Autor de correspondencia
Excitotoxicidad y muerte neuronal en la epilepsia
Lourdes Lorigados
1, Sandra Orozco
2, Lilia Morales
3, Bárbara Estupiñán
3,
Iván García
4, Luisa Rocha
5 1 Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Cirén Ave. 25, No. 15805 e/ 158 y 160 Playa, CP 11300, La Habana, Cuba 2 Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, IMSS México 3 Departamento de Neurofisiología, Cirén 4 Servicio de Neurocirugía, Cirén 5 Laboratorio Farmacobiología, Centro de Investigación y de Estudios Avanzados, CinvestavSede Sur, DF, México E-mail: [email protected]
RESUMEN
La epilepsia es una afección neurológica de evolución crónica, recurrente, casi siempre progresiva, que afecta del 1 al 2 % de la población mundial. Modelos experimentales y estudios de imágenes neurológicas de pacientes con este padecimiento muestran que las crisis recurrentes provocan estrés oxidativo, relacionado fundamentalmente con la excitabilidad neuronal. La estimulación excesiva de los receptores de glutamato induce neurotoxicidad, un proceso que se ha defi nido como excitotoxicidad. Se considera que este puede ser el principal mecanismo de muerte celular en numerosas afecciones del sistema nervioso central, incluida la epilepsia. Desde los años 70 se han estudiado con profundidad las vías de señalización, los mecanismos moleculares y los sitios de acción relacionados con la excito-toxicidad; aunque de forma muy limitada en las enfermedades del sistema nervioso central. En particular, deberán evaluarse con especial cuidado la función crucial de la muerte neuronal y los mecanismos que se potencian con la sobreactivación de los receptores de glutamato, principalmente los relativos a las enfermedades neurológicas, con el fi n de intervenir de manera oportuna para retardar el desarrollo de estas afecciones neurológicas. Se repasan las evidencias clínicas y experimentales sobre las alteraciones del sistema glutamatérgico, las vías de muerte celular, la activación de las caspasas y de la familia de genes Bcl-2 involucrados, como moduladores de la muerte celular en la epilepsia. Tales hallazgos sustentan que en la epilepsia farmacorresistente convergen procesos excitotóxicos y de muerte neuronal apoptótica y necrótica.
Palabras clave: excitotoxicidad, apoptosis, necrosis, epilepsia
Biotecnología Aplicada 2013;30:1-8
ABSTRACT Excitotoxicity and neuronal death in epilepsy. Epilepsy is a recurrent, often progressive neurological disorder with a chronic evolution, affecting 1 to 2 % of the world population. Research with experimental models and imag-ing analysis of diseased patients have been used to show that recurrent episodes produce oxidative stress, most of which is related to neuronal excitability phenomena. It is known that the excessive stimulation of glutamate receptors results in neurotoxicity; a process that, under the denomination of excitotoxicity, is thought to constitute the principal cellular death mechanism behind different disorders of the central nervous system, including epilepsy. Paradoxically, although the signaling pathways, molecular mechanisms and sites of action of excitotoxicity have received consider-able attention since the 1970s, little is known about their relevance to CNS disorders. Further detail is necessary about the fundamental role of neuronal death and the mechanisms, particularly those relevant to neurological pathogenesis, that are engaged whenever glutamate receptors are excessively stimulated, as the results would aid considerably the development of timely clinical interventions delaying the evolution of these disorders. We review clinical and experimental data on the relevant alterations of the glutamatergic system, cell death pathways, and the activation of caspases and members of the Bcl-2 gene family involved in the process as modulators of cell death during epilepsy. The fi ndings support the hypothesis that excitotoxic processes as well as both apoptotic and necrotic neuronal cell death phenomena converge in drug-resistant epilepsy.
Keywords: excitotoxicity, apoptosis, necrosis, epilepsy
I
ntroducción
La excitotoxicidad mediada por el receptor de glutama-to ejerce una función importante en el desarrollo neu-ral, la diferenciación y la plasticidad sinápticas [1, 2]. Este proceso se considera el principal mecanismo de la muerte celular en numerosas enfermedades del sis-tema nervioso central (SNC) como el trauma cerebral, los desórdenes neurodegenerativos y la epilepsia [3-6].
En el SNC de los mamíferos, el glutamato es el neu-rotransmisor excitador por excelencia. La regulación de la neurotransmisión glutamatérgica es crítica, no
solo por las propiedades de señalización atribuidas a los niveles y la actividad del glutamato y su receptor, sino también por la muerte celular excitotóxica [2]. Ini-cialmente se describió que la muerte celular inducida por excitotoxicidad se caracteriza por el aumento del volumen de la célula, la vacuolización del citoplas-ma y la ruptura de las membranas, características que apuntan a un mecanismo de muerte celular necrótica [7-10]. Luego se defi nió que la degradación internu-cleosomal del ADN y la activación de las caspasas son
REVISIÓN
1. Yang JL, Sykora P, Wilson DM, 3rd, Mattson MP, Bohr VA. The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resil-iency. Mech Ageing Dev. 2011;132(8-9): 405-11.
indicativas de muerte neuronal apoptótica [11-13]. Y recientemente se sugiere que la autofagia puede ser un mecanismo de muerte celular no apoptótica inducida por excitotoxicidad, lo que revela que la autofagia es una estrategia de supervivencia ante el estrés [2]. De esta forma, el incremento de la actividad del receptor del glutamato podría inducir la expresión de proteínas proapoptóticas tales como la p53, lo cual provocaría el daño y la muerte neuronales por apoptosis y autofagia [14-16]. La autofagia se activa como respuesta a un daño excitotóxico agudo [17, 18].
A pesar de que se ha estudiado ampliamente la excitotoxicidad en cuanto a sus vías de señalización y acción, aún es muy escaso el conocimiento sobre su función en el SNC, así como sus mecanismos mo-leculares de acción. No obstante, teniendo en cuen-ta cuen-tales hallazgos se puede considerar que la muerte excitotóxica en el cerebro no es un evento uniforme; más bien es un proceso continuo que va de necrosis a apoptosis y a autofagia. En este trabajo se discuten los mecanismos celulares y moleculares de la excitotoxi-cidad y su efecto en la muerte neuronal que ocurre en la epilepsia.
C
oncepto de excitotoxicidad
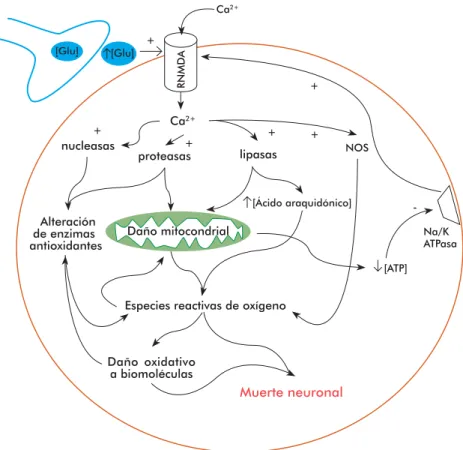
Diversos hallazgos experimentales y clínicos relacio-nados con la posible toxicidad de los aminoácidos ex-citadores han dado lugar a la teoría excitotóxica, que postula que los niveles excesivos de glutamato endó-geno o la hipersensibilidad de sus receptores se rela-cionan con la degeneración neuronal [19]. La excito-toxicidad es el mecanismo que promueve la muerte celular mediante la sobreactivación de los receptores glutamatérgicos o de cualquiera de sus análogos. Esta provoca la entrada excesiva de calcio (Ca2+) a la célu-la, que es secuestrado por la mitocondria. Ello provo-ca un incremento del provo-calcio mitocondrial, que provoprovo-ca la disfunción metabólica, la producción de radicales libres, la activación de proteasas, fosfolipasas, la óxi-do nítrico sintasa y enóxi-donucleasas, y la inhibición de la síntesis de proteínas [20].
La pérdida de la homeostasis del calcio se debe a la sa-turación de los mecanismos de regulación como la bom-ba de calcio, el intercambiador sodio/calcio (Na+/Ca2+) y las proteínas amortiguadoras de calcio. Una vez satu-rados estos sistemas, la mitocondria captura el exceso de calcio que se acumula en la matriz mitocondrial. Ello induce la despolarización de la membrana mito-condrial por dos mecanismos: el abatimiento parcial del potencial quimiosmótico por la acumulación de cargas positivas en la matriz mitocondrial; y ante una sobre-carga sostenida ocurre una despolarización irreversible por la activación del poro de transición mitocondrial. El colapso del potencial quimiosmótico mitocondrial reduce la síntesis de trifosfato de adenosina (ATP) y la activación del poro de transición, que constituye una vía por donde el calcio retorna al citosol [21, 22]. El aumento sostenido de las concentraciones de cal-cio promueve la generación de radicales libres, que inducen la peroxidación de lípidos de la membrana, la síntesis de óxido nítrico y la activación de enzimas involucradas en el catabolismo de proteínas, fosfolípi-dos y ácifosfolípi-dos nucleicos. Además, el oxido nítrico puede actuar como mensajero retrógrado y potenciar el efecto excitotóxico del glutamato al aumentar su liberación desde las terminales presinápticas [23] (Figura 1).
Una vía de daño celular es la activación de la óxido nítrico sintasa, cuyo producto reacciona con el superóxi-do y forma el peroxinitrito. Otra vía es la activación de
la poli-adenosina difosfato ribosa-polimerasa (PARP), como respuesta al daño del ADN mediado por los radi-cales libres [24, 25].
E
xcitotoxicidad y epilepsia
Algunas evidencias sustentan la hipótesis de que los cambios neurodegenerativos asociados con la epilep-sia humana resultan de la actividad de las descargas persistentes en la vía del glutamato. El mecanismo es relativamente simple: la liberación del exceso de glutamato provoca la despolarización y repolariza-ción repetitiva de las terminales del glutamato, lo que conduce a su concentración tóxica, y fi nalmente ori-gina la degeneración excitotóxica de la neurona pos-sináptica [26, 27].
Estudios de microdiálisis en humanos y modelos animales documentan la asociación entre la actividad convulsiva prolongada y la duración del estado epilép-tico por la elevación signifi cativa del glutamato [28]. Se conoce que la sobrexcitación de las neuronas por glutamato puede provocar descargas epilépticas y que la aplicación directa de glutamato en la amígdala indu-ce un efecto similar al de la activación propagada [29]; mientras que el empleo de antagonistas del receptor ácido α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico
[Glu] [Glu] +
RNMD
A
Ca2+
+
+ +
NOS +
+
Ca2+
proteasas nucleasas
Alteración de enzimas antioxidantes
Daño mitocondrial lipasas
[Ácido araquidónico]
-Na/K ATPasa
[ATP]
Especies reactivas de oxígeno
Daño oxidativo a biomoléculas
Muerte neuronal
Figura 1. Mecanismo de excitotoxicidad. La activación sostenida del receptor N-metil-D aspartato (RNMDA) por concentraciones incrementadas de glutamato (Glu) provoca la entrada masiva de calcio a la célula que activa a las enzimas líticas y la óxido nítrico sintasa (NOS). El daño mitocondrial y el aumento de la concentración de ácido araquidónico es uno de los factores implicados en el incremento de la generación de especies reactivas de oxígeno, que conducen a la muerte neuronal provocada por el daño a las biomoléculas y la activación de programas de muerte apoptóticos. El défi cit energético contribuye a la perpetuación del proceso degenerativo porque favorece la despolarización de la membrana por el défi cit en el funcionamiento de la bomba sodio/potasio (Na/K ATPasa) y mantiene el estado activo del RNMDA. Esto sensibiliza a la célula a la aferencia glutamatérgica normal proce-dente de la corteza cerebral.
3. Severino PC, Muller Gdo A, Van-dresen-Filho S, Tasca CI. Cell sig-naling in NMDA preconditioning and neuroprotection in convulsions induced by quinolinic acid. Life Sci. 2011;89(15-16):570-6.
4. Araujo IM, Carreira BP, Carvalho CM, Carvalho AP. Calpains and de-layed calcium deregulation in ex-citotoxicity. Neurochem Res. 2010; 35(12):1966-9.
5. Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA recep-tor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104(6):3551-6.
Lourdes Lorigados et al. Excitotoxicidad y muerte neuronal en la epilepsia
3
Biotecnología Aplicada 2013; Vol.30, No.1retarda el desarrollo de la activación propagada amig-dalina en ratones [30].
La activación del receptor de N-metil-D-aspartato media la muerte celular durante el estado epiléptico [31], y el uso de un antagonista de este receptor, el MK-801, previene la aparición de las crisis espontá-neas en modelos animales [32]. En la epileptogénesis participan los receptores de kainato, especialmente la subunidad GluR6 como inductora [33, 34].
En general, el daño neuronal excitotóxico celular en pacientes epilépticos es mediado por la entrada ex-cesiva de calcio en las células durante las convulsio-nes [35]. Los niveles elevados de calcio desatan una secuencia de eventos como la activación de la óxido nítrico sintasa, que interfi ere con el metabolismo oxi-dativo y genera radicales libres y daño de la membra-na neuromembra-nal; igualmente se activan las procaspasas, y ocurre la muerte neuronal, ya sea por necrosis, apop-tosis o autofagia.
E
xcitotoxicidad en modelos
experimentales de epilepsia
Por las limitaciones para estudiar la epilepsia humana, se han desarrollado modelos experimentales que la asemejan. Sin embargo, aún es imposible evaluar las manifestaciones conductuales, sobre todo la conducta motora.
Estos modelos se clasifi can en agudos y crónicos. Los primeros se logran por la aplicación de fárma-cos convulsivantes o por estimulación eléctrica; los segundos reproducen mejor la fi siopatología de la epilepsia en humanos. Ambos pueden padecer crisis parciales o generalizadas. Sin embargo, debido a que la epilepsia se caracteriza por la recurrencia de mani-festaciones ictales, solamente los modelos que repro-ducen esa condición se consideran verdaderos mode-los de epilepsia. El último desafío en cualquier estudio experimental de la epilepsia es determinar cuál de los muchos cambios como respuesta a un daño cerebral está relacionado de forma causal con el subsecuente desarrollo de la epilepsia.
El estrés oxidativo inducido por las crisis recurren-tes contribuye grandemente al daño y la muerte celu-lares. Los radicales libres derivados del estrés oxida-tivo son componentes de la excitotoxicidad. En parte, ello se sustenta en que las crisis prolongadas inducen daño celular en las macromoléculas y se relacionan sobre todo con la excitabilidad neuronal.
En los modelos animales se han estudiado dos tipos de daño en particular: crisis febriles prolongadas (20 a 30 min) y estado epiléptico prolongado (5 a 8 h). Este último es inducido por la inyección sistémica de agonistas colinérgicos (pilocarpina) o por la inyección unilateral de agonistas glutamatérgicos en el hipocam-po de ratas (ácido kaínico, análogo del glutamato). En los modelos de crisis febriles, hipoxia neonatal y espasmos se ha demostrado que las neuronas en de-sarrollo son menos vulnerables al daño neuronal y la pérdida celular que las neuronas adultas. Por ejemplo, las neuronas hipocampales de animales cuyos cere-bros son inmaduros continúan respondiendo a estímu-los sinápticos, y en un ambiente totalmente anóxico se requiere mucho tiempo para destruir los circuitos de manera irreversible [36]. El cerebro inmaduro pa-rece ser también más resistente a los efectos tóxicos del glutamato que el cerebro maduro [37]. Mark et al. [38] demostraron que la cantidad de calcio que entra a una neurona piramidal está directamente relacionada
con la edad del individuo: en los tres primeros días de vida, el glutamato incrementa el calcio mínimamente; en cambio, entre los días 21 al 25 ocurre un marca-do incremento en el calcio intracelular, aumento del volumen del soma y retracción de las dendritas. Esta resistencia relativa se debe a la menor densidad de las sinapsis activas, al bajo consumo de energía y en ge-neral a la relativa inmadurez de las cascadas bioquí-micas que llevan a la muerte celular. Por consiguiente, los animales jóvenes son menos vulnerables que los adultos a la pérdida celular después de las crisis epi-lépticas prolongadas [39-41].
Los modelos de excitotoxicidad en animales adul-tos más utilizados son el del ácido kaínico y la pi-locarpina: modelos de epilepsia del lóbulo temporal, inducidos por la inyección unilateral o sistémica de uno de estos compuestos, en dosis convulsionantes que provocan daño excitotóxico en las neuronas pi-ramidales del hipocampo y en la región hilar. El daño depende de la dosis, la especie y la cepa animal; pero el resultado es la muerte de las neuronas en las re-giones vulnerables, la proliferación de astrocitos y el aumento de las fi bras gliales. Por ese motivo, los mo-delos de administración sistémica de ácido kaínico o pilocarpina se consideran adecuados para el estudio de las convulsiones tónico-clónicas generalizadas o estado epiléptico, cuyo sustrato neuroanatómico es la esclerosis temporal mesial [42-46].
Uno de los primeros cambios que ocurren después de la administración de ácido kaínico es la inducción de ARN mensajero (ARNm) y la expresión de proteí-nas de choque térmico de diferentes pesos molecula-res (HSP27, HSP70 y HSP72). Esta última se expmolecula-resa constitutivamente en el cerebro de los mamíferos, y se sobreexpresa en las poblaciones neuronales sensibles del hipocampo [47]. La expresión de estas proteínas parece prevenir el plegamiento anormal de proteínas de nueva síntesis en las poblaciones vulnerables al ácido kaínico. Durante las dos semanas siguientes a la administración, esas proteínas se transportan por el árbol dendrítico y a lo largo de los axones hacia las zonas más distales. La HSP70 y la HSP72 tienen una función protectora, aunque no consiguen rescatar a las células de la muerte excitotóxica. La sobreexpresión de HSP27 y HSP70 in vivo protege del daño exci-totóxico [47, 48], mientras que los niveles excesiva-mente altos de la HSP72 pueden ser nocivos para las células [49-51].
Luego de tres a cinco horas de la inyección de áci-do kaínico también se induce la síntesis de ARNm y la sobreexpresión de las proteínas cFos y cJun en las regiones vulnerables del hipocampo y en el giro denta-do [52]. La inmunorreactividad contra cFos decrece a las seis horas en el giro dentado, pero permanece alta en el hipocampo. Ello sugiere que la muerte celular puede asociarse con los niveles altos de cFos. Además, el incremento prolongado de cFos no tiene un carácter predictivo y no es preciso para que ocurra daño neu-ronal excitotóxico [53, 54]. También se ha observado un aumento de la expresión de cJun en el hipocampo y en la circunvolución dentada, 24 horas después de las crisis epilépticas. El signifi cado del aumento de los niveles de cJun es contradictorio, ya que se considera marcador de la muerte celular retardada, secundaria a crisis epilépticas, o como posible marcador de supervi-vencia neuronal frente al daño excitotóxico [52].
Con respecto a la señalización a través de la mem-brana celular, el activador tisular del plasminógeno
7. Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures pro-duce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98(1):41-53.
8. Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000; 41 Suppl 6:S9-13.
9. Ebert U, Brandt C, Loscher W. Delayed sclerosis, neuroprotection, and limbic epileptogenesis after status epilepticus in the rat. Epilepsia. 2002;43 Suppl 5:86-95.
10. Kubova H, Druga R, Lukasiuk K, Su-chomelova L, Haugvicova R, Jirmanova I, et al. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J Neu-rosci. 2001; 21(10):3593-9.
11. Bengzon J, Mohapel P, Ekdahl CT, Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog Brain Res. 2002;135:111-9.
12. Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, et al. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22(19):8458-65.
13. Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69(2):103-42.
14. Dong XX, Wang Y, Qin ZH. M o l e c u l a r m e c h a n i s m s o f e x-citotoxicity and their relevance to pathogenesis of neurodegenerative dis-eases. Acta Pharmacol Sin. 2009;30(4): 379-87.
15. Qin ZH, Tao LY, Chen X. Dual roles of NF-kappaB in cell survival and implications of NF-kappaB in-hibitors in neuroprotective therapy. Acta Pharmacol Sin 2007;28(12):1859-72.
16. Zhang XD, Wang Y, Zhang X, Han R, Wu JC, Liang ZQ, et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5(3):339-50.
17. Shacka JJ, Lu J, Xie ZL, Uchi-yama Y, Roth KA, Zhang J. Kainic acid induces early and transient au-tophagic stress in mouse hippocampus. Neurosci Lett. 2007;414(1):57-60.
18. Wang Y, Dong XX, Cao Y, Liang ZQ, Han R, Wu JC, et al. p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur J Neurosci. 2009; 30(12):2258-70.
(tPA), serina proteasa extracelular, parece ser necesario para que ocurra la muerte celular, ya que los ratones nulos para tPA o para plasminógeno son relativamente resistentes al daño excitotóxico. Este efecto parece me-diado por la interacción de tPA con laminina, una pro-teína de la matriz extracelular [55]. También el aumen-to de la expresión del ligando específi co del recepaumen-tor Fas (FasL) en el hipocampo y en las células granulares del giro dentado, tres horas después de la inyección de ácido kaínico, está relacionado con la señalización a través de la membrana celular [56]. Mientras, en esta última región, la expresión de FasL decrece seis horas después de la inyección de ácido kaínico, y la inmuno-rreactividad contra FasL se mantiene en el hipocampo. Este aspecto es relevante, porque la unión de FasL a Fas activa el dominio de muerte de este último, al que se une el dominio de muerte asociado a Fas (FADD), y a su vez activa la caspasa 8 que actúa sobre las caspasas efectoras, que provocan la muerte por apoptosis. La uti-lización de ratones transgénicos para Fas aporta datos sustanciales para conocer el papel del sistema Fas/FasL en la señalización de muerte celular excitotóxica [56].
Aún no se ha precisado la función de los miem-bros de la familia de genes bcl-2. Un estudio preli-minar mostró la reducción de la proteína Bcl-2 y el aumento del ARNm para la proteína Bax en el hipo-campo de ratones después de la inyección sistémica de ácido kaínico [57]. Estudios más precisos mediante Northern blot han mostrado una inducción del ARNm de la proteína Bax (pero no de las proteínas Bcl-2 y Bcl-x) desde seis a 24 horas en el hipocampo de ra-tas inyectadas con ácido kaínico. La expresión de las proteínas Bcl-2, Bcl-x y Bax en el hipocampo, ana-lizada mediante Western blot e inmunohistoquímica, es similar en las células destinadas a morir y en las células que sobreviven [58]. Es posible que los efectos de los miembros de la familia Bcl-2 no dependan de cambios globales de las proteínas; pero sí su locali-zación subcelular. La señalilocali-zación de muerte apoptó-tica por la vía mitocondrial se desencadena por una unión de la proteína Bax a la membrana mitocondrial y por una liberación de citocromo c al citosol. Esta liberación comprende la unión al factor de activación de proteasas apoptóticas Apaf1 en presencia de ATP y la activación de la caspasa 9, que a su vez activa distintas caspasas efectoras o ejecutoras. El modo en que ocurre la salida del citocromo c de la mitocondria al citosol no está claro, pero parece establecerse una interacción entre Bcl-2, Bcl-x y Bax, y los canales ió-nicos dependientes de voltaje que controlan la salida del citocromo c. Se ha propuesto que el balance entre Bax y Bcl-2 en la célula es esencial para determinar si una célula experimentará apoptosis [59].
La excitotoxicidad provocada con ácido kaínico por vía intraperitoneal también induce la expresión del ARNm de la caspasa 3 y el aumento de la ex-presión de procaspasa 3 en algunas neuronas de las regiones vulnerables del hipocampo [60, 61]. Unas pocas neuronas expresan el fragmento activo (escin-dido) de 17 kDa de la caspasa 3 [62]. Ello indica la participación de la vía de las caspasas en algunas neuronas del hipocampo, lo que revela muerte ce-lular con componente apoptótico en subpoblaciones del hipocampo. Sin embargo, los estudios por Wes-tern blot señalan la presencia de bandas de PARP de 89 kDa y de otras de menor tamaño. Esto demuestra que la fragmentación de PARP no ocurre exclusiva-mente por la activación de las caspasas, sino también
por la acción de otras proteasas, lo cual presagia la muerte indiscriminada por necrosis [61].
Como resultado del daño excitotóxico que afecta de manera preferencial las células de la región hilar [63], las fi bras musgosas procedentes de las células granulares del giro dentado se desconectan de sus dia-nas. Esta diferenciación da lugar a la producción de ramifi caciones axonales musgosas que progresan en la región supragranular y en toda la capa molecular del giro dentado [64, 65]. Sin embargo, el recrecimiento de las fi bras producido por las crisis convulsivas es menos evidente en animales jóvenes [10, 66]. La for-mación de ramifi caciones o racimos se asocia con un incremento de la expresión de la proteína GAP-43 en la capa supragranular durante la primera semana, y en toda la capa molecular a partir de un mes [67]. Se ha detectado el incremento de la expresión de la proteína asociada con los sinaptosomas de 25 kDa (SNAP25) en las neuronas y en la capa molecular del giro denta-do, así como en las fi bras musgosas del hipocampo en los días siguientes a la lesión excitotóxica por ácido kaínico [68, 69]. Parece probable la intervención de señales trófi cas específi cas en el desarrollo de estas conexiones aberrantes, aunque todavía es discutible la función de los factores trófi cos. El factor neurotrófi co derivado de cerebro y el receptor TrkB en las neuro-nas del giro dentado posiblemente infl uyen sobre el trofi smo de estas células en la construcción de ramifi -caciones plásticas dirigidas a reinervar zonas destrui-das por el ácido kaínico. También se ha sugerido que el factor neurotrófi co derivado de cerebro confi ere protección al atenuar el estrés oxidativo [70, 71].
M
uerte neuronal y epilepsia
La apoptosis es una forma característica de muerte ce-lular, regida por un programa genético común en va-rios tipos celulares. Usualmente afecta más a células individuales, que a todas las células de un tejido. La condensación del citoplasma y la reducción del volu-men celular, acompañado de cambios en la estructura del núcleo, son de los primeros cambios morfológicos que exhiben las células al inicio del proceso apoptóti-co. La cromatina se condensa y forma cúmulos densos adosados a la membrana, seguido por invaginaciones de la membrana nuclear, y lleva a la fragmentación del núcleo en estructuras membranosas con cantidades variables de cromatina. De manera análoga, la mem-brana celular presenta invaginaciones que terminan por fragmentar la célula en racimos de vesículas de tama-ño variable que contienen orgánulos intactos que no se fusionan con los lisosomas. A estas vesículas se les denominan cuerpos apoptóticos, que rápidamente son fagocitadas por células vecinas. Por lo tanto, una de las consecuencias fi siológicas más relevantes de la muerte neuronal por apoptosis es que no se libera material in-tracelular al medio intersticial [72].
La muerte neuronal inducida por convulsiones no ex-ceptúa la complejidad molecular de la muerte neuronal por neurodegeneraciones. Hay una gran controversia sobre si la muerte neuronal es apoptótica o necrótica. Basándose en la defi nición clásica y los criterios mor-fológicos de la necrosis, esta constituye el mecanismo más frecuente por el que las células del cerebro mueren después de una convulsión [7-10, 13, 73].
Varios autores sustentan que la muerte neuronal indu-cida por un estado epiléptico no es apoptótica sino ne-crótica, con la necrosis como mecanismo dominante de muerte celular después de una crisis epiléptica [73, 74].
20. Haglid KG, Wang S, Qiner Y, Ham-berger A. Excitotoxicity. Experimental correlates to human epilepsy. Mol Neurobiol. 1994;9(1-3):259-63.
21. Nicholls DG, Ward MW. Mito-chondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23(4):166-74.
22. Murchison D, Griffith WH. Mi-tochondria buffer non-toxic calcium loads and release calcium through the mitochondrial permeability transition pore and sodium/calcium exchanger in rat basal forebrain neurons. Brain Res. 2000;854(1-2):139-51.
23. Almeida A, Heales SJ, Bolanos JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathi-one depletion. Brain Res. 1998;790(1-2): 209-16.
24. Olney JW. New insights and new is-sues in developmental neurotoxicology. Neurotoxicology. 2002;23(6):659-68.
25. Struzynska L. A glutamatergic com-ponent of lead toxicity in adult brain: the role of astrocytic glutamate trans-porters. Neurochem Int. 2009;55(1-3): 151-6.
26. Pereno GL. Fisiopatología de la epilepsia del lóbulo temporal: revisión del proceso de muerte neuronal a la neuroplasticidad. Rev Argentina Cienc Comportamiento. 2010;2(1):46-57.
27. Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes-key players in human me-sial temporal lobe epilepsy? Epilepsia. 2008;49 Suppl 2:42-52.
28. Ueda Y, Yokoyama H, Nakajima A, Tokumaru J, Doi T, Mitsuyama Y. Glutamate excess and free radical formation during and following kainic acid-induced status epilepticus. Exp Brain Res. 2002;147(2):219-26.
29. Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus mod-els of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73(1):1-60.
30. Rogawski MA, Kurzman PS, Yamaguchi SI, Li H. Role of AMPA and GluR5 kainate receptors in the development and expression of amygdala kindling in the mouse. Neuropharmacology. 2001;40(1): 28-35.
31. Deshpande LS, Lou JK, Mian A, Blair RE, Sombati S, Attkisson E, et al. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: role of NMDA receptor activation and NMDA depen-dent calcium entry. Eur J Pharmacol. 2008;583(1):73-83.
Lourdes Lorigados et al. Excitotoxicidad y muerte neuronal en la epilepsia
5
Biotecnología Aplicada 2013; Vol.30, No.1Sin embargo, se ha demostrado la presencia de un com-ponente apoptótico. Estos estudios se basan en hallazgos bioquímicos que establecen la participación de algunos miembros de la familia del gen Bcl-2 y las caspasas en el proceso de muerte celular después de las convulsio-nes. Otros factores que sustentan tales hallazgos son la detección de fragmentos múltiples de 180 a 200 pares de bases con activación temprana de endonucleasas y la fragmentación del ADN en células destinadas a morir (originariamente descrita como indicador de apoptosis), la acumulación nuclear de p53 en neuronas vulnerables al ácido kaínico y el aumento de receptores de muerte celular y sus ligandos. Ello evidencia la intervención de mecanismos apoptóticos en el proceso de muerte celular [11-13, 75-78].
Se han descrito alteraciones en la familia de proteí-nas Bcl-2 y en el corte proteolítico de las procaspasas 1 y 3. Además se han detectado varios marcadores de muerte celular apoptótica en diferentes modelos expe-rimentales de epilepsia: las caspasas se activan por con-vulsiones así como los receptores de muerte neuronal y las proteínas de la familia del Bcl-2 [62, 76, 78-83]. Los niveles elevados de Bcl-2 en el suero de los pacientes con epilepsia del lóbulo temporal, se relacionan con la duración de la enfermedad, la frecuencia de las crisis y la severidad de la epilepsia [84].
El gen p53 fue el primer elemento regulador de la apoptosis identifi cado como dañado por la actividad convulsiva [85]. Se ha descrito su sobreexpresión, tanto de su ARNm como de la proteína, sustentada funcionalmente en que 1) la unión del ADN de p53 ocurre después de las convulsiones [86], y 2) la expre-sión de Bax aumenta con las convulsiones [57, 87]. Un inhibidor de la síntesis de p53 protege contra la excitotoxicidad provocada por el ácido kaínico [88]. Las neuronas de ratón defi cientes del p53 son resis-tentes a las convulsiones y a la apoptosis inducida por excitotoxinas [89]. No obstante, las consecuencias de las alteraciones en el p53 en la muerte neuronal indu-cida por convulsiones no están del todo esclareindu-cidas, debido sobre todo a las múltiples vías funcionales en las que el p53 está implicado. En general, los datos apuntan a que las caspasas, el Bcl-2 y el p53 ejercen alguna función después de las convulsiones.
Se ha discutido la clásica división apoptosis y ne-crosis, procesos que pueden ocurrir de forma indepen-diente, secuencial e incluso simultáneamente [79, 90]; muchas veces determinados por el tipo de estímulo y su intensidad. Se sugiere un modelo que explica la continuidad entre la vía clásica de apoptosis mediada por caspasas y la necrosis o lisis celular [91]. Los pa-sos intermedios que se plantean son 1) la muerte ce-lular programada, similar a la apoptosis, 2) la muerte celular independiente de las caspasas y 3) la muerte celular programada, similar a la necrosis. Este criterio es importante, en especial en el análisis de la muerte celular que ocurre en los procesos neurológicos [92].
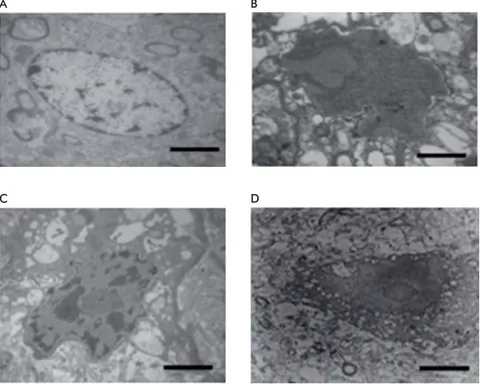
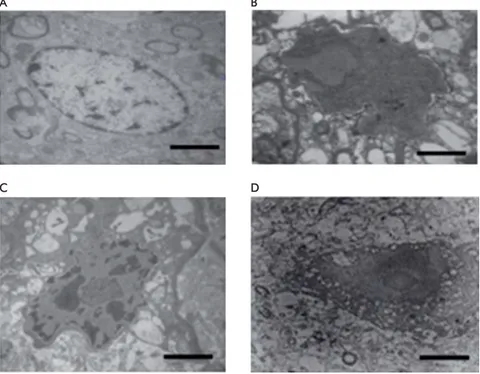
Un estudio de nuestro grupo de trabajo en pacien-tes con epilepsia del lóbulo temporal farmacorresis-tentes, avala la participación de los dos procesos de muerte neuronal (necrosis y apoptosis). Se evidenció el incremento de la inmunodetección con anexina V y ensayo Túnel en el tejido neocortical (Figura 2). Ello indicó la presencia de un proceso de muerte neuronal en esta área cerebral que podría ser apoptó-tica, sin descartar la posibilidad de muerte necróapoptó-tica, ya que el marcador Túnel+ se asocia con ambos ti-pos de muerte. También valoramos la ti-posibilidad de
una fase intermedia o de continuidad entre ambos tipos de muerte [93]. Adicionalmente, describimos la presencia de un desequilibrio del sistema redox en estos pacientes [94], que explicaría la muerte por disfunción mitocondrial, causada por la despolariza-ción de la membrana mitocondrial que ocasiona la muerte celular. En estudios posteriores por micros-copía electrónica en estos tejidos detectamos células tanto en proceso de muerte necrótica como apoptó-tica (Figura 3). Estas evidencias podrían ayudar en el desarrollo de estrategias neuroprotectoras contra los procesos de muerte celular que se desencadenan por epilepsia.
0 10 20 30 40 50
60 ***
Células Túnel + (%)
Paciente Control 0
10 20 30 40 50
60 **
Células anexina V+ (%)
Paciente Control
Figura 2. Determinación de inmunorreactividad por ensayo Túnel y contra anexina V, en pacientes epilépticos y controles. A) Comparación entre el porcentaje de células inmunorreactivas al ensayo Túnel en la capa IV de la neocorteza de pacientes con epilepsia del lóbulo temporal y un grupo control. El porcentaje de células Tunel+ se calculó en relación con el total de células teñidas con yoduro de propidio por milímetro cúbico visualizadas por doble tinción y microscopía confocal (prueba de Mann-Whitney, *** p ≤ 0.001). B) Comparación entre el porcentaje de células inmunorreactivas a la anexina V en la capa IV de la neocorteza de pacientes con epilepsia del lóbulo temporal y un grupo control (no epilépticos). El porcentaje de células anexina V+ se calculó en relación con el total de células teñidas con yoduro de propidio por milímetro cúbico, visualizadas por doble tinción y microscopía confocal (prueba Mann-Whitney, ** p ≤ 0.01).
33. Ullal G, Fahnestock M, Racine R. Time-dependent effect of kainate-induced seizures on glutamate receptor GluR5, GluR6, and GluR7 mRNA and Protein Expression in rat hippocampus. Epilepsia. 2005;46(5):616-23.
34. Vincent P, Mulle C. Kainate recep-tors in epilepsy and excitotoxicity. Neuroscience. 2009;158(1):309-23.
35. Lado FA, Laureta EC, Moshe SL. Seizure-induced hippocampal damage in the mature and immature brain. Epileptic Disord. 2002;4(2):83-97.
A B
C D
A B
C
onclusiones
Son múltiples los hallazgos en torno a la epilepsia, debido fundamentalmente a la diversidad de mode-los experimentales que se utilizan y a la difi cultad de reproducir fi elmente todas las características de la enfermedad. Los estudios en humanos se han efectua-do en diferentes localizaciones del foco epileptogé-nico, tiempo de evolución, tipo y edad de inicio de las crisis, entre otros aspectos. Se requieren nuevos estudios para caracterizar completamente la acción de estos mecanismos de muerte celular en los procesos convulsivos, y establecer una vía de interacción que atenúe el daño ocasionado por la epilepsia. La fi gura 4 resume los mecanismos de excitotoxicidad propuestos para las enfermedades neurológicas.
Las vías de señalización y la función de la excito-toxicidad se han estudiado exhaustivamente desde los años 70. Sin embargo, aún parece limitado el conoci-miento sobre la excitotoxicidad en el SNC, los meca-nismos moleculares y los sitios de acción. Con el fi n de
una intervención oportuna para retardar el desarrollo de afecciones como la epilepsia, deben ser cuidadosa-mente evaluados tanto la función esencial de la muerte neuronal como los mecanismos que se potencian con la sobreactivación de los receptores para glutamato en las enfermedades neurológicas. Los actuales hallazgos revelan que en la epilepsia farmacorresistente con-vergen procesos excitotóxicos y de muerte neuronal apoptótica y necrótica.
R
econocimientos
Agradecemos a los licenciados Leticia Neri Bazán y Héctor Vázquez Espinosa y a la Unidad de Inves-tigación Médica en Enfermedades Neurológicas del Hospital de Especialidades, Centro Médico Nacional Siglo XXI, perteneciente al Instituto Mexicano del Seguro Social (IMSS), de México, por su estimable contribución. Los resultados de nuestro grupo de tra-bajo han sido fi nanciados por el Conacyt (proyecto 98386).
36. Cherubini E, Ben-Ari Y, Krnjevic K. An-oxia produces smaller changes in synaptic transmission, membrane potential, and input resistance in immature rat hippocam-pus. J Neurophysiol. 1989;62(4):882-95.
37. Stafstrom CE, Holmes GL. Effects of uncontrolled seizures. Neural changes in animal models. Adv Exp Med Biol. 2002;497:171-94.
38. Marks JD, Friedman JE, Haddad GG. Vulnerability of CA1 neurons to glutamate is developmentally regulated. Brain Res Dev Brain Res. 1996; 97(2):194-206.
39. Albala BJ, Moshe SL, Okada R. Kainic-acid-induced seizures: a
devel-opmental study. Brain Res. 1984;315(1): 139-48.
40. Bender R, Baram TZ. Do prolonged fe-brile seizures injury hippocampal neurons? Insights from animal models. In: Baram TZ, Shinnar S. editors. Febrile seizures. San Diego, Academic Press. 2002. p. 583-7.
41. Berger ML, Tremblay E, Nitecka L, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat. III. Postnatal development of kainic acid binding sites in the limbic system. Neuroscience. 1984;13(4):1095-104.
42. Ben-Ari Y, Tremblay E, Riche D, Ghilini G, Naquet R. Electrographic, clinical and
pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping us-ing the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience. 1981;6(7):1361-91.
43. Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14(2):375-403.
44. Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injec-tions of kainic acid: a histological study. Neuroscience. 1980;5(6):991-1014.
Excitotoxicidad
Activación de receptores a glutamato
Estrés / retículo
endoplasmático Movilización de Ca Inestabilidad lisosomal
2+,
Na+, K+, Cl- ions mitochondrial Disfunción
Generación de
radicales libres quinasas y genes tempranosActivación de proteínas Activación de factores transcripcionales Liberación de proteasas
Autofagia Apoptosis Necrosis
Muerte neuronal Neurogénesis Gliosis
Plasticidad axonal y dendrítica Angiogénesis
Infl amación
Reorganización molecular
Figura 4. Mecanismos de excitotoxicidad en desórdenes neurológicos como la epilepsia. Modifi cado de Wang y Qin [2] y Pitkanen [95]. Las fl echas horizontales indican transiciones de procesos y las fl echas verticales dobles indican circuitos de amplifi cación de los procesos desencadenantes. Los receptores de glutamato incluyen la sobreactivación del receptor N-metil-D aspartato.
Lourdes Lorigados et al. Excitotoxicidad y muerte neuronal en la epilepsia
7
Biotecnología Aplicada 2013; Vol.30, No.145. Sloviter RS. The neurobiology of temporal lobe epilepsy: too much infor-mation, not enough knowledge. C R Biol. 2005;328(2):143-53.
46. Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: dose-relation-ship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338(2):289-95.
47. Anguelova E, Smirnova T. Differ-ential expression of small heat shock protein 27 in the rat hippocampus and septum after fimbria-fornix lesion. Neurosci Lett. 2000;280(2):99-102.
48. Valentim LM, Geyer AB, Tavares A, Cimarosti H, Worm PV, Rodnight R, et al. Effects of global cerebral ischemia and preconditioning on heat shock protein 27 immunocontent and phosphoryla-tion in rat hippocampus. Neuroscience. 2001;107(1):43-9.
49. Planas AM, Soriano MA, Estrada A, Sanz O, Martin F, Ferrer I. The heat shock stress response after brain lesions: induction of 72 kDa heat shock protein (cell types involved, axonal transport, transcriptional regula-tion) and protein synthesis inhibition. Prog Neurobiol. 1997;51(6):607-36.
50. Planas AM, Soriano MA, Ferrer I, Rodriguez Farre E. Kainic acid-induced heat shock protein-70, mRNA and pro-tein expression is inhibited by MK-801 in certain rat brain regions. Eur J Neurosci. 1995;7(2):293-304.
51. Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, et al. Gene therapy with HSP72 is neuroprotective in rat mod-els of stroke and epilepsy. Ann Neurol. 1998;44(4):584-91.
52. Pozas E, Ballabriga J, Planas AM, Fer-rer I. Kainic acid-induced excitotoxicity is associated with a complex c-Fos and c-Jun response which does not preclude either cell death or survival. J Neurobiol. 1997;33(3):232-46.
53. Gass P, Herdegen T. Neuronal ex-pression of AP1 proteins in excitotoxic neurodegenerative disorders and fol-lowing nerve fiber lesions. Progr Neurobiol. 1995;47(4-5):257-90.
54. Kasof GM, Mandelzys A, Maika SD, Hammer RE, Curran T, Morgan JI. Kainic acid-induced neuronal death is associated with DNA damage and a unique immediate early gene response in c-fos-lacZ transgenic rats. J Neurosci. 1995;15(6):4238-49.
55. Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91(7):917-25.
56. Tan Z, Levid J, Schreiber SS. Increased expression of Fas (CD95/APO-1) in adult rat brain after kainate-induced seizures. Neuroreport. 2001;12(9):1979-82.
57. Gillardon F, Wickert H, Zimmermann M. Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci Lett. 1995;192(2): 85-8.
58. Lopez E, Pozas E, Rivera R, Ferrer I. Bcl-2, Bax and Bcl-x expression follow-ing kainic acid administration at con-vulsant doses in the rat. Neuroscience. 1999;91(4):1461-70.
59. Gillardon F, Klimaschewski L, Wickert H, Krajewski S, Reed JC, Zimmermann M. Expression pattern of candidate cell death
effector proteins Bax, Bcl-2, Bcl-X, and c-Jun in sensory and motor neurons following sciatic nerve transection in the rat. Brain Res. 1996;739(1-2):244-50.
60. Faherty CJ, Xanthoudakis S, Smeyne RJ. Caspase-3-dependent neuronal death in the hippocampus following kainic acid treatment. Brain Res Mol Brain Res. 1999;70(1):159-63.
61. Ferrer I, Lopez E, Blanco R, Rivera R, Krupinski J, Marti E. Differential c-Fos and caspase expression following kainic acid excitotoxicity. Acta Neuropathol. 2000;99(3):245-56.
62. Henshall DC, Chen J, Simon RP. Involvement of caspase-3-like protease in the mechanism of cell death fol-lowing focally evoked limbic seizures. J Neurochem. 2000;74(3):1215-23.
63. Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation-induced status epilepticus in the mouse. J Comp Neurol. 2009;515(2):181-96.
64. Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fi ber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J Comp Neurol. 2006;494(6):944-60.
65. Tauck DL, Nadler JV. Evidence of func-tional mossy fi ber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5(4): 1016-22.
66. Bender R, Dubé C, Baram TZ. Mossy fiber sprouting into the inner molecu-lar layer of the dentate gyrus follows prolonged febrile seizures in immature rat model. Epilepsia. 2000;41(suppl 7): 76-9.
67. Bendotti C, Pende M, Samanin R. Expression of GAP-43 in the granule cells of rat hippocampus after seizure-induced sprouting of mossy fi bres: in situ hybridiza-tion and immunocytochemical studies. Eur J Neurosci. 1994;6(4):509-15.
68. Boschert U, O’Shaughnessy C, Dick-inson R, Tessari M, Bendotti C, Catsicas S, et al. Developmental and plasticity-related differential expression of two SNAP-25 isoforms in the rat brain. J Comp Neurol. 1996;367(2):177-93.
69. Geddes JW, Hess EJ, Hart RA, Kes-slak JP, Cotman CW, Wilson MC. Le-sions of hippocampal circuitry define synaptosomal-associated protein-25 (SNAP-25) as a novel presynaptic marker. Neuroscience. 1990;38(2):515-25.
7 0 . N u m a k a w a T, M a t s u m o t o T, Numakawa Y, Richards M, Yamawaki S, Kunugi H. Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration. J Toxicol. 2011;2011:405194.
71. Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF func-tion and intracellular signaling in neurons. Histol Histopathol. 2010;25(2):237-58.
72. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516.
73. Fujikawa DG, Shinmei SS, Zhao S, Aviles ER, Jr. Caspase-dependent programmed cell death pathways are not activated in generalized seizure-induced neuronal death. Brain Res. 2007;1135(1):206-18.
74. Uysal H, Cevik IU, Soylemezoglu F, Elibol B, Ozdemir YG, Evrenkaya T, et al. Is the cell death in mesial temporal scle-rosis apoptotic? Epilepsia. 2003;44(6): 778-84.
75. Uysal H, Cevik IU, Soylemezoglu F, Elibol B, Ozdemir YG, Evrenkaya T, et al. Is the cell death in mesial temporal sclerosis apop-totic? Epilepsia. 2003;44(6):778-84.
76. Narkilahti S, Nissinen J, Pitkanen A. Administration of caspase 3 inhibitor during and after status epilepticus in rat: effect on neuronal damage and epilepto-genesis. Neuropharmacology. 2003;44(8): 1068-88.
77. Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63(1):7-18.
78. Sloviter RS, Dean E, Sollas AL, Good-man JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366(3): 516-33.
79. Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. Neuroreport. 1995;7(1):61-4.
80. Roy M, Hom JJ, Sapolsky RM. HSV-mediated delivery of virally derived anti-apoptotic genes protects the rat hippocam-pus from damage following excitotoxicity, but not metabolic disruption. Gene Ther. 2002;9(3):214-9.
81. Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, et al. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuro-pathol Exp Neurol. 2006; 65(3):217-25.
82. Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, et al. Caspase-3 cleavage and nuclear localization of cas-pase-activated DNase in human temporal lobe epilepsy. J Cereb Blood Flow Metab. 2006;26(4):583-9.
83. Yamamoto A, Schindler CK, Murphy BM, Bellver-Estelles C, So NK, Taki W, et al. Evidence of tumor necrosis factor receptor 1 signaling in human temporal lobe epilepsy. Exp Neurol. 2006;202(2):410-20.
84. Kilany A, Raouf ER, Gaber AA, Aloush TK, Aref HA, Anwar M, et al. Elevated serum Bcl-2 in children with temporal lobe epi-lepsy. Seizure. 2012;21(4):250-3.
85. Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associ-ated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91(16):7525-9.
86. Liu H, Cao Y, Basbaum AI, Mazarati AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachy-kinin A gene. Proc Natl Acad Sci USA. 1999;96(21):12096-101.
87. Lopez-Meraz ML, Wasterlain CG, Rocha LL, Allen S, Niquet J. Vulnerability of postna-tal hippocampal neurons to seizures varies regionally with their maturational stage. Neurobiol Dis. 2010;37(2):394-402.
89. Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16(4):1337-45.
90. Zeiss CJ. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol. 2003;40(5): 481-95.
91. Schmechel DE. Apoptosis in neu-rodegenerative disorders. In: Hannum
YA, Boustany RM, editors. Apoptosis in neurobiology. Washington DC: CRC Press; 1999. p. 23-48.
92. Martin LJ. Neuronal cell death in ner-vous system development, disease, and injury (Review). Int J Mol Med. 2001;7(5): 455-78.
93. Lorigados L, Orozco S, Morales L, García I, Estupiñán B, Bender JE, et al. Muerte neuronal en la neocorteza de pacientes con epilepsia del lóbulo
tem-poral resistente a fármacos. Neurología. 2008;23(9):555-65.
94. Lopez J, Gonzalez ME, Lorigados L, Morales L, Riveron G, Bauza JY. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin Biochem. 2007;40(5-6):292-8.
95. Pitkanen A. Drug-mediated neuropro-tection and antiepileptogenesis: animal data. Neurology. 2002;59(9 Suppl 5): S27-33.
Corresponding author
Excitotoxicity and neuronal death in epilepsy
Lourdes Lorigados
1, Sandra Orozco
2, Lilia Morales
3, Bárbara Estupiñán
3,
Iván García
4, Luisa Rocha
5 1 Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Cirén Ave. 25, No. 15805 e/ 158 y 160 Playa, CP 11300, La Habana, Cuba 2 Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, IMSS México 3 Departamento de Neurofisiología, Cirén 4 Servicio de Neurocirugía, Cirén 5 Laboratorio Farmacobiología, Centro de Investigación y de Estudios Avanzados, CinvestavSede Sur, DF, México E-mail: [email protected]
ABSTRACT
Epilepsy is a recurrent, often progressive neurological disorder with a chronic evolution, affecting 1 to 2 % of the world population. Research with experimental models and imaging analysis of diseased patients have been used to show that recurrent episodes produce oxidative stress, most of which is related to neuronal excitability phenomena. It is known that the excessive stimulation of glutamate receptors results in neurotoxicity; a process that, under the denomination of excitotoxicity, is thought to constitute the principal cellular death mechanism behind different dis-orders of the central nervous system, including epilepsy. Paradoxically, although the signaling pathways, molecular mechanisms and sites of action of excitotoxicity have received considerable attention since the 1970s, little is known about their relevance to CNS disorders. Further detail is necessary about the fundamental role of neuronal death and the mechanisms, particularly those relevant to neurological pathogenesis, that are engaged whenever gluta-mate receptors are excessively stimulated, as the results would aid considerably the development of timely clinical interventions delaying the evolution of these disorders. We review clinical and experimental data on the relevant alterations of the glutamatergic system, cell death pathways, and the activation of caspases and members of the Bcl-2 gene family involved in the process as modulators of cell death during epilepsy. The fi ndings support the hy-pothesis that excitotoxic processes as well as both apoptotic and necrotic neuronal cell death phenomena converge in drug-resistant epilepsy.
Keywords: excitotoxicity, apoptosis, necrosis, epilepsy
Biotecnología Aplicada 2013;30:9-16
RESUMEN Excitotoxicidad y muerte neuronal en la epilepsia. La epilepsia es una afección neurológica de evolución crónica, recurrente, casi siempre progresiva, que afecta del 1 al 2 % de la población mundial. Modelos experimentales y estudios de imágenes neurológicas de pacientes con este padecimiento muestran que las crisis recurrentes provo-can estrés oxidativo, relacionado fundamentalmente con la excitabilidad neuronal. La estimulación excesiva de los receptores de glutamato induce neurotoxicidad, un proceso que se ha defi nido como excitotoxicidad. Se considera que este puede ser el principal mecanismo de muerte celular en numerosas afecciones del sistema nervioso central, incluida la epilepsia. Desde los años 70 se han estudiado con profundidad las vías de señalización, los mecanis-mos moleculares y los sitios de acción relacionados con la excitotoxicidad; aunque de forma muy limitada en las enfermedades del sistema nervioso central. En particular, deberán evaluarse con especial cuidado la función crucial de la muerte neuronal y los mecanismos que se potencian con la sobreactivación de los receptores de glutamato, principalmente los relativos a las enfermedades neurológicas, con el fi n de intervenir de manera oportuna para retardar el desarrollo de estas afecciones neurológicas. Se repasan las evidencias clínicas y experimentales sobre las alteraciones del sistema glutamatérgico, las vías de muerte celular, la activación de las caspasas y de la familia de genes Bcl-2 involucrados, como moduladores de la muerte celular en la epilepsia. Tales hallazgos sustentan que en la epilepsia farmacorresistente convergen procesos excitotóxicos y de muerte neuronal apoptótica y necrótica.
Palabras clave: excitotoxicidad, apoptosis, necrosis, epilepsia
I
ntroduction
Glutamate receptor-mediated excitotoxicity not only plays an important role in neural development, differ-entiation and synaptic plasticity [1, 2], but is regarded as the principal mechanism for cell death in a num-ber of disorders of the central nervous system (CNS), including brain trauma, neurodegenerative disorders and epilepsy [3-6].
Glutamate is the ultimate excitatory neurotrans-mitter of mammalian CNS. Accurate control of
glu-tamatergic neurotransmission is of paramount impor-tance, due to its involvement in both excitotoxic cell death and neural signaling [2]. Early descriptions of excitotoxicity-mediated cell death mentioned increas-es in cell volume, vacuolization of the cytoplasm and loss of membrane integrity; all of which are consistent with a necrotic mechanism for this event [7-10]. Later evidence, however, has demonstrated that this process can also be associated with apoptotic hallmarks such
REVIEW
1. Yang JL, Sykora P, Wilson DM, 3rd, Mattson MP, Bohr VA. The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resil-iency. Mech Ageing Dev. 2011;132(8-9): 405-11.
s
as the degradation of DNA at internucleosomal sites and the activation of caspases [11-13]. In addition, re-cent publications have pointed at autophagy, induced as the result of sustained cellular stress, as the mecha-nism behind excitotoxicity-induced non-apoptotic cell death [2]. Increased glutamate receptor activity levels would, therefore, induce the expression of pro-apop-totic proteins such as p53, leading to cell damage and death mediated by apoptosis or autophagy [14-16]. The latter would be induced as a response to acute excitotoxic damage [17, 18].
Despite the large body of knowledge accrued on the signaling pathways and the sequence of events taking place during excitotoxicity, little is known about its role in the CNS or the molecular mechanisms underly-ing its effects. It is clear, however, that far from beunderly-ing a uniform process, excitotoxic cell death in the brain actually represents a continuum going from necrosis to apoptosis and autophagy. This review discusses the cellular and molecular mechanisms of excitotoxicity and its effect on the process of neuronal death taking place during epilepsy.
C
oncept of excitotoxicity
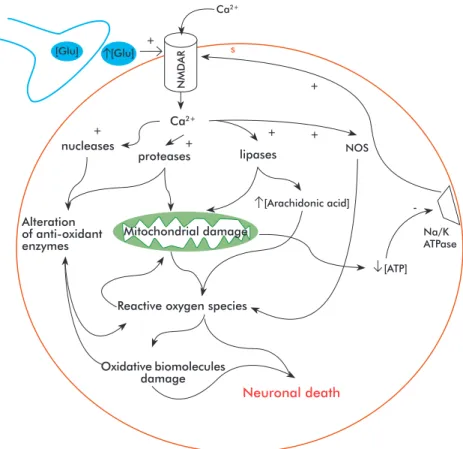
A number of different experimental and clinical find-ings on the potential toxicity of excitatory amino acids have provided the foundation for excitotoxic theory, which postulates the existence of a direct link between neuronal degeneration and glutamate receptor hyper-sensitivity or excessive levels of endogenous gluta-mate [19]. Excitotoxicity is therefore a mechanism promoting cell death through the hyper activation of glutamatergic receptors or its analogues. This hyper activation leads to excess calcium (Ca2+) inflow to the cell, where this ion is sequestered inside mitochon-dria, leading to metabolic dysfunction, the generation of free radicals, the activation of proteases, phospho-lipases, endonucleases, nitric oxide synthase, and the inhibition of protein synthesis [20].
For calcium homeostasis to be lost, regulatory mechanisms for this ion, including the calcium pump, the sodium/calcium (Na+/Ca2+) exchanger and calci-um buffering proteins, must first be overflowed. Once these systems saturate, excess calcium accumulates inside the mitochondrial matrix. This accumulation depolarizes the mitochondrial membrane by two dif-ferent mechanisms: first, the increased concentration of positive ions in the mitochondrial matrix decreases the chemo-osmotic potential across the membrane (leading in turn to reduced rates of adenosine triphos-phate (ATP) synthesis), and second, the activation of mitochondrial transition pores (a mechanism normally used to shunt calcium back to the cytosol), which can lead to irreversible membrane depolarization if cal-cium unbalance is prolonged [21, 22]. High calcal-cium concentrations in the mitochondrial matrix also pro-mote the generation of free radicals, which propro-mote the peroxidation of membrane lipids, the synthesis of nitric oxide and the activation of enzymes involved in the catabolism of proteins, phospholipids and nucleic acids. In addition, nitric oxide can act as a retrograde messenger, further contributing to the excitotoxic ef-fect of glutamate by enhancing its release from pre-synaptic terminals [23] (Figure 1).
Additional contributions to cellular damage are pro-vided by the activation of nitric oxide synthase, whose
reaction products react with superoxide anions to yield peroxynitrite, and the activation of poly-adenos-ine diphosphate ribose-polymerase (PARP), triggered by free radical-mediated DNA damage [24, 25].
E
xcitotoxicity and epilepsy
There is evidence supporting the hypothesis that the neurodegenerative changes associated with human epilepsy arise from persistent discharges in the glu-tamate pathway. The mechanism is relatively simple: excess glutamate release leads to repeated depolariza-tion-repolarization cycles in glutamate terminals, un-til glutamate reaches toxic concentrations and, finally, the excitotoxic degeneration of post-synaptic neurons takes place [26, 27].
Micro-dialysis studies in humans and animal mod-els have demonstrated an association between pro-longed convulsive activity and the duration of the epileptic episode due to significant increases in gluta-mate levels [28]. It is well known that neuronal over excitation by glutamate can trigger epileptic seizures, and that the effect of directly applying glutamate to the amygdala is similar to that of propagated activa-tion [29]. Using agonists of the α-amino-3-hydroxy-5-methyl-4-isoxazolpropionic acid receptor has been
[Glu] [Glu] +
NMD
AR
Ca2+
+
+ +
NOS +
+
Ca2+
proteases nucleases
Alteration of anti-oxidant enzymes
Mitochondrial damage lipases
[Arachidonic acid]
-Na/K ATPase
[ATP]
Reactive oxygen species
Oxidative biomolecules damage
Neuronal death
Figure 1. Mechanism for excitotoxicity. The sustained activation of N-methyl-D aspartate receptors (NMDAR) due to abnormally high glutamate (Glu) concentrations leads to a massive inflow of calcium to the cell, which activates lytic enzymes as well as nitric oxide synthase (NOS). Mitochondrial damage, together with increased concentrations of arachidonic acid, increases the generation of reactive oxygen species, eventually leading to cell death due to biomolecule damage and the activation of apoptotic death programs. Energy deficits also contribute to the degenerative process, perpetuating membrane depolarization by cutting short energy supply to the sodium/potassium pump (Na/K ATPase) and keeping NMDAR in an active state, thus sensitizing the cell to the normal glutamatergic afferences of the brain cortex.
3. Severino PC, Muller Gdo A, Van-dresen-Filho S, Tasca CI. Cell sig- naling in NMDA preconditioning and neuroprotection in convulsions induced by quinolinic acid. Life Sci. 2011;89(15-16):570-6.
4. Araujo IM, Carreira BP, Carvalho CM, Carvalho AP. Calpains and de-layed calcium deregulation in ex-citotoxicity. Neurochem Res. 2010; 35(12):1966-9.
5. Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA recep-tor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104(6):3551-6.
6. Farooqui AA, Ong WY, Horrocks LA. Glutamate receptors and their
association with other neurochemical
parameters in excitotoxicity. In: Farooqui AA, Ong WY, Horrocks LA, editors.
Neu-rochemical aspects of excitotoxicity. New
Lourdes Lorigados et al. Excitotoxicity and neuronal death in epilepsy
11
Biotecnología Aplicada 2013; Vol.30, No.1shown to delay the development of propagated activa-tion at the amygdala in mice [30].
The activation of N-methyl-D-aspartate receptors is a mediator of cell death during the epileptic state [31], and the use of MK-801, an antagonist for this receptor, prevents the occurrence of spontaneous sei-zures in animal models [32]. Kainate receptors,
spe-cifi cally the GluR6 subunit, are known to participate
in epileptogenesis as inducers [33, 34].
In general, excitotoxic damage to the neurons of epileptic patients is mediated by excessive calcium
in-fl ow during seizures [35]. The resulting high levels of
calcium trigger a sequence of events that includes the activation of nitric oxide synthase, thereby interfering with oxidative metabolism and generating free radi-cals that ultimately damage the neuronal membrane. Pro-caspases are activated likewise, and neuronal death eventually takes place by necrosis, apoptosis or autophagy.
E
xcitotoxicity in experimental models
of epilepsy
The limitations for studying epilepsy in humans have led to the development of experimental models repro-ducing this condition. It should be noticed, however, that existing models fail to accurately reproduce the behavioral manifestations of this disease, especially in the case of motor alterations.
Animal epilepsy models are classifi ed as either
acute or chronic. The former are implemented through the delivery of convulsant drugs or the application of electrical stimulation; the latter, while harder to im-plement, provide a closer approximation to the phys-iopathology of this disorder in humans, although both are capable of producing partial and generalized sei-zures. In addition, true epilepsy models reproduce the recurrence of ictal manifestations characterizing this disease in humans. The ultimate challenge when using experimental models to study epilepsy is to determine which, of the many alterations stemming from a
spe-cifi c brain injury, is causally linked to the subsequent
development of epilepsy.
Oxidative stress plays an important role in cellu-lar damage and death induced by recurrent seizures. The free radicals generated during oxidative stress have long been acknowledged as part and parcel of excitotoxicity, due in part to the fact that prolonged, sustained seizures generate macromolecular damage in the cells that is related, above all, to neuronal excit-ability.
Animal models have been used to study two specif-ic types of epileptspecif-ic events: prolonged (20 to 30 min) febrile crises and long epileptic seizures (5 to 6 h), the latter induced through the systemic delivery of cho-linergic agonists (pilocarpine) or by unilateral injec-tion of glutamatergic agonists into the hippocampus of experimental rats (kainic acid, a glutamate analog). Using models of febrile crises, neonatal hypoxia and spasms, it has been possible to demonstrate that devel-oping neurons are less vulnerable to cellular damage and survive much better than fully grown neurons. For instance, the hippocampal neurons of animals with immature brains placed under an anoxic environment continue to react to synaptic stimuli for a longer time and require more prolonged exposures to completely
and irreversibly destroy their neural circuits [36]. Immature brains also appear to be more resistant to the toxic effects of glutamate than mature ones [37].
Mark et al. [38] demonstrated that the amount of
cal-cium entering a pyramidal neuron is directly related
to the animal’s age: during the fi rst three days of life,
glutamate increases calcium concentration only by marginal amounts; however, in days 21 to 25 there is a marked increase in intracellular calcium concentra-tion and soma volume, while dendrites retract. This relatively higher resistance stems from a lower densi-ty of active synapses, lower energy requirements and, in general, the relative immaturity of the biochemical cascades leading to cell death, explaining why young individuals are less vulnerable than adults to the cell loss taking place after prolonged epileptic seizures [39-41].
The most popular models of excitotoxicity em-ploying adult animals are those based on the use of kainic acid and pilocarpine. These are models of epi-lepsy of the temporal lobe, induced by the unilateral or systemic injection of these compounds at convul-sant doses, causing excitotoxic damage at the pyra-midal neurons of hippocampus and the hilar region. Damage depends on dosage, species and line of the animal, but the result in all cases is neuronal death at vulnerable regions, the proliferation of astrocytes and
increased glial fi bers. For these reasons, the models
based on systemically administering kainic acid or pilocarpine are widely used for studying generalized tonic-clonic convulsions or the epileptic state, whose neuroanatomical substrate is temporal mesial sclero-sis [42-46].
One of the fi rst changes taking place after the
in-jection of kainic acid is the induction of messenger RNA (mRNA) coding for heat shock proteins (HSPs) of varying molecular weights (HSP27, HSP70 and HSP72), whose expression levels increase conse-quently. HSP72, particularly, is constitutively ex-pressed in the mammalian brain, and exhibits in-creased concentrations among sensitive neuronal populations of the hippocampus [47]. The expression of these chaperones seems to prevent the misfolding of newly synthesized proteins in kainic acid-vulner-able populations. During the two weeks following administration of the convulsant, these proteins are transported to the most distal zones along dendrites and axons. Both HSP70 and HSP72 have been shown to play a protective role in this process, although they are unable to rescue damaged cells from excito-toxic death. While the overexpression of HSP27 and
HSP70 in vivo protects from excitotoxic damage [47,
48], excessively high levels of HSP72 can be noxious to the cell [49-51].
Three to fi ve hours after the injection of kainic acid,
mRNA coding for cFos and cJun are also induced, increasing the concentrations of their respective pro-teins at vulnerable regions of the hippocampus and dentate gyrus [52]. cFos immunoreactivity at the den-tate gyrus disappears after six hours, but remains high at the hippocampus, suggesting an association of cell death with high cFos levels. However, a prolonged increase in cFos is poorly predictive, and is not a nec-essary condition for neuronal excitotoxic damage to take place [53, 54]. cJun levels have also been found
7. Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures pro-duce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98(1):41-53.
8. Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000; 41 Suppl 6:S9-13.
9. Ebert U, Brandt C, Loscher W. Delayed sclerosis, neuroprotection, and limbic epileptogenesis after status epilepticus in the rat. Epilepsia. 2002;43 Suppl 5:86-95.
10. Kubova H, Druga R, Lukasiuk K, Su-chomelova L, Haugvicova R, Jirmanova I, et al. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J Neurosci. 2001;21(10):3593-9.
11. Bengzon J, Mohapel P, Ekdahl CT, Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog Brain Res. 2002;135:111-9.
12. Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, et al. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22(19):8458-65.
13. Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69(2):103-42.
14. Dong XX, Wang Y, Qin ZH. M o l e c u l a r m e c h a n i s m s o f e x-citotoxicity and their relevance to pathogenesis of neurodegenerative dis-eases. Acta Pharmacol Sin. 2009;30(4): 379-87.
15. Qin ZH, Tao LY, Chen X. Dual roles of NF-kappaB in cell survival and implications of NF-kappaB in-hibitors in neuroprotective therapy. Acta Pharmacol Sin 2007;28(12):1859-72.
16. Zhang XD, Wang Y, Zhang X, Han R, Wu JC, Liang ZQ, et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5(3):339-50.
17. Shacka JJ, Lu J, Xie ZL, Uchi-yama Y, Roth KA, Zhang J. Kainic acid induces early and transient au-tophagic stress in mouse hippocampus. Neurosci Lett. 2007;414(1):57-60.
18. Wang Y, Dong XX, Cao Y, Liang ZQ, Han R, Wu JC, et al. p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur J Neurosci. 2009; 30(12):2258-70.
![Figura 4. Mecanismos de excitotoxicidad en desórdenes neurológicos como la epilepsia. Modifi cado de Wang y Qin [2] y Pitkanen [95]](https://thumb-us.123doks.com/thumbv2/123dok_es/6481217.219052/6.918.64.666.414.808/figura-mecanismos-excitotoxicidad-desórdenes-neurológicos-epilepsia-modifi-pitkanen.webp)
![Figure 4. Excitotoxicity mechanisms in neurological disorders such as epilepsy, modified from Wang & Qin [2] and Pitkanen [95]](https://thumb-us.123doks.com/thumbv2/123dok_es/6481217.219052/14.918.63.665.349.748/figure-excitotoxicity-mechanisms-neurological-disorders-epilepsy-modified-pitkanen.webp)
