Enero-Abril 2006 Págs. 27-32
Artículo de revisión
edigraphic.com
1 Médico residente de la Especialidad de Dermatología.2 Médico residente de la Subespecialidad de Cirugía Dermatológica. 3 Médico adscrito de la División de Dermatología.
Hospital General “Dr. Manuel Gea González”, Secretaría de Salud. México, D.F.
Correspondencia:
Dra. Diana Sugey Vera-Izaguirre.
División de Dermatología, Hospital General “Dr. Manuel Gea González”. Calzada de Tlalpan 4800, Col. Toriello Guerra, Delegación Tlalpan, 14000 México, D.F.
Tel: 56653511. Extensión 168. E-mail: [email protected]
Penfigoide ampolloso
Diana Sugey Vera-Izaguirre,1 Marcia Karam-Orantes,2 Elisa Vega-Memije3
RESUMEN
Antecedentes: El penfigoide ampolloso (PA) es una enfermedad
autoinmune frecuentemente observada en pacientes de edad avanzada. Su diagnóstico se establece con la combinación de características clínicas, histológicas e inmunopatológicas. El PA se caracteriza por la presencia de ampollas tensas asentadas en piel normal o eritematosa que tienen predilección por las extremida-des y frecuentemente la enfermedad tiende a generalizarse. En el estudio histológico se observa una ampolla subepidérmica, con predominio de eosinófilos. Los estudios de inmunofluorescencia (IF) directa de piel perilesional muestran depósitos lineales de IgG y/o C3 en la zona de la membrana basal (ZMB). Pueden utilizarse corticosteroides tópicos para el penfigoide localizado; para el generalizado se utilizan dosis altas de corticosteroides y otras drogas inmunosupresoras, como dapsone y azatioprina. El PA es una enfermedad autolimitada, pero su curso y pronóstico va a depender del estado general del paciente geriátrico.
Palabras clave: Penfigoide ampolloso (PA), ampolla
subepidér-mica, inmunofluorescencia (IF), corticosteroides (CE).
ABSTRACT
Background: Bullous pemphigoid (BP) is an autoimmune bullous disease commonly seen in the elderly. The diagnosis is established in combination of clinical, histologic and immunopa-thologic features. BP is characterized by the presence of tense bullae arising on normal skin or on an erythematous base, that have a predilection for the extremities and often tends to be generalized. The direct immunofluorescence (IF) studies on peri-lesional skin reveal linear basement membrane zone (BMZ) deposits of IgG and/or C3. Topical corticosteroids can be used for localized bullous pemphigoid and they may also be considered for generalized pemphigoid. Other immunosuppressive drug therapy is considered for patients who require high maintenance doses of corticosteroid, cases of adverse effects, and for patients whose disease does not respond completely to corticosteroid therapy. The course and prognosis of BP are variable but in general is a self-limiting disease.
Key words: Bullous pemphigoid (BP), immunoflorescence (IF), corticosteroids (CE.)
INTRODUCCIÓN
El término “penfigoide” es utilizado para un grupo de enfermedades que comparten características clínicas de vesículas y ampollas, que en la histopatología muestran una ampolla subepidérmica, con infiltrado
inflamatorio rico en eosinófilos y en menor proporción neutrófilos, así como anticuerpos IgG tanto circulantes como unidos a piel, contra proteínas específicas de la zona de la membrana basal (ZMB). Hay tres tipos principales de penfigoide: 1) Penfigoide ampolloso (primariamente enfermedad cutánea). 2) Penfigoide de membranas mucosas (primariamente enfermedad de mucosas). 3) Penfigoide (herpes) gestacional (pri-mariamente una enfermedad cutánea en mujeres embarazadas.1
Este artículo está enfocado a revisar al penfigoide ampolloso, con lesiones dermatológicas. La enferme-dad se considera una enfermeenferme-dad ampollosa autoin-mune. La base de la autoinmunidad del PA fue sugeri-da por Jordon et al (1967) con la identificación de los depósitos de IgG y componentes del complemento en la piel de los pacientes con PA.2
edigraphic.com
sustraídode-m.e.d.i.g.r.a.p.h.i.c cihpargidemedodabor
:rop odarobale FDP VC ed AS, cidemihparG
arap acidémoiB arutaretiL :cihpargideM sustraídode-m.e.d.i.g.r.a.p.h.i.c
occidental,1,3 con una incidencia estimada de 10 casos
por millón2 y con una incidencia anual estimada de seis
a siete casos por millón en poblaciones de Francia y Alemania.4 Es principalmente una enfermedad de
pacientes de edad avanzada, primariamente indivi-duos mayores de 60 años de edad. Raramente se ha reportado en infantes y niños, sin embargo, se han reportado casos en pacientes de todas las edades. Afecta ambos sexos por igual y no existe una fuerte asociación con raza o localización geográfica. No se conoce asociación con ningún gen HLA clase II y PA1,3-6
y no han podido ser identificados los factores etiológi-cos. Se han reportado eventos como trauma cutáneo local (cirugía), exposición a radiaciones ionizantes o ultravioleta, vacunación y medicamentos sistémicos asociados con el inicio de PA como furosemida, ben-zotiazidas, espironolactona y diazepam.
Muchos pacientes presentan una erupción prodró-mica, erupción pre-ampollosa que consiste en máculas eritematosas, pápulas urticariformes y placas o, con menor frecuencia, lesiones eccematosas.4 Las vesículas
y ampollas tienden a diseminarse y ocasionalmente forman rosetas.
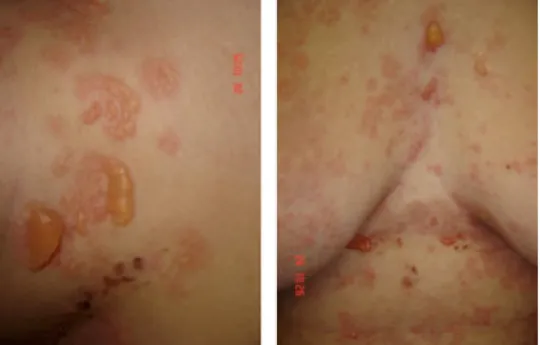
Las lesiones típicas son ampollas tensas y grandes, las cuales tienen predilección por las extremidades (cara interna de muslos y áreas flexurales de brazos y pier-nas), área inguinal, axilas, abdomen y cuello1,5,6 (Figuras
1 y 2). Se involucra de forma menos común cara y piel
cabelluda.7 El líquido de la ampolla es comúnmente
claro, pero ocasionalmente puede ser hemorrágico. Estas ampollas frecuentemente permanecen intactas y no se deforman con la presión, su ruptura conduce a la formación de erosiones que evolucionan a costras y ocasionalmente pueden ser invadidas por bacterias patógenas. La dermatosis puede ser leve o intensamen-te pruriginosa o incluso ser asintomática. Pueden pre-sentarse lesiones en mucosas,5,6 la frecuencia varía del
8% al 50%1,4 y es la mucosa oral la más afectada. Dentro
de otras membranas mucosas involucradas se incluyen faringe, uretra y conjuntiva; las lesiones tienden a ser leves y transitorias y son generalmente observadas en pacientes con enfermedad cutánea extensa.1
Existen múltiples variantes clínicas de la enferme-dad: penfigoide dishidrosiforme, penfigoide vesicular, penfigoide nodular, penfigoide vulvar localizado de la infancia, penfigoide localizado pretibial y penfigoide vegetante.6,8
Las características histológicas de la biopsia de una ampolla reciente son las mismas para el penfigoide ampolloso o gestacional e incluyen la presencia de ampolla subepidérmica con infiltración moderada a densa de eosinófilos y otras células inflamatorias den-tro de la cavidad de la ampolla. No hay necrosis de la epidermis subyacente, excepto de la porción media
Figuras 1 y 2. Penfigoide
ampo-lloso. Dermatosis generalizada, constituida por ampollas tensas con líquido claro y exulceraciones.
edigraphic.com
del techo de la ampolla, donde la ampolla tiene más tiempo de aparición. En algunos casos, los eosinófilos pueden ser observados en una dirección en aproxima-ción con la ZMB. Los linfocitos también se observan frecuentemente y, en casos raros, predominan los neutrófilos. El infiltrado inflamatorio usualmente es confinado a la dermis papilar y a la porción superficial de la dermis reticular. Puede haber también edema en dermis papilar (Figura 3). Los diagnósticos diferenciales histológicos incluyen casos de dermatitis herpetiforme, dermatosis lineal por IgA cuando presentan el número incrementado de esosinófilos. Contrariamente, de
for-ma ocasional existen casos de PA que presentan un número incrementado de neutrófilos. En estas situa-ciones, la inmunofluorescencia (IF) directa es proba-blemente la forma más fácil de distinguirlos.8
Los procedimientos diagnósticos convencionales para el PA, como la (IF) directa e indirecta, e inmuno-blot (IB); se han complementado con el método sensi-ble y específico de ELISA.9
La (IF) es la investigación más notable para realizar el diagnóstico; la IF directa muestra depósitos de autoanticuerpos (IgG) en la unión dermo-epidérmica
(Figura 4) y la IF indirecta muestra autoanticuerpos
Figura 3. A. Formación de ampollas subepidérmicas e infiltrado inflamatorio compuesto por neutrófilos y eosinófilos en la dermis
y cavidad de la ampolla, por microscopia de luz. B. Un acercamiento muestra el infiltrado rico en eosinófilos (tinción de
hematoxilina-eosina 4 y 60x).
Figura 4. Penfigoide ampolloso. A. Estudios de inmunofluorescencia directa de piel perilesional que demuestran depósitos continuos
edigraphic.com
circulantes directamente dirigidos contra las proteínas de la membrana basal que se localizan en el lado epidérmico de la piel en la técnica de SALT split.3
Una biopsia perilesional para la IF directa muestra depósitos lineales de IgG y/o C3 en la ZMB en cerca del 100% de los casos; se pueden observar adicionalmente otras inmunoglobulinas (IgM, IgA, IgD, o IgE) en la misma distribución.4-6,8 La IgG es la inmunoglobulina
más comúnmente observada en un 60% a 90% de los casos en donde la IgG4 es la subclase predominante.8
La IF indirecta puede ser positiva en más del 90% de los pacientes con PA.
Los autoanticuerpos reaccionan con 2 componen-tes de los hemidesmosomas del epitelio estratificado: el antígeno del PA 230 (BP230) y el antígeno 180 (BP180).9,10 El antígeno del PA 230 ha sido implicado
en la organización de los filamentos de queratina y es localizado en la placa de los hemidesmosomas de forma intracelular. El antígeno del PA 180, está involu-crado en el anclaje del epitelio estratificado a la membrana basal subyacente, es una proteína trans-membrana tipo II.
El PA es una enfermedad que resulta de una res-puesta autoinmune anormal (autoanticuerpos) con una respuesta inflamatoria prominente (infiltrado celu-lar). Las terapias del PA tienen como finalidad suprimir la inflamación y/o respuesta inmune. Antes de elegir la terapia, se deben considerar las variables relacionadas con la enfermedad (extensión del involucro y los síntomas) y con el paciente (edad; otras enfermedades como diabetes, hipertensión). El objetivo del trata-miento es la cicatrización de las lesiones existentes y la prevención de nuevas lesiones. La aparición de una lesión no requiere del incremento de la dosis del tratamiento.1,4,11
En la literatura se menciona el uso de los corticoste-roides tópicos potentes para el PA localizado. Un estudio reciente sugiere que incluso deben considerar-se como tratamiento para el PA generalizado.12 Para la
enfermedad leve a moderada, las tetraciclinas y nico-tinamida son una buena opción terapéutica.3,13
La mayoría de los pacientes con PA generalizado requieren de terapia sistémica. Los corticosteroides son los agentes sistémicos más comúnmente utiliza-dos.1 Los corticosteroides tienen tanto efectos
anti-inflamatorios como inmunosupresores que resultan en una disminución de los linfocitos circulantes, eosinófi-los, monocitos y basófilos. Los neutrófilos circulantes aumentan porque existe una liberación incrementada
de la médula ósea, una disminución en su remoción de la circulación y un aumento en su acumulación de la pared de los vasos.1 La prednisona es el agente más
frecuentemente utilizado y es suficiente como mo-noterapia en la mayoría de los casos. La dosis es de 0.5-1 mg/kg/día dependiendo de la severidad de la enfermedad. Se han utilizado los pulsos de corticoste-roides con metilprednisolona intravenosa 1 mg diario por 3 días consecutivos para el control inicial de una enfermedad severa.14 Se debe pensar en la utilización
de otros agentes inmunosupresores sólo si la dosis de los corticosteroides no se puede reducir a niveles acepta-bles, presencia de efectos adversos y en los pacientes en quienes su enfermedad no responde completamente con la terapia con corticosteroides. La azatioprina es el principal agente establecido, seguido del metotrexa-te.3,13 Otros inmunosupresores también frecuentemente
utilizados son la ciclofosfamida, ciclosporina y micofe-nolato de mofetilo.1,4,5,15
La azatioprina es un antimetabolito de las purinas y un imidazol derivado de la mercaptopurina; interactúa con los compuestos sulfihidrilo como el glutatión. Es utilizada a dosis de 2-5 mg/kg/día.1,16 El metotrexate es
un antimetabolito y es un análogo del ácido fólico. La dihidrofolato reductasa (DHFR) reduce el folato a tetrahidrofolato, se utiliza a dosis de 10-25 mg sema-nalmente. La ciclofosfamida es un agente alquilante; los tejidos de proliferación y con altos rangos de mitosis son más susceptibles,1 es utilizada a dosis de 1-2 mg/kg/
día.17 La ciclosporina suprime la inmunidad celular
significativamente y en menor grado la humoral, se utiliza a dosis de 6 mg/kg/día (en dos dosis iguales).18 El
micofenolato de mofetilo es un inhibidor de la síntesis de purina en células T y B activadas. Se ha utilizado exitosamente como terapia coadyuvante, por ser aho-rrador de corticosteroides, y también como monotera-pia. El efecto terapéutico puede ser esperado después de 6 a 8 semanas de la administración continua de 0.5-1 g diariamente.4
Con terapias alternativas, el tratamiento puede llevar-se a cabo con 50-200 mg/día de dapsona o 500-1,500 mg/día de sulfapiridina (asociada a menor hemólisis en comparación con la dapsona). La respuesta se obtiene después de 2 a 12 semanas. La dapsona y sulfapiridina se han utilizado de forma efectiva en pocos casos,19-22
edigraphic.com
mecanismo de acción en enfermedades inflamatorias no está bien entendido, se cree que inhibe la mielo-peroxidasa mediada por el sistema citotóxico, el cual probablemente juega un papel en la destrucción de tejido por la quimiotaxis de neutrófilos.11 Los efectos
anti-inflamatorios de la dapsona se observan particular-mente en las dermatosis con acumulación de neutrófi-los, como la dermatitis herpetiforme y enfermedad lineal por IgA. En PA el rango de respuesta es alrededor del 45%, a pesar de que en unas series fue sólo del 15%.13 Se debe excluir la presencia de la deficiencia de
glucosa-6-fosfato deshidrogenasa, la cual predispone al paciente a los efectos adversos hematológicos. Casi siempre puede observarse cierto grado de meta-hemo-globinemia (en personas jóvenes, un 15-20% es bien tolerado), siendo severa en pacientes deficientes de la enzima. La anemia hemolítica que se observa en pa-cientes dosis-dependientes se presenta en las primeras semanas de tratamiento. La hemólisis ocurre en el 100% de los pacientes, quienes reciben dosis de 200 a 300 mg diarios. Ambos efectos pueden reducirse con el trata-miento con cimetidina 400 mg de dos a tres veces al día o dosis altas de vitamina E 800 IU/día. El Probenecid reduce la excreción de dapsona, con lo que incrementa los efectos adversos.4,11
Otros tratamientos se han reportado efectivos en el manejo del PA, incluyéndose la plasmaféresis,23,24 altas
dosis de IgG intravenosa,25,26 tetraciclinas o
eritromici-na con o sin nicotieritromici-namida,27 clorambucil,28
tacroli-mus29 y talidomida.1
La plasmaféresis consiste en el vaciamiento de la sangre del paciente, utilizando un filtro que separa los elementos celulares del plasma y regresando sólo los elementos celulares al paciente. Este procedimiento da como resultado la capacidad de remover los au-toanticuerpos patogénicos. Altas dosis de IgG intrave-nosa es una fuente humana purificada de IgG del plasma. Las tetraciclinas y nicotinamida se utilizan usualmente juntas para el tratamiento de dermatosis ampollosas autoinmunes. Las tetraciclinas tienen un efecto anti-inflamatorio así como propiedades modu-ladoras inmunes. El mecanismo no es claro, pero inhiben la quimiotaxis de neutrófilos y eosinófilos. La nicotinamida es una vitamina cuyo mecanismo de acción en las enfermedades ampollosas y otras derma-tosis es desconocido. El clorambucil es también un agente alquilante del tipo de la mostaza nitrogenada que inhibe la síntesis del DNA.1 El tacrolimus reduce
aparentemente las lesiones urticariformes.4
El curso y pronóstico del PA es variable. A diferencia del pénfigo vulgar, el penfigoide ampolloso es una enfermedad que se autolimita y que usualmente se resuelve dentro de los 5 años posteriores al diagnóstico. Exacerbaciones siguen a remisiones y son usualmente leves.1,4 A pesar de la introducción de los
corticosteroi-des sistémicos, la mortalidad del PA no se ha mejorado y varía entre 0% y 40%.4 El riesgo de muerte aumenta
con incremento de la edad en el inicio de la enferme-dad y en la dosis de corticosteroides así como con la disminución de la albúmina sérica. La muerte ocurre frecuentemente dentro de las primeras 12 semanas de inicio del tratamiento.30 Las causas de muerte incluyen
efectos adversos de la terapia, debilidad generalizada y enfermedad subyacente asociada.
CONCLUSIONES
El PA es la enfermedad ampollosa autoinmune más frecuente del mundo occidental. El objetivo del trata-miento es suprimir los signos clínicos del PA lo suficien-te para hacerlo tolerable para el paciensuficien-te. La mayoría de los pacientes con PA generalizado requieren de terapia sistémica. Los efectos inmunomoduladores de los corticosteroides usualmente conducen a una rápida supresión de la formación de ampollas; de hecho son el tratamiento principal desde su desarrollo, sin embar-go, a pesar de su introducción de forma sistémica, la mortalidad del PA no se ha mejorado; por lo que aun cuando el penfigoide ampolloso es una enfermedad que se autolimita hay que brindarle la atención nece-saria de forma cautelosa y precisa.
REFERENCIAS
1. Mutasim DF. Autoimmune bullous dermatoses in the elderly. diagnosis and management. Drugs Aging 2003; 20: 664-78. 2. Colbert RL, Allen DM, Eastwood D, Fairley JA. Mortality rate of
bullous pemphigoid in a US Medical Center. J Invest Dermatol 2004; 122: 1091-95.
3. Khumalo NP, Wojnarowska F et al. A systematic review of treat-ments for bullous pemphigoid. Br J Dermatol 2002; 138: 385-89. 4. Khumalo NP. Management of bullous pemphigoid. recommenda-tions for immunomodulatory treatments. Am J Clin Dermatol 2004; 5: 319-26.
5 Fitzpatrick’s et al. Dermatology in General Medicine. McGraw-Hill Sixth Edition; Volume I: 574-79.
6. Bologna JL, Jean L, Jorrizzo JL, Joseph L, Rapini RP. Dermatology Mosby 2003: 463-470.
edigraphic.com
8. Barnhill RL, Crouson AN. Textbook of dermatopathology.McGraw-Hill Second edition. 1998: 181-83.
9. Mariotti FG, Terracina M, Ruffelli M., Cordiali-Fei P, Sera F, Zambruno G, Mastrogiacomo A, Di Zenzo G. Development of a novel ELISA system for detection of anti-BP 180 IgG and charac-terization of autoantibody profile in bullous pemphigoid patients. British Journal of Dermatology 2004; 151: 1004-10.
10. Diaz LA et al. Isolation of a human epidermal cDNA correspon-ding to the 180-kD autoantigen recognized by bullous pemphi-goid and herpes gestations sera. Immunolocalization of this pro-tein to the hemidesmosome. J Clin Invest 1990; 86: 1088-94. 11. Diya F, Mutasim M. Management of autoinmmune bullous
disea-ses: Pharmacology and therapeutics. J Am Acad Dermatol 2004; 51: 859-877.
12. Roijeau JP, Benichou J et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 2002; 346: 321-27.
13. Wojnarowska F, Kirtschig G, Highet AS et al. Guidelines for the management of bullous pemphigoid. Br J Dermatol 2002; 147: 214-21. 14. Eaglitein WH, Siegel J. High-dose methylprednisolone in the treatment of bullous pemphigoid. Arch Dermatol 1984; 120: 1157-65.
15. Fine JD. Management of acquired bullous skin disease. New Engl J Med 1995; 333: 1475.
16. Ahmed AR. Azathioprine. Int J Dermatol 1981; 20: 461-69. 17. Ahmed AR. Cyclophosphamide review of relevant pharmacology
and clinical use. J Am Acad Dermatol 1984; 6: 1115-1126. 18. Thiovolet J, Bartelemy H, Rigot-Muller G et al. Effects of
cyclos-porine on bullous pemphigoid and pemphigus. Lancet 1985; I: 334-335.
19. Person JR. Bullous pemphigoid responding to sulfapyridine and the sulfones. Arch Dermatol 1997; 113: 610-615.
20. Venning VA et al. Dapsone as first line therapy for bullous pemphigoid. Br J Dermatol 1989; 120: 83-87.
21. Bouscarat F et al. Treatment of bullous pemphigoid with dapso-ne: retrospective study of thirty-six cases. J Am Acad Dermatol 1996; 34: 686.
22. Jeffes EW, Ahmed AR. Adjuvant therapy of bullous pemphigoid with dapsone. Clin Exp Dermatol 1991; 25: 691-697.
23. Goldberg NS, Roenigk Jr HH et al. Plasmapheresis therapy for bullous pemphigoid. Arch Dermatol 1985; 121: 1484-85. 24. Roujeau JC, Guillaune JC, Morel P et al. Plasma ex change in
bullous pemphigoid. Lancet 1984; II: 486-89.
25. Enginneer L, Ahmed AR. Role of intravenous immunoglobulin in the treatment of bullous pemphigoid: analysis of current data. J Am Acad Dermatol 2001; 44: 83-88.
26. Ahmed AR. Intravenous immunoglobulin therapy for patients with bullous pemphigoid unresponsive to conventional immuno-suppressive treatment. J Am Acad Dermatol 2001; 45: 825-35. 27. Thomas I, Khorenian S, Arbesteld DM. Treatment of generalized
bullous pemphigoid with oral tetracycline. J Am Acad Dermatol 1993; 28: 74-77.
28. Milligan A, Hutchinson P. The use of chlorambucil in the treatment of bullous pemphigoid. J Am Acad Dermatol 1990; 22: 769-01. 29. Chu J, Bradley M, Marinkovich MP. Topical tacrolimus is a useful
adjunctive therapy for bullous pemphigoid. Arch Dermatol 2003; 139: 813-15.