Anticuerpos antinucleares específicos de la esclerodermia como determinantes del pronóstico y de los diferentes patrones clínicos de la enfermedad
151
0
0
Texto completo
(2) TESIS DOCTORAL ANTICUERPOS ANTINUCLEARES ESPECÍFICOS DE LA ESCLERODERMIA COMO DETERMINANTES DEL PRONÓSTICO Y DE LOS DIFERENTES PATRONES CLÍNICOS DE LA ENFERMEDAD. Tesis presentada para optar al grado de Doctor Doctorando:. Alejandra Fernández Luque Directora:. Dra. Carmen Pilar Simeón Aznar Tutor:. Dr. Vicent Fonollosa Plá. UNIVERSITAT AUTÒNOMA DE BARCELONA Programa de Doctorat en Medicina Departament de Medicina. BARCELONA 2016.
(3)
(4) A Manolo, Sofía y Lucas. A mis padres. A la memoria de Nines..
(5) ■■. AGRADECIMIENTOS Y DEDICATORIAS Han sido muchas las personas que me han acompañado durante esta etapa de mi formación que ahora concluye y que han contribuido, ya sea directa o indirectamente, a la realización de esta tesis. A todas ellas me gustaría mostrar mi más sincero agradecimiento.. Gracias especialmente a la directora de esta tesis, Carmen Pilar Simeón, no sólo por haberme enseñado todo lo que sé sobre la esclerodermia, un mundo del que lo conoce todo, sino por su calidad personal infinita, por demostrarme que la excelencia no está reñida con la humildad. Gracias por tu paciencia y tu cariño, y por haber ejercido también de “mami” en los momentos de crisis.. Al Dr. Vicent Fonollosa, protagonista principal de la historia de la esclerodermia en nuestro país, por su capacidad de liderazgo y haber sabido transmitirnos la pasión e interés por esta enfermedad.. A Alfredo, Edu y Adrián, por su compañerismo y su ayuda desinteresada acompañados siempre de la mejor sonrisa.. A Maria José Rodrigo y Ana Marín, porque sin su contribución desde el laboratorio, esta tesis no podría haberse llevado a cabo.. A todos los compañeros del servicio de Enfermedades autoinmunes del Hospital Vall d’Hebron (“la tercera”), mi segundo hogar durante algunos años y donde crecí como médico. Gracias a todos y cada uno de ellos por haberme enseñado tanto y haberme hecho sentir como en una gran familia. Quería hacer mención especial al Dr. Miquel Vilardell, por ser un ejemplo y transmitirnos sus valores, por recordarnos constantemente que el paciente siempre es lo más importante. Gracias por la cálida acogida en el servicio, por tu cercanía y por tu. preocupación constante por el bienestar de los residentes..
(6) AGRADECIMIENTOS Y DEDICATORIAS ■ ■ Al Dr. Albert Selva, por ser fuente inagotable de motivación. Por contagiarnos su pasión y entusiasmo por la medicina, y, ahora en la distancia, seguir siendo una referencia cuando tengo dudas sobre casos raros con los que se desenvuelve como nadie.. A Ane y a Gemma, mis compañeras y hermanas de fatigas, que espero que sigan siéndolo a pesar de no tenerlas tan cerca.. A mis compañeros del Hospital de Mollet, por las risas, por el compañerismo, por hacer que ir a trabajar cada día sea un lujo.. A mis compañeros de residencia del Hospital Vall d’Hebron, muchos ya más amigos que compañeros, con los que he compartido tanto. Gracias sobre todo a “los malignos”, por conseguir que, incluso algo tan duro como las guardias se convirtiera en un recuerdo feliz.. A mis amigos fuera de las fronteras hospitalarias: La Gesta, a los Lomos, los portonoveses, los catalano-mallorquines y las Berlinburguesas, por ser una parte esencial en mi vida.. A mi familia, que cada vez es más grande: a mis padres y hermanas, por vuestro estímulo y vuestro ejemplo, por inculcarme los valores del trabajo, y por vuestro apoyo y respeto en cada decisión que he ido tomando en la vida. Os quiero. Y a "las nuevas adquisiciones", especialmente a ti, Nines, por tu cariño sin límites y por ser un ejemplo constante. Nunca dejaré de echarte de menos.. Y, por último, el agradecimiento más especial para Manolo, Sofía y Lucas, por hacerme sentir cada día la más afortunada y por ser para mí, simplemente, todo..
(7) ■■. ABREVIATURAS Y SÍMBOLOS. A. ACA: Anticuerpos anticentrómero. ACR: American College of Rheumatology. AMA: Anticuerpos antimitocondriales. ANA: Anticuerpos antinucleares. ANu: Anticuerpos antinucleolares. ATA-I: Antitopoisomerasa I / anti-Scl-70.. B C. BNP: Péptido natriurético cerebral. CENP: Proteínas centroméricas. CF: Clase funcional. CREST: Calcinosis, Raynaud, Esclerodactilia, Esófagopatia, Telangiectasias CRE: Crisis renal esclerodérmica. CSGR: Grupo de investigación canadiense sobre Esclerodermia. CTGF: Factor de crecimiento del tejido conectivo. CVF: Capacidad vital forzada.. D E. DLCO: Capacidad de difusión de monóxido de carbono ECG: Electrocardiograma. ELISA: Enzimoinmunoanálisis. EMG: Electromiograma. EMTC: Enfermedad Mixta del Tejido Conectivo ENA: Antígenos extraíbles del núcleo. EPI: Enfermedad pulmonar intersticial ES: Esclerosis Sistémica / Esclerodermia ES sine ES: ES sine esclerodermia ET-1: Endotelina-1. EULAR: European League Against Rheumatism EUSTAR: European League Against Rheumatism Scleroderma Trials and Research. F. FGF: Factor de crecimiento de fibroblastos.. G H. GAVE: Ectasias vasculares del antro gástrico.. FR: Fenómeno de Raynaud.. HAP: Hipertensión Arterial Pulmonar. HLA: Complejo mayor de histocompatibilidad. HR: Hazard ratio.
(8) ABREVIATURAS Y SÍMBOLOS ■ ■. I. IB: Inmunoblot. IC: Intervalo de confianza. IECA: Inhibidores del enzima convertidor de la angiotensina. IFI: Inmunofluorescencia indirecta. IFN: Interferón. IMC: Índice de masa corporal. ISRCS: Investigational Scleroderma Renal Crisis Survey. IP: Inmunoprecipitación.. L M N. LES: Lupus eritematoso sistémico. mRSS: Modified Rodnan skin score. NINE: Neumonía intersticial no específica. NIU: Neumopatía intersticial usual. NT-proBNP: Porción amino terminal del pro-péptido natriurético cerebral. NYHA: New York Heart association.. O P. OR: Odds ratio. PAPm: Presión de la arteria pulmonar media. PDGF: Factor de crecimiento derivado de las plaquetas. PDGFR: Receptor del factor de crecimiento derivado de las plaquetas. PCP: Presión de enclavamiento capilar pulmonar. PET: Tomografía por emisión de protones. PFR: Pruebas de función respiratoria. PM: Polimiositis. Pre-ES: Preesclerodermia.. R. RESCLE: Grupo español de estudio de la Esclerodermia. RGE: Reflujo gastroesofágico. RNAp: Anticuerpos anti RNA polimerasa III. RNP: Ribonucleoproteínas nucleolares. RVP: Resistencias vasculares pulmonares.. S T. SPECT: Tomografía computarizada de emisión monofotónica. TACAR: Tomografía axial computarizada de alta resolución pulmonar. TEGD: Tránsito esófagogastroduodenal. TGF-B: Factor de crecimiento transformante beta.. U V. UD: Úlceras digitales. VSG: Velocidad de sedimentación globular..
(9)
(10) ÍNDICE ■ ■ AGRADECIMIENTOS Y DEDICATORIAS ABREVIATURAS Y SÍMBOLOS 1. INTRODUCCIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1. 1.1. GENERALIDADES ESCLERODERMIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 1.1.1. DEFINICIÓN/EPIDEMIOLOGÍA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 1.1.2. PATOGENIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 1.1.3. CRITERIOS DE CLASIFICACIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 1.1.4. MANIFESTACIONES CLÍNICAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 1.1.5. PRONÓSTICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24 1.1.6. TRATAMIENTO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 1.2. LOS ANTICUERPOS Y LA ESCLERODERMIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 1.2.1. ASPECTOS CLÍNICOS DE LOS ANTICUERPOS EN LA ESCLERODEMIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 1.2.2. CONSIDERACIONES EPIDEMIOLÓGICAS Y GENÉTICAS DE LOS ANTICUERPOS EN LA ES . . . . . . . . . . 29 1.2.3. ANTICUERPOS Y PATOGÉNESIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 1.2.4. CARACTERÍSTICAS DE LOS ANTICUERPOS DE LA ESCLERODERMIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 1.2.4.1. ANAs específicos de Esclerodermia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 1.2.4.2. ANAs asociados a Esclerodermia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 1.2.4.3. Otros anticuerpos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32 1.2.5. MÉTODOS DE DETECCIÓN DE AUTOANTICUERPOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 1.2.6. PERFILES CLÍNICOS DE LOS ANTICUERPOS ESPECÍFICOS DE LA ES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 1.2.6.1. Anticentrómero (ACA) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 1.2.6.2. Antitopoisomerasa I o Scl70 (ATA-I) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37 1.2.6.3. Anti RNA polimerasa III (RNAp) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39 1.2.6.4. Anti U3-RNP o fibrilarina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42 1.2.6.5. Anti Th/To . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43 1.2.6.6. Anti U1-RNP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 1.2.6.7. Anti Pm/Scl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 1.2.6.8. AntiKu . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47 1.2.6.9. Otros (Anti Ro 52, Anti U11/U12 RNP, Anti Nor 90) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 1.2.7. IMPLICACIÓN DE LOS ANTICUERPOS EN LA SUPERVIVENCIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50 1.3. DETERMINACIÓN DEL PODER PRONÓSTICO DE LOS SUBTIPOS CUTÁNEOS VERSUS PERFIL INMUNOLÓGICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53. 2. OBJETIVOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61. 2.1. JUSTIFICACIÓN DEL ESTUDIO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 2.2. OBJETIVOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64. 3. PACIENTES Y MÉTODOS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67. 3.1. PACIENTES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69 3.2. DEFINICIÓN DE LAS VARIABLES CLÍNICAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 3.3. ANÁLISIS INMUNOLÓGICOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .72 3.4. MÉTODOS ESTADÍSTICOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .72. 4. RESULTADOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75. 4.1. PRIMER ESTUDIO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 4.2. SEGUNDO ESTUDIO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88 4.3. TERCER ESTUDIO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92. 5. DISCUSIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 6. CONCLUSIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .121 7. BIBLIOGRAFÍA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .125.
(11)
(12) Introducción. 1. INTRODUCCIÓN.
(13)
(14) INTRODUCCIÓN ■ ■ 1 1.1. GENERALIDADES ESCLERODERMIA 1.1.1. Definición/Epidemiología La esclerosis sistémica o esclerodermia (ES) es una enfermedad autoinmune y sistémica de causa desconocida que afecta al tejido conectivo. Se caracteriza por alteraciones vasculares a nivel de la microcirculación, activación del sistema inmunológico con producción de autoanticuerpos y por una producción excesiva de la matriz extracelular colágena que condiciona fibrosis en diferentes órganos internos y, en el caso de la piel, endurecimiento o esclerosis cutánea 1.. La ES es una enfermedad poco frecuente, con una prevalencia muy variable según el área geográfica o etnia estudiada, que oscila entre 7 y 489 casos por millón de habitantes 2. La incidencia estimada varía entre 3.7 y 22 casos/millón de habitantes/año 3,4.. Afecta predominantemente a mujeres (3/1 a 10/1) y la edad media del diagnóstico es de 40 años 5.. 1.1.2. Patogenia Actualmente se desconoce el proceso que desencadena la enfermedad, pero se considera que es una mezcla de factores genéticos y ambientales que lleva al desarrollo de tres procesos patogénicos fundamentales e interrelacionados entre sí: lesión endotelial y daño vascular, desregulación de la producción de tejido conectivo y activación de la inmunidad y autoinmunidad.. Se han postulado diferentes agentes ambientales como posibles desencadenantes de la ES, como infecciones víricas (citomegalovirus, parvovirus B19), bacterias como el Helicobacter pylori, pesticidas o exposición a metales pesados; sin embargo, ninguno se ha definido de forma contundente como agente responsable del desarrollo de la enfermedad.. Se sabe que los factores genéticos juegan un papel esencial en la patogenia, tanto en su desarrollo como en la expresión clínica. No hay un claro patrón hereditario, pero sí una clara. 3.
(15) 1. ■■. INTRODUCCIÓN. agregación familiar, con un riesgo relativo estimado de padecer la enfermedad 15 veces mayor en hermanos de pacientes con ES y 13 veces mayor en familiares de primer grado, si bien el riesgo absoluto es bajo (1.6 a 1.7%) 6 . Además, en gemelos monocigóticos se ha encontrado una concordancia en la presencia de anticuerpos específicos de la ES que puede llegar hasta el 90% 6,7.. Por otro lado, hay diferencias fenotípicas entre diferentes razas, grupos étnicos o regiones geográficas 8, lo que también apoya la hipótesis de que los factores genéticos contribuyen al desarrollo de la enfermedad.. Se han descrito alteraciones en la expresión de genes fundamentalmente implicados en la autoinmunidad, la producción de colágeno y la respuesta vascular, pero siempre en estudios con cohortes de pacientes muy pequeñas y con resultados que además no se han podido reproducir 7. En los últimos años se han comenzado a utilizar estudios de asociación del genoma completo (GWAS, genome wide association studies) que han dado importantes resultados identificando nuevos marcadores genéticos y nuevas rutas involucradas en la patogenia de diferentes enfermedades autoinmunes 9.. A pesar de que se han realizado importantes avances en el conocimiento de la base genética de la ES 10-12, todavía el número de loci conocidos hasta el momento que explican su componente genético es limitado en comparación con otras patologías autoinmunes como la enfermedad de Crohn, la colitis ulcerosa o el LES 13.. La asociación de ciertos complejos mayores de histocompatibilidad (HLA) tipo II con fenotipos clínicos y autoanticuerpos determinados 14 ha sido establecida en diferentes trabajos. Se ha observado una mayor susceptibilidad de padecer la enfermedad según los genes del sistema HLA, con resultados heterogéneos según la población estudiada. Así, mientras que en estudios realizados en raza caucásica demuestran que los haplotipos HLA-DR5 y DR-3 confieren una mayor susceptibilidad 15,16, en la población japonesa son el HLA-DR1*1502 y HLA-DQB1*0601 17.. 4.
(16) INTRODUCCIÓN ■ ■ 1 A continuación se detallan los 3 procesos patogénicos de la enfermedad: ◀ La vasculopatía de la ES afecta a todas las capas de los pequeños vasos y capilares. Se produce vasoconstricción, hiperplasia de las capas íntima y adventicia, inflamación y trombosis, siendo el hallazgo fundamental la hiperplasia fibrótica de la capa íntima de los vasos 18. Se cree que el daño sobre la célula endotelial es el factor inicial desencadenante de la enfermedad 19. Éste produce un desbalance entre sustancias vasoconstrictoras y vasodilatadores y una alteración de los fenómenos reparativos vasculares (vasculogénesis y angiogénesis). Así aumentan las sustancias vasoconstrictoras como la Endotelina 1 y disminuyen vasodilatadores como el Óxido nítrico. La Endotelina 1 es crucial en la patogenia de la ES: promueve la proliferación celular, la hipertrofia de la pared arterial, estimula la fibrosis y la cascada inflamatoria de la enfermedad 20,21. A parte de tener un potente efecto vasoconstrictor también disminuye los niveles de Óxido nítrico, que tiene un papel crucial en la relajación vascular 22. Por otro lado, por el daño de la célula endotelial se produce una pérdida de capilares desde fases precoces en la enfermedad 23 y en la ES todos los factores que intervienen en la reparación del daño vascular están alterados 24. ◀ La fibrosis en la ES es un proceso muy complejo y no del todo conocido, que básicamente consiste en una activación de los fibroblastos y una diferenciación y sobreproducción de colágeno, que conlleva una acumulación de éste en la matriz extracelular. Todo este mecanismo depende de diferentes citoquinas (TGF-B, CTGF, ET-1 y FGF, principalmente) 25,26. ◀ La inmunidad en la ES está alterada, tanto la innata como la adquirida. La inmunidad innata no se conoce en profundidad, si bien hay evidencia en diferentes trabajos de la implicación de mastocitos y células natural killer en la patogenia de la enfermedad 27,28, así como de diferentes citocinas, especialmente el interferón (IFN). Recientes estudios demuestran un aumento de la expresión de IFN de tipo I en la ES 29, al igual que en otras enfermedades autoinmunes como el lupus eritematoso sistémico (LES). El papel de la inmunidad adquirida es más conocido en la ES. Hay un amplio espectro de autoanticuerpos específicos de la enfermedad que tienen una probable conexión con el daño tisular. Además, el linfocito T actúa en las fases iniciales de la enfermedad como inductor de la fibrosis posterior 30,31.. 5.
(17) 1. ■■. INTRODUCCIÓN. 1.1.3. Criterios de clasificación La ES es una enfermedad que presenta una gran variabilidad clínica con un pronóstico que difiere mucho dependiendo del subtipo clínico. Mientras la mayor parte de enfermos alcanzan una larga supervivencia, otros experimentan un curso tórpido y de forma rápidamente progresiva.. Por este motivo, la consecución de unos criterios de clasificación homogéneos es de gran importancia tanto para el diagnóstico como para el pronóstico de la enfermedad. La clasificación de la ES ha sido y sigue siendo en la actualidad un motivo de discusión e investigación.. Criterios de Clasificación oficiales del Colegio Americano de Reumatología (AC R): El primer intento de clasificación de la enfermedad data de 1980. Los primeros criterios los desarrolló el Colegio Americano de Reumatología (Tabla 1), y son los que se han utilizado durante años para clasificar la enfermedad. Nació como un intento de establecer unas directrices que permitiesen comparar grupos de pacientes de diferentes centros para poder desarrollar ensayos clínicos 32. Sin duda, supusieron un avance muy importante en la definición de la enfermedad, sin embargo, a pesar de presentar una sensibilidad del 92% y una especificidad del 94%, tienen una baja sensibilidad para detectar a enfermos con poca afección cutánea 33,34, y un número no menospreciable de enfermos con ES limitada establecida o con el subtipo sine esclerosis cutánea que no reúnen ni el criterio mayor (esclerosis proximal a metacarpofalángicas) ni 2 de los criterios menores, quedan excluidos de los estudios. Tampoco permiten realizar un diagnóstico precoz de la enfermedad en pacientes que presenten fenómeno de Raynaud (FR), alteraciones capilaroscópicas y autoanticuerpos positivos y que requieren un control médico similar a la de los enfermos con ES ya establecida para detectar la evolución hacia ésta, ni sirven para establecer grupos clínicos y pronósticos 1.. Además, enfermos con síndromes esclerodermiformes como la fascitis eosinofílica o esclerodermia localizada como la morfea generalizada pueden cumplir los criterios de clasificación de ES, por lo que pierden especificidad.. 6.
(18) INTRODUCCIÓN ■ ■ 1 Criterio Mayor Esclerosis cutánea proximal: Induración de la piel proximal a articulaciones metacarpofalángicas o metatarsofalángicas, afectando otras partes de las extremidades, fascies, cuello, tronco, habitualmente bilateral, simétrica y casi siempre incluye esclerodactilia. Criterios menores 1. Esclerodactilia 2. Cicatrices puntiformes en pulpejos, o pérdida de substancia en pulpejos de los dedos 3. Fibrosis pulmonar bibasal. Tabla 1: Criterios de clasificación de la ES (ACR 1980) La presencia del criterio mayor y dos o más criterios menores dan una sensibilidad del 92% y una especificidad del 94% para el diagnóstico de ES. Clasificación de la ES en subtipos cutáneos: En las últimas décadas se han intentado dividir a los pacientes con ES en diferentes subtipos según características clínicas e inmunológicas determinadas, y así identificar subgrupos de pacientes que tengan una evolución clínica y pronóstico similar.. Se han establecido diferentes criterios de clasificación, todos ellos considerando como característica clínica definitoria la extensión de la esclerosis cutánea.. Una revisión realizada por Johnson et al. 35. de diferentes criterios de clasificación de la. ES sugeridos entre 1966 y 2005 por diferentes autores, concluyeron que la propuesta de LeRoy 1 era la que tenía más validez, fiabilidad y un buen valor predictivo, además de ser la más utilizada hasta el momento y referenciada.. 7.
(19) 1. ■■. INTRODUCCIÓN. En la clasificación de LeRoy (Tabla 2) los enfermos se dividen en 2 subgrupos: - ES cutánea difusa, pacientes con esclerosis cutánea distal y proximal a codos y rodillas. - ES cutánea limitada, pacientes con esclerosis cutánea distal a codos o rodillas o sin esclerosis. Además de la extensión de la esclerosis cutánea, los autores definen una serie de características clínicas, capilaroscópicas e inmunológicas que acompañan a los diferentes subtipos.. Los pacientes con ES difusa presentan FR generalmente el primer año o de manera concomitante a la afección cutánea, mayor riesgo de compromiso articular, más frecuencia de enfermedad pulmonar intersticial (EPI), crisis renal esclerodérmica (CRE), compromiso gastrointestinal o miocárdico y presentan peor supervivencia que los pacientes con ES limitada. En los pacientes con ES limitada, el FR se manifiesta generalmente mucho antes que las otras afecciones orgánicas de la enfermedad, además de presentar calcinosis, telangiectasias y mayor frecuencia de Hipertensión arterial pulmonar (HAP). El pronóstico de los pacientes con ES limitada en general es mejor que el de los pacientes con ES difusa.. Los dos subtipos se diferencian además desde el punto de vista serológico: en la ES difusa hay una mayor presencia de anticuerpos antitopoisomerasa / anti-Scl-70 (ATA-I), mientras que los pacientes con ES limitada expresan más frecuentemente anticuerpos anticentrómero (ACA), con una especificidad serológica del 82% y 93% respectivamente 36.. Tras esta clasificación se han dejado atrás los antiguos términos de “CREST” y “esclerosis sistémica progresiva” que se correspondían con la forma limitada y difusa de la enfermedad, respectivamente. El término síndrome de CREST propuesto por Winterbauer et al en 1964 37, incluía pacientes con calcinosis, FR, afección esofágica, esclerodactilia y telangiectasias, y eran considerados enfermos con buen pronóstico, a pesar de que no todos presentan un curso benigno, y el término esclerosis sistémica progresiva se atribuía a un mal pronóstico, si bien hay pacientes englobados en este grupo que no presentaban un curso rápidamente progresivo de la enfermedad ni afecciones orgánicas graves.. 8.
(20) INTRODUCCIÓN ■ ■ 1 Esclerodermia. Limitada. Difusa. Raynaud. Años. < 1 año. Afectación cutánea. Distal a codos y rodillas, cara o ausente. Proximal y distal a codos y rodillas. Afección visceral. - HAP Tardía. - EPI temprana, CRE. - Telangiectasias. - GI difusa. - Calcinosis. - Roces tendinosos. Anticentrómero. 70-80 %. Excepcional. Antitopoisomerasa I. Raro. 30 %. Capilaroscopia. Megacapilares y escasa pérdida. Pérdida importante y escasa dilatación. Tabla 2: Clasificación de la ES según criterios de LeRoy 1 EPI:enfermedad pulmonar intersticial; CRE: crisis renal esclerodérmica; GI: gastrointestinal; HAP: hipertensión pulmonar.. Sin embargo, la división de pacientes con esclerodermia en 2 grupos, difusa y limitada en los últimos años se ha considerado incompleta y con limitaciones, ya que no incluye pacientes con ES en fases iniciales o preesclerodermia (pre-ES), que presentan alteraciones capilaroscópicas, inmunología específica y FR; e incluye dentro del subtipo limitada aquellos pacientes con esclerodermia sine esclerosis (ES sine ES), que no presentan evidencia de afección cutánea pero sí afección orgánica característica de ES (afección esofágica distal o hipomotilidad del intestino delgado, EPI, HAP, afección miocárdica o CRE ), FR o cambios vasculares equivalentes ( úlceras digitales, cicatrices puntiformes en pulpejos, alteraciones capilaroscópicas) y presencia de anticuerpos antinucleares (ANA) 38.. Se consideraba que los pacientes con ES sine ES presentan una evolución similar a aquellos pacientes con ES limitada 39,40, pero en trabajos recientes se han observado diferencias en cuanto a la manifestaciones respiratorias y cardíacas 41, lo que supone un infradiagnóstico de esta entidad.. Actualmente, se considera que la clasificación más adecuada de la ES es en 4 subtipos: ES difusa, ES limitada, sine esclerodermia y preesclerodermia (Tabla 3). Se definen por la extensión de la esclerosis cutánea y se asocian a determinadas afecciones viscerales, capilaroscópicas e inmunológicas.. 9.
(21) 1. ■■. INTRODUCCIÓN Esclerodermia cutánea limitada • Fenómeno de Raynaud durante años, ocasionalmente décadas . • Esclerosis limitada distal a codos y rodillas pudiendo afectarse cara y cuello • Alteraciones capilaroscópicas: dilatación con escasa pérdida • Incidencia tardía de hipertensión arterial pulmonar, calcinosis, enfermedad gastrointestinal, telangiectasias o enfermedad pulmonar intersticial difusa • Enfermedad renal muy rara • Anticuerpos anticentroméricos (ACA) en el 70 a 80%. Esclerodermia cutánea difusa • Fenómeno de Raynaud, menos de un año de evolución • Esclerosis proximal a codos y rodillas . Presencia de roces tendinosos • Alteraciones capilaroscópicas: pérdida capilar • Incidencia precoz y significativa de enfermedad renal, intersticial pulmonar, gastrointestinal difusa y miocárdica . Desarrollo de hipertensión pulmonar • Anticuerpos ATA-I (30%) y anti-RNA polimerasa I,II ó III (RNAp) (12-15%). Esclerodermia sine esclerodermia • Fenómeno de Raynaud o equivalentes (úlceras, alteraciones capilaroscópicas) • No esclerosis cutánea • Anticuerpos antinucleares . Pueden estar presentes: ATA-I, ACA o RNAp . • Afecciones viscerales típicas de esclerodermia: hipomotilidad esofágica distal o intestinal, enfermedad pulmonar intersticial, hipertensión pulmonar, enfermedad cardiaca, crisis renal esclerodérmica .. Pre-Esclerodermia • Fenómeno de Raynaud • Alteraciones capilaroscópicas • Autoanticuerpos antinucleares (ANAs) específicos (ATA-I, ACA, RNAp, U3 RNP, Pm-Scl, Th/To) o ANAs positivos con patrón de inmunofluorescencia nucleolar. Tabla 3: Clasificación en subtipos cutáneos de la ES. Clasificación de la Esclerodermia en estadíos iniciales: En 1996 se propuso incluir en la clasificación de ES un grupo de enfermos que presentaban FR, alteraciones capilaroscópicas y/o anticuerpos antinucleares (ATA-I, ACA o patrón IFI nucleolar), y que incluso habían presentado úlceras digitales 39.. En el 2001, LeRoy y Medsger 42 modificaron su clasificación para incluir a estos pacientes, porque consideraban que presentaban una alta probabilidad de desarrollar una enfermedad definida, sobre todo en forma de ES limitada (Tabla 4). La llamaron esclerodermia limitada, término que. 10.
(22) INTRODUCCIÓN ■ ■ 1 se ha cambiado más recientemente por el de preesclerodermia o esclerodermia temprana o precoz, para evitar confusiones con el término esclerodermia cutánea limitada 43,44. A. Esclerodermia limitada I. Fenómeno de Raynaud objetivo documentado por: 1) Observación directa de 2 de las siguientes manifestaciones: palidez acra, cianosis o sufusión o 2) Respuesta a la exposición al frío por: Test de Nielssen asociado a: IIa. Alteraciones capilaroscópiccas: dilatación o pérdida capilar IIb. Presencia de anticuerpos selectivos de la ES: ACA, ATA-I, antifibrilarina, anti Pm-Scl, RNAp. Si el fenómeno de Raynaud es subjetivo tienen que cumplir los 2 criterios ii) para tener ES limitada definida. ES limitada puede formar parte de un síndrome overlap con otras conectivopatías. B. Esclerodermia cutánea limitada: Criterios de ES limitada y cambios cutáneos distales.. C. Esclerodermia cutánea difusa: Criterios de ES limitada y cambios proximales cutáneos. D. Fascitis difusa con eosinofilia: Cambios cutáneos proximales sin criterios de ES limitada ni de ES cutánea limitada.. Tabla 4: Clasificación de la ES según criterios de LeRoy and Medsger 42. A su vez, dentro de los pacientes con preesclerodermia, se observó que algunos de ellos presentaban afecciones viscerales incipientes sugestivas de ES cuando se realizaban exploraciones específicas 45, por lo que, para una mejor clasificación de la ES se debería dividir el subgrupo preesclerodermia en 2: preesclerodermia o ES muy temprana, cuando se descartan afecciones viscerales incipientes, y ES temprana o inicial, cuando en las exploraciones complementarias correspondientes se detectan afecciones viscerales leves que no se pueden diagnosticar de afecciones establecidas de ES o bien que presenten úlceras digitales o telangiectasias.. Estas alteraciones orgánicas subclínicas serían por ejemplo una DLCO disminuida en las pruebas funcionales respiratorias (PFR) o un esfínter esofágico inferior hipotenso.. 11.
(23) 1. ■■. INTRODUCCIÓN. Más adelante se hablará de los últimos criterios de clasificación de la ES propuestos en 2013 de forma conjunta por la ACR y el grupo EULAR (European League Against Rheu-. matism), que son los que hasta ahora se consideran más sensibles y específicos, y consiguen acercarse a los criterios diagnósticos. Cabe destacar que mientras los pacientes con pre-ES o ES muy temprana nunca cumplen estos criterios, algunos pacientes con ES inicial o temprana sí pueden hacerlo en aquellas casos en que desarrollan alteraciones vasculares en forma de lesiones en los pulpejos de los dedos o telangiectasias.. Clasificación basada en la combinación de subtipos cutáneos y anticuerpos: La clasificación de la ES por subtipos cutáneos, sin embargo, también presenta limitaciones, ya que pacientes con un mismo subtipo cutáneo pueden presentar gran variabilidad en las afecciones viscerales y la gravedad de las mismas.. Esto ha llevado a que se hayan propuesto subclasificaciones dentro de los subgrupos cutáneos según el perfil inmunológico. Así Steen 46 considera que son los diferentes anticuerpos específicos de la enfermedad los que marcan el comportamiento clínico y pronóstico de la misma, observando una diferente gravedad de las afecciones viscerales según el anticuerpo presentado. Propone, por tanto, una clasificación de la enfermedad en al menos 7 grupos diferentes en función del perfil inmunológico, repartidos en 2 grandes subtipos cutáneos.. De esta forma el subtipo ES limitada se subclasificaría en 4 grupos inmunológicos según la positividad de los siguientes autoanticuerpos: ACA, Th/To, Pm/Scl y U1-RNP, y el subtipo ES difusa en 3 subgrupos por los anticuerpos ATA-I, RNAp o U3-RNP.. Más recientemente, el Profesor Medsger 47 también recomienda la utilización de los anticuerpos para subclasificar los pacientes de los 2 grandes subtipos cutáneos (Tabla 5).. Parece evidente que la combinación del subtipo cutáneo y el perfil inmunológico aporta mayor especificidad en la predicción del desarrollo de las diferentes afecciones. 12.
(24) INTRODUCCIÓN ■ ■ 1 orgánicas y de su gravedad. Sin embargo, la utilización de esta clasificación está limitada por la dificultad de determinar todos los diferentes autoanticuerpos específicos de la ES, cuya técnica no está disponible en todos los centros. Patrón IFI ANA. Frecuencia aproximada. Subtipo clínico. Afección orgánica. ATA-I. Moteado. 10-40%. ES difusa. EPI. RNAp. Moteado fino Nucleolar. 4-25%. ES difusa. Antígeno. - CRE - Esclerosis cutánea - Cardiomiopatía. U3 RNP. Nucleolar. 1-5%. ES difusa. - Miopatía - HAP. Pm-Scl. Nucleolar. 3-6%. Overlap. Miopatía. U1 RNP. Moteado. 5-35%. Overlap. Miopatía - HAP,. ACA. Centromérico. 15-40%. ES limitada. - Afección esofágica - Protector EPI - HAP. Th/To. Nucleolar. 1-7%. ES limitada. - EPI - Afección intestinal. Tabla 5: Asociaciones clínico serológicas en ES. Nuevos criterios de clasificación de Esclerosis sistémica de la ACR/EULAR 2013: La última propuesta de clasificación de la ES ha surgido recientemente mediante la valoración de manera conjunta de una serie de ítems por parte de la ACR y el European League. Against Rheumatism (EULAR), con el objetivo de incluir a todos los pacientes que realmente tienen ES, y poder reconocerlos precozmente. Estos criterios han sido testados en diferentes cohortes de enfermos con ES, otras enfermedades sistémicas autoinmunes y síndromes esclerodermiformes, observándose una sensibilidad y especificidad para el diagnóstico de la enfermedad del 91% y 92% respectivamente 48.. Estos criterios superan la mayor parte de limitaciones de los establecidos en 1980 en relación a sensibilidad y especificidad, consiguiendo el práctico solapamiento entre los criterios de clasificación y diagnósticos. Sin embargo, no distinguen entre los diferentes subtipos de la enfermedad. 13.
(25) 1. ■■. INTRODUCCIÓN. y únicamente determinan la presencia de ES, por lo que para los subtipos clínicos se tendrá que utilizar la clasificación en subtipos cutáneos: ES limitada, ES difusa y ES sineES para la enfermedad definida, y preesclerodermia y esclerodermia inicial o precoz, para las fases iniciales.. Los criterios de clasificación de la ACR/EULAR 2013 están recogidos en la Tabla 6. Ítem. Subítem. Puntuación. Endurecimiento cutáneo de dedos de ambas manos que se extiende a región proximal de articulaciones metacarpofalángicas Endurecimiento de la piel de los dedos. Lesiones en pulpejos. 9. Dedos edematosos. 2. Esclerodactilia. 4. Úlceras digitales. 2. Cicatrices puntiformes en pulpejos. 3. Telangiectasia. 2. Alteraciones en la capilaroscopia periungueal. 2. Hipertensión arterial pulmonar y/o Enfermedad pulmonar intersticial. 2. Autoanticuerpos específicos. ACA. 3. ATA-I RNAp. Tabla 6: Criterios ACR/EULAR para la clasificación de la ES de 2013 Pacientes con una puntuación igual o mayor a 9 se pueden catalogar como pacientes con ES definida. 1.1.4. Manifestaciones clínicas La ES es una enfermedad sistémica muy heterogénea que puede afectar a múltiples localizaciones y con diferente gravedad.. Mientras la clínica cutánea es la más característica y representativa de la enfermedad, es la afección orgánica la que condiciona la mortalidad y mayor morbilidad y, por tanto, el pronóstico. Según el tipo de manifestaciones orgánicas y su gravedad, la ES puede ir desde una enfermedad con evolución lenta y relativamente indolente, a presentar un deterioro rápidamente progresivo que conlleva un pronóstico infausto.. 14.
(26) INTRODUCCIÓN ■ ■ 1 Las características clínicas más relevantes se detallan a continuación:. Afección cutánea La afección cutánea es la manifestación más característica de la enfermedad y es clave para la clasificación de los pacientes con ES.. Se manifiesta como una induración de la piel que presenta tres fases clínicas evolutivas: una primera fase edematosa, que se sigue de una segunda en forma de endurecimiento cutáneo y, finalmente, una última fase de atrofia cutánea 49.. El grado de extensión de afección cutánea varía según el subtipo de ES y puede cambiar en un mismo paciente a lo largo de la enfermedad.. El único método validado del que disponemos para cuantificar el grado de afección cutánea es el índice de Rodnan (modified Rodnan skin score-mRSS), en el que se utiliza una escala del 0 al 3 (según sea piel normal hasta induración extrema) que se aplica en 17 áreas anatómicas diferentes 50.. El grado de afección cutánea se relaciona con la presencia de mayor afección orgánica, lo que explica que la ES difusa tenga peor pronóstico que la ES limitada 51. Así, el mRSS presenta un valor pronóstico dentro de la enfermedad 52 .. Característicamente, cuando la fibrosis cutánea afecta a la cara puede producir disminución de las arrugas y una limitación de la apertura bucal (microstomía) por endurecimiento de la piel y mucosa peribucal. A nivel de los dedos se produce la esclerodactilia pudiendo ocasionar tanto a nivel de manos sobre todo, y de pies, contracturas en flexión de las articulaciones con gran limitación funcional secundaria.. Dentro de las manifestaciones cutáneas se encuentran las telangiectasias y la calcinosis, que no son infrecuentes en pacientes con ES.. 15.
(27) 1. ■■. INTRODUCCIÓN. Las primeras son dilataciones post-capilares de las vénulas o neo-vascularización. 53,. que aparecen inicialmente en la cara y en las manos y se extienden en el transcurso de la enfermedad a tronco y raramente a extremidades inferiores.. La calcificación se produce por precipitación de cristales de fosfato cálcico en piel y partes blandas que afectan fundamentalmente a partes acras (dedos, antebrazos).. Fenómeno de Raynaud, úlceras digitales y vasculopatía digital El FR es la manifestación clínica más frecuente de la ES, siendo además el primer síntoma en más del 90% de enfermos 54.. Se define como episodios recurrentes de vasoespasmo en partes acras desencadenados generalmente por un estímulo, que suele ser el frío.. Mientras en pacientes con ES limitada puede preceder en varios años a otras manifestaciones de la enfermedad, en la ES difusa aparece normalmente a la vez que otras afecciones 55.. El FR no es sólo una alteración funcional. Traduce una alteración estructural de la microcirculación, con proliferación de la íntima, fibrosis y trombosis intraluminal.. Estas alteraciones de la pared vascular pueden producir hasta en casi un 50% de los pacientes complicaciones vasculares 56; la más frecuente es la afección vascular digital en forma de úlceras, localizadas predominantemente en el pulpejo de los dedos o en prominencias óseas, seguidas de isquemia, necrosis digital e incluso infecciones de partes blandas y osteomielitis.. Afección gastrointestinal La afección gastrointestinal se puede dar en cualquier parte del aparato digestivo alterando las funciones de motilidad, digestión, absorción y excreción. El tracto digestivo superior es el más frecuentemente afectado: entre un 70 y un 90% de los pacientes desarrollan alteración. 16.
(28) INTRODUCCIÓN ■ ■ 1 esofágica 57,58, que no siempre es sintomática. Típicamente se manifiesta con una disminución de presión del esfínter esofágico inferior y una alteración de la motilidad de los 2/3 inferiores del esófago que traduce una pérdida de ondas peristálticas 59, lo que condiciona reflujo gastroesofágico, disfagia, pirosis y síntomas respiratorios secundarios al reflujo.. El compromiso de otras áreas del tracto intestinal es mucho menos frecuente en la ES.. La afección del estómago se manifiesta característicamente como retraso del vaciamiento y, en algunos pacientes en forma de ectasias vasculares del antro gástrico (GAVE), también conocido como estómago “en sandía” 60. La GAVE se podría considerar como un componente del espectro de alteraciones vasculares de la ES debido a las similitudes anatomopatológicas con los otros cambios vasculares de la enfermedad.. A nivel del intestino delgado la alteración de la motilidad favorece el sobrecrecimiento bacteriano 61. Finalmente es relativamente frecuente la incontinencia fecal por sustitución del esfínter anal interno por colágeno, que afecta al 38% de los pacientes 62 .. Afección pulmonar La afección pulmonar es la segunda en orden de frecuencia en la ES. Más de un 70% de los pacientes la presentan 63, dividida en enfermedad pulmonar intersticial (EPI) e hipertensión arterial pulmonar (HAP). Actualmente es la principal causa de mortalidad de la enfermedad 64. ◀ Hipertensión arterial pulmonar: Es la afección vascular más grave de la enfermedad y se presenta en aproximadamente el 9% de los pacientes 65. Se manifiesta clínicamente por disnea y en los estadios finales con signos de insuficiencia cardiaca derecha. El diagnóstico de la HAP es exclusivamente hemodinámico. Se realiza mediante cateterismo cardiaco derecho, y se define como una presión arterial pulmonar media (PAPm) en reposo ≥25mmHg asociada a una presión capilar pulmonar (PCP) ≤15mmHg y unas resistencias vasculares pulmonares (RVP) > a 3 unidades Wood 66. Histopatológicamente los cambios observados en la arteria pulmonar son similares a los de la vasculopatía digital, con presencia de hiperplasia de la íntima, proliferación. 17.
(29) 1. ■■. INTRODUCCIÓN de adventicia, fenómenos de trombosis in situ y diversos grados de inflamación y arteriopatía plexiforme 18. Todos los pacientes con ES están en riesgo de desarrollar HAP a lo largo del transcurso de la enfermedad, pero hay un subgrupo en los que este riesgo es mayor, que son los que presentan: afección cutánea limitada de larga evolución, telangiectasias y ACA o ANAs con patrón nucleolar por IFI y sin la presencia de ATA-I 67,68. Además, existen otros factores asociados a mayor riesgo de HAP: valores desproporcionadamente bajos de capacidad de difusión de monóxido de carbono (DLCO) en las pruebas de función respiratoria (PFR), ratio de la capacidad vital forzada (CVF) / porcentaje esperado de la DLCO >1.6. 68,69,. diferentes parámetros ecocardiográficos. 68,70,. valores elevados del péptido natriurético cerebral (BNP) o su porción amino terminal del pro-péptido natriurético cerebral (NT-proBNP) 71 e incluso la presencia de pérdida de densidad capilar en la capilaroscopia 72 . El cribado precoz de la HAP se realiza mediante Ecocardiograma y PFR, y es de vital importancia, ya que los pacientes diagnosticados en clases funcionales bajas de la NYHA (I y II) tienen mejor pronóstico a largo plazo que los diagnosticados con clase funcional más evolucionada (III y IV) 73. ◀ Enfermedad Pulmonar Intersticial: La EPI es la afección pulmonar más frecuente y relevante de la enfermedad con una prevalencia aproximada del 60% clínica y 90% en las autopsias realizadas 74. Se cree que se produce por una combinación de factores inflamatorios y lesiones fibróticas. La EPI actualmente es la principal causa de mortalidad de la ES 64. En un 45-55% de los pacientes con ES y EPI se produce un deterioro en las PFR y, de estos, un 16% desarrollará una EPI grave que se define como una CVF <55% 51. Los principales factores pronósticos de desarrollar una EPI grave son: CVF o DLCO <70% en las PFR, una extensión >20% de cambios de EPI en la Tomografía axial computerizada de alta resolución pulmonar (TACAR) o el deterioro rápidamente progresivo de la función pulmonar (definido como un descenso de 10% de CVF o 15% de la DLCO en los 12 meses previos). Los pacientes que cumplan estas características serían tributarios de recibir tratamiento específico 75. La presencia de ACA o la ausencia de lesiones en TACAR en el momento del diagnóstico se han relacionado con un riesgo menor de presentar EPI grave 76,77.. 18.
(30) INTRODUCCIÓN ■ ■ 1 Respecto al patrón histológico, el más prevalente es el de neumonía intersticial no específica (NINE) que se presenta en el 76% de los casos, seguido por la neumopatía intersticial usual (NIU) en el 11% de los pacientes 78. Dado el gran impacto de la EPI en la ES, al igual que ocurre con la HAP, es fundamental una detección precoz de la misma. Se recomienda realizar una TACAR pulmonar a todos los pacientes con ES al diagnóstico de la enfermedad así como PFR completas periódicas tanto para evaluar la gravedad de la EPI como para monitorizarla. Afección renal El riñón se puede afectar de varias formas dentro de la ES. La más característica y relevante es la crisis renal esclerodérmica (CRE), que se define por los criterios de Traub 79, como la presencia de hipertensión arterial grave de inicio brusco y/o insuficiencia renal oligoanúrica. Son comunes la microangiopatía trombótica, la anemia y la insuficiencia cardiaca, y algunos pacientes desarrollan encefalopatía hipertensiva y retinopatía 80.. En hasta un 10% de los casos la CRE cursa sin hipertensión (CRE normotensiva) y parece asociarse al uso previo de antagonistas del calcio o inhibidores del enzima convertidor de la angiotensina (IECA) 80,81.. Normalmente aparece en los primeros años de diagnóstico de la ES e incluso en algunos casos es la clínica que lleva al diagnóstico de la enfermedad 82.. Su incidencia ha disminuido en los últimos años hasta ser actualmente en Europa de 5%. Afecta a un 10% de pacientes con ES difusa y a un 2% con ES limitada 81.. Antes de la introducción de los IECA, la CRE era la primera causa de muerte y presentaba una incidencia mucho mayor que la actual, de hasta un 20%. Actualmente justifica sólo el 4% de las muertes de los pacientes con ES 83.. 19.
(31) 1. ■■. INTRODUCCIÓN. Los factores que aumentan el riesgo de sufrir CRE son: una duración de la enfermedad menor de 4 años, ES difusa con afectación cutánea rápidamente progresiva, presencia de anticuerpos RNAp, la exposición previa a dosis altas de corticoides, los roces tendinosos y nuevos eventos cardiacos 81,84,85. Steen, además, demostró que la reducción del flujo renal debido a sepsis, deshidratación y disfunción cardiaca es un potencial desencadenante de CRE 80, y recientemente estudios genéticos han evidenciado que los antígenos de histocompatibilidad HLA-DRB1*0407 y *1304 son un factor de riesgo independiente para el desarrollo de CRE 86.. Por otro lado, se ha considerado el uso profiláctico de IECAs para intentar prevenir la CRE, teniendo en cuenta sus buenos resultados en el tratamiento de la misma 87. Sin embargo, el hecho de que existan casos de CRE en pacientes con exposición previa a IECAs, unido a la teoría de que el tratamiento con IECAs puede enmascarar la hipertensión arterial que acontece en una CRE y, con ello, dificultar el diagnóstico, pone en serias dudas el beneficio de su uso de forma preventiva 88. Además, no hay una explicación fisiopatológica racional que lo apoye, ya que la mayor parte de pacientes con ES no presentan hiperreninemia antes del desarrollo de una CRE 89.. De hecho, hay datos extraídos de estudios retrospectivos que sugieren que la exposición previa a IECAs no sólo no reduce el riesgo de CRE, sino que se asocia con una peor evolución y mayor riesgo de requerir diálisis permanente 90-92 .. En un reciente estudio de ISRCS (Investigational Scleroderma Renal Crisis Survey) se encontró que el uso de IECAs previo al debut de la CRE se relacionaba con un riesgo dos veces mayor de mortalidad (2.42, 95% CI 1.0-5.75, p=0.046) 93.. Por tanto, se puede afirmar que, en el momento actual, el papel de los IECAs en la prevención de la CRE no está resuelto.. 20.
(32) INTRODUCCIÓN ■ ■ 1 Las características histopatológicas de la CRE consisten en una vasculopatía obliterativa de las arteriolas renales que provocan una isquemia del glomérulo.. Por otro lado, la afección renal subclínica afecta aproximadamente al 50% de pacientes con ES y se puede asociar con otras manifestaciones vasculares 94. Esta afección subclínica raramente progresa a una insuficiencia renal terminal, sin embargo se ha asociado en estudios recientes con un peor pronóstico de otras complicaciones vasculares, como la HAP 81,95.. Afección cardiaca La afección cardiaca primaria debida directamente a la ES es muy heterogénea. Puede afectar al miocardio, pericardio, al sistema de conducción, endocardio y menos frecuentemente, a las válvulas cardiacas 96. Hasta hace unos años se considerada que la ateroesclerosis y las lesiones coronarias macrovasculares no estaban aumentadas en la ES, al contrario de lo que ocurría en otras enfermedades sistémicas como el LES o la artritis reumatoide donde predomina más la inflamación 97, pero recientemente se ha evidenciado un riesgo aumentado de enfermedad coronaria en pacientes con ES respecto al de la población general 98.. Así, Man et al concluyeron que la ES se asocia con un mayor riesgo de presentar infarto de miocardio, infarto cerebral o patología vascular periférica 99. De la misma manera en una amplia cohorte australiana se observó una prevalencia de enfermedad coronaria tres veces mayor en los pacientes con ES que en la población general 100.. Sin embargo, al contrario que en otras enfermedades autoinmunes, los mecanismos potenciales de una ateroesclerosis acelerada en el curso de la ES todavía no están claros. Se necesitan más estudios para demostrar la posible contribución de la inflamación activa y el daño vascular fibroproliferativo.. La prevalencia de la patología cardiaca en la ES es variable y difícil de determinar por la diversidad de las manifestaciones clínicas y los diferentes métodos diagnósticos utilizados.. 21.
(33) 1. ■■. INTRODUCCIÓN. Actualmente, el uso de técnicas como el ecocardiograma, la tomografía cardiaca, la tomografía computarizada de emisión monofotónica (SPECT) miocárdica, la resonancia magnética cardiaca, la tomografía por emisión de positrones (PET) o la ventriculografía nuclear, que presentan una alta sensibilidad para su diagnóstico, permite una detección precoz tanto de las patologías cardiacas estructurales como funcionales asociadas a la ES 101.. Con frecuencia, la afección cardiaca es silente y hay una clara disparidad entre su prevalencia determinada por estudios clínicos y la encontrada tras resultados histológicos. Así, en algún estudio se recoge una prevalencia clínica global del 10%, siendo ésta en la autopsia mayor del 50% 102.. Los hallazgos histopatológicos típicos incluyen un compromiso del miocardio con fibrosis en placas sin afección de epicardio pero con presencia de hipertrofia de la capa íntima arteriolar de las arterias intramurales coronarias 103.. Cada vez hay mayor evidencia de que la afección miocárdica es debida a procesos isquémicos focales repetidos que llevan a fibrosis miocárdica con lesiones irreversibles, con una alteración de la microcirculación y vasorreactividad alterada, con o sin anormalidades vasculares estructurales. Esto lleva a una disfunción del ventrículo izquierdo sistólica o diastólica y también del ventrículo derecho 104.. Recientemente se ha demostrado que el uso de vasodilatadores, incluyendo antagonistas del calcio y IECAs, mejoran tanto la perfusión como la función, lo que apoya el papel del daño de la microcirculación 97.. Cuando se manifiesta clínicamente, la patología cardiaca asociada a la ES supone un factor pronóstico importante 97. En los últimos años, ante la mejoría del pronóstico de la CRE con el uso de IECAs, la afección cardiaca y la pulmonar son las principales causas de muerte asociadas a la enfermedad en los pacientes con ES 96.. 22.
(34) INTRODUCCIÓN ■ ■ 1 En una cohorte de 953 pacientes con ES y subtipo cutáneo difusa se encontró una prevalencia del 15% de afección cardiaca basada en hallazgos clínicos, ecocardiografía, electrocardiografía o Holter. Además, se estudió la distribución de las muertes en estos pacientes en función del tiempo transcurrido entre el debut de la enfermedad y el momento de la muerte, y se observó que de las muertes asociadas a la ES un 20% eran debidas a patología cardiaca, con un claro mayor impacto de esta afección en la mortalidad en los primeros 5 años 51.. La afección cardiaca se presenta tanto en la forma limitada como en la forma difusa de la enfermedad, si bien su prevalencia es mayor en esta última. Se ha encontrado asociación entre los anticuerpos ATA-I y RNAp, la presencia de miopatía o afección cutánea rápidamente progresiva y el desarrollo de afección cardiaca 105,106.. Afección osteoarticular Las artralgias son muy frecuentes en la ES, al contrario que la artritis que se presenta con un porcentaje de 16% 107. Se puede observar solapamiento con artritis reumatoide en el 1-5% de los pacientes con ES 108, que es más frecuente en pacientes con ES limitada.. La presencia de roces tendinosos es del 11% de los pacientes europeos con ES, asociándose a mayor afección vascular digital, muscular, renal y una peor supervivencia 107. Además, el 20% de los enfermos presentan acroosteolisis o resorción ósea del penacho distal de las falanges 109, hallazgo muy característico de la enfermedad.. Afección muscular En la ES se pueden presentar 2 tipos de miopatías: la no inflamatoria descrita como debilidad muscular unida a alteraciones de los enzimas musculares y patrón miógeno en EMG, presente hasta en un 53% y la inflamatoria, menos frecuente, que afecta al 17% de pacientes con ES que clínicamente es indistinguible a la presentada en entidades como la polimiositis o dermatomiositis 110. Se asocia a presencia de anticuerpos anti Pm/Scl 111 o antiKu 112.. 23.
(35) 1. ■■. INTRODUCCIÓN. Los hallazgos histológicos de los pacientes con ES y miopatía inflamatoria son muy heterogéneos e incluyen fenómenos de necrosis, fibrosis, anomalías vasculares e inflamación.. Otras manifestaciones clínicas Otras manifestaciones menos frecuentes de la enfermedad son el síndrome seco asociado o no al síndrome de Sjögren, la cirrosis biliar primaria, neuropatía periférica (neuralgia del trigémino), atrapamiento nervioso (túnel carpiano), disfunción eréctil en los varones y afectación tiroidea.. 1.1.5. Pronóstico La ES es una enfermedad muy heterogénea con un pronóstico muy variable. Presenta una elevada mortalidad debida generalmente a las propias complicaciones orgánicas específicas.. Hace unos años se publicaron las causas de muerte de la cohorte de pacientes con ES del grupo EUSTAR 83 que incluía un total de 5860 pacientes. El 55% de todas las muertes registradas estaban directamente relacionadas con la ES y el 54.6% de los pacientes fallecidos pertenecían al subtipo con ES difusa. Las principales causas de muerte eran la EPI (19%) y la HAP (14%). Un 14% de las muertes se atribuyeron a afección cardíaca (la mayoría por arritmias) y sólo un 4% fueron secundarias a CRE. Los porcentajes de las causas de muerte no relacionadas con la ES fueron las siguientes: 13% infecciones, 13% neoplasias y 12% causas cardiovasculares.. Resultados similares hemos observado posteriormente en el estudio realizado por el Grupo español (RESCLE) 40 en el que un 55% de las muertes se debían a una causa directamente relacionada con la ES, de las cuales la más frecuente fue la HAP aislada (16.6%), seguida de la EPI (13%) y HAP relacionada con EPI en el 12.3%. La CRE fue una causa poco frecuente de muerte (8.7%).. 24.
(36) INTRODUCCIÓN ■ ■ 1 De las causas no relacionadas con la ES el cáncer suponía el 11.6%, seguido por la insuficiencia cardiaca (no atribuida a la ES) en 8.7%.. También un metanálisis realizado recientemente que incluye los datos de 17 estudios, estimó un 47.6% de muertes debidas a complicaciones directas de la ES, y, de éstas, un 73% eran por causa cardiopulmonar. Por otro lado, este estudio objetivó que las afecciones gastrointestinal y renal grave se habían reducido en las últimas 2 décadas considerablemente, representando actualmente sólo un 18% de las muertes 113.. Estudios recientes evidencian una mejoría del pronóstico en los últimos años, probablemente debido a la detección precoz de algunas complicaciones orgánicas, con un mejor manejo y tratamiento de las mismas, especialmente de la CRE 64.. En el metanálisis previamente citado, se analizó la supervivencia de la ES antes y después de 1990, encontrando que a los 10 años del debut de la enfermedad, ésta era mayor en la época actual que antes de 1990; sin embargo no se consiguió la significación estadística que sí se encontró en la supervivencia a los 5 años de seguimiento de los pacientes. A raíz de estos datos, el autor sugiere que los nuevos tratamientos utilizados para la patología pulmonar parenquimatosa y vascular han mejorado la esperanza de vida, pero fracasan en mantener su efecto de forma prolongada.. Ferri et al publicaron recientemente un interesante estudio donde revisaron la literatura médica y encontraron que había un incremento considerable en la supervivencia de los enfermos con ES en las últimas décadas a los 10 años, que pasaba del 54% en los pacientes reclutados entre 1955 y 1985 al 83.5% en pacientes reclutados entre 1999 y 2011 114. Los autores, en su cohorte de pacientes reclutados entre el 2000 y el 2011, objetivaron una supervivencia a los 10 años calculada desde la aparición del FR del 95%, similar a la de Simeón-Aznar et al (93%) 40.. 25.
(37) 1. ■■. INTRODUCCIÓN. Nihtyanova et al nos muestran en otro trabajo reciente unas tasas de supervivencia en una cohorte de enfermos en el Reino Unido del 94%, 81.7% y 69.2% a los 5, 10 y 15 años, respectivamente para la ES limitada y del 85.5%, 71.6% y 55.1% para la ES difusa 115.. Respecto a los factores pronósticos de mortalidad de la ES, se han identificado los siguientes en los estudios multivariables: - Extensión cutánea: el subtipo cutáneo difuso, sobre todo con rápida progresión cutánea; - Afección orgánica: CVF <70%, HAP, CRE, derrame pleural, arritmia clínicamente significativa en ECG ; - Autoinmunidad: presencia de ATA-I y ausencia de ACA; - Epidemiológicos: edad al diagnóstico >60 años, raza no caucásica, sexo masculino; - Genéticos: presencia de alelos HLA-DRB*0802 y DQA1*0501; - Serológicos: VSG >15, hemoglobina <12.5; - Capilaroscópicos: patrón activo 116.. 1.1.6. Tratamiento En la actualidad, todavía no disponemos de un tratamiento curativo ni que pueda modificar el curso de la ES, por lo que el abordaje de la enfermedad se basa en tratar las complicaciones orgánicas e intentar impedir la progresión de éstas, así como el tratamiento sintomático de las diferentes manifestaciones clínicas.. En la siguiente tabla se recogen las recomendaciones terapéuticas de las complicaciones más frecuentes de la ES, publicadas por el grupo EUSTAR en el 2009 117. Este grupo publicará próximamente una actualización de las mismas.. 26.
(38) INTRODUCCIÓN ■ ■ 1. Número. I. II. Recomendación. Evidencia. Vasculopatía Digital Grave 1. Un meta-análisis de antagonistas de los receptores de la endotelina y un meta-análisis de prostanoides indica que nifedipino e iloprost reducen la frecuencia y severidad de los ataques del FR-ES. Los antagonista del calcio dihidropiridínicos, usualmente el nifedipino oral, deben ser considerados para el tratamiento de primera línea del FR-ES y el Iloprost intravenoso u otro prostanoide intravenoso para el tratamiento del FR-ES severo.. A. 2. Dos ECA indican que los prostanoides intravenosos (particularmente iloprost) son eficaces para curar las UD-ES. Los prostanoides intravenosos se deben de considerar para el tratamiento de UD activas en pacientes con ES.. A. 3. Bosentan no ha confirmado su eficacia en el tratamiento de UD activas. Bosentan ha confirmado eficacia en dos ECA para prevenir las UD en ES, especialmente en pacientes con múltiples UD. Se puede considerar el uso de Bosentan en pacientes con UD múltiples después de fallo a antagonistas del calcio o terapia prostanoide. A. Hipertensión arterial pulmonar 4. Dos ECA de alta calidad indican que bosentan mejora la capacidad de ejercicio, la CF y algunos parámetros hemodinámicos en la HAP. Bosentan debe ser fuertemente considerado para el tratamiento de la HAP-ES.. A/B. 5. Dos ECA de alta calidad indican que sitaxsentan mejora la capacidad de ejercicio, la CF y algunos parámetros hemodinámicos en la HAP. Sitaxsentan puede ser considerado para tratar la HAP-ES. A/B. 6. Un ECA de alta calidad indica que sildenafilo mejora la capacidad de ejercicio, CF y algunos parámetros hemodinámicos en HAP. Sildenafilo se puede considerar para tratar la HAP-ES.. A/B. 7. Un ECA de alta calidad indica que epoprostenol intravenoso en infusión continua mejora la capacidad de ejercicio, CF y parámetros hemodinámicos en HAP . La retirada repentina del fármaco puede ser peligrosa para la vida. Epoprostenol debe ser considerado para el tratamiento de la HAP-ES grave. III. Compromiso cutáneo 8. Dos ECA han mostrado que MTX mejora la afección cutánea en la ES inicial. Otros efectos beneficiosos en otros órganos no se han observado . MTX debe ser considerado para el tratamiento de las manifestaciones cutáneas de la ES inicial.. IV. VI. A. Enfermedad pulmonar intersticial 9. Tras el resultado de 2 ECA y a pesar de su conocida toxicidad, CFM debe ser considerada para el tratamiento de la EPI-ES. V. A. A. CRE 10. A pesar de la ausencia de ECA, los expertos consideran que los IECA tiene que usarse en el tratamiento de la CRE. C. 11. Cuatro estudios retrospectivos sugieren que los esteroides están asociados con un riesgo elevado de CRE . Hay que controlar de manera estricta la presión arterial y la función renal en pacientes con corticoterapia. C. Compromiso gastrointestinal 12. A pesar de la ausencia de ECA específicos en la ES, el comité de expertos considera que los IBP deben usarse para la prevención de el RGE asociado a la ES, úlceras esofágicas y estenosis esofágicas. B. 13. A pesar de la ausencia de ECA específicos, el comité de expertos considera que el uso de fármacos proquinéticos debe usarse para el manejo de las alteraciones de la motilidad sintomáticas (disfagia, dispepsia, pseudo-obstrución, etc .) .. C. 14. A pesar de la ausencia de ECA específicos, el comité de expertos cree que cuando la malabsorción está causada por el sobrecrecimiento bacteriano, el uso de antibióticos rotatorio puede ser útil. D. Tabla 7: Recomendaciones EULAR 2009 para el tratamiento de las complicaciones de la Esclerosis Sistémica. ECA: ensayo clínico aleatorizado; CF: Clase funcional de la NYHA; MTX: metotrexato; CFM: ciclofosfamida; IECA: inhibidores del enzima convertidor de la angiotensina; IBP: inhibidores de la bomba de protones; RGE: reflujo gastroesofágico.. 27.
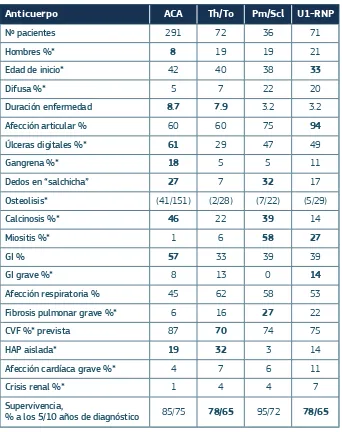
(39) 1. ■■. INTRODUCCIÓN. 1.2. LOS ANTICUERPOS Y LA ESCLERODERMIA 1.2.1. Aspectos clínicos de los anticuerpos en la Esclerodemia Los anticuerpos antinucleares son la marca de las enfermedades autoinmunes y en la ES están presentes es más del 95% de pacientes 46. Son biomarcadores atractivos de la enfermedad debido a su alta especificidad, la exclusividad mutua, su persistencia durante el curso la enfermedad y, sobre todo, la relación de cada uno de ellos con perfiles clínicos determinados 118.. Los ANA específicos de la ES normalmente son excluyentes entre sí, de tal manera que es excepcional que en un paciente se detecten 2 ó más tipos. Así, en un estudio de 5423 pacientes, en sólo 28 (0.52%) coexistían el ATA-I y el ACA. 119,. los 2 anticuerpos más. frecuentes.. Están presentes normalmente al principio de la enfermedad y se mantienen durante todo el curso de la misma, y, por otro lado, es muy infrecuente su presencia en otras enfermedades autoinmunes y excepcional en patologías no inmunomediadas 118.. Los primeros autoanticuerpos relacionados con la ES fueron identificados en 1960. 120,. y se asociaron con los 2 tipos de esclerodermia que se conocían. Los anticuerpos ATA-I se relacionaron clínicamente con la ES difusa y los anticuerpos ACA, con la ES Limitada. En la década de los 90, se identificaron nuevos anticuerpos específicos de esta enfermedad, que se relacionaron con diferencias significativas en la frecuencia y la especificidad de la afectación de los diferentes órganos 121,122 .. Actualmente se conocen siete anticuerpos antinucleares específicos para la ES. Así, pacientes con ES limitada presentan generalmente uno de los siguientes cuatro anticuerpos: ACA, anti Th/To, Pm/Scl y U1-RNP; y los pacientes con ES difusa pueden presentar ATA-I, RNAp y U3-RNP 46.. 28.
Figure




+7





Outline
CARACTERÍSTICAS DE LOS ANTICUERPOS DE LA ESCLERODERMIA
Antitopoisomerasa I o Scl70 (ATA-I)
Anti RNA polimerasa III (RNAp)
Anti Th/To
Anti Pm/Scl
Otros (Anti Ro 52, Anti U11/U12 RNP, Anti Nor 90)
IMPLICACIÓN DE LOS ANTICUERPOS EN LA SUPERVIVENCIA
DETERMINACIÓN DEL PODER PRONÓSTICO DE LOS SUBTIPOS
JUSTIFICACIÓN DEL ESTUDIO
Documento similar
You may wish to take a note of your Organisation ID, which, in addition to the organisation name, can be used to search for an organisation you will need to affiliate with when you
Where possible, the EU IG and more specifically the data fields and associated business rules present in Chapter 2 –Data elements for the electronic submission of information
The 'On-boarding of users to Substance, Product, Organisation and Referentials (SPOR) data services' document must be considered the reference guidance, as this document includes the
In medicinal products containing more than one manufactured item (e.g., contraceptive having different strengths and fixed dose combination as part of the same medicinal
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
Package Item (Container) Type : Vial (100000073563) Quantity Operator: equal to (100000000049) Package Item (Container) Quantity : 1 Material : Glass type I (200000003204)
