Inclusion complexes of a bichromophoric diester containing anthracene
and naphthalene groups with
␣
- and

-cyclodextrins:
thermodynamics and molecular mechanics
Leticia Serna, Antonio Di Marino, Francisco Mendicuti
∗Departamento de Qu´ımica F´ısica, Universidad de Alcal´a, 28871 Alcal´a de Henares, Madrid, Spain
Received 25 June 2004; accepted 28 July 2004
Abstract
Fluorescence and molecular mechanics have been used to study the inclusion complexes of the (9-anthryl) COO (CH2)2 OOC
(2-naphthyl) bichromophoric compound with␣- and-cyclodextrins. Emission spectra upon excitation of the naphthalene group denote the presence of non-radiative energy transfer from naphthalene to anthracene, which is influenced by the type of CD. Naphthalene emission also shows two peaks whose ratio of intensities R is sensitive to the medium polarity. The stoichiometry, the formation constants and the changes of enthalpy and entropy upon inclusion of complexes formed were obtained from the change of R with CD concentration and temperature. Both complexes, in agreement with Job’s plots, show 1:1 stoichiometry. Quenching, fluorescence depolarization and the analysis of R when all the guests are complexed permit us to explain the possible location of CDs in the complexes formed. Molecular mechanics calculations were also employed to study the formation of 1:1 complexes with both␣- andCDs. The study was mainly performed in the presence of water as a solvent. Results seem to explain the stoichiometries and geometry for both complexes.
© 2004 Elsevier B.V. All rights reserved.
Keywords: Cyclodextrins; Inclusion complex; Fluorescence; Energy transfer; Molecular mechanics
1. Introduction
Cyclodextrins (CDs) are naturally occurring container type host molecules made up glucopyranose units connected at the carbon atoms one and four. They are well-known for their ability to form inclusion complexes with small molecules and polymers [1–4]. Complexation is reversible in solution and depends on the polarity, size and shape of the guest molecule relative to the host hydrophobic inter-nal cavity. However, many uncertainties about the driving forces of the inclusion process nowadays remain unclear [1,2]. Since many guest compounds present fluorescent prop-erties, the change of these properties upon complexation can be used to obtain stoichiometry, association constants and thermodynamics parameters accompanying the complexa-tion[1,2,5–16]. Molecular modeling calculations[15,17–26]
∗Corresponding author. Tel.: +34 91 8854672; fax: +34 91 8854763.
E-mail address: [email protected] (F. Mendicuti).
strengthens experimental results, clarifies the complexation mechanism and provides information on the complexation driving forces.
Steady-state fluorescence was used in a previous study on the energy transfer from N to A in AxMN bichromophoric compounds, where A and N are 9-anthroate and 2-naphthoate chromophores; xM is an spacer formed by x = 2–6 (or 1–4) methylene[27](or oxyethylene)[28]groups. Dilute solution experiments on these compounds, demonstrated the presence of non-radiative singlet intramolecular energy transfer (IET) whose efficiency, which decreases with x, depends on the type of spacer and the solvent used. The first member of both series, A2MN, when measured in different media always showed efficiencies for IET that were higher than 0.8. The combination of the experimental results and the theoretical analysis gave F¨orster radius for the IET from N to A of 1.6± 0.2 nm. More recently we also reported thermodynamics and molecular mechanics (MM) studies on the complexation of 2-methyl naphthoate (MN) with␣-,- and␥CDs[14,19,21]
and 9-methyl anthroate (MA) withCD[15]. The analysis always revealed the existence of complexes of 1:1 stoichiom-etry with different stabilities. MN and MA are model com-pounds for the chromophores placed at both ends of AxMN compounds.
In this work we investigate the complexation of the A2MN bichromophoric compound with␣- andCDs. Stoichiome-tries, association constants, enthalpy and entropy changes upon complexation were obtained from analysis of fluores-cence measurements. Experimental results were discussed together with the MM calculations performed in the pres-ence of water. Quenching and fluorescpres-ence depolarization measurements contributed to elucidating the geometry of the complexes formed.
2. Materials and methods
2.1. Reagents
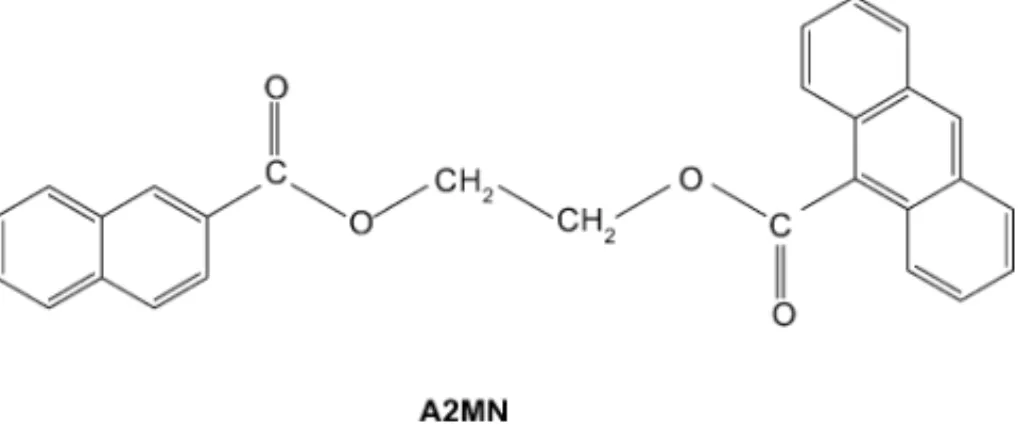
The synthesis and characterization of M2MN, depicted in Fig. 1, was carried out as described elsewhere [27,28]. The preparation of 2-methyl naphthoate (MN) and 9-methyl-anthroate (MA) model compounds was also reported[27]. Both CDs were purchased from Aldrich. The␣CD was used as received and the CD was purified by recrystallization (2×) in deionized water (Milli-Q). Karl Fisher analysis for␣ -andCD reveals water contents by mass of 10% and 12.5%, respectively. Deionized water and other solvents, n-alcohols H(CH2)nOH with n = 1–6 (Aldrich spectrophotometric grade or higher than 98%) were checked for impurities by fluores-cence before using.
2.2. CD solutions
All A2MN/CD solutions were prepared (by weighting) in the same quartz cuvettes employed to perform the spectro-scopic measurement, using an A2MN guest saturated aque-ous solution. This was done by vigoraque-ously stirring the guest for 48 h in water and then filtering it (2×) through Teflon fil-ters (Millipore,∅1m size) giving a [A2MN]≈10−7M. The cuvettes were then sealed with Teflon stoppers and the con-tents were stirred for another 24 h. Concentrations of␣- and
Fig. 1. Structure of the bichromophoric compound denoted by A2MN.
CD ranged from 0 to 20.43 mM and from 0 to 16.16 mM, respectively.
2.3. Apparatus
Steady-state fluorescence measurements were performed by using an SLM 8100 AMINCO spectrofluorometer equipped with a Xenon lamp, a double (single) concave grat-ing monochromator at the excitation (emission) path, two Glan-Thompson polarizers in both paths (fixed at the magic angle, except for polarization measurements) and a photo-multiplier cooled by a Peltier system. Slit widths were 8 nm for excitation and emission. Measurements were made with right angle geometry. Most of the experiments were carried out in the 5–45◦C temperature range, at 10◦C intervals (Hu-ber, ministat and Pt100 probe).
2.4. Computational details
Molecular mechanics calculations (MM) were performed with Sybyl 6.9[29]and the Tripos Force Field[30]. The sum of six contributions, bond stretching, angle bending, torsion, van der Waals, electrostatics and out-of-plane were used to calculate potential energy. A relative permittivityε= 3.5 and a function of the distance,ε= ε0r (whereε0 = 1 and r is the interatomic distance) were used for electrostatics interac-tions in the vacuum and in the presence of water, respectively. A2MN geometry and charges were obtained by MOPAC[31]. Geometry and charges for water and CDs were identical to those previously used[19–26]. Non-bonded cut-off distances for van der Waals and electrostatics interactions were set at 8 ˚A. Minimization was performed by the simplex algorithm and the conjugate gradient was used as a termination method with gradients of 0.2 kcal/mol ˚A (3.0 kcal/mol ˚A) for the cal-culations performed in vacuum (water) respectively[32,33]. Water solvation was achieved by using the molecular sil-verware algorithm (MS)[34]. Periodic boundary conditions (PBC) were also employed. Binding energy or any contribu-tion to this energy were obtained as the difference between the potential energy of the guest:host system and the sum of the potential energies of the isolated guest and host in the same structure. The strain energy of CDs was obtained as the sum of torsional, stretching and bending energies. An hydro-gen bonds (HB) is assumed when the distance between the hydrogen (H) bonded to a donor (D) and the acceptor (A) is in the 0.8–2.8 ˚A range and the angle D–H–A is larger than 120◦.
3. Results and discussion
3.1. Fluorescence of A2MN and an equimolecular MN and MA mixture
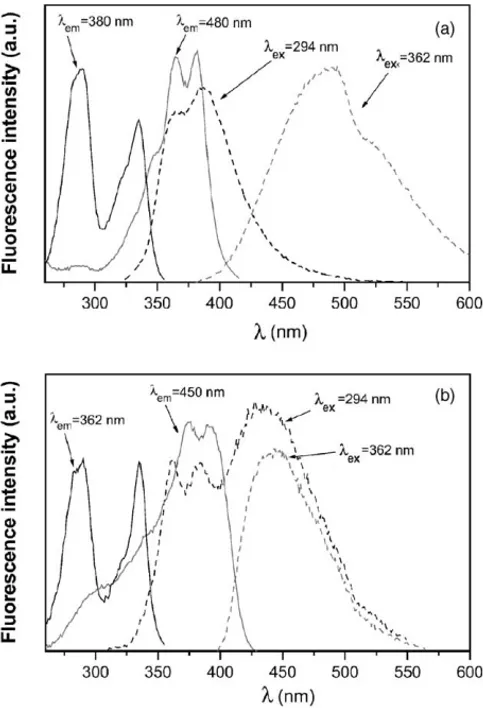
Fig. 2. (a) Excitation and emission (dashed lines) spectra at 25◦C, for an aqueous dilute solution of an approximately equimolecular MA + MN mix-ture monitored at the wavelengths of emission and excitation showed. (b) Idem for an aqueous dilute solution of A2MN in the absence of CDs.
were recorded upon excitation where the naphthoate (N) and anthroate (A) groups are preferentially excited, i.e., at 294 and 362 nm, respectively, showed obviously typical bands from N (peaks centered at∼380 and∼360 nm) and A (sin-gle band at∼480 nm), respectively. Emission spectra from a water solution of A2MN, as depicted inFig. 2(b), exhibit two main features with respect to the previous system: (a) band from A is shifted to the blue and is subsequently centered at
∼450 nm upon 362 nm of excitation; (b) the emission is a combination of bands from N and A when the excitation is selected at 294 nm. This is good evidence of the intramolec-ular energy transfer (IET) from naphthalene to anthracene in A2MN. The top panel ofFig. 2 also depicts the excita-tion spectra for the MA and MN mixture, monitored at 380 and 480 nm, corresponding to the maximum of the direct emission from N and A groups. Spectra are similar to those observed for isolated MN and MA with peaks (p) and shoul-ders (s) centered at 294 nm (p),∼320 nm (s) and∼335 nm (p) for MN and∼335 nm (s),∼360 nm (p),∼380 nm (p) for MA. Excitation spectra for A2MN in the same solvent upon selecting the emission of N (362 nm), as illustrated in the bottom panel, showed characteristics similar to those of MN
aqueous solution. However, a broadening of the typical bands of MA to the blue appears upon selection of the wavelength of the maximum of A emission (450 nm). This broadening corresponds to the excitation of MN, which constitutes ad-ditional evidence that IET takes place in the naphthalene to anthracene direction.
3.2. Fluorescence of A2MN in the presence of CDs
Excitation spectra of water A2MN guest solutions in the presence of ␣- or CD upon emission of 362 or 450 nm show characteristics similar to those of spectra for isolated A2MN. The excitation spectra obtained in the presence of CD upon selecting 450 nm also denotes the typical broad-ening to the blue which suggests the occurrence of IET. No significant shifts in the wavelengths of peaks and shoulders were observed upon changing [CD] or the type of CD. Nev-ertheless, changes in the ratios of intensity of the bands were observed.
Fig. 3shows the emission spectra uponλexc= 294 nm for aqueous A2MN and A2MN/CD solutions at different [CD] at 5◦C. Both groups of spectra illustrated bands due to N and A, which means that IET is also present when CD is added to the medium. The intensity of the broad band centered at
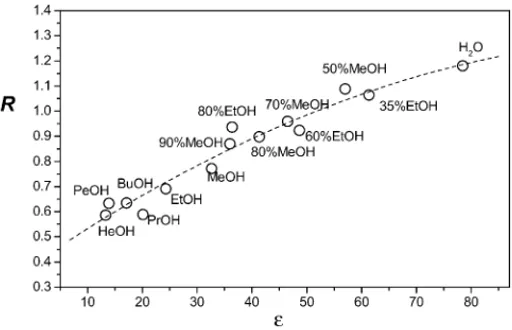
∼450 nm (and also the efficiency of energy transfer) does not change monotonically with [CD]. The intensity of this band also seems to depend on the time of exposure to the excitation lamp, slightly decreasing with time. However, a characteristic of this band is that, with respect to the isolated A2MN solution, it seems to increase in the presence of␣CD and to decrease upon CD addition. These results indicate a growth in the efficiency of IET relative to the efficiency in the absence of CD with␣CD and a decrease withCD. Paying much attention to the high energy region, where N emission occurs, peaks around 360 and 385 nm are shown. These are also characteristic in the emission spectrum of MN and isolated A2MN. Small shifts to the blue of both peaks and significant changes in their relative intensities with [CD] and temperature are also observed. At each temperature, the ratio of intensities, denoted by R and measured as I (385 nm)/I (360 nm) decreases as the␣- orCD concentration increases. The amount of this decrease, however, depends on the CD used and the temperature. The change in R, observed pre-viously for several naphthoate derivatives[14,16,25,26], is associated with the change in the polarity of the microen-vironment surrounding the N group during complexation. In factFig. 4depicts R values from N emission for A2MN dilute solutions of several hydroxylated solvents covering a wide range of effective dielectric constants. R decreases monoton-ically as the solvent polarity decreases. The dependence at 25◦C can be fitted to a simple function such as, R = 0.2881 + 1.5×10−2ε−6.2×10−5ε2.
Fig. 5shows the effect on R of the increasing [␣CD] or [CD] at several temperatures. The shape of the variation of
R with [CD] and the [CD] at which the curves level off for
Fig. 3. Uncorrected emission spectra of A2MN (- - -) and A2MN-CD (␣- orCD) aerated aqueous solutions at different CD concentrations at 25◦C upon
λexc=294 nm. Left: [␣CD] = 0, 0.31, 1.42, 2.79, 4.14, 6.37, 10.41, 13.51, 15.76, 18.35 and 20.43 mM; right: [CD] = 0, 0.72, 1.09, 1.48, 1.87, 2.58, 4.95, 8.34, 10.82, 12.58 and 16.16 mM. [A2MN]≈10−7M was held constant.
3.3. Association constants
For a A2MN:CDncomplex, the equilibrium can be written as
A2MN+nCDA2MN:CDn (1) and the association constant K expressed as
K = [A2MN:CDn]
[A2MN][CD]n (2)
Assuming that [CD]0, the initial analytical concentration of CD, is [CD]0[A2MN:CDn][A2MN:CDn−1] for equi-librium 1 and that R is the weighted average from the guest complexed fraction f (=[A2MN:CDn]/[A2MN]0) evaluated
Fig. 4. Plot of R vs. the solvent dielectric constantobtained from the emis-sion spectra for dilute solutions of guest in different solvents at 25◦C upon 294 nm of excitation. Solvents are methanol–water and ethanol–water mix-tures (% volume) and a series of n-alcohols (MeOH, EtOH, PrOH, BuOH, PeOH and HeOH).
as[14,16,25,26]
f = R0−R
R0−R∞
(3)
By combining 2 and 3, the R parameter can be related to the association constant by means of
R=R0+R∞K[CD]n0
1+K[CD]n0 (4)
which can be rearranged as a linear relationship, origin of the so-called double-reciprocal linear plot, as
1
R0−R = 1
R0−R∞+
1
K(R0−R∞)[CD]n0
(5)
where R0, R∞and R are the values of the ratio defined in the previous section for the isolated A2MN guest, extrapolated at [CD]→ ∞and at a particular [CD]. Both representations,
R versus [CD]n0and (R0−R)−1versus [CD]−0nderived from Eqs. (4) and (5), respectively, should provide the association constants and stoichiometry of the complexes formed. Lin-ear plots, however, weigh more values of (R0−R)−1which are accompanied by larger uncertainties and that correspond to the lowest [CD]0. A way to improve the linear analysis consists in modifyingEq. (5)as
[CD]n0
R0−R =
1
K(R0−R∞) +
[CD]n0
R0−R∞
(6)
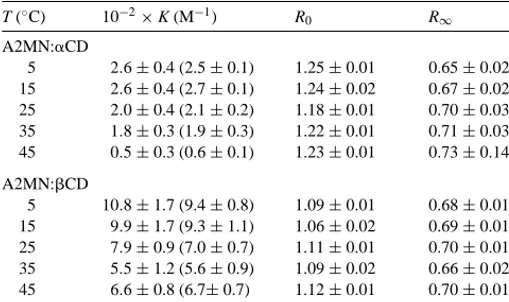
The representation [CD]n0/(R0−R) versus [CD]n0is also lin-ear and the values a more equally weighted. The curves and lines depicted inFigs. 5 and 6were obtained by adjusting the experimental data toEqs. (4) and (6), respectively for n = 1[35]. Both complexes show 1:1 stoichiometry. The cal-culated association constants at different temperatures are collected inTable 1. This table also summarizes the values of parameters R0 and R∞. R0 at 25◦C are 1.11 and 1.18, very similar to the value of 1.19 obtained for MN in the same conditions[14]. The presence of A at the other end of the chain does not substantially modify the ratio of intensity of peaks from N emission of free A2MN. The value of R∞will be a measure of the polarity surrounding the naphthoate, N, group. The results at 25◦C give R∞values close to 0.7 for both complexes, substantially lower than R0when the guest
Table 1
Equilibrium constants K, R0 and R∞at different temperatures for A2MN complexes with␣−andCDs of 1:1 stoichiometry, determined by using nonlinear regression fits and linear ones (in parentheses)
T (◦C) 10−2×K (M−1) R0 R
∞
A2MN:␣CD
5 2.6±0.4 (2.5±0.1) 1.25±0.01 0.65±0.02 15 2.6±0.4 (2.7±0.1) 1.24±0.02 0.67±0.02 25 2.0±0.4 (2.1±0.2) 1.18±0.01 0.70±0.03 35 1.8±0.3 (1.9±0.3) 1.22±0.01 0.71±0.03 45 0.5±0.3 (0.6±0.1) 1.23±0.01 0.73±0.14 A2MN:CD
5 10.8±1.7 (9.4±0.8) 1.09±0.01 0.68±0.01 15 9.9±1.7 (9.3±1.1) 1.06±0.02 0.69±0.01 25 7.9±0.9 (7.0±0.7) 1.11±0.01 0.70±0.01 35 5.5±1.2 (5.6±0.9) 1.09±0.02 0.66±0.02 45 6.6±0.8 (6.7±0.7) 1.12±0.01 0.70±0.01
is surrounded by water molecules (ε≈78). The value corre-sponds to a microenvironment that is quite hydrophobic and that, more importantly, is very similar in both complexes. Ac-cording toFig. 3, naphthoate groups for both complexes are in a medium ofε≈23.
Association constants are accompanied by relatively large uncertainties, most of which are due to the low fluorescence signals of the A2MN guest ([A2MN]≈10−7M). The asso-ciation constants, as usually occurs with naphthalene deriva-tives, are larger for the complex formed withCD than with ␣CD. These values are 205 and 740 M−1 (weighted aver-age) for 1:1 A2MN:␣CD and A2MN:CD complexes at 25◦C. At this temperature and at [CD] = 16 mM, close to the plateau in Fig. 5, the fraction of the complexed guest with CD is slightly larger than 0.92. Reaching this frac-tion for the A2MN:␣CD complex would require a [␣CD]
≈58 mM, which is in the solubility limit of␣CD. At 25◦C A2MN:CD shows smaller stability than the MN:CD com-plex (∼1960 M−1) [14] and larger than the MA:CD one (∼190 M−1)[15]. The A2MN:␣CD complex, however, ex-hibits a stability that is similar to that of MN:␣CD[14].
Fig. 7. Job’s plot for the formation of A2MN complexes with␣CD () or CD ().
3.4. Job’s plots
Stoichiometries of the complexes were also confirmed by the continuous variation method known as Job’s plot[36,37]. Fig. 7depicts the Job’s plots as (I0−I)[G] versus q, where
I is the fluorescence intensity at each [CD] (I0for [CD] = 0) and q the ratio [CD]/([A2MN] + [CD]) for each measured sample. The sum [CD] + [A2MN] was kept constant for all samples. For both complexes q is 0.5 at the maximum, which strengthens the 1:1 stoichiometry for both complexes.
3.5. Fluorescence depolarization
Anisotropies of the fluorescence, r were obtained from depolarization measurements by using the L-Format [38]. Emission wavelengths 362 and 450 nm upon excitation of 294 and 362 nm, respectively, were used. These wavelengths correspond to the emission-excitation pairs for N and A, re-spectively.
The top ofFig. 8shows, as an example, the variation of r with [␣CD] and [CD] for both systems at 5 and 45◦C select-ing 362 nm, upon excitation of 294 nm. r increases with [CD] due to the larger amount of complex, which has a larger rota-tional relaxation time than the free A2MN. r also decreases with temperature probably due to the decrease in the amount of the complexed form ( H < 0) and the temperature effect
on the rotational diffusion rate of the components of the sys-tem. The values of r at a [CD] are larger for the A2MN/CD system than for the A2MN/␣CD one, owing to the larger size and association constant of the A2MN:CD complex relative to the A2MN:␣CD complex.
The bottom ofFig. 8illustrates the variation of r under ob-servation of A (362 and 450 nm). Values of r for A2MN/␣CD and A2MN/CD systems are larger than those obtained from N inspection and they are accompanied by larger uncertain-ties, but they also do not show any monotonic behavior upon CD addition. The anisotropy of A and thus its mobility is
Fig. 8. (a) Variation of the anisotropy, r with [␣CD] (open symbols) and [CD] (filled symbols) monitored at 362 nm of emission upon excitation of 294 nm and measured at 5◦C (squares) and 45◦C (circles). (b) Idem, but monitored at 450 nm of emission upon 362 nm of excitation.
hardly altered upon␣- orCD addition. Perhaps the CD is placed far enough from A in both the A2MN:␣CD and the A2MN:CD complexes.
3.6. Quenching of fluorescence
Measurements were performed on water solution of A2MN, free and in the presence of CDs, by using a KBr aqueous solution (0.8519 M) as a quencher. Water guest so-lutions in the presence of ␣- and CDs were prepared at a [CD] for which the fraction of the complexed guest was
∼0.7. Data were collected at 362 and 385 nm (N emission) and 450 nm (A emission) uponλexc= 294 and 362 nm, re-spectively. Throughout these experiments, the value of R is almost constant upon quencher addition, which means that the medium surrounding N (complexed or not) hardly change. Quenching data, in the range used ([Q] = 0–50 mM), can be fitted linearly to the known Stern–Volmer equation[39]. Val-ues of Stern–Volmer constants, KSV, are collected inTable 2.
Table 2
Stern–Volmer constants, KSVfor the quenching of free A2MA and for its CD complexes with a KBr solution at 5◦C
System KSV(M−1)
λexc= 294 nm,λem= 362 nm λexc= 294 nm,λem= 386 nm λexc= 362 nm,λem= 450 nm
A2MN/␣CD 4.4±0.8 7.1±0.3 0.0±3.8
A2MN/CD 5.7±0.4 7.7±0.4 0.0±1.4
A2MN 10.8±1.2 15.6±1.1 0.9±2.5
Table 3
Values of the enthalpy ( H◦) and entropy ( S◦) changes of the (1:1) for the complexation of N2MN with CD hosts
Host H◦(kJ mol−1) S◦(J K−1mol−1) ␣CD −24.3±9.6 −38.9±32.5
CD −9.4±3.1 +23.4±10.6
more effective than for the complexed one and it does also not depend on the CD type. This evidence will agree with the fact that CDs in both complexes are probably located close to N and that a similar portion of N would be exposed to the quencher effects for both complexes.
The KBr solution does not seem to be an effective quencher of anthroate groups just as diacetyl, NaNO2 and KSCN were not either.
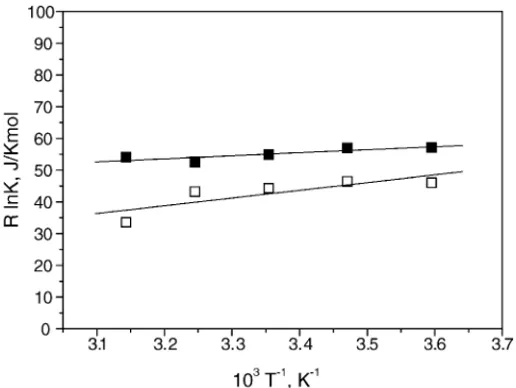
3.7. Thermodynamic parameters
H◦and S◦, collected inTable 3, were obtained by us-ing van’t Hoff plots depicted inFig. 9, from the weighted average of K’s, collected inTable 1. The data can be reason-ably fitted linearly. Both inclusion processes are enthalpically governed, H◦< 0, but the formation of A2MN:␣CD is more favored by this term than the formation of the A2MN:CD one. Negative signs of enthalpy changes are the character-istics of complexation of hydrophobic species that involve mainly attractive van der Waals (VDW) and/or
intermolecu-Fig. 9. van’t Hoff plots of R ln K vs. T−1for the formation of A2MN:␣CD () and A2MN:CD () complexes.
lar hydrogen bonding (HB) interactions. Van der Waals forces usually increase as the CD cavity size relative to the guest molecule decreases. The S◦ signs during association are usually the balance of two opposite effects, the change in the rotational and translational degrees of freedom of the system ( S◦< 0) and the variation in the solvating shells of the guest or those included inside the host during complexation ( S◦> 0). The A2MN:CD formation is entropically favored while the formation of the A2MN:␣CD, even though accompanied by a large uncertainty, is disfavored. Despite the smaller neg-ative enthalpy term the entropy change makes the association constant at 25◦C larger for the A2MN:CD formation than for the A2MN:␣CD one. Entropy variation signs are usually associated to the relative host/guest location. If the guest size is so that it cannot penetrate totally inside the cavity a neg-ative S◦is expected, as usually happens with naphthalene derivatives 1:1 complexation with␣CD[14,16,21]. However, if the guest penetrates totally inside a relatively wide cavity where the guest motion is only moderately hindered, com-plexation should be accompanied by positive S◦, which is the case of MN complexes withCD[14,16,21].
3.8. Molecular mechanics calculations
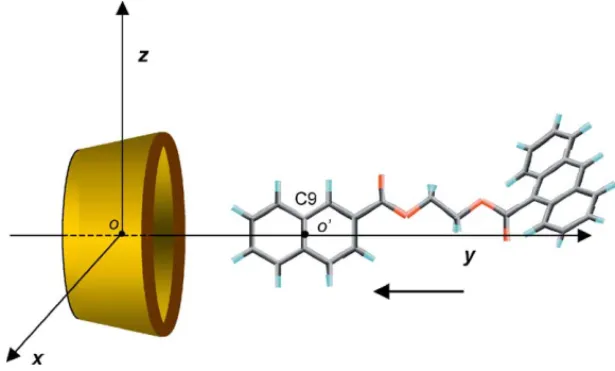
The␣- andCD initial structures were constructed in the non-distorted form, as previously[19–26]. The A2MN guest was placed at one of the equivalent conformations of minima energy coming after performing a grid search followed by a minimization (0.2 kcal/mol ˚A) over the torsional angles of the spacer between naphthalene and anthracene groups. For this conformation, depicted in Fig. 10, all torsional angles were in trans, the naphthalene and ester groups in the same plane and the ester group of A was separated by∼55◦from the plane of the aromatic ring.
To describe the inclusion process, the center of mass of the glycosidic oxygen atoms of the host (denoted by ‘o’ in Fig. 10) was located at the origin of a coordinate system. The
y-axis refers to the six- or seven-fold rotation host axis. The
Fig. 10. Coordinate systems used to emulate the (1:1) A2MN complexation process with a CD. the y-coordinate from 12 up to−12 ˚A, for an initially fixed
pair of most favorable values θ andε. Each of the struc-tures generated was solvated (MS), optimized (PBC, gradi-ent 3.0 kcal/mol ˚A) and saved for further analysis. Initially, the most favorableθandεangles were estimated by critical inspection of binding energies obtained from the structures generated by scanning the three parameters at regular inter-vals in the vacuum. Values ofε,θpairs obtained were 100◦, 5◦and 90◦, 10◦for A2MN to ␣CD andCD approaches, respectively.
Fig. 11depicts binding energy for the approach of A2MN to both CDs by the naphthoate side. The complexation pro-cesses seem to be energetically favorable. Thus the binding energy decreases monotonically upon A2MN approaching theCD. The most feasible structure, named as (1) inFig. 11,
Fig. 11. Binding energies as a function of y-coordinate ( ˚A) for A2MN complexation with␣CD () andCD (). Superimposed are the structures (1), (2) and (3) for the complexes corresponding to minima binding energy.
with the conclusions derived from anisotropy and quenching measurements and with the values of R∞, which were very similar for both complexes. The different location of␣- and CDs with respect to N in both complexes, on the cavity en-trance or shielding the naphthoate groups and a little portion of the spacer, respectively, should be related to the IET effi-ciency. TheCD placement relative to the guest for (1) may restrict those conformations of spacer for which anthracene and naphthalene groups come together where the IET is more efficient. In addition, the fact that theCD cavity is totally occupied by the guest molecule in the complex could sup-port that the disruption of the water shells initially solvating the CD cavity (and also around the guest) upon complexa-tion could be mainly responsible for the increase in entropy. On the contrary, the entropy decrease upon complexation of A2MN with␣CD should come from the large exposure of both guest and host to the solvent. The decreasing should mostly due to the loss of translational and rotational freedom degrees of both host and guest.
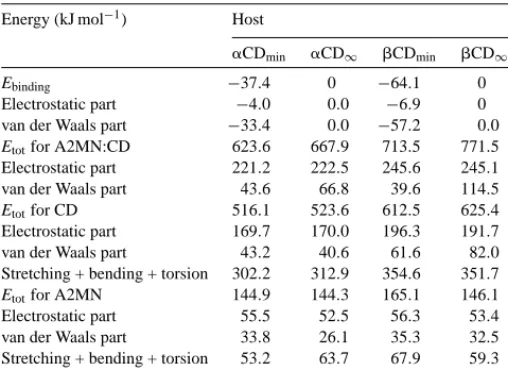
Table 4collects contributions to binding energy and the most important components to the total energy for the com-plexes, hosts and guest at the minimum and at the separation of 20 ˚A, where host and guest hardly interact. For the struc-tures of minima binding energy most of the contribution to this energy comes from non-bonded van der Waals interac-tions. Electrostatics only represents approximately 10% of the total Ebinding. This van der Waals contribution to the sta-bilization could account for the negative H◦values obtained for both complexes, which experimentally is more favorable for the A2MN:␣CD complex. As we inferred previously, the possibility of guest–host HBs interactions should contribute in the same direction. According to the criteria established for HB formation, an HB is formed between the carboxylic oxygen from the naphthoate group and one of the primary OH(6) for the structure (1) of A2MN:CD complex. This
Table 4
Ebindingand selected components (kJ mol−1) at the minimum binding energy (subscript min) at y = 20 ˚A (subscript∞) for A2MN:host 1:1 complexes in the presence of water
Energy (kJ mol−1) Host
␣CDmin ␣CD∞ CDmin CD∞
Ebinding −37.4 0 −64.1 0
Electrostatic part −4.0 0.0 −6.9 0 van der Waals part −33.4 0.0 −57.2 0.0 Etotfor A2MN:CD 623.6 667.9 713.5 771.5 Electrostatic part 221.2 222.5 245.6 245.1 van der Waals part 43.6 66.8 39.6 114.5 Etotfor CD 516.1 523.6 612.5 625.4 Electrostatic part 169.7 170.0 196.3 191.7 van der Waals part 43.2 40.6 61.6 82.0 Stretching + bending + torsion 302.2 312.9 354.6 351.7 Etotfor A2MN 144.9 144.3 165.1 146.1 Electrostatic part 55.5 52.5 56.3 53.4 van der Waals part 33.8 26.1 35.3 32.5 Stretching + bending + torsion 53.2 63.7 67.9 59.3
is not possible for any of the structures, (2) or (3), of the A2MN:␣CD complex.
Both complexation processes are accompanied by a de-crease in total potential energy, which is mainly due to van der Waals contributions. Complexation also produces a slight decrease in the total potential energy of CDs. In spite of the different size due to the guest location for both complexes, CD macrorings hardly strain upon complexation.
4. Conclusions
The present study demonstrates experimentally that the A2MN guest can form complexes with 1:1 stoichiometries with both ␣- andCDs. The stability for the A2MN:CD is larger than for the A2MN:␣CD. Both processes are en-thalpically favored. Nevertheless, the formation with ␣CD is also governed enthalpically as the entropy contribution is negative. However, for the A2MN:CD complexation both, enthalpic and entropic terms, contribute to increasing the stability of such complexes. Intramolecular energy transfer, which takes place for the free A2MN guest, also occurs in the complexes but with an efficiency that depends on the host type.␣CD appears to increase such efficiency andCD to decrease it. Molecular mechanics calculations prove the experimental evidence that 1:1 complex formations are pos-sible. The non-bonded van der Waals interactions are mainly responsible for the stability of both complexes. The most feasible structures are those for which CDs are placed close to naphthoate group, either shielding it from the solvent as for theA2MN:CD or occupying only a little portion of the cavity as for the A2MN:␣CD. Both structures, in agreement with the anisotropy, naphthoate quenching measurements and with the studies on the polarity surrounding naphthoate in the complex, may explain the differences in IET efficiencies.
Acknowledgements
This research was supported by MCYT (BQU2001/1158). We also express our thanks to M.L. Heijnen for assistance with the preparation of the manuscript.
References
[1] J. Szejtli, T. Osa, Comprehensive Supramolecular Chemistry, vol. 3, Cyclodextrins, Elsevier, Oxford, 1996.
[2] V.T. VD’Souza, K.B. Lipkowitz, Chem. Rev. 98 (5) (1998) 1741–2076.
[3] A. Harada, Adv. Polym. Sci. 133 (1998) 141–201. [4] A. Harada, Acc. Chem. Res. 34 (6) (2001) 456–464.
[5] G. Nelson, G. Patonay, I.M. Warner, Appl. Spectrosc. 41 (7) (1987) 1235–1238.
[6] K.W. Street Jr., W.E. Acree Jr., Appl. Spectrosc. 43 (7) (1988) 1315–1318.
[9] L.J. Flamigni, Phys. Chem. 97 (1993) 9566–9572.
[10] A.Y. Will, A. Mu˜noz de la Pe˜na, T.T. Ndou, I.M. Warner, Appl. Spectrosc. 47 (3) (1993) 277–282.
[11] J.M. Schuette, I.M. Warner, Talanta 41 (5) (1994) 647–649. [12] E.K. Fraiji Jr., T.R. Cregan, T.C. Werner, Appl. Spectrosc. 48 (1)
(1994) 79–84.
[13] A. Nakamura, S. Sato, K. Hamasaki, A. Ueno, F. Toda, J. Phys. Chem. 99 (1995) 10952–10959.
[14] J.M. Madrid, F. Mendicuti, Appl. Spectrosc. 51 (1997) 1621–1627. [15] J.M. Madrid, M. Villafruela, R. Serrano, F. Mendicuti, J. Phys.
Chem. B 103 (1999) 4847–4853.
[16] A. Di Marino, F. Mendicuti, Appl. Spectrosc. 56 (2002) 1579–1587. [17] M.J. Sherrod, in: J.E.D. Davies (Ed.), Spectroscopic and Computa-tional Studies of Supramolecular Systems, Kluwer Academic Pub-lishers, Dordrecht, The Netherlands, 1992, p. 187.
[18] V.T. VD’Souza, K.B. Lipkowitz, Chem. Rev. 98 (5) (1998) 1829–1874.
[19] J.M. Madrid, J. Pozuelo, F. Mendicuti, W.L. Mattice, J. Colloid Interf. Sci. 193 (1997) 112–120.
[20] J. Pozuelo, F. Mendicuti, W.L. Mattice, Macromolecules 30 (1997) 3685–3690.
[21] J.M. Madrid, F. Mendicuti, W.L. Mattice, J. Phys. Chem. B 102 (1998) 2037–2044.
[22] J. Pozuelo, F. Mendicuti, W.L. Mattice, Polym. J. 30 (1998) 479–484. [24] J. Pozuelo, A. Nakamura, F. Mendicuti, J. Incl. Phenom. Macroc.
Chem. 35 (3) (1999) 467–485.
[25] M. Cervero, F. Mendicuti, J. Phys. Chem. B 104 (7) (2000) 1572–1580.
[26] I. Pastor, A. Di Marino, F. Mendicuti, J. Phys. Chem. B 106 (8) (2002) 1995–2003.
[27] J. Bravo, F. Mendicuti, E. Saiz, W.L. Mattice, Macromol. Chem. Phys. 195 (1994) 3411–3424.
[28] J. Bravo, F. Mendicuti, E. Saiz, W.L. Mattice, Macromol. Chem. Phys. 197 (1996) 1349–1360.
[29] Sybyl 6.9, Tripos Associates, St. Louis, MO, USA, 2002. [30] M. Crark, R.C. Cramer III, N. van Opdenbosch, J. Comput. Chem.
10 (1989) 982–1012.
[31] MOPAC (AM1), Included in the Sybyl 6.9 package, 2002. [32] Y. Brunel, H. Faucher, D. Gagnaire, A. Rassat, Tetrahedron (1975)
31.
[33] W.H. Press, B.P. Flannery, S.A. Teukolski, W.T. Vetterling, Numeri-cal Recipes: The Art of Scientific Computing, Cambridge University Press, 1988, p. 312.
[34] M. Blanco, J. Comput. Chem. 12 (1991) 237–247.
[35] MicroCalTM OriginTM, Version 6.0, MicroCal Software, Inc., Northampton, MA, USA, 1998.
[36] P. Job, Ann. Chim. 9 (1928) 113–134.
[37] Y.L. Loukas, J. Phys. Chem. B 101 (1997) 4843–4866.