Tema 2
TÉCNICAS DE SEPARACIÓN CROMATOGRÁFICA
1. Introducción
La etimología de la palabra cromatografía es curiosa. Por las palabras griegas que la forman significaría: “escritura en colores”.
El botánico ruso Mikhail S. Tsvet formalizó el uso de la cromatografía en los estudios científicos -por el año 1910-, dio ese nombre a la técnica y la aplicó a la separación de los pigmentos naturales que se encuentran en las plantas (conocidos como carotenoides y clorofilas).
Tsvet empacó material adsorbente en una columna de vidrio vertical (de unos cuantos centímetros de diámetro). Posteriormente, por dicha columna vertical vertió una disolución que contenía la mezcla de pigmentos provenientes de las hojas molidas de una planta. Pasados unos minutos, el material empacado en la columna había adquirido una coloración diferente por segmentos. Es decir, se había logrado la separación de los pigmentos naturales de la planta. En cada segmento de color definido había un pigmento diferente.
Como su nombre indica las técnicas de separación sirven para separar componentes de una muestra.
Desde el punto de vista de la caracterización de los materiales, conocer la composición de los mismos es imprescindible puesto que las propiedades de los materiales dependen fundamentalmente, por un lado, de los componentes que lo forman y, por otro, de la proporción existente entre ellos.
Otra de las aplicaciones importantes en cromatografía es realizar el seguimiento de una síntesis que podría ir asociada a la preparación de un material (una polimerización por ejemplo). A medida que se produce la reacción en el matraz de reacción tendremos productos y reactivos, cuya proporción irá variando en función de la conversión.
Existen al menos tres puntos importantes de incidencia de la cromatografía en la Ciencia y Tecnología de Materiales:
i) Por la aplicación como técnica de separación y caracterización de compuestos relacionados con la síntesis de materiales. Así, muchos sólidos se preparan a partir de precursores moleculares organometálicos o de complejos de coordinación, y éstos a su vez a partir de compuestos orgánicos. De aquí la necesidad de controlar el grado de pureza de los compuestos de partida, de efectuar el seguimiento de las reacciones de síntesis en que intervienen los mencionados compuestos y de evaluar rendimientos y detectar procesos secundarios mediante aplicación cromatográfica. Además, el empleo de columnas convencionales (columnas de gravedad) sigue todavía teniendo interés preparativo para el aislamiento y purificación de reactivos, con potencial interés en la obtención de materiales sólidos.
ii) Por la necesidad analítica de identificación y cuantificación de compuestos transformados por la acción de materiales sobre sustancias orgánicas. Las técnicas cromatográficas son imprescindibles para una correcta evaluación de la actividad de materiales catalizadores y para el estudio de soportes sólidos de reacciones químicas de gran importancia en petroquímica y en la elaboración de compuestos orgánicos de alto valor añadido (química fina). Pero las técnicas cromatográficas no sólo tienen aplicación en catálisis convencional donde intervienen transformaciones de compuestos orgánicos; podemos también citar la separación e identificación de gases simples como el hidrógeno y el oxígeno producidos por la acción de catalizadores que actúan en la fotodescomposición del agua.
iii) Por el desarrollo de nuevas fases estacionarias para cromatografía (rellenos de columnas cromatográficas). La extensión cada vez mayor de las técnicas de análisis cromatográfico llevan a su vez a una demanda de materiales utilizables como fases estacionarias que permitan, ya sea resolver algunas deficiencias de las fases utilizadas tradicionalmente, tales como la existencia de interacciones secundarias no deseadas, o bien realizar de forma específica análisis que de otra forma resultarían muy complejos por la gran cantidad de compuestos interferentes en las mezclas a utilizar o por la enormemente compleja preparación de la muestra que es necesaria en algunos casos.
2.
Lugar que ocupa la cromatografía en la caracterización de materialesEsquema 1.- Pasos a seguir en cuanto a la manipulación de una muestra hasta su caracterización.
3. La cromatografía. Conceptos y definiciones
a) Cromatografía
La cromatografía es un método físico de separación en el que los componentes a separar se distribuyen entre dos fases, una de las cuales está en reposo (fase estacionaria o lecho estacionario) mientras que la otra (fase móvil) se mueve en una dirección definida. El proceso cromatográfico se da como resultado de la mayor o menor capacidad para ser retenidos los diferentes componentes de una muestra por la fase estacionaria, es decir, se basa en los repetidos procesos de interacción durante el movimiento de los componentes de una mezcla arrastrados por la fase móvil a lo largo de la fase estacionaria (elución), produciéndose la separación debido a las diferencias en las constantes de distribución de los componentes de la mezcla entre la fase estacionaria y la móvil.
b) Fase estacionaria
Es una de las dos fases que forman un sistema cromatográfico. Puede ser un sólido, un
M Muueessttrraa
¿Hay que solubilizarla?
¿Es volátil? ¿Descompone térmicamente?
¿Forma plasma?
Otros métodos
¿Es soluble? Digestión
GAS DISOLUCIÓN
¿Hay que separar
componentes? Cromatografía
¿Hay que separar componentes?
T
TééccnniiccaassyyMMééttooddoossddeeaannáálliissiiss NO
NO NO
NO
NO NO
SI
SI SI SI
SI
SI SI
SI SI
desarrollo inicial de la cromatografía líquida se usó el término "fase líquida" para referirse a la fase móvil, frente a la "fase sólida", es decir, la estacionaria. Debido a esta ambigüedad, se desaconseja utilizar el término "fase liquida". Si se debe expresar el estado físico de la fase estacionaria, se propone el uso de adjetivos, tales como fase estacionaria líquida, fase estacionaria sólida, fase ligada sólida o fase inmovilizada sólida.
c) Fase ligada
Una fase estacionaria que está unida de forma covalente a las partículas de soporte o a la pared interior de la columna (lugar donde se produce la separación).
d) Fase inmovilizada
Una fase estacionaria que está inmovilizada sobre las partículas del soporte o sobre la pared interior de la columna, por ejemplo por polimerización in situ (entrecruzamiento químico) tras un recubrimiento.
e) Fase móvil
Fluido que se filtra a través o a lo largo del lecho estacionario, en una dirección definida. Puede ser un líquido (cromatografía cíquida, LC), un gas (cromatografía de gases, GC) o un fluido supercrítico (cromatografía con fluido supercrítico). En la cromatografía de gases se puede usar la expresión gas portador para la fase móvil. En la cromatografía de elución se usa también para la fase móvil la expresión eluyente.
f) Eluir
Proceso en el que la fase móvil se desplaza a lo largo de la fase estacionaria transportando los componentes a separar. El proceso de elución se puede detener mientras todos los componentes de la muestra están aún en el lecho cromatográfico, o continuarse hasta que lo hayan abandonado (se prefiere el término "Eluir" a "Desarrollar", término usado en nomenclaturas anteriores de cromatografía plana).
g) Efluente
La fase móvil que abandona la columna.
h) Muestra
Mezcla consistente en cierto número de componentes, cuya separación se pretende en el lecho cromatográfico al ser arrastrados o eluidos por la fase móvil.
i) Componentes de la Muestra
j) Soluto
Término que se refiere a los componentes de la muestra en la cromatografía de reparto.
k) Disolvente
Término que en ocasiones se refiere a la fase estacionaria líquida en la cromatografía de reparto.
Nota: En la cromatografía líquida, el término "disolvente" se ha usado a menudo para la fase móvil. Este uso no se recomienda.
l) Zona
Región del lecho cromatográfico donde se localizan uno o más componentes de la muestra. Se puede usar para ello también el término banda.
m) Cromatograma
Una gráfica u otro tipo de presentación de la respuesta de un detector (la concentración de un analito en el efluente u otra magnitud usada como medida de la concentración en el efluente) frente al volumen de efluente o al tiempo. En la cromatografía plana, "cromatograma" puede referirse al papel o capa con las zonas separadas.
n) Cromatografiar
Separar por cromatografía.
o) Cromatógrafo
El instrumento empleado para realizar una separación cromatográfica.
3.1.
Mecanismos de separación
3.1.1. Cromatografía de Adsorción
La separación se basa principalmente en la diferente afinidad en términos de adsorción de los componentes de la muestra sobre la superficie de un sólido activo.
3.1.2. Cromatografía de Reparto
La separación se basa principalmente en la diferente solubilidad de los componentes de la muestra en la fase estacionaria (caso de la cromatogafía de gases), o en las diferentes solubilidades de los componentes en las fases móvil y estacionaria (caso de la cromatografía líquida).
La mayor o menor migración de un componente en este tipo de cromatografía será función del coeficiente de reparto de éste entre la fase estacionaria y la fase móvil:
ia estacionar fase en A de ión Concentrac móvil fase en A de ión Concentrac Kd ====
La cromatografía de reparto se utiliza para la separación de mezclas de compuestos de polaridad media y alta. Algunos ejemplos de este tipo de cromatografía son las cromatografías sobre papel, sílice hidratada y fase inversa en la cromatografía líquida de alta eficacia.
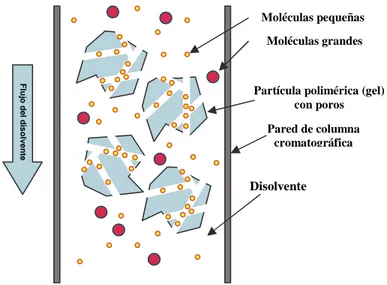
3.1.3. Cromatografía de Exclusión
La separación se basa principalmente en efectos de exclusión, tales como las diferencias de tamaño molecular o de forma, o las diferencias de carga (Figura 2.1). Se puede usar también el término cromatografía de exclusión por tamaños, SEC, cuando la separación se basa en el tamaño molecular. Anteriormente se usaban para esto los términos filtración en gel y cromatografía de permeación en gel (GPC), cuando la fase estacionaria es un gel hinchado. El término cromatografía de exclusión de iones se usa específicamente para la separación de iones en una fase acuosa.
La cromatografía de filtración en gel se realiza empleando unas matrices formadas por unas esferas porosas. El volumen de los poros es muy elevado y su diámetro se conoce. Cuando penetran en el lecho de la columna dos macromoléculas de tamaños tales que una penetra en los poros de las bolas de gel y la otra no, la primera se reparte entre el espacio entre las bolas y el interior de los poros, reduciéndose la concentración en la fase libre entre las bolas. La segunda macromolécula, por tamaño, sólo puede encontrarse entre las bolas. El flujo de disolvente es más elevado entre las bolas que en el interior de los poros de éstas, por lo que el efecto neto es el de acelerar el desplazamiento de las macromoléculas de mayor peso molecular respecto al de las de menor peso molecular. En esta cromatografía se eluyen primero las macromoléculas mayores, y en segundo lugar las menores, y tiene una gran influencia en el resultado la longitud de la columna y el volumen de la misma. Columnas largas aseguran separaciones de mayor calidad. Empleando patrones de macromoléculas de masa molecular conocida se pueden construir curvas de calibrado que posteriormente permitan la determinación de la masa molecular de macromoléculas de tamaño desconocido.
fases es su límite de exclusión que se define como la masa molecular a partir de la cual los compuestos pasarán a través del lecho estacionario sin experimentar retención.
Figura 2.1.- Esquema del proceso de separación por cromatografía de exclusión por tamaños.
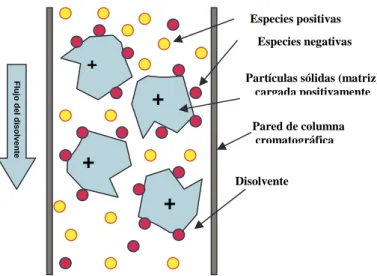
3.1.4. Cromatografía de Intercambio Iónico
La separación se basa principalmente en la diferente afinidad para el intercambio de iones de los componentes de la muestra (Figura 2.2). Actualmente, cuando se lleva a cabo sobre columnas de partículas pequeñas y elevada eficiencia, y empleando habitualmente detectores conductimétricos o espectroscópicos, se denomina cromatografía de iones (IC).
La cromatografía de intercambio iónico se realiza sobre matrices que tienen una carga neta (positiva en el esquema de la Figura2.2). La carga de la matriz de la columna así como la carga de las moléculas dependerá del pH del disolvente y de su fuerza iónica (proporcional a la concentración de iones). En unas condiciones determinadas serán retenidas en la columna las moléculas o especies que tengan una carga complementaria a la de la matriz del gel (las moléculas cargadas negativamente serán retenidas por una matriz cargada positivamente), siendo eluidas las restantes. Para eluir las moléculas retenidas se puede variar la carga iónica del disolvente o su pH de forma que se alcance el punto isoeléctrico de la moléculas o especies de interés o el de la matriz, neutralizando de este modo la fuerza que retiene a las moléculas en la columna.
Partícula polimérica (gel) con poros Moléculas grandes Moléculas pequeñas
Pared de columna cromatográfica
Disolvente
F
lu
jo
d
e
l d
is
o
lv
e
n
Figura 2.2. Esquema del proceso de separación por cromatografía de intercambio iónico.
3.1.5. Cromatografía de Afinidad
Esta expresión caracteriza una variedad particular de cromatografía en la que se utiliza para la separación la especificidad biológica singular respecto a la interacción entre analito y ligando.
3.2.
Métodos principales de análisis cromatográfico
3.2.1. Análisis por desarrollo
Es el método utilizado en los experimentos iniciales de cromatografía. El cromatograma se desarrolla hasta que el frente del disolvente alcanza el final del lecho estacionario, de forma que los constituyentes de la mezcla una vez separados permanecen sobre el lecho al finalizar la separación. La técnica de análisis por desarrollo presenta la ventaja de que es analizable la totalidad de la muestra, incluyendo los compuestos de muy baja o muy alta retención. Se utiliza fundamentalmente en cromatografía de papel y capa fina (Figura 2.3).
Figura 2.3.- Análisis por desarrollo. Esquema de sistema para hacer cromatografía en capa fina. F a se M ó v il Cromatografía
Pared de columna cromatográfica Partículas sólidas (matriz)
-3.2.2. Análisis por elución (Cromatografía de elución)
Procedimiento en el que la fase móvil pasa de forma continua a través o a lo largo del lecho cromatográfico y la muestra se suministra al sistema de forma discreta, como una pequeña cantidad en un tiempo breve. Esta técnica presenta la desventaja de que los componentes con retenciones muy altas pueden no ser observados.
Figura 2.4.- Esquema de dispositivo para realizar cromatografía con análisis por elución.
Es la técnica más utilizada en casi todas las separaciones cromatográficas analíticas y en muchas preparativas.
3.2.3. Análisis frontal (cromatografía frontal)
Procedimiento en el que la muestra (líquida o gasesosa) se alimenta de forma continua al lecho cromatográfico. No se utiliza ninguna fase móvil adicional. Se utiliza la diferencia de afinidad del adsorbente por cada una de las sustancias a separar. Se utiliza una pequeña columna, que se satura sucesivamente por cada una de las sustancias a separar, emergiendo de ella el primer componente puro hasta que la columna se satura del segundo componente, momento en el que empezará a emerger éste mezclado con el primero. El análisis frontal tiene su principal aplicación como técnica preparativa en la purificación de sustancias.
Volumen de fase móvil
Cromatograma
Muestra Fase Móvil
Sorbente
Placa Porosa
Figura 2.5.-
3.2.4. Análisis por Desplazamiento (Cromatografía por desplazamiento)
Procedimiento en el cual la fase móvil contiene un compuesto (el desplazante) que es retenido más fuertemente que el resto de los componentes de la muestra analizada. La muestra se alimenta al sistema en forma discreta, como una pequeña cantidad en un intervalo breve. Este método se utiliza tanto para separaciones analíticas como preparativas, siendo muy utilizada en cromatografía de cambio iónico.
3.3.
Clasificación de acuerdo a la forma del lecho cromatográfico
3.3.1. Cromatografía en columna
Técnica de separación en la que el lecho estacionario está contenido dentro de un tubo. Las partículas de fase estacionaria sólida, o de soporte recubierto con una fase estacionaria líquida, pueden llenar por completo el tubo (columna empaquetada) o estar concentradas sobre o a lo largo de su pared interna, dejando una ruta abierta, no restringida, para el paso de la fase móvil por el centro del tubo (columna tubular abierta).
3.3.2. Cromatografía Plana
Técnica de separación en la que la fase estacionaria está en forma de plano o sobre un plano. Éste puede ser un papel, que sirva como tal o que esté impregnado con una sustancia que actúe de fase estacionaria (cromatografía en papel), o una capa de partículas sólidas extendida sobre un soporte, tal como una placa de vidrio (cromatografía en capa fina, TLC). A veces a la cromatografía plana se la llama también cromatografía de lecho abierto.
3.4.
Clasificación de acuerdo al estado físico de la fase móvil
Fase Móvil Con la muestra A+B+C
A
Placa Porosa
Efluente A+B
3.4.1. Técnicas cromatográficas
Las técnicas cromatográficas se clasifican a menudo especificando el estado físico de las dos fases empleadas. En consecuencia, se utilizan los siguientes términos:
• Cromatografía gas-líquido (GLC) • Cromatografía gas-sólido (GSC) • Cromatografía líquido-líquido (LLC) • Cromatografía líquido-sólido (LSC)
Se puede encontrar también en la bibliografía el término cromatografía de reparto gas-líquido (GLPC). Sin embargo, a menudo la distinción entre estos modos no es sencilla. Por ejemplo, en cromatografía de gases se puede usar un líquido para modificar una fase estacionaria sólida de tipo adsorbente.
3.4.2. Cromatografía de Gases (GC)
Técnica de separación en la que la fase móvil es un gas. Se lleva siempre a cabo en columna.
3.4.3. Cromatografía Líquida (LC)
Técnica de separación en la que la fase móvil es un líquido. Puede desarrollarse en una columna o sobre un plano. La cromatografía líquida en la actualidad emplea generalmente partículas muy pequeñas y una presión de entrada relativamente alta, denominándose entonces cromatografía líquida de alta eficacia o de alta presión, cuyas siglas provenientes del inglés son HPLC.
3.4.4. Cromatografía con fluido supercrítico (SFC)
Técnica de separación en la que la fase móvil es un fluido por encima y relativamente cerca de sus temperatura y presión críticas. En general, los términos y definiciones usados en la cromatografía de gases y líquida son aplicables igualmente a la de fluido supercrítico.
3.5.
Técnicas especiales
3.5.1. Cromatografía con fase invertida
Procedimiento de elución empleado en cromatografía líquida en el cual la fase móvil es significativamente más polar que la estacionaria; por ejemplo, un material microporoso de base silícea con cadenas alquilo unidas químicamente.
Procedimiento en el que la composición de la fase móvil permanece constante a lo largo del proceso de elución.
3.5.4. Elución con Gradiente
Procedimiento en el que la composición de la fase móvil cambia, de forma continua o en etapas, a lo largo del proceso de elución.
3.5.5. Elución en Etapas o Escalonada
Proceso de elución en el que se cambia la composición de la fase móvil en etapas o escalones durante un sólo desarrollo de la cromatografía.
3.5.6. Cromatografía Bidimensional
Procedimiento en el que parte o todos los componentes de la muestra se someten, una vez separados, a etapas adicionales de separación. Esto se puede hacer, por ejemplo, conduciendo a una fracción en particular que eluye de la columna hacia otra columna (o sistema) que tenga diferentes características de separación. Cuando se combina con etapas de separación adicionales, se puede hablar de cromatografía multidimensional. En la cromatografía plana, cromatografía bidimensional se refiere al proceso cromatográfico que hace que los componentes migren primero en una dirección y luego en otra perpendicular a ella; las dos eluciones se llevan a cabo con eluyentes diferentes.
3.6.
Parámetros básicos de cromatografía
3.6.1. Retención y factor de capacidad
Cuando se introduce un compuesto en la corriente de la fase móvil de un sistema cromatográfico, de no existir interacción entre éste y la fase estacionaria, la banda que forma se desplazaría a la misma velocidad que la fase móvil y emergería de la fase estacionaria cuando el volumen total de la fase móvil utilizado fuese igual al volumen vacío o intersticial del lecho estacionario (volumen muerto). Sin embargo, como las moléculas de la muestra interaccionan con la fase estacionaria, necesitarán para su elución un volumen de fase móvil mayor que se denomina volumen de retención de la muestra. Dado que en un sitema cromatográfico que opere a caudal constante el volumen eluido y el tiempo de elución son proporcionales, los conceptos de volumen muerto y volumen de retención son directamente intercambiables por los de tiempo muerto (tM) y tiempo de retención (tR).
Según lo anterior se puede decir que la “retención” es la capacidad real de la fase estacionaria para retardar la salida de un componente. El tiempo muerto, tM, es el
tiempo que tarda en salir un componente que no se retiene (o el propio eluyente). El tiempo de retención, tR, es el tiempo medio que tarda en salir un determinado
componente.
En la Figura 2.6 se muestran los parámetros de retención para el caso de una única banda cromatográfica. Se observa que el tiempo de retención de la banda puede dividirse en dos partes: i) el tiempo muerto, tM; ii) y el tiempo transcurrido a partir de
Figura 2.6. Cromatograma de un componente y parámetros más característicos (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
.
A partir de la definición de tiempo muerto se induce que el segundo, tR’, es el tiempo
real que la fase estacionaria ha retrasado el avance del compuesto y se conoce con el nombre de tiempo de retención corregido. Se define el factor de capacidad, K’, como:
M M R
M R
t
t
t
t
t
K
'
=
'
=
−
(2.1)Este factor adimensional expresa la retención de un compuesto por la fase estacionaria independientemente del caudal de la fase móvil. En la práctica, los compuestos de interés deben presentar valores de K’ comprendidos entre 1 y 15 con el fin de no alargar excesivamente los tiempos de separación.
3.6.2. Dispersión de bandas
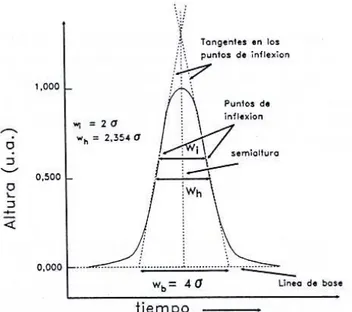
Figura 2.7. Parámetros característicos de un pico cromatográfico (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
3.6.3. Eficacia
Para hacer máxima la operatividad de una separación cromatográfica debe evitarse en lo posible el ensanchamiento de las bandas (debido a la dispersión térmica). En efecto, cuanto más anchas sean las bandas, menor número de ellas se podrán resolver en un espacio de tiempo dado, en otras palabras, cuanto más agudos sean los picos que emergen de una columna mejor estará actuando dicha columna. Las anchuras de las bandas cromatográficas dependen de las características de la columna (tamaño de las partículas de la fase estacionaria, homogeneidad de ésta, etc.), así como de otros factores como por ejemplo, la velocidad de la fase móvil. En consecuencia, para expresar la calidad de un pico, se utiliza un parámetro comparativo relativo como es el cociente entre el tiempo de retención y la desviación estándar, σ, de la banda cromatográfica. En la práctica, se define la eficacia de una columna como el cuadrado del cociente entre el tiempo de retención y σ, y se expresa como el número de platos teóricos:
2
=
σ
Rt
n
(2.2)Así, el número de platos teóricos de una columna puede calcularse utilizando la anchura de uno de sus picos, normalmente el último que pueda medirse directamente sobre el cromatograma (Figuras 2.7 y 2.8) utilizando las expresiones:
2
16
= b R
W t
2
545 .
5
=
h R
W t
n (2.4)
El número de platos de una columna cromatográfica depende lógicamente de su longitud, por lo que para comparar la eficacia de columnas con diferente longitud se introduce otro término, denominado altura equivalente de un plato teórico, que relaciona la eficacia de una columna con su longitud y que se define como el cociente entre la longitud de la columna, L, y su número de platos teóricos, n.
n L
h= (2.5)
“Es de destacar que al considerar la eficacia, solamente se han tenido en cuenta los efectos propios de la columna, aunque existen otros parámetros del sistema (anchura de banda inicial, velocidad de la fase móvil, etc.) que contribuyen al ensanchamiento de las bandas cromatográficas. De esta manera, no siempre la baja eficacia de un sistema cromatográfico es atribuible a la columna, sino a otros factores que será preciso optimizar”
3.6.4. Separación y resolución
Hasta el momento, únicamente se ha considerado el caso en que existe una sola banda cromatográfica; sin embargo, el objeto de la cromatografía es separar mezclas de varios componentes y es de suma importancia considerar la separación entre ellos. Debe recordarse siempre que, independientemente del número de componentes que pueda tener la mezcla, el problema de separación de todos ellos queda siempre reducido al de las dos bandas adyacentes más difíciles de separar. La separación entre dos bandas cromatográficas (Figura 2.8) puede estudiarse en función de dos parámetros:
- La retención relativa, α, definida como el cociente entre los tiempos de retención corregidos de las dos bandas
1 2
'
'
R R
t
t
=
α
(2.6)- La resolución, Rs, definida como el cociente de la distancia entre los centros de las
Figura 2.8.- Cromatograma de dos bandas y parámetros para su resolución. (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
En consecuencia, una separación total de dos bandas requerirá valores de α y Rs lo más
elevados posible. Es de tener en cuenta que mientras que un valor elevado de Rs puede
conseguirse aumentando la eficacia de la columna (mayor longitud de columna, menores dispersiones de banda, etc.), el valor de la retención relativa, α, es constante para cada sistema cromatográfico y solamente podrá variar alterando la fase estacionaria o la fase móvil; en otras palabras, cambiando el sistema cromatográfico. La solución de un problema real de separación entre dos bandas deberá darse en función de los valores de α y Rs que presente. Así, para valores altos de retención relativa y bajos de
resolución, la separación podrá conseguirse mediante un aumento de eficacia, mientras que para valores bajos de α no es práctico intentar la separación aumentando la eficacia y debe procederse a un cambio del sistema.
4. Técnicas cromatográficas no instrumentales
De las técnicas clásicas de cromatografía no instrumentales, solamente continúan teniendo utilización amplia las de columna y capa fina, dado que la cromatografía sobre papel ha sido prácticamente relegada por esta última. Estos dos métodos son particularmente útiles a escala preparativa, aunque en el caso de la cromatografía de capa fina sigue utilizándose ampliamente como método analítico.
4.1.
Cromatografía en columna
estacionaria esté distribuida de forma homogénea, sin fracturas ni burbujas de aire, siendo también fundamental que los extremos de la columna sean planos para evitar eluciones simultáneas de más de una fracción, especialmente si la capacidad de separación de la columna no es muy grande. El método más recomendable de llenado consiste en añadir al tubo, una vez colocado en posición vertical, una pasta formada con la fase estacionaria y un disolvente de menor actividad que la fase móvil a utilizar, dejando salir el disolvente sobrante a medida que la fase estacionaria va sedimentando.
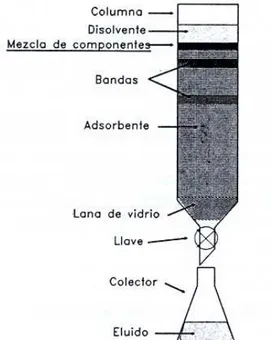
Figura 2.9.- Esquema de una columna de gravedad (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
La capacidad de separación de las columnas de gravedad no es muy grande ya que su eficacia se encuentra limitada por el tamaño de las partículas de la fase estacionaria que debe ser bastante alto (70-230 mallas ASTM) para permitir un caudal razonable de la fase móvil. La cantidad de fase estacionaria y el tamaño de la columna también limitan su capacidad de separación. Como norma general, se utilizan de 20 a 30g de fase estacionaria por gramo de mezcla a separar, pudiéndose llegar hasta relaciones de 100:1 para separaciones especialmente difíciles. La relación entre el diámetro y la longitud de la columna es también importante, ya que columnas demasiado cortas no proporcionarán espacio suficiente para lograr la separación. En general, las relaciones longitud/diámetro para este tipo de columnas deben oscilar entre 8:1 y 10:1.
Las fases estacionarias utilizadas normalmente para cromatografía en capa fina son, alúmina o gel de sílice para cromatografía de adsorción y, celulosa para cromatografía de adsorción o de reparto.
En la cromatografía en capa fina, se utiliza normalmente el análisis por desarrollo. El proceso se lleva a cabo en recipientes cerrados, por ascensión del disolvente, debiendo colocarse en el interior del recipiente cubriendo una de sus paredes una capa de papel de filtro impregnado en la fase móvil (disolvente) para conseguir una atmósfera saturada en ella. Una vez desarrollada la placa, la visualización de los cromatogramas se realiza, caso de no tratarse de compuestos coloreados, mediante iluminación con luz ultravioleta, si la fase estacionaria lleva un indicador de fluorescencia, o bien mediante pulverizado de la placa con reveladores de carácter general (yodo, ácido sulfúrico, etc.) si se quieren visualizar únicamente compuestos de un tipo determinado.
La cromatografía en capa fina puede utilizarse para separaciones analíticas o preparativas:
i) Aplicaciones analíticas. Las muestras se aplican en forma de manchas circulares sobre uno de los extremos de la placa. Tras el desarrollo, la identificación de cada componente de la mezcla se realiza midiendo el desplazamiento relativo de cada mancha en la placa respecto a un compuesto patrón o al frente del disolvente, definiéndose para cada banda sus valores Rx o Rf respectivamente (Figura 2.10)
como:
patrón del
ento desplazami
compuesto del
ento desplazami
Rx = (2.8)
disolvente del
frente del
ento desplazami
compuesto del
ento desplazami
Figura 2.10.- Determinación de Rf y Rx en cromatografía en capa fina (Figura tomada
de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
ii) Aplicaciones preparativas. La muestra se sitúa en la placa en forma de banda a lo largo de uno de los extremos de la placa, realizándose el revelado mediante luz ultravioleta (método no destructivo) o por pulverización con un revelador de uno de los laterales del cromatograma que servirá como guía para conocer la posición de las bandas (método destructivo). La recuperación de las muestras se realiza por raspado de la fase estacionaria y extracción con algún disolvente adecuado.
5. Métodos instrumentales en cromatografía
5.1.
Cromatografía de gases
En la cromatografía de gases (GC), la muestra se volatiliza y se inyecta en la cabeza de una columna cromatográfica. La elución se produce por el flujo de una fase móvil de un gas inerte y, a diferencia de la mayoría de los equipos de cromatografía, la fase móvil no interacciona con las moléculas del analito, su única función es la de transportar al componente a través de la columna. Existen dos tipos de cromatografía de gases:
- Cromatografía gas-sólido (GSC)
- Cromatografía gas-líquido (GLC). Tiene gran aplicación en todos los campos de la ciencia y su denominación se abrevia normalmente como cromatografía de gases (GC).
5.1.1. Cromatografía Gas-Líquido
La cromatografía gas-líquido se basa en la distribución del componente a separar entre una fase móvil gaseosa y una fase líquida inmovilizada sobre la superficie de un sólido inerte.
Los principios generales en esta cromatografía coinciden con lo ya explicado en secciones anteriores y las relaciones matemáticas son aplicables con sólo ligeras modificaciones que resultan de la compresibilidad de las fases móviles gaseosas.
5.1.1.1. Instrumentos para la cromatografía Gas-Líquido
Figura 2.11.- Esquema de un sistema cromatográfico Gas-Líquido. Componentes básicos (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
i) Gas portador
He, Ar, N2, CO2, H2. La elección del gas está con frecuencia determinada por el tipo de
detector que se utilice. Los caudales se controlan mediante un regulador de presión de dos niveles colocado en el cilindro de gas y algún tipo de regulador de presión o regulador de flujo instalado en el cromatógrafo. Los caudales pueden determinarse mediante un rotámetro en la cabeza de la columna y un medidor de pompas de jabón al final de la columna.
ii) Sistema de inyección de muestra
El método más común de inyección de muestra implica el uso de una microjeringa para inyectar una muestra líquida o gaseosa a través de un diafragma o “septum” de goma de silicona, en una cámara de vaporización instantánea situada en la cabeza de la columna. Las muestras sólidas se introducen como disoluciones o, alternativamente, en viales cerrados herméticamente de paredes delgadas que pueden colocarse junto a la cabeza de la columna y ser perforados o aplastados desde el exterior.
iii) Configuraciones de columna y hornos
En cromatografía de gases se usan dos tipos generales de columnas, las empaquetadas o de relleno y las tubulares abiertas o capilares.
Figura 2.12.- Mejora de un cromatograma por programación de temperatura en columna (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
En general, la resolución óptima se asocia con una menor temperatura; sin embargo, la consecuencia de una reducción de temperatura es un aumento en el tiempo de elución y por tanto, del tiempo que se necesita para completar el análisis. Las Figuras 2.12a y 2.12b ilustran este principio.
iv) Detectores
CARACTERÍSTICAS DEL DETECTOR IDEAL - Adecuada sensibilidad
En un quemador, el efluente de la columna se mezcla con hidrógeno y con aire para luego encenderse eléctricamente. La mayoría de los compuestos orgánicos cuando se pirolizan a la temperatura de una llama de hidrógeno/aire, producen iones y electrones que pueden conducir la electricidad a través de la llama. Entre el extremo del quemador y un electrodo colector situado por encima de la llama, se aplica una diferencia de potencial de unos pocos cientos de voltios y para la medición de la corriente que resulta se utiliza un amplificador operacional de alta impedancia.
La ionización en la llama de los compuestos que contienen carbono no es un proceso bien establecido, aunque se observa que el número de iones que se produce es aproximadamente igual al de átomos de carbono transformados en la llama. Por tanto, este detector es sensible a la masa. Grupos funcionales como –C-OH, -C=O, -X, -NH2,
originan pocos iones o prácticamente ninguno. Además, el detector es insensible al H2O, CO2, SO2 y NOx. Estas propiedades hacen del detector de ionización de llama uno
de los detectores generales más utilizados para el análisis de la mayoría de los compuestos orgánicos.
b) DETECTOR DE CONDUCTIVIDAD TÉRMICA
Se basa en los cambios de la conductividad térmica del gas portador ocasionados por la presencia de las moléculas del componente a analizar. El sensor consiste en un elemento calentado eléctricamente cuya temperatura, a una potencia eléctrica constante, depende de la conductividad térmica del gas circundante. El elemento calentado puede ser un hilo fino de platino, oro o wolframio, o también, un termistor semiconductor. La resistencia del hilo o del termistor da una medida de la conductividad térmica del gas. Este detector posee las siguientes características:
- Simple
- Amplio rango dinámico lineal (~ 105)
- Respuesta universal (especies orgánicas e inorgánicas) - Carácter no destructivo
- Sensibilidad relativamente baja (~ 10-8 g de soluto/ml de gas portador) c) DETECTOR TERMOIÓNICO
El detector termoiónico (TID) es un detector selectivo de los compuestos orgánicos que contienen fósforo y nitrógeno. En este caso el efluente de la columna se mezcla con hidrógeno, pasa a través de una llama y se quema. El gas caliente fluye alrededor de una bola de silicato de rubidio calentada eléctricamente, la cual se mantiene a unos 180 V con respecto al colector. La bola caliente forma un plasma que alcanza una temperatura de 600 a 800 ºC. Lo que ocurre exactamente en el plasma, que hace que se produzcan insólitamente una gran cantidad de iones a partir de las moléculas que contienen fósforo o nitrógeno, realmente no está bien establecido, pero el resultado es una gran corriente de iones, la cual se utiliza para la determinación de compuestos que contienen P y N. d) DETECTOR DE CAPTURA DE ELECTRONES
platino o titanio). Un e- del emisor provoca la ionización del gas portador (con frecuencia N2) y la producción de una ráfaga de e-. De este proceso de ionización, en
ausencia de especies orgánicas, resulta una corriente constante entre un par de electrodos. Sin embargo, la corriente disminuye en presencia de moléculas orgánicas que tiendan a capturar electrones.
Este detector es de respuesta selectiva, siendo sensible a grupos funcionales electronegativos: halógenos, peróxidos, quinonas y grupos nitro. En cambio, no es sensible a aminas, alcoholes e hidrocarburos.
e) DETECTOR DE EMISIÓN ATÓMICA
En este dispositivo el eluyente se introduce en un plasma de helio obtenido con microondas que se acopla a un espectrómetro de emisión con series de diodos. El plasma es suficientemente energético como para atomizar todos los elementos de una muestra, excitarlos y así obtener sus espectros de emisión característicos.
5.1.2. Acoplamiento de la cromatografía de gases con métodos espectroscópicos La cromatografía de gases a menudo se combina con otras técnicas selectivas de análisis como la espectroscopia y la electroquímica. Los métodos de análisis que resultan se denominan métodos acoplados y proporcionan unas potentes herramientas para la identificación de los componentes de una mezcla compleja.
- Cromatografía de gases/espectrometría de masas - Cromatografía de gases/espectroscopia infrarroja 5.1.3. Cromatografía Gas-Sólido
Esta cromatografía se basa en una fase estacionaria sólida en la cual se produce la retención de los analitos como consecuencia de la adsorción física. Los coeficientes de distribución son generalmente mucho mayores que en el caso de la cromatografía gas-líquido. En consecuencia, la cromatografía gas-sólido es útil para la separación de especies que no se retienen en columnas de gas-líquido, tales como los componentes del aire, H2S, CS2, óxidos de nitrógeno, CO, CO2 y los gases nobles. Se suelen utilizar dos
tipos de adsorbentes: tamices moleculares y polímeros porosos.
5.2.
Cromatografía de líquidos de alta resolución
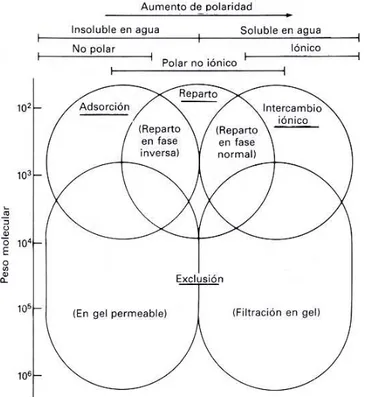
La Figura 2.13 pone de manifiesto que los distintos procedimientos que utilizan la cromatografía de líquidos tienden a ser complementarios por lo que a sus campos de aplicación se refiere. Así, para solutos con masas moleculares superiores a 10000, a menudo se utiliza la cromatografía de exclusión, aunque ahora también es posible tratar estos compuestos con cromatografía de reparto de fase inversa. Para especies iónicas de baja masa molecular, se utiliza con frecuencia la cromatografía de intercambio iónico. Los métodos de reparto se aplican a las especies polares pero no iónicas. Además, este procedimiento se utiliza muchas veces para la separación de los integrantes de una serie homóloga. La cromatografía de adsorción se elige con frecuencia para separar especies no polares, isómeros estructurales y grupos de compuestos como, por ejemplo, los hidrocarburos alifáticos de los alcoholes alifáticos.
Figura 2.13. Distintos procedimientos que se utilizan en la cromatografía de líquidos así como su ámbito de utilización en función del tipo de especies a separar (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
5.2.1. Instrumentación para cromatografía de líquidos
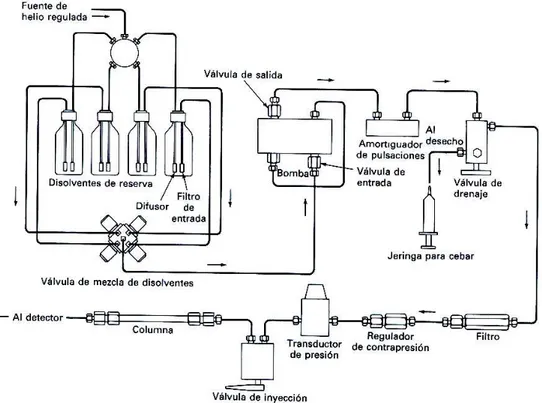
La figura 2.14 muestra un esquema de los componentes fundamentales de un cromatógrafo de líquidos de alta resolución típico.
i) Recipientes para la fase móvil y sistemas para el tratamiento de los disolventes (eliminación de O2 y N2)
Figura 2.14.- Esquema de los componentes fundamentales de un cromatógrafo de líquidos de alta resolución (Figura tomada de: ALBELLA, J.M.; CINTAS, A.M.; MIRANDA, T. y SERRATOSA, J.M.: "Introducción a la ciencia de materiales". C.S.I.C., 1993).
ii) Sistemas de bombeo
Se utilizan tres tipos de bombas: bombas recíprocas, bombas de jeringa o de desplazamiento y bombas neumáticas o de presión constante.
iii) Sistemas de inyección de muestra
iv) Columnas para cromatografía de líquidos v) Precolumnas
vi) Termostatos
responden a alguna de las propiedades del soluto, como la absorbancia en el UV-VIS, la fluorescencia, o la intensidad de dispersión que no son propias de la fase móvil.
- Detectores de absorbancia UV-Vis con filtros
- Detectores de absorbancia UV-Vis con monocromadores - Detectores de absorbancia en el IR
- Detectores de fluorescencia - Detectores de índice de refracción - Detector de dispersión de luz - Detectores electroquímicos
8.- Bibliografía
1. D.A. Skoog, J.J. Leary, “Análisis Instrumental”, McGraw-Hill, Madrid (1996)
2. H.H. Willard, L.L. Merritt Jr.,J.A. Dean, F.A. Settle Jr., “Métodos Instrumentales de análisis”, Grupo Editorial Iberoamericana S.A. de C.V., México (1991).
3. TECHNIQUES in liquid chromatography Simpson, Colin F. John Wiley & Sons (1984).
4. Liquid chromatography column theory Scott, Raymond P.W. John Wiley & Sons (1992).
5. Chromatography of polymers : characterization by SEC and FFF American Chemical Society (1993).
6. Handbook of size exclusion chromatographyMarcel Dekker (1995).
7. Cromatografía de exclusión por tamaños [Vídeo] Pérez Dorado, Angel,
CEMAV, Universidad Nacional de Educación a Distancia [1997].
8. Manual de cromatografía Loro Ferrer, Juan Francisco Gobierno de Canarias, Dirección General de Universidades e Investigación (2001).
9.- Direcciones URL útiles
Nomenclatura IUPAC para cromatografía
Cromatografía de gases
http://www.uib.es/depart/dqu/dquo/pau/Cromatograf%92a/chrom10/chrom/GC/concept/ main.htm
http://www.relaq.mx/RLQ/tutoriales/cromatografia/Gas.htm http://ciencias.ucv.cl/micro/croma/cro1.htm