL). After filtration and removal of the solvent, complex 11 was isolated as a brown solid. (3.20 g, 6.34 mmol, 99%). 1H NMR (C6D6, 20 °C): δ ⫽ 0.40 (s, 6 H, SiMe2), 0.42 (s, 6 H, SiMe2), 1.27 (s, 18 H, NtBu), 2.12 (s, 2 H, CH2Ph), 6.14 (d, 2 H, C5H3), 6.53 (t, 1 H, C5H3) 6.9⫺7.23 (m, 5 H, C6H5) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.4 (SiMe2), δ ⫽ 2.4 (SiMe2), 35.8 (NtBu), 55.8 (CH2Ph), 57.1 (NtButert), 116.5 (C5H3ipso), 120.7 (C5H3), 126.4 (C5H3), 128.5 (C6H5), 129.8 (C6H5), 132.7 (C6H5), 150.6 (C6H5ipso) ppm. C24H40N2Si2Zr (503.97): calcd. C 57.20, H 8.00, N 5.56; found C 57.01, H 7.65, N 5.48.. [Ti{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}(NMe2)] (8): When a C6D6 solution of the diamido complex 4 was heated at 160 °C for 12 h in a sealed NMR tube, a slow reaction took place with elimination of NHMe2 to give 8, identified by its 1H and 13C NMR spectra, together with the starting complex 4. When the reaction mixture was cooled to room temperature, the reaction proved to be irreversible. 1H NMR (C6D6, 20 °C): δ ⫽ 0.50 (s, 6 H, SiMe2), 0.52 (s, 6 H, SiMe2), 1.38 (s, 18 H, NtBu), 2.83 (s, 6 H, NMe2), 6.17 (d, 2 H, C5H3), 6.71 (t, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.7 (SiMe2), 2.8 (SiMe2), 35.2 (NtBu), 51.3 (NMe2), 59.3 (NtButert), 117.7 (C5H3ipso), 121.3 (C5H3), 131.9 (C5H3) ppm.. [Ti{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}]ⴙ [(CH2Ph)B(C6F5)3]ⴚ (12): A toluene (20 mL) solution of the monobenzyl complex 10 (0.125 g, 0.25 mmol) was treated at room temperature with B(C6F5)3 (0.126 g, 0.25 mmol), and the mixture was stirred for 30 min and cooled to ⫺35 °C. The solvent was filtered off from the resulting insoluble residue, which was then dried under vacuum to give 12 (0.13 g, 60% yield) as an orange, crystalline solid. 1H NMR (C6D6, 20 °C): δ ⫽ 0.19 (s, 6 H, SiMe2), 0.38 (s, 6 H, SiMe2), 1.12 (s, 18 H, NtBu), 3.49 (s, 2 H, BCH2), 5.03 (d, 2 H, C5H3), 5.86 (t, 1 H, C5H3), 6.21⫺7.10 (m, 5 H, C6H5) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 0.6 (SiMe2), 1.4 (SiMe2), 34.6 (NtBu), 59.3 (NtButert), 122.1 (C5H3ipso) 126.1 (C5H3), 126.2 (C5H3), 128.3 (C6H5), 128.7 (C6H5), 132.9 (C6H5), 135.1 (C6F5), 140.1 (C6F5), 145.8 (C6F5), 150.9 (C6F5) ppm. 19F NMR (300 MHz, C6D6, 20 °C, CCl3F): δ ⫽ 132.1 (m, 2 F, o-C6F5), 163.7 (m, 1 F, p-C6F5), 167.3 (m, 2 F, m-C6F5) ppm. C42H40BF15N2Si2Ti (972.63): calcd. C 51.87, H 4.15, N 2.88; found C 51.98, H 4.04, N 3.08.. [Zr{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}(NMe2)] (9). Method A: When a C6D6 solution of the diamido complex 5 was heated at 120 °C for 12 h in a sealed NMR tube, a slow reaction took place with elimination of NHMe2 to give 9, identified by its 1H and 13C NMR spectra. However, when the reaction mixture was cooled to room temperature, a reversible reaction with the free NHMe2 present in solution once more yielded the starting complex 5. Method B: A 1:1 molar ratio of the benzyl derivative 11 (1.46 g, 3.1 mmol) and the diamido derivative 5 (1.45 g, 3.1 mmol) in toluene was heated at 120 °C in a Teflon-valved Schlenk tube for 12 h. The solvent was removed under vacuum and the residue was extracted into pentane (50 mL). After filtration and removal of the solvent, complex 9 (1.32 g, 2.9 mmol, 94%) was isolated as a yellow solid. 1H NMR (C6D6, 20 °C): δ ⫽ 0.51 (s, 6 H, SiMe2), 0.55 (s, 6 H, SiMe2), 1.31 (s, 18 H, NtBu), 2.77 (s, 6 H, NMe2), 6.34 (d, 2 H, C5H3), 6.78 (t, 1 H, C5H3) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.9 (SiMe2), 3.0 (SiMe2), 35.6 (NtBu), 45.1 (NMe2), 55.8 (NtButert), 117.7 (C5H3ipso) 119.8 (C5H3), 133.2 (C5H3) ppm. C19H39N3Si2Zr (456.93): calcd. C 49.94, H 8.60, N 9.20; found C 50.28, H 8.31, N 8.86. [Ti{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}(CH2Ph)] (10): A solution of Ti(CH2Ph)4 (5.34 g, 12.9 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (4.21 g, 12.9 mmol) was added by syringe. The resulting deep red solution was warmed at 70 °C for 5 h. The solvent was removed under vacuum and the residue was extracted into pentane (70 mL). After filtration and removal of the solvent, complex 10 was isolated as a waxy, red solid, which was recrystallized from hexane to give orange crystals (5.88 g, 12.7 mmol, 98%). 1H NMR (C6D6, 20 °C): δ ⫽ 0.39 (s, 6 H, SiMe2), 0.40 (s, 6 H, SiMe2), 1.42 (s, 18 H, NtBu), 2.59 (s, 2 H, CH2Ph), 6.12 (d, 2 H, C5H3), 6.39 (t, 1 H, C5H3), 6.89 (m, 1 H, C6H5), 6.95 (m, 2 H, C6H5), 7.22 (m, 2 H, C6H5) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 2.1 (SiMe2), 2.2 (SiMe2), 35.6 (NtBu), 59.3 (NtButert), 69.6 (CH2Ph), 117.7 (C5H3ipso), 121.5 (C5H3), 126.3 (C5H3), 128.5 (C6H5), 130.3 (C6H5), 132.6 (C6H5), 152.4 (C6H5ipso) ppm. C24H40N2Si2Ti (460.63): calcd. C 62.58, H 8.75, N 6.08; found C 62.68, H 8.68, N 5.76. [Zr{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}(CH2Ph)] (11): A solution of Zr(CH2Ph)4 (2.93 g, 6.43 mmol) in toluene (70 mL) was cooled to 0 °C, and 1 (2.09 g, 6.43 mmol) was added by syringe. The resulting yellow solution was heated at reflux for 5 h. The solvent was removed under vacuum and the residue was extracted into pentane 2472. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. [Zr{η5-C5H3-1,3-[SiMe2(η1-NtBu)]2}]ⴙ [(CH2Ph)B(C6F5)3]ⴚ (13): A toluene (20 mL) solution of the monobenzyl complex 11 (0.116 g, 0.25 mmol) was treated with B(C6F5)3 (0.126 g, 0.25 mmol) at room temperature and the mixture was stirred for 30 min and cooled to ⫺35 °C. The solvent was filtered off from the resulting insoluble residue, which was then dried under vacuum to give 13 (0.21 g, 83% yield) as a dark brown oil. After recrystallization, a suitable orange monocrystal of 13 was separated for X-ray diffraction studies. 1H NMR (C6D6, 20 °C): δ ⫽ 0.17 (s, 6 H, SiMe2), 0.36 (s, 6 H, SiMe2), 0.99 (s, 18 H, NtBu), 3.42 (s, 2 H, BCH2), 5.20 (d, 2 H, C5H3), 6.01 (t, 1 H, C5H3), 6.10 (m, 1 H, p-C6H5), 6.34 (m, 2 H, m-C6H5), 6.87 (m, 2 H, o-C6H5) ppm. 13C NMR (C6D6, 20 °C): δ ⫽ 1.5 (SiMe2), 1.6 (SiMe2), 34.8 (NtBu), 58.6 (NtButert), 121.4 (C5H3), 123.5 (C5H3ipso), 127.4 (C5H3), 127.9 (C6H5), 128.1 (C6H5), 128.3 (C6H5), 136.5 (C6F5), 138.3 (C6F5), 147.8 (C6F5), 149.8 (C6F5), 162.0 (C6H5ipso) ppm. 19F NMR (300 MHz, C6D6, 20 °C, CCl3F): δ ⫽ 132.1 (m, 2 F, o-C6F5), 163.6 (m, 1 F, p-C6F5), 167.2 (m, 2 F, m-C6F5) ppm. C42H40BF15N2Si2Zr (1015.97): calcd. C 49.65, H 3.97, N 2.76; found C 50.51, H 3.71, N 3.35. Crystal Structure Determination for Compound 10: Crystals of 10 were grown from a hexane solution. An orange fragment in perfluorinated ether was selected and transferred to a glass capillary, which was mounted in a cold N2 stream. Preliminary examination and data collection were carried out with a Nonius KappaCCD device at the window of a rotating anode X-ray generator with use of graphite-monochromated Mo-Kα radiation (λ ⫽ 0.71073 Å), controlled by the Collect software package.[25] Collected images were processed by use of Denzo. The unit cell parameters were obtained by full-matrix, least-squares refinements of 4895 reflections.[26] A total number of 41477 reflections were integrated. After merging, 4739 reflections remained, and these were used for all further calculations. Absorption effects were corrected during the scaling procedure. The structure was solved by direct methods[27] and refined by standard difference Fourier techniques.[28] All nonhydrogen atoms of the asymmetric unit were refined with aniso-. www.eurjic.org. Eur. J. Inorg. Chem. 2003, 2463⫺2474.
(11) Neutral and Cationic [Bis(η1-amidosilyl)-η5-cyclopentadienyl]titanium and -zirconium Complexes tropic thermal displacement parameters. All hydrogen atoms were found in the difference Fourier map and refined freely with individual isotropic thermal displacement parameters, except for those located at a disordered tert-butyl group, which were placed in calculated positions (riding model). Full-matrix, least-squares refinements were carried out by minimization of Σw(Fo2⫺Fc2)2 by use of the SHELXL-97 weighting scheme and stopped at a maximum shift/err ⬍ 0.001.[28,29,30] One tert-butyl group in 10 appears to be disordered over two positions (73:27). Crystal Structure Determination for Compound 13: Crystals of 13 were grown from a C6D6 solution. An orange plate in perfluorinated ether was selected and transferred to a glass capillary. Preliminary examination and data collection were carried out with a Nonius KappaCCD device at the window of a rotating anode Xray generator by use of graphite-monochromated Mo-Kα radiation (λ ⫽ 0.71073 Å), controlled by the Collect software package.[26] Collected images were processed by use of Denzo. The unit cell parameters were obtained by full-matrix, least-squares refinements of 8080 reflections.[27] A total number of 44162 reflections were integrated. After merging, 8099 reflections remained and were used for all further calculations. Absorption effects were corrected during the scaling procedure. The structure was solved by direct methods[27] and refined by standard difference Fourier techniques.[28] All non-hydrogen atoms of the asymmetric unit were refined with anisotropic thermal displacement parameters. All hydrogen atoms were found in the difference Fourier map and refined freely with individual isotropic thermal displacement parameters. Full-matrix, least-squares refinements were carried out by minimization of Σw(Fo2⫺Fc2)2 by use of the SHELXL-97 weighting scheme and stopped at a maximum shift/err ⬍ 0.001. A correction for extinction effects was applied.[28⫺30] CCDC-200139 (10) and -200140 (13) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (internat.) ⫹ 44-1223/336033; E-mail: [email protected]]. Computational Details: Gradient corrected density functional calculations were carried out, with corrections for exchange and correlation according to Becke[31] and Perdew,[32] respectively (BP86). Geometries were optimized by use of the TURBOMOLE[33] program system within the framework of the RI-J approximation.[34] For the model systems, a triple-ζ valence basis plus polarization TZVP[35] was employed. For the geometry optimization of the real molecules, the main group elements were described by a split-valence basis set[36] with one set of polarization functions for the nonH atoms, and Ti was treated with the TZVP basis. For the determination of bond energies, final energies were obtained in a singlepoint calculation using the TZVP basis set for all atoms.. Table 4. Crystallographic data for complexes 10 and 13. Empirical formula Formula mass Colour/shape Crystal size [mm] Crystal system Space group a [Å] b [Å] c [Å] α [°] β [°] γ [°] V [Å3] Z T [K] ρcalcd. [g cm⫺3] µ [mm⫺1] F000 θ range [°] Data collected (h,k,l) No. of reflns. collected No. of independent reflns./Rint No. of obsd. reflns. (I ⬎ 2σ(I)] No. of params. refined R1 (obsd./all) wR2 (obsd./all) GOF (obsd./all) max./min. ∆ρ [e·Å⫺3]. [1] [1a]. P. C. Möhring, N. J. Coville, J. Organomet. Chem. 1994, 479, 1⫺29. [1b] H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger, R. M. Waymouth, Angew. Chem. Int. Ed. Engl. 1995, 34, 1143⫺1170. [1c] G. G. Hlatky, Coord. Chem. Rev. 1999, 181, 243⫺296. [1d] H. G. Alt, A. Köppl, Chem. Rev. 2000, 100, 1205⫺1222. [1e] L. Resconi, L. Cavallo, A. Fait, F. Piemontesi, Chem. Rev. 2000, 100, 1253⫺1345.. Eur. J. Inorg. Chem. 2003, 2463⫺2474. www.eurjic.org. 10. 13. C24H40N2Si2Ti 460.63 orange/fragment 0.36 ⫻ 0.23 ⫻ 0.20 monoclinic P21/c (No. 14) 10.6962(1) 16.0412(2) 15.3339(2) 90 99.6928(7) 90 2593.43(5) 4 123 1.180 0.435 992 1.85⫺25.34 ⫾12, ⫾19, ⫾18 41477 4739 (all)/0.033. C42H40BF15N2Si2Zr 1015.97 orange/plate 0.56 ⫻ 0.30 ⫻ 0.10 triclinic P1̄ (No. 2) 11.7349(1) 13.4253(1) 14.4024(1) 77.4374(3) 88.2620(4) 86.9316(5) 2211.12(3) 2 293 1.526 0.399 1028 1.45⫺25.38 ⫾14, ⫾16, ⫾17 44162 8099 (all)/0.033. 4193 (obsd.). 7177 (obsd.). 414 0.0333/0.0394 0.0855/0.0895 1.045/1.045 ⫹0.32/⫺0.27. 729 0.0306/0.0370 0.0752/0.0788 1.040/1.040 ⫹0.30/⫺0.27. [2] [2a]. [3]. Acknowledgments The authors acknowledge the MCyT (project MAT2001-1309) for financial support and the EC (project COST-D12/0016/98). J. C. acknowledges CAM for a fellowship.. FULL PAPER. [4]. P. J. Shapiro, E. E. Bunel, W. E. Piers, J. E. Bercaw, Synlett 1990, 2, 74⫺84. [2b] P. J. Shapiro, E. Bunel, W. P. Schäfer, J. E. Bercaw, Organometallics 1990, 9, 867⫺869. [2c] P. J. Shapiro, W. E. Cotter, W. P. Schäfer, J. A. Labinger, J. E. Bercaw, J. Am. Chem. Soc. 1994, 116, 4623⫺4640. [2d] J. Okuda, Chem. Ber. 1990, 123, 1649⫺1651. [2e] A. K. Hughes, A. Meetsma, J. H. Teuben, Organometallics 1993, 12, 1936⫺1945. [2f] W. A. Herrmann, M. J. A. Morawietz, J. Organomet. Chem. 1994, 482, 169⫺181. [2g] S. Ciruelos, T. Cuenca, R. Gómez, P. Gómez-Sal, A. Manzanero, P. Royo, Organometallics 1996, 15, 5577⫺5585. [2h] R. Gómez, P. Gómez-Sal, A. Martı́n, A. Núñez, P. A. del Real, P. Royo, J. Organomet. Chem. 1998, 564, 93⫺100. [2i] A. L. Mcknight, R. M. Waymouth, Chem. Rev. 1998, 98, 2587⫺2598. [2j] J. Klosin, W. J. Kruper, Jr., P. N. Nickias, G. R. Roof, P. DeWaele, K. A. Abboud, Organometallics 2001, 20, 2663⫺2665. [2k] K. Kunz, G. Erker, S. Döring, R. Fröhlich, G. Kehr, J. Am. Chem. Soc. 2001, 123, 6181⫺6182. [3a] M. Bochmann, J. Chem. Soc., Dalton Trans. 1996, 255⫺270. [3b] C. P. Casey, D. W. Carpenetti II, H. Sakurai, Organometallics 2001, 20, 4262⫺4265. [3c] C. G. Brandow, A. Mendiratta, J. E. Bercaw, Organometallics 2001, 20, 42534261. [4a] Y.-X. Chen, P.-F. Fu, C. L. Stern, T. J. Marks, Organometallics 1997, 16, 5958⫺5963. [4b] B. E. Bosch, G. Erker, R. Fröhlich, O. Meyer, Organometallics 1997, 16, 5449⫺5456. [4c] Y.-X. Chen, T. J. Marks, Organometallics 1997, 16, 3649⫺3657. [4d] A. Bertuleit, C. Fritze, G. Erker, R. Fröhlich, Organometallics 1997, 16, 2891⫺2899. [4e] G. Lanza, I. L. Fragalà, T. J. Marks, J. Am. Chem. Soc. 1998, 120, 8257⫺8258. [4f] F. Amor, A. Butt, K. E. Du Plooy, T. B. Spaniol, J. Okuda, Organometallics 1998, 17, 5836⫺5849. [4g] R. Gómez, P. Gómez-Sal, P. A. del Real, P. Royo, J. Organomet. Chem. 1999, 588, 22⫺27. [4h] R. J. Keaton, K. C. Jayaratne, J. C. Fettinger, L. R. Sita, J. Am. Chem. Soc. 2000, 122, 12909⫺12910. [4i] J.-F. Carpentier, V. P. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2473.
(12) FULL PAPER [5]. [6] [7]. [8]. [9] [10]. [11]. [12]. [13]. [14]. [15]. [16]. [17]. [18]. [19]. [20]. Maryin, J. Luci, R. F. Jordan, J. Am. Chem. Soc. 2001, 123, 898⫺909. [5a] J. C. Stevens, F. J. Timmers, D. R. Wilson, G. F. Schmidt, P. N. Nickias, R. K. Rosen, G. W. Knight, S. Y. Lai, Eur. Patent Appl. EP 1991, 416815 (Dow Chemical Co.) (Chem. Abstr. 1991, 115, 93163). [5b] J. M. Canich, Eur. Patent Appl. EP 1991, 420436 (Exxon Chemical Co) (Chem. Abstr. 1991, 115, 184145). V. C. Gibson, J. Chem. Soc., Dalton Trans. 1994, 1607⫺1618. [7a] J. Cano, P. Royo, A. Tiripicchio, M. A. Pellinghelli, M. Lanfranchi, Angew. Chem. Int. Ed. 2001, 40, 2495⫺2497. [7b] P. Royo, J. Cano, M. A. Flores, B. Peña, EP 2001, 1225179 A1; US 2002, 0115560 A1. [8a] B. E. Bursten, L. F. Rhodes, J. Strittmatter, J. Am. Chem. Soc. 1989, 111, 2758⫺2766. [8b] T. Brackemeyer, G. Erker, R. Fröhlich, Organometallics 1997, 16, 531⫺536. [8c] H. Jacobsen, H. Berke, S. Doring, G. Kehr, G. Erker, R. Fröhlich, O. Meyer, Organometallics 1999, 18, 1724⫺1735. [8d] N. Kliegrewe, T. Brakemeyer, R. Fröhlich, G. Erker, Organometallics 2001, 20, 1952⫺1955. J. M. Rozell, P. R. Jones, Organometallics 1985, 4, 2206⫺2210. [10a] Y. A. Ustynyuk, A. V. Kisin, J. M. Pribytkova, A. A. Zarkin, N. D. Antonova, J. Organomet. Chem. 1972, 42, 47⫺63. [10b] P. Jutzi, Chem. Rev. 1986, 86, 983⫺996. G. M. Diamond, R. F. Jordan, J. L. Petersen, J. Am. Chem. Soc. 1996, 118, 8024⫺8033. U. Zucchini, E. Albizzati, U. Giannini, J. Organomet. Chem. 1971, 26, 357⫺372. Y. Mu, W. E. Piers, L. R. MacQuarrie, M. Zaworotko, J. Young, Organometallics 1996, 15, 2720⫺2726. D. M. P. Mingos, Essential Trends in Inorganic Chemistry, Oxford Univ. Press, Oxford, 1998, p. 368. [15a] W. A. Nugent, B. L. Haymore, Coord. Chem. Rev. 1980, 31, 123⫺175. [15b] D. E. Wigley, Prog. Inorg. Chem. 1994, 42, 239⫺482. [15c] M. J. Humphries, M. L. H. Green, M. A. Leech, V. C. Gibson, M. Jolly, D. N. Williams, M. R. J. Elsegood, W. Clegg, J. Chem. Soc., Dalton Trans. 2000, 4044⫺4051. J. Okuda, T. Eberle, T. P. Spaniol, Chem. Ber./Recueil 1997, 130, 209⫺215. J. Okuda, S. Verch, R. Sturmer, T. P. Spaniol, J. Organomet. Chem. 2000, 605, 55⫺67. A. Martı́n, M. Mena, C. Yelamos, R. Serrano, P. R. Raithby, J. Organomet. Chem. 1994, 467, 79⫺84. D. W. Carpenetti, L. Kloppenburg, J. T. Kupec, J. L. Petersen, Organometallics 1996, 15, 1572⫺1581. W. S. Seo, Y. J. Cho, S. C. Yoon, J. T. Park, Y. Park, J. Organomet. Chem. 2001, 640, 79⫺84.. 2474. 2003 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. J. Cano, P. Royo, H. Jacobsen, O. Blacque, H. Berke, E. Herdtweck [21]. [22]. [23]. [24]. [25]. [26]. [27]. [28]. [29]. [30] [31] [32] [33]. [34]. [35]. [36]. Y. Mu, W. E. Piers, L. R. MacQuarrie, M. Zaworotko, Can. J. Chem. 1996, 74, 1696⫺1703. J. Okuda, F. J. Schattenmann, S. Wocadlo, W. Massa, Organometallics 1995, 14, 789⫺795. [23a] K. Shelly, D. C. Finster, Y. J. Lee, W. R. Scheidt, C. A. Reed, J. Am. Chem. Soc. 1985, 107, 5955⫺5959. [23b] M. Laguna, M. D. Villacampa, M. Contel, J. Garrido, Inorg. Chem. 1998, 37, 133⫺135. [23c] A. S. Batsanov, S. P. Crabtree, J. A. K. Howard, C. W. Lehmann, M. Kilner, J. Organomet. Chem. 1998, 550, 59⫺61. A. G. Massey, A. J. Park, J. Organomet. Chem. 1964, 2, 245⫺250. COLLECT, Data Collection Software for Nonius Kappa CCD Devices, Hooft, R.Nonius B. V., Delft, The Netherlands, 1998. Z. Otwinowski, W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode, Methods in Enzymology, vol. 276: Macromolecular Crystallography, part A (Eds.: C. W. Carter, Jr., R. M. Sweet), Academic Press, New York, 1997, p. 307⫺326. A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori, M. Camalli, J. Appl. Crystallogr. 1994, 27, 435⫺436. G. M. Sheldrick, SHELXL-97, Universität Göttingen, Germany 1998. International Tables for Crystallography, vol. C, Tables 6.1.1.4 (pp. 500⫺502), 4.2.6.8 (pp. 219⫺222), and 4.2.4.2 (pp. 193⫺199) (Ed.: A. J. C. Wilson), Kluwer Academic Publishers, Dordrecht, 1992. A. L. Spek, Acta Crystallogr., Sect. A 1990, 46, C34. A. D. Becke, Phys. Rev. 1988, A38, 3098⫺3100. J. P. Perdew, Phys. Rev. 1986, B33, 8822⫺8824. [33a] R. Ahlrichs, M. Bär, M. Häser, H. Horn, C. Kölmel, Chem. Phys. Lett. 1989, 162, 165⫺169. [33b] O. Treutler, R. Ahlrichs, J. Chem. Phys. 1995, 102, 346⫺354. [33c] R. Ahlrichs, M. von Arnim, Methods and Techniques in Computational Chemistry: METECC-95 (Eds.: E. Clementi, G. Corongiu), STEF, Cagliari, 1995, chapter 13.[33d]R. Ahlrichs, M. von Arnim, J. Comput. Chem. 1998, 19, 1746⫺1757. [34a] K. Eichkorn, O. Treutler, H. Öhm, M. Häser, R. Ahlrichs, Chem. Phys. Lett. 1995, 242, 652⫺660. [34b] K. Eichkorn, F. Weigand, O. Treutler, R. Ahlrichs, Theor. Chem. Acc. 1997, 97, 119⫺124. A. Schäfer, C. Huber, R. Ahlrichs, J. Chem. Phys. 1994, 100, 5829⫺5835. A. Schäfer, H. Horn, R. Ahlrichs, J. Chem. Phys. 1992, 97, 2571⫺2577. Received December 20, 2002. www.eurjic.org. Eur. J. Inorg. Chem. 2003, 2463⫺2474.
(13)
Figure
![Table 2. Selected bond lengths [A ˚ ] and bond angles [°] for related complexes](https://thumb-us.123doks.com/thumbv2/123dok_es/7327073.357173/5.892.488.786.320.425/table-selected-bond-lengths-bond-angles-related-complexes.webp)
![Figure 3. Optimized geometry for the model cation [Ti{C 5 H 3 (SiH 2 NH) 2 }] ⫹ and a main empty frontier MO](https://thumb-us.123doks.com/thumbv2/123dok_es/7327073.357173/7.892.83.426.155.312/figure-optimized-geometry-model-cation-sih-main-frontier.webp)
+3
![Figure 4. Relative energies and selected bond lengths for meta (a) and para coordination (b) of [Ti{η 5 -C 5 H 3 (SiMe 2 NtBu) 2 }{η 1 - -C 6 H 5 CH 2 B(C 6 F 5 ) 3 }]; comparison with X-ray analysis](https://thumb-us.123doks.com/thumbv2/123dok_es/7327073.357173/8.892.193.693.102.303/figure-relative-energies-selected-lengths-coordination-comparison-analysis.webp)
Documento similar
Basic properties include monotonicity, so dimension 1 classes are maximal and dimension 0 ones are minimal, and the fact that dimension is defined for every class X, making
Unemployment duration longer than 6 months reduces first birth hazards by about 20 percent for the primary and secondary education groups, while its effect is
MD simulations in this and previous work has allowed us to propose a relation between the nature of the interactions at the interface and the observed properties of nanofluids:
The Global Compact for Safe, Orderly and Regular Migration (Global Compact for Migration hereinafter) has been considered the first instrument to comprehensively address all
The expansionary monetary policy measures have had a negative impact on net interest margins both via the reduction in interest rates and –less powerfully- the flattening of the
• It is imperative that conciliation should no longer exclusively focus on women and it should become definitely detached from those aspects related to the
The concept of breakdown probability profile is useful to describe the break- down properties of any estimate obtained through resampling, not just the bagged median or the
Relative energies are given in eV with respect to the final product in each mechanism: (a) neutral 71 and cationic 71 + fragments producing cAA + ; (b) neutral 71 and cationic 71
