Aplicación de un algoritmo bioinspirado para optimizar los parámetros de los métodos scan
57
0
0
Texto completo
(2) Declaración de autoría Hago constar que el presente trabajo fue realizado en la Universidad Central “Marta Abreu” de Las Villas como parte de la culminación de los estudios de la especialidad de Ciencia de la Computación, autorizando a que el mismo sea utilizado por la institución, para los fines que estime conveniente, tanto de forma parcial como total y que además no podrá ser presentado en eventos ni publicado sin la autorización de la Universidad.. ______________________________ Firma del autor. Los abajo firmantes, certificamos que el presente trabajo ha sido realizado según acuerdos de la dirección de nuestro centro y el mismo cumple con los requisitos que debe tener un trabajo de esta envergadura referido a la temática señalada.. __________________________ Firma del tutor. ___________________________ Firma del jefe del Laboratorio.
(3) Dedicatoria.
(4) Agradecimientos.
(5) Pensamiento.
(6) Resumen Los métodos Scan clásicos y borrosos se utilizan ampliamente para la detección de conglomerados sobre una secuencia linear o circular de datos. Generalmente la secuencia ha sido procesada previamente y transformada en una secuencia binaria. El valor uno representa la categoría de interés y el valor cero representa lo demás. El objetivo de los métodos es detectar un conglomerado de unos. Estos métodos dependen de varios parámetros: el ancho de la ventana móvil que recorre la secuencia de entrada, el paso con el que la ventana se mueve y el tamaño de la parte borrosa en las variantes borrosas de los métodos Scan. En este trabajo se muestra como la unión de un algoritmo bioinspirado y el método de simulación de Monte Carlo, sirven de ayudan en la búsqueda de valores adecuados para los parámetros de las técnicas Scan. Además se muestran aplicaciones con datos simulados y se resuelve un problema de bioinformática..
(7) Abstract The classic and fuzzy Scan methods are widely used for the detection of conglomerates on a linear data or on a circular data. Generally, the sequence has been processed previously and transformed into a binary sequence. The value one represents the category of interest and value zero represents the others. The objective of the methods is to detect conglomerates of the values one. These methods depend on several parameters: the length of the moving window that scan the binary sequence, the step to be used by the window and the size of the fuzzy part, in the fuzzy variants of the Scan methods. In this work it is shown how the union of a Particle Swarm Optimization algorithm (PSO) and the Monte Carlo Simulation cooperate in the search of the correct values for the parameters of the Scan techniques. Besides, applications with simulated data are discussed and a bioinformatics’ problem is solved..
(8) Índice INTRODUCCIÓN........................................................................................................................................... 1 CAPÍTULO 1. MÉTODOS MATEMÁTICOS............................................................................................. 5 1.1 LOS MÉTODOS SCAN CLÁSICOS PARA LA DETECCIÓN DE CONGLOMERADOS ............................................ 5 1.1.1 El método Scan sobre una Línea ...................................................................................................... 5 1.1.2 El método Scan sobre un Círculo ..................................................................................................... 7 1.1.3 Algunas consideraciones sobre los métodos Scan............................................................................ 8 1.2 LOS MÉTODOS SCAN GENERALIZADOS ..................................................................................................... 8 1.3 LOS MÉTODOS SCAN BORROSOS ............................................................................................................. 10 1.3.1 Elementos de lógica borrosa .......................................................................................................... 10 1.3.2 El método Scan Borrosos sobre una Línea..................................................................................... 13 1.3.3 El método Scan Borroso sobre un Círculo ..................................................................................... 17 1.3.4 Algunas consideraciones sobre los métodos Scan Borrosos .......................................................... 18 1.4 ALGORITMOS BIOINSPIRADOS: EL PSO................................................................................................... 19 1.5 ALGORITMO PSO APLICADO A LOS MÉTODOS SCAN ............................................................................... 21 1.6 MÉTODOS DE MONTE CARLO ................................................................................................................. 21 1.7 MÉTODOS DE MONTE CARLO COMBINADO CON EL PSO Y LOS MÉTODOS SCAN ..................................... 22 CONSIDERACIONES FINALES DEL CAPÍTULO ................................................................................................. 23 CAPÍTULO 2. DETALLES SOBRE LA IMPLEMENTACIÓN DEL SISTEMA.................................. 25 2.1 ESTRUCTURA DE LAS CLASES ................................................................................................................. 25 2.2 MANUAL DE USUARIO ............................................................................................................................ 25 CONSIDERACIONES FINALES DEL CAPÍTULO ................................................................................................. 36 CAPÍTULO 3. APLICACIONES ................................................................................................................ 37 3.1 GENERACIÓN DE SECUENCIAS ................................................................................................................ 37 3.1.1 Bases de la simulación realizada ................................................................................................... 37 3.2 RESULTADOS Y DISCUSIÓN CON DATOS SIMULADOS ............................................................................... 38 3.2.1 Análisis con verdaderos conglomerados ........................................................................................ 39 3.2.2 Análisis con falsos conglomerados................................................................................................. 40 3.3 ESTUDIO DEL GENOMA DE LA ESCHERICHIA COLI .................................................................................. 41.
(9) 3.3.1 Análisis de la E. Coli usando los métodos Scan ............................................................................. 41 3.3.2 Análisis de la E. Coli usando los métodos Scan y el PSO .............................................................. 42 CONSIDERACIONES FINALES DEL CAPÍTULO ................................................................................................. 42 CONCLUSIONES ......................................................................................................................................... 44 RECOMENDACIONES ............................................................................................................................... 45 REFERENCIAS BIBLIOGRÁFICAS ........................................................................................................ 46.
(10) Introducción La secuenciación de genomas ha generado millones de secuencias de pares de bases nucleotídicas de ADN (Ácido desoxirribonucleico), o moléculas de la vida. Se conocen las secuencias de más de un millón y medio de proteínas, de más de cien genomas, la estructura tridimensional de más de 20 mil proteínas, entre otras. Gracias a los experimentos de matrices de ADN o micro arreglos se sabe cuándo y cómo se expresan muchos genes. Además se dispone de muchos datos que indican si dos proteínas interactúan entre sí o no. Todo el conocimiento científico acumulado a lo largo de las últimas décadas se encuentra disperso en más de 12 millones de artículos Uno de los mayores retos de los científicos de hoy es poder procesar de manera eficiente este enorme volumen de datos. Las técnicas estadísticas tradicionales no dan abasto para resolver problemas de tal magnitud y complejidad. Es por ello que la estadística como ciencia se ha desarrollado de manera vertiginosa en los últimos años. Su intersección con técnicas de inteligencia artificial (aprendizaje automatizado), aumenta cada vez más. Es muy común encontrar libros actuales de “Estadística Computacional”. Por sólo mencionar un ejemplo, la versión 16 del SPSS (Statistical Package for the Social Sciences), que es sin dudas, uno de los paquetes estadísticos más utilizados en la actualidad, incluye un módulo de redes neuronales, técnicas clásica de inteligencia artificial. En este sentido pueden mencionarse además, algunos métodos de aprendizaje supervisado, como árboles de decisión, y otros de aprendizaje no supervisado, como las técnicas de detección de conglomerados. Existen numerosos métodos estadísticos para la detección de conglomerados. Algunos son clásicos, como los métodos que conforman conglomerados jerárquicos, o los de las k medias, mientras que otros surgieron para resolver problemas específicos de una rama de las ciencias médicas: la epidemiología. Cualquier enfermedad nueva que exhiba un patrón epidemiológico marcado será rápidamente reconocida como tal. Características típicas de las transmisibles pueden ser la tendencia de que los casos aparezcan en grupos familiares o laborales, la existencia de un intervalo de tiempo relativamente bien definido entre enfermos (intervalos seriales) y la posibilidad de encontrar contactos de cada caso con un caso previo. En enfermedades no transmisibles pudiera tenerse información acerca del comportamiento de ciertos. 1.
(11) determinantes de la enfermedad tales como la exposición a tóxicos y condiciones ambientales adversas entre otros. Cuando no están disponibles todas estas observaciones, diversos tipos de tablas y gráficos ayudan a detectar posibles patrones. Así por ejemplo, en una serie de casos en un poblado puede notarse que el gráfico de fechas de diagnóstico muestre picos regularmente espaciados que deben corresponderse con los intervalos seriales. El uso de gráficos y en especial de mapas mostrando la distribución geográfica son algunas de las técnicas más utilizadas en el ámbito epidemiológico. Los epidemiólogos tienen sus propios métodos de detección de epidemias, que de hecho han probado ser eficientes en numerosas ocasiones; les permiten detectar con cierta precisión la aparición de focos infecciosos, pero no son totalmente confiables y en ocasiones conllevan a cometer errores. Los matemáticos están interesados en redefinir y hacer más precisos esos procedimientos mediante el uso de alguna prueba de significación. Las mayores dificultades surgen cuando los datos tienen una naturaleza anecdótica. No se trata en estos casos de que no puedan aplicarse pruebas estadísticas para arrojar un resultado, más bien lo que ocurre es que tales pruebas quedan invalidadas porque los datos pueden estar sesgados o parcializados en algún sentido. La formulación rigurosa de técnicas estadísticas ayuda así a los epidemiólogos también en un sentido metodológico, con el fin de lograr datos correctos o al menos seguir un esquema o diseño preconcebido. Si ello se logra, aunque el proceso de recolección no sea perfecto, será posible extraer conclusiones más fidedignas en la medida en que se utilice el aparato matemático más amplia y consecuentemente. En la práctica suele ocurrir que la información disponible no es tan satisfactoria y los datos, aunque quizás sugieran una epidemia, no descartan una incidencia puramente al azar. Es en estos casos en los que se debe esperar que algún test de significación estadística ayude al proceso de toma de decisiones. Por otra parte, la mayoría de las investigaciones matemáticas sobre epidemias asumen que ya esta fue reconocida como tal y comienzan sus estudios buscando un modelo matemático suficientemente sencillo que no se aleje demasiado de la realidad epidemiológica. Por lo general, para probar si un modelo es adecuado se realizan pruebas con diversos tests de bondad de ajuste, (Bailey 1975). 2.
(12) En numerosos trabajos se aborda matemáticamente la detección de focos epidémicos buscando “conglomerados”. Se denomina conglomerado, aglomeración o cluster de enfermos a un exceso de casos diagnosticados con respecto a cierto patrón previamente predefinido. En casi la totalidad de los casos, los métodos de detección de conglomerados dependen de parámetros. En ocasiones resulta difícil para un investigador, que puede ser incluso un especialista en el tema que se aborda, determinar a priori los valores óptimos para esos parámetros. Valores incorrectos pueden conducir a resultados erróneos: detectar falsos conglomerados, o lo que es mucho peor no detectar a tiempo los verdaderos. Por otra parte, existen nuevos algoritmos de optimización que han demostrado ser altamente eficientes en la solución de numerosos problemas. Pueden citarse por ejemplo los algoritmos que usan heurísticas aleatorias bioinspiradas como los algoritmos genéticos, la optimización basada en enjambres de partículas (PSO) de sus siglas en inglés: Particle Swarm Optimization y la optimización basada en las colonias de hormigas. Hasta aquí se han mencionado dos líneas de trabajo aparentemente paralelas: los métodos de detección de conglomerados, entre los que se encuentran las técnicas Scan, y los algoritmos bioinspirados que resuelven problemas de optimización. Ello nos lleva a plantear la siguiente pregunta de investigación: ¿Podrán los algoritmos bioinspirados ayudar a la selección de los parámetros adecuados de los métodos de detección de conglomerados? En el presente trabajo se pretende combinar el uso de varios métodos matemáticos con el objetivo general de determinar valores adecuados de los parámetros de los métodos Scan. Para ello se utiliza un algoritmo bioinspirado, (PSO), y el método de Monte Carlo. Los objetivos específicos se desglosan como sigue: ¾ Diseñar un sistema computacional que “optimice” los valores de los parámetros de los métodos Scan utilizando el PSO. ¾ Utilizar el método de Monte Carlo para generar secuencias similares a la original y aplicar los algoritmos anteriores sobre ellas. ¾ Implementar el sistema. ¾ Validar el sistema con datos simulados. 3.
(13) ¾ Mostrar ejemplos reales en el campo de la Bioinformática. La tesis está estructurada en tres capítulos. El primero de ellos constituye el marco teórico. En él se detalla una revisión bibliográfica sobre los métodos Scan para la detección de conglomerados y se mencionan algunas aplicaciones de los mismos para resolver problemas específicos en el campo de la Epidemiología y de la Bioinformática. Se describe además en detalle el algoritmo PSO y el método de Monte Carlo. Se muestran igualmente aplicaciones bioinformáticas. El capítulo dos está dedicado a explicar el sistema computacional que se elabora. Se muestran detalles de su análisis y diseño. En el tercer capítulo se realiza la validación, se enuncia la metodología creada y se muestran aplicaciones concretas en Bioinformática. Finalmente se enuncian las conclusiones y las recomendaciones del trabajo.. 4.
(14) Capítulo 1. Métodos matemáticos En este capítulo se exponen los fundamentos matemáticos desarrollados en el presente trabajo. Primeramente se brinda una breve panorámica acerca de los métodos de detección de conglomerados, haciendo énfasis en los métodos Scan clásicos y borrosos. Posteriormente, para determinar los valores óptimos de sus parámetros se utiliza un algoritmo bioinspirado: optimización basada en enjambre de partículas vinculada con una técnica de simulación por Monte Carlo.. 1.1 Los métodos Scan Clásicos para la detección de conglomerados Según la literatura especializada se denomina conglomerado o cluster a un exceso de casos de enfermos diagnosticados en un área geográfica determinada (conglomerado espacial), en un período de tiempo limitado (conglomerado temporal), o considerando dominios espacio temporales (conglomerado espacio-temporal). En los últimos años han surgido numerosos métodos capaces de detectar clusters en espacio, en tiempo o considerando ambos escenarios a la vez. No existen técnicas globales que puedan aplicarse a todas las situaciones, por eso hay gran diversidad de métodos con la misma finalidad.. 1.1.1 El método Scan sobre una Línea Los métodos Scan se utilizaron inicialmente para detectar aglomeraciones dentro de períodos de tiempo consecutivos, pues puede suceder que un conglomerado temporal se extienda por dos o más intervalos. (Jacquez, Waller et al. 1996). Todos los casos diagnosticados deben estar ordenados cronológicamente de acuerdo con la fecha de primeros síntomas o de diagnóstico de la enfermedad, de muerte o cualquier otro evento de salud que se considere. Sean X1, X2, … Xn variables aleatorias independientes e idénticamente distribuidas que denotan las fechas de ocurrencias de n eventos en el intervalo (0,T]. Se quiere probar la hipótesis nula de que los eventos están uniformemente distribuidos contra la alternativa de que existe un conglomerado dentro de algún subintervalo de (0,T],(Nagarwilla 1996) Se define un intervalo o una ventana de tamaño fijo de acuerdo con la duración esperada de la epidemia, (esto debe hacerse antes de inspeccionar los datos recolectados). La ventana.
(15) seleccionada se desplaza a lo largo de la línea del tiempo y se determinan en cada caso, la cantidad de enfermos asociados a ella, (Aldrich and Wanzer 1993). Para la formulación sean: t : amplitud de la ventana, T : período de tiempo total que se analiza, L = T t : fracción que representa el período de tiempo total que se analiza con relación al. ancho de la ventana, n : cantidad de enfermos diagnosticados en T,. λ : número esperado de casos por unidad de tiempo en un proceso de Poisson, w y , y +t : cantidad de enfermos en la ventana [y, y+t). Hipotéticamente el estadístico: w = w t (T ) = max {w y , y +t } representa el número de casos 0 ≤ y ≤ T −t. que aparecen en una ventana cuando se mueve continuamente a lo largo del tiempo. En la práctica, la ventana [y, y+t) se mueve discretamente a partir de una sucesión de puntos equidistantes y1, y2,…yk que cubren todo el período de análisis de amplitud T. Se denomina paso del Scan o paso del desplazamiento a Δy = y k − y k −1 . Realmente, el estadístico anterior se estima por su versión discreta:. {. w = w t , Δt (T ) = max w y i , y i + t 1 ≤ i ≤kt. }. La idea del método es que, si existe un conglomerado, el número máximo de casos hallados en una ventana debe ser grande. El test estadístico depende de varios de los parámetros explicados con anterioridad y en esencia calcula la probabilidad p de que aparezcan w o más casos en una ventana. La fórmula que se utilizó para p es la propuesta en (Naus 1982):. p = P * (w, λ L, 1 L ) =1 − Q * (w, λ L, 1 L ). (1.1). donde Q * puede ser aproximado para cualquier L>2 a partir de sus valores con L = 2 y L = 3.. [. ]. Q * (w, λ L,1 L ) ≈ Q * (w, 2 λ , 1 2) Q * (w, 3 λ , 1 3) Q * (w, 2 λ , 1 2). L−2. (1.2). 6.
(16) La aproximación (1.2) es fácilmente calculable usando una microcomputadora personal. El cálculo exacto de Q* (w, 2λ ,1 2) y Q * (w, 3 λ , 1 3) se basa en un teorema demostrado también en (Naus 1982)y cuya esencia se resume aquí: w. Para w>2, pi = e − λ λi i ! , Fw = ∑ pi , λ > 0 , se tiene que: i =0. Q * (w, 2 λ ,1 2) = Fw2−1 − (w − 1) p w p w− 2 − (w − 1 − λ ) p w Fw−3 Q * (w, 3 λ ,1 3) = Fw3−1 − A 1 + A2 + A3 − A4 donde:. A 1 = 2 p w Fw−1 ((w − 1) Fw−2 − λ Fw−3 ). (. A2 = 0.5 p w2 (w − 1)(w − 2)Fw−3 − 2(w − 2) λ Fw− 4 + λ2 Fw−5. ). w −1. A3 = ∑ p 2 w− r Fr2−1 r =1. w −1. A4 = ∑ p 2 w− r p r ((r − 1)Fr − 2 − λ Fr −3 ) r =2. donde Fi = 0 para todo i<0. La aproximación (1.2) puede calcularse para valores no enteros de L. Esto la diferencia de otras expresiones matemáticas que se usaban con estos fines anteriormente. Además de ser menos restrictiva, varios autores demuestran que (1.2) es mucho más precisa, (Naus 1982; Glaz 1993; Sahu, Bendel et al. 1993).. 1.1.2 El método Scan sobre un Círculo Este método es una variación del anterior y se utiliza para enfermedades que tengan un comportamiento estacional. Los datos se encuentran ordenados cronológicamente a lo largo de la línea del tiempo y el círculo se forma uniendo la última fecha con la primera. La ventana se desplaza sobre el círculo y se determina en cada una, la cantidad de enfermos asociados a ella. Con este desplazamiento circular se pretende incorporar al análisis la cercanía de posibles casos a “finales del último período considerado” con los del principio del “primer período considerado”, como si fueran “los del siguiente período”.. 7.
(17) La probabilidad de observar w o más casos en un intervalo o ventana de tamaño fijo se estima por:. p = Pc* (w, λ L, 1 L ) = 1 − Qc* (w, λ L,1 L ). (1.3). donde ahora:. [. Qc* (w, λ L,1 L ) ≈ Q * (w, 4λ ,1 4 ) Q * (w, 3λ ,1 3). ] [Q (w, 2λ ,1 2) ] L−2. *. L −1. (1.4). Para hallar Q * (w, 4λ ,1 4) se utiliza L=4 en (1.2). Después de simplificar se obtiene:. [. ]. Q * (w , 4λ ,1 4) ≈ Q * (w, 3λ ,1 3). 2. Q * (w, 2λ ,1 2 ). (1.5). Luego Q * (w, 4λ ,1 4) también queda en función de Q * (w, 2λ ,1 2) y de Q * (w, 3λ ,1 3) . Estos últimos valores se calculan en forma exacta a partir de las fórmulas anteriores, (Naus 1982).. 1.1.3 Algunas consideraciones sobre los métodos Scan Como se ha visto la probabilidad p hallada para un conjunto particular de casos, depende del ancho de la ventana y del paso del Scan seleccionados por el investigador. Resulta imposible determinar los valores ideales para cada enfermedad, por lo que se recomienda realizar varias repeticiones del método utilizando amplitudes diferentes, (Aldrich and Wanzer 1993; Kulldorff 2001) Algunos autores han tratado de generalizar el método Scan a dos dimensiones para que pueda utilizarse en la detección de aglomeraciones espaciales de enfermos, (Kulldorff 1997; Kulldorff 1999; Kulldorff 2001). En vez de un intervalo de longitud fija se mueve un rectángulo de dimensiones fijas sobre un área también rectangular, aunque se ha estado estudiando la posibilidad de que el conjunto a mover tenga una forma general cualquiera al igual que el área total a analizar, (Erick 1997). También se han hecho generalizaciones al caso espacio – temporal, (Kulldorff 1998).. 1.2 Los métodos Scan Generalizados En epidemiología, un conglomerado o “cluster” temporal de enfermos es un exceso de casos diagnosticados muy cercanos en el tiempo (Barrera 2000).. 8.
(18) En los métodos originales de detección, la variable de interés es el tiempo en que ocurre el evento. Dicho evento pudiera ser la fecha de diagnóstico de la enfermedad o incluso, la fecha en la que aparecieron los primeros síntomas si esta es suficientemente precisa (Burra 2002). El primer paso del algoritmo consiste en ordenar cronológicamente los datos obtenidos. Posteriormente se divide el eje que representa el tiempo total considerado en intervalos fijos que puede ser años, meses o días. A partir de este punto cada método sigue sus propios pasos para determinar si existen conglomerados. Estos algoritmos pueden transformarse para ampliar su campo de aplicación. La idea que se defiende en este trabajo es generalizarlos de manera que ellos puedan utilizarse para detectar conglomerados en un sentido más universal. Para lograrlo se propone ordenar los datos por algún criterio determinado que depende del campo de aplicación. Si se trabaja con fechas, los datos se ordenan cronológicamente, si se trabaja con secuencias de bases que representan algún gen completo, o una porción de este, sería correcto asumir que tal juego de datos ya está ordenado. El segundo paso consiste en transformar dicha secuencia en una secuencia análoga, pero dicotómica. El valor uno se colocará cada vez que aparezca la categoría de interés: una base, un aminoácido o una subsecuencia determinada dentro de una secuencia del ADN o de proteínas una fecha y otro evento que se considere. El valor cero se asociará a todas las demás categorías, (Rodríguez, Casas et al. 2006; Rodríguez, Casas et al. 2008). Los datos transformados se representan en una línea. El nuevo problema que surge es el de determinar si en la secuencia formada por ceros y unos existen conglomerados de unos. Por ejemplo, supóngase que se tiene una determinada secuencia de un gen y que dentro de ella resulta de interés determinar si existen conglomerados de la subsecuencia GCG. La transformación de la secuencia original en una dicotómica se realiza como se muestra en la figura 1.. Figura 1.1: Ejemplo de la conversión de una porción de la secuencia de un gen. 9.
(19) Obsérvese que la categoría de interés: subsecuencia GCG, se sustituyó por un uno, mientras que el resto de los casos considerados se sustituyó por el valor cero.. 1.3 Los métodos Scan Borrosos En este epígrafe se describen los fundamentos matemáticos de los métodos San borrosos.. 1.3.1 Elementos de lógica borrosa La lógica borrosa (o difusa) es algo que ha venido desarrollándose desde siglos atrás, hace 2500 años Aristóteles consideraba que existían ciertos grados de veracidad y falsedad y Platón había trabajado con grados de pertenencia. (Buckley 2006). Un conjunto borroso es aquel que no está formado por números sino por etiquetas lingüísticas. Una etiqueta lingüística es una palabra o conjunto de palabras, que representan los nombres de los conjuntos borrosos. En los conjuntos clásicos se sabe si un elemento de un universo de discurso pertenece o no a él acudiendo a la lógica booleana. Es decir, estos conjuntos se pueden definir con un predicado que asigne a cada elemento del conjunto el valor 0 ó 1, en función de su pertenencia al conjunto. En los conjuntos borrosos esto no es posible. Así, cada elemento tendrá un valor asociado dentro del conjunto que indicará en qué “cantidad” pertenece a dicho conjunto. Esto es lo que se define como grado de pertenencia. Por ello, un conjunto borroso es la unión de los grados de pertenencia de todos aquellos elementos que forman parte de su universo de discurso. El universo de discurso de un conjunto borroso es el intervalo en el que se incluyen los posibles valores que pueden tomar los elementos del conjunto. Con independencia de los valores que formen este universo, debe indicarse que siempre estará normalizado al intervalo [0,1]. Entonces un conjunto borroso A definido sobre un universo X es un par de la forma (x, ϕA(x)). Donde ϕA(x) es llamada la función de pertenencia o membresía (MF) para el conjunto A. La MF asigna a cada elemento de X un grado de pertenencia en el intervalo [0,1]. A X seria el universo de discurso y puede ser un espacio discreto o continuo.. 10.
(20) La existencia del grado de pertenencia para saber si un elemento pertenece a un conjunto o no, puede utilizarse para tratar problemas de imprecisión o incertidumbre en bases de datos, reconocimiento de patrones, clasificación, entre otras (Buckley, 2006). 1.3.1.1 Funciones de pertenencias. A continuación se mostrarán algunas funciones de pertenencia típicas: Función de pertenencia triangular. Se define por sus límites inferior a y superior b, y el valor modal m, tal que a < m < b.. Figura 1.2 Función de pertenencia triangular También puede representarse así: A(x;a,m,b) = máx { mín{ (x-a)/(m-a), (b-x)/(b-m) }, 0 } Función de pertenencia trapezoidal. Definida por sus límites inferior a y superior d, y los límites de su soporte, b y c, inferior y superior respectivamente.. Figura 1.3 Función de pertenencia trapezoidal Función de pertenencia Gausiana. Definida por su valor medio m y el valor k>0. Es la típica campana de Gauss. Cuanto mayor es el valor de k, más estrecha es la campana:. 11.
(21) Figura 1.4 Función de pertenencia Gausiana Función de pertenencia S. La Función S está definida por sus límites inferior a y superior b, y el valor m, o punto de inflexión tal que a < m < b. Un valor típico es: m=(a+b) / 2. El crecimiento es más lento cuanto mayor sea la distancia a-b.. Figura 1.5 Función de pertenencia S Función de pertenencia Gamma. Está definida por su límite inferior a y el valor k>0.. Figura 1.6 Función de pertenencia Gamma Esta función se caracteriza por un rápido crecimiento a partir de a. Mientras más grande sea el valor de k, el crecimiento es más rápido. La primera definición tiene un crecimiento más rápido. No llegan a tomar el valor 1, aunque tienen una asíntota horizontal en él. 1.3.1.2 Operaciones con conjuntos borrosos 12.
(22) Subconjunto:. El conjunto borroso A está contenido en el conjunto borroso B (es un subconjunto de B) o A es menor o igual que B, si y solo si, ϕB(x) ≥ϕA(x) para todo x, (Martín del Brio and Sánchez 2005). Unión (disyunción):. El resultado es un conjunto en el que se encuentren todos aquellos elementos de ambos conjuntos. En la representación gráfica, serían aquellas zonas en las que la superposición de ambas funciones de pertenencia, cuya función de membresía se define como: ϕC(x)= máx (ϕA(x), ϕB(x)), ver Figura 1.3. Intersección (conjunción):. El resultado de esta operación entre dos conjuntos borrosos, será un conjunto borroso en el que se encuentren aquellos elementos que están en ambos conjuntos. Si lo pensamos según sus funciones de pertenencia, se puede afirmar que: si se superponen ambas gráficas, la intersección es la zona en la que coinciden ambas funciones, cuya función de membresía se define como: ϕC(x)= mín (ϕA(x), ϕB(x)), ver Figura 1.2. Negación:. Esta es una operación válida sobre un único conjunto. La negación se define como todos aquellos elementos si forman parte de su universo de discurso, pero no forman parte de él, ϕ¬A(x)= 1-ϕA(x), ver Figura 1.4.. 1.3.2 El método Scan Borrosos sobre una Línea En este epígrafe se propone una modificación de los métodos Scan clásicos. Se trata de modificar la ventana de tamaño fijo por una ventana que tenga en cada extremo una función de pertenencia. De esa forma, se suavizan los extremos. (Rodríguez, Casas et al. 2009), Este método se utilizará en principio sobre secuencias binarias. Así, en cada uno de los extremos de la ventana, se incorpora una función de pertenencia que incorpora la información acerca de la magnitud de participación de los valores próximos a la ventana dentro de la secuencia. La ventana borrosa se define de la siguiente manera:. 13.
(23) s i -k ⎧ ⎪i * (g + 1) i ⎪⎪ Fuzzy Window k = ⎨s i ⎪ s ⎪(1 - i ) * i- k ⎪⎩ (g i + 1). i = k - g i ,..., g i i = k,..., k + t i = k + t + 1,..., k + t + g i. donde:. s1 , s2 , …, sn : es la secuencia binaria,. t : longitud de la ventana fija, gi : longitud de la parte borrosa de la nueva ventana. A esta parte se le llamará “suavizado”. La formulación matemática del test es esencialmente la misma: el método “escanea” los datos usando una ventana móvil borrosa. Ahora, el número máximo de casos reportados en una ventana no es necesariamente un valor entero, sino real. La figura 1.7 muestra una representación gráfica de ambas ventanas.. Figura 1.7 Ventanas clásica y borrosa en el método Scan sobre una línea. Obsérvese en el epígrafe 1.1.1 que el valor de la significación del método Scan sobre una Línea se basa en distribuciones de Poisson. Esta distribución está definida para variables aleatorias discretas, luego hay que realizar transformaciones al cálculo de la significación en el método clásico. Concretamente se proponen tres formas diferentes de calcular la significación: 1. Aproximar el valor real al valor entero más próximo. Las distribuciones de probabilidad y de distribución de Poisson se utilizan con bastante frecuencia dentro de las expresiones que aparecen en 1.1.1 y que se utilizan para hallar el valor de p. De aquí se deduce que la propagación del error pudiera no ser tan pequeña. Nos referiremos a este método como aproximación borrosa 1, ver figura 1.8. 14.
(24) Poisson distribution. Probability. 0.25 0.2 0.15 0.1 0.05 0 k.f f<0.5. k’.f’ f’≥0.5. Value. Figura 1.8: Función de probabilidad de Poisson ajustada usando la aproximación al valor entero más próximo, (aproximación borrosa 1) 2. Aproximar el valor real usando una combinación de dos distribuciones: Poisson hasta el valor entero inferior y uniforme para estimar la parte decimal. Nos referiremos a este método como aproximación borrosa 2, ver figura 1.9.. Figure 1.9: Función de probabilidad de Poisson ajustada usando la aproximación basada en dos distribuciones: Poisson y uniforme, (aproximación borrosa 2) Las fórmulas originales de Naus (Naus 1982), necesitan ser modificadas de la manera siguiente: k. λn. n =0. n!. - P[ x <= k.f ] = ∑. e -λ + f *. e - λ λk +1 (k + 1)!. -λ k -λ k - λ k +1 - P[ x = k.f ] = e λ + f * ⎛⎜ e λ - e λ ⎞⎟ ⎜ ⎟. k!. - A3 =. ⎝ k!. (k + 1)! ⎠. k.f. ∑ P[ x = 2 * k.f - r ] * P[ x <= r - 1] 2. r =1+ f. - A4 =. k.f. ∑ P [ x = 2 * k.f - r ] * P [ x = r ] ((r - 1) P [ x <= r - 2] - λ P [ x <= r - 3]). r =2+f. 15.
(25) 3. Aproximar el valor real utilizando funciones de interpolación. La interpolación es un método matemático de “construcción” de nuevos datos a partir de los ya existentes. En nuestro caso, los datos ya existentes se corresponden con las funciones de probabilidad y de distribución de Poisson respectivamente. Se utilizó un polinomio de interpolación de grado 4. Nos referiremos a este método como aproximación borrosa 3, ver figura 1.10. 8.5. 8.5. 0.14. 1.0. 0.12. 0.8. 0.10. 0.6. 0.08 0.06. 0.4. 0.04. 0.2 0.02 5. 10. 15. 20. 25. 30. 5. 10. 15. 20. 25. 30. Figura 1.10: Funciones de interpolación para las funcionees de probabilidad y de distribución de Poisson, (aproximación borrosa 3) Finalmente, la respuesta del método se “particiona” en dos conjuntos borrosos con las etiquetas: “significativo” y “no significativo”. Cada uno de ellos tiene una función de pertenencia S de la manera siguiente:. No significativo:. ⎧0 ⎪ ⎪ 2 * u - 0.05 ⎪ 0.0252 S(u ,0.05,0.0625,0.075) = ⎨ ⎪ 1 - 2 * u - 0.075 ⎪ 0.0252 ⎪ ⎩1. ⎧1 ⎪ u - 0.05 ⎪ 1- 2 * Significativo: ⎪ 0.025 2 S (u ,0.075,0.0875,0.1) = ⎨ ⎪ 2 * u - 0.075 ⎪ 0.0252 ⎪0 ⎩. 0.05. 0.075. u ≤ 0.05 0.05 < u < 0.0625 0.0625 ≤ u < 0.075 u ≥ 0.075. u ≤ 0.05 0.05 < u < 0.0625 0.0625 ≤ u < 0.075 u ≥ 0.075. 1. 16.
(26) Figure 1.11: Funciones de pertenencia borrosas: “significantivo” y “no significativo” Se aplica el método del máximo para eliminar el término borroso y obtener una respuesta dura (Martín del Brio and Sánchez 2005).. 1.3.3 El método Scan Borroso sobre un Círculo El método Scan Borroso sobre un círculo, se obtiene de una forma similar a su equivalente lineal. Este método se aplica sobre su variante generalizada, es decir la cadena de entrada es una secuencia binaria y el objetivo es determinar la existencia de conglomerados de “unos”. El círculo se forma uniendo los dos extremos de la secuencia. De esta forma, los primeros valores de la secuencia se analizan también al final. Esta técnica es particularmente útil para la detección de enfermedades que tienen un comportamiento estacional y en problemas de bioinformática para el análisis de conglomerados en secuencias de organismos que tienen su ADN circular. Al igual que su variante lineal, la ventana móvil se “suaviza” con una función de pertenencia en cada uno de sus extremos. El método borroso, usa estas funciones de pertenencia como factores de peso que influyen en la cantidad de casos repostados en cada una de las ventanas. Analíticamente, la ventana se define como: si ⎧ ⎪(i - k + g + 1) * (g + 1) ⎪⎪ Ventana móvil k = ⎨s i ⎪ s ⎪(k + t + g - i ) * i ⎪⎩ (g + 1). i = k - g,..., g i = k ,..., k + t - 1. (6). j = k + t ,..., k + t + g - 1. donde: S1, S2, S3, . . . . . . . . . . . . , Sn, Sn +1, Sn + 2, . . . . , Sn + t – 1 es la secuencia binaria. S n + i = Si. para i = 1 to t -1. Si = Sn −i. •. If i<1 then. •. If i > n + t -1 then. Si = Sn −i. g: longitud de la parte borrosa de la nueva ventana.. 17.
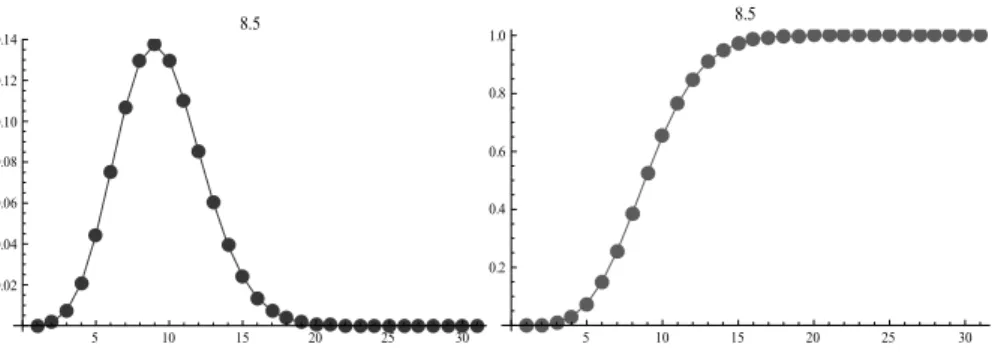
(27) La formulación matemática del test es esencialmente la misma: el método “escanea” la secuencia circular y contabiliza la cantidad de “unos” en cada ventana. Como que la ventana es ahora borrosa, el máximo número de casos reportados en una ventana no es necesariamente un valor entero, sino real. De la misma forma que para el método linear, se definen tres formas diferentes de calcular la significación del test: •. Aproximar el valor real al valor entero más próximo.. •. Aproximar el valor real usando una combinación de dos distribuciones: Poisson hasta el valor entero inferior y uniforme para estimar la parte decimal.. •. Aproximar el valor real utilizando funciones de interpolación.. La explicación de estas formas es básicamente la misma que se explicó en el epígrafe anterior. Igualmente, la respuesta del método se “particiona” en dos conjuntos borrosos con las etiquetas: “significativo” y “no significativo”. Cada uno de ellos tiene una función de pertenencia S como muestra la figura 1.5. Se aplica el método del máximo para eliminar el término borroso y obtener una respuesta dura.. 1.3.4 Algunas consideraciones sobre los métodos Scan Borrosos En estudios de simulación realizados en (Rodríguez, Casas et al. 2007) se ha demostrado la superioridad de los métodos Scan borrosos en relación con los clásicos. No obstante, la detección del parámetro óptimo para el tamaño de la ventana, o al menos la detección de un parámetro adecuado, sigue siendo un problema no resuelto. En algunas aplicaciones epidemiológicas, en las que se conoce bien el comportamiento de una determinada enfermedad, la selección del ancho de la ventana puede no ser un problema tan grave. Sin embargo, en la mayoría de los estudios bioinformáticos, esta selección a priori no resulta ser tan sencilla. La selección de parámetros no adecuados, puede conllevar a falsas conclusiones. En el siguiente epígrafe, se explican los fundamentos de un algoritmo de optimización que pretende ayudar a resolver el problema anterior.. 18.
(28) 1.4 Algoritmos bioinspirados: el PSO En la actualidad los modelos bioinspirados se muestran eficientes en la solución de problemas prácticos de diversas áreas. Dentro de los algoritmos bioinspirados usados para la selección de rasgos, la Inteligencia de Enjambres (Swarm Intelligence, SI) ha sido objeto de estudio, investigación y de mucha aplicación por su simplicidad y robustez. En particular se puede mencionar el uso de la técnica PSO en la búsqueda de la estructura de una red bayesiana (Chávez, Silveira et al. 2007; Chávez, Casas et al. 2008) La metaheurística PSO, fue desarrollada por Kennedy y Eberhart (Kennedy and Eberhart 1995; Kennedy 1997; Kennedy, Spears et al. 1998) y está inspirada en el comportamiento social observado en grupos de individuos tales como bandadas de pájaros, enjambres de insectos o bancos de peces. Un enjambre se define como una colección estructurada de organismos (agentes) que interactúan. La inteligencia no está en los individuos sino en la acción de todo el colectivo. Tal comportamiento social se basa en la transmisión del éxito de cada individuo a los demás del grupo, lo cual resulta en un proceso “sinergético” que permite a los individuos satisfacer de la mejor manera posible sus necesidades más inmediatas, tales como la localización de alimentos o de un lugar de cobijo. Cada organismo (partícula) se trata como un punto en un espacio N dimensional el cual ajusta su propio “vuelo” de acuerdo a su propia experiencia y la experiencia del resto de la banda. La banda (swarm) “vuela” por el espacio de búsqueda localizando regiones o partículas prometedoras (Kennedy and Eberhart 1995; Kennedy, Spears et al. 1998) Fundamentos generales del Algoritmo. Sean: xik. – Posición de la partícula i en la iteración k.. vik. – Velocidad de la partícula i en la iteración k.. pik. – Mejor posición de la partícula i.. pgk. – Mejor posición del grupo.. fik. – Valor de la función objetivo evaluada en xik.. fibest. – Mejor valor de la función objetivo evaluada en la partícula i.. fgbest. – Mejor valor de la función objetivo evaluada en el grupo.. 19.
(29) c1, c2 – Parámetros sociales y cognoscitivos. r1, r2 – Números aleatorios entre 0 y 1.. A continuación se describen los pasos del algoritmo: 1. Inicializar. a. Darle valores a las variables kmax, c1, c2. b. Inicializar aleatoriamente la posición de las partículas xi0 Є D en Rn for i = 1, .., p. c. Inicializar aleatoriamente la velocidad de las partículas 0 ≤ vi0 ≤ vmax for i = 1,.., p. d. k = 1 2. Optimizar. a. Calcular los valores de fik. b. Si fik ≤ fibest entonces fibest = fik , pik = xik. c. Si fik ≤ fgbest entonces fgbest = fik , pgk = xik. d. Si se cumple la condición de parada entonces ir a 3. e. Actualizar la velocidad de las partículas como sigue vik+1 = vik + c1 r1(pik xik ) + c2 r2 (pgk - xik).. f. Actualizar la posición de las partículas como sigue xik+1= xik + vik+1. g. Incrementar k. h. Ir a 2(a). 3. Terminar.. La fórmula de velocidad se explica de la siguiente forma: vik+1 = velocidad anterior de esa partícula + influencia personal+influencia social. Matemáticamente queda de la forma: vik+1 = vik+ c1 r1(pik - xik )+c2 r2 (pgk - xik).. Siguiendo recomendaciones de la literatura consultada: c1=2.08 y c2=2.08 20.
(30) 1.5 Algoritmo PSO aplicado a los métodos Scan Para la aplicación del PSO a la solución del problema de la detección de un parámetro adecuado en el método Scan se siguen los siguientes pasos: Cada partícula se define por: xik. – Es el vector (window, step, fuzzy) en la iteración k. con las siguientes restricciones: • 1 ≤ Window ≤ Sequence Size • 1 ≤ Step ≤ Window • 0 ≤ Fuzzy ≤ Window/2 pik. – Es el mejor vector (window_best, step_best, fuzzy_best) de la partícula i, hasta la iteración k. pgk. – Es el mejor vector (window_bestg, step_bestg, fuzzy_bestg) hasta la iteración k. vik. – Velocidad de la partícula i en la iteración k.. Como se explicó anteriormente, la velocidad se define por: vik+1 = vik + c1 r1(pik - xik )+ c2 r2 (pgk - xik).. fik. – Valor de la función objetivo evaluada en xik.. fibest. – Mejor valor de la función objetivo evaluada en la partícula i.. fgbest. – Mejor valor de la función objetivo evaluada en el grupo.. 1.6 Métodos de Monte Carlo Informalmente, la definición de método de Monte Carlo es cualquier técnica que use números aleatorios. Una definición más formal afirma que los métodos de Monte Carlo son un conjunto de algoritmos computacionales que basan sus resultados en el uso de un muestreo aleatorio con reposición. (Buckley and Jowers 2007). 21.
(31) Se utilizan con frecuencia para simular el comportamiento de sistemas físicos o matemáticos complejos. Debido a su uso intensivo, a partir de la generación de números aleatorios (o pseudo-aleatorios), los métodos de Monte Carlo se utilizan para realizar sus cálculos con ayuda de microcomputadoras. La invención del método de Monte Carlo se asigna a Stan Ulam y a John von Neumann. En 1946, Ulam ha explicado cómo se le ocurrió la idea mientras jugaba un solitario durante una enfermedad en 1946. A principios de 1947 Von Neumann envió una carta a Los Álamos en la que expuso de modo influyente tal vez el primer informe por escrito del método de Monte Carlo. El método fue llamado así por el principado de Mónaco por ser “la capital del juego de azar”, al tomar una ruleta como un generador simple de números aleatorios. El uso real de los métodos de Monte Carlo como una herramienta de investigación, viene del trabajo de la bomba atómica durante la Segunda Guerra Mundial. Bajo el nombre de “Método de Monte Carlo” o “Simulación Monte Carlo” se agrupan una serie de procedimientos que analizan distribuciones de variables aleatorias usando simulación de números aleatorios. De manera general, este método da solución a una gran variedad de problemas matemáticos haciendo experimentos con muestreos estadísticos en una computadora. Es aplicable a cualquier tipo de problema, ya sea estocástico o determinístico. Generalmente en estadística los modelos aleatorios se usan para simular fenómenos que poseen algún componente aleatorio. Pero en el método de Monte Carlo, por otro lado, el objeto de la investigación es el objeto en sí mismo, un suceso aleatorio o pseudo-aleatorio se usa para estudiar el modelo.. 1.7 Métodos de Monte Carlo combinado con el PSO y los métodos Scan En este epígrafe se explica el uso de la simulación de Monte Carlo combinada con los algoritmos presentados con anterioridad. A partir de la secuencia binaria original (la que se lee en el fichero) se pueden “generar” tantas secuencias “similares” como el usuario desee, por ejemplo 20. La generación se 22.
(32) hace introduciendo “mutaciones” en la secuencia original, es decir cambiando los valores en algunas de sus posiciones, (Buckley and Jowers 2007). El usuario controla la cantidad de secuencias mutantes a generar (variable: Cantidad_de_secuencias_a_generar) y el “grado de similaridad” con la secuencia original (variable: Cantidad_de_posiciones_a_modificar, por defecto 3%). La elección de las posiciones que cambiarán su valor, se realiza al azar, como lo muestra el algoritmo siguiente: 1. Repetir a. Calcular Cantidad_de_posiciones_a_modificar {Este valor lo introduce el usuario, 3% por defecto} b. Para i=1 hasta Cantidad_de_posiciones_a_modificar hacer: i. Generar Posición_a_cambiar {Generar un número aleatorio con distribución uniforme entre 1 y el largo de la secuencia} ii. Secuencia[Posición_a_cambiar]=1- Secuencia[Posición_a_cambiar] {Si en Secuencia[Posición_a_cambiar]= había un 0, ahora habrá un 1 o viceversa} 2. Hasta Cantidad_de_secuencias_a_generar De esta forma se garantiza que las secuencias generadas sean similares a la original, pues se diferencian de ella en un porcentaje pequeño de sus valores. Ante secuencias similares, el resultado de cualquiera de los métodos Scan, y del algoritmo PSO para optimizar los parámetros del Scan, no debe diferenciarse demasiado. Para resumir, se muestran medidas descriptivas que resumen los valores encontrados. La aplicación del método de Monte Carlo fortalece los resultados que el PSO puede hallar, pero aumenta de manera notable el tiempo de ejecución de los algoritmos, sobre todo en caso de secuencias largas.. Consideraciones finales del capítulo En este capítulo se realiza una revisión de la literatura acerca de varios temas aparentemente independientes:. 23.
(33) 1. Métodos Scan para la detección de conglomerados: Se explica el surgimiento de los métodos Scan, sus variantes para la detección de aglomeraciones sobre una línea o sobre un círculo, su generalización y sus variantes borrosas. Para comprender este último tema, se incluye un epígrafe con algunos elementos básicos de la lógica borrosa. 2. Algoritmos bioinspirados: se explican brevemente los fundamentos matemáticos del algoritmo de optimización basado en enjambres de partículas y conocido en la literatura como PSO. 3. Método de Monte Carlo: Se mencionan algunos datos relacionados con el surgimiento de la simulación de Monte Carlo y se explica muy brevemente en qué consiste el método. Una vez expuestos estos métodos matemáticos, se explica la forma en la que el algoritmo PSO tendrá como función objetivo el cálculo de la significación de cualquiera de los métodos Scan, para de esta forma “optimizar” los valores de sus parámetros. Para minimizar la probabilidad de errores, se complementa el análisis con el método de Monte Carlo que genera “secuencias mutantes” similares a la original y sobre ellas se repite todo el cálculo anterior. Para resumir los resultados se muestran algunos estadígrafos descriptivos. Como resultado de la revisión de la literatura se propone para este trabajo las siguientes hipótesis de investigación: “Los algoritmos bioinspirados, en particular el PSO sirven de ayuda para la determinación de parámetros adecuados de los métodos Scan en la detección de conglomerados. El método de Monte Carlo se aplica opcionalmente para corroborar los resultados obtenidos”.. 24.
(34) Capítulo 2. Detalles sobre la implementación del sistema El sistema elaborado: Optimus 1.0 se implementó en Borland Delphi 7. Se ejecuta sobre Windows y brinda al usuario un ambiente cómodo. El sistema consta de un fichero: Optimus.exe. Lee los datos a partir de un fichero texto que contiene una secuencia binaria. Para la implementación del sistema, el algoritmo PSO tendrá como función objetivo el cálculo de la significación de cualquiera de los métodos Scan, para de esta forma “optimizar” los valores de sus parámetros, y la posición de las partículas representa los valores de los parámetros del método Scan que se trata de optimizar.. 2.1 Estructura de las clases El sistema utiliza adecuadamente las facilidades de las componentes visuales del lenguaje en aras de brindar un ambiente cómodo y sencillo para el usuario. Se elaboró según el paradigma de la programación orientado a objetos. La figura 2.1 muestra el diagrama de clases utilizado.. Figura 2.1 Diagrama de clases utilizado en Optimus.. 2.2 Manual de usuario 25.
(35) En este epígrafe se muestra el manual de usuario pare el sistema Optimus. Empezar un nuevo proyecto. Este es el Menú principal:. Figura 2.2 Menú principal de Optimus Para empezar un nuevo proyecto, se debe dar clic sobre “Start new project”.. Figura 2.3 Comenzando un nuevo proyecto Para abrir el fichero que contiene la secuencia binaria a tratar se puede dar clic sobre “None” o presionar ctrl+O.. 26.
(36) Figura 2.4 Abriendo un fichero de secuencias binarias (.gen) Para seleccionar el método Scan que se quiere utilizar.. Figura 2.5 Seleccionando el método Si no desea usar el PSO para optimizar los parámetros del Scan entonces marque la opción de “Do not use PSO”. En este caso se ejecutará sólo el método Scan con los parámetros que especifique el usuario. Los valores de los parámetros del Scan se ajustan según lo mostrado en la figura 2.6:. 27.
(37) Figura 2.6 Seleccionando los parámetros del Scan (en este caso no se usa PSO) Si usted desea optimizar los parámetros de Scan usando el PSO deje la opción de “Do not use PSO” sin marcar, como se muestra en la figura 2.7.. Figura 2.7 Si se usa PSO, no hay parámetros a seleccionar A continuación deberá seleccionar la condición de parada del algoritmo PSO: •. Número de iteraciones: las decide el usuario, por defecto 20. •. Valor menor que un epsilon: el epsilon también lo decide el usuario. Esta condición de parada significa que el algoritmo termina cuando el valor de la significación asociada al método Scan es menor que un epsilon especificado por el usuario.. La figura 2.8 muestra gráficamente la forma de elegir entre una u otra condición de parada:. 28.
(38) Figura 2.8 Seleccionando los parámetros del PSO En esta pestaña el usuario puede ajustar todos los parámetros del PSO a su gusto. •. Ajustar los límites de posición. Por omisión los límites de posición están entre 1 y el tamaño del genoma considerado para el ancho de la ventana. Para los demás parámetros: paso y longitud de la parte borrodade la ventana, los límites son los explicados en le epígrafe 1.5. •. Ajustar los límites de velocidad. Por omisión estos valores se encuentran entre -5 y 5.. Si el usuario no desea usar el método del Monte Carlos desmarcar “Use Monte Carlo”, como se muestra en la figura 2.8.. Figura 2.8 Opción para seleccionar el uso del método de Monte Carlo Si desea usar el método de Monte Carlos marcar “Use Monte Carlos”. En ese caso aparecerán los valores del mismo para que el usuario pueda modificarlo si así lo desea: 29.
(39) Figura 2.9 Seleccionando los parámetros del Monte Carlo Además se puede ajustar el número de dígitos usados para buscar la moda y los demás estadísticos descriptivos que se calculan sobre la significación de la simulación. En caso de tener que corregir algún dato, presionar el botón “Back” hasta que pueda corregirlo.. Figura 2.10 Resumen de todos los parámetros involucrados en los cálculos Cuando todos los datos estén correctos presionar el botón “Next”. En la pestaña Results aparecen todos los resultados obtenidos, ver figura 2.11.. 30.
(40) Figura 2.11 Pestaña de resultados Salvar los parámetros del proyecto Para salvar los parámetros del proyecto se debe: •. Ir al menú principal,. •. Dar clic sobre “Save project parameters”, ver figura 2.12.. Figura 2.12 Salvar los parámetros del proyecto. •. A continuación se escribe el nombre del fichero donde se van salvar los parámetros del proyecto.. Abrir un proyecto existente 31.
(41) Para abrir un proyecto existente se debe: •. ir al menú principal,. •. Dar clic sobre “Open a project”, ver figura 2.13.. Figura 2.13 Abrir un proyecto existente. •. Seleccionar el fichero que contiene los parámetros del proyecto, ver figura 2.14.. Figura 2.14 El proyecto tiene extensión .par. 32.
(42) En caso de tener que corregir algún dato, presionar el botón “Back” hasta que pueda corregirlo. Salvar los resultados del proyecto. Para abrir un proyecto existente se debe: •. ir al menú principal,. •. Dar clic sobre “Save project results”, ver figura 2.15.. Figura 2.15 Salvar los resultados del proyecto. •. Escribir el nombre del fichero donde va a salvar los resultados del proyecto, ver figura 2.16.. 33.
(43) Figura 2.16 Escribiendo en el fichero resultados Salir de la aplicación. Para salir de la aplicación se puede: •. Presionar el botón “Quit” siempre que esté visible, como se muestra en la figura 2.17. Figura 2.17 Paso 1 para salir de la aplicación. O de lo contrario ir al menú principal, •. Dar clic sobre “Exit the program”, ver figura 2.18.. 34.
(44) Figura 2.18 Paso 2 para salir de la aplicación. 2.3 Guía para la reutilización de las bibliotecas. 1. Crear una nueva clase de entrada.. 1.1 Crear una nueva clase. 1.2 Heredar de TInputList. 1.3 Implementar la función Get para que devuelva un elemento de la entrada. 1.4 Implementar el procedimiento Put para que cambie un elemento de la entrada. 1.5 Implementar la función Count para que devuelve la cantidad de elementos en la entrada. 1.6 Implementar la función Copy para copiar la entrada. 1.7 Implementar el procedimiento Refresh para refrescar la entrada. 2. Modificar la clase de partícula.. 2.1 Ir a la clase de TPraticle. 2.2 Volver a implementar el método CheckRestrictions para que satisfaga las restricciones de su partícula. 3. Crear una nueva clase de conveniencia.. 3.1 Crear una nueva clase. 3.2 Heredar de TFitnessFunction.. 35.
(45) 3.3 Implementar el método Execute para que devuelva el valor de conveniencia que se desea. 4. Crear una nueva clase de generador.. 4.1 Crear una nueva clase. 4.2 Heredar de TGenerator. 4.3 Implementar el método Execute para que devuelva la partícula deseada. 5. Hacer uso normal de las clases TGenome, TSwan y TMonteCarlos.. Consideraciones finales del capítulo En este capítulo aparecen detalles de la implementación del sistema, como el diagrama de clases utilizado. Además se muestra en detalles el manual de usuario, que posibilita que cualquier persona no necesariamente especialista en el tema, pueda ejecutar exitosamente Optimus.. 36.
(46) Capítulo 3. Aplicaciones En este capítulo se muestra el uso del sistema creado con todas sus variantes. Se presentan aplicaciones con datos simulados y con datos reales de una aplicación bioinformática.. 3.1 Generación de secuencias Los avances de la Computación en las últimas décadas han contribuido notablemente al fortalecimiento de la matemática y esta a su vez ha sido utilizada como una poderosa herramienta de trabajo por otras ciencias. Varios modelos de epidemias han mostrado su utilidad en la prevención y en la aplicación de diversos procedimientos de control de enfermedades; sin embargo ninguno de ellos es infalible, todos tienen sus ventajas y limitaciones y ambas deben conocerse a fin de evitar aplicaciones incorrectas que pueden ser muy perjudiciales.. 3.1.1 Bases de la simulación realizada La simulación se realizó con ayuda del paquete Mathematica® por razones de velocidad, facilidad de generación y precisión de los cálculos. Se generaron secuencias de datos de diversos tamaños para analizar su influencia en la capacidad de detección de las diferentes variantes. Se generaron secuencias que contienen conglomerados (a ellas se les conglomerados verdaderos) y otras que no lo tienen (conglomerados falsos). Con todas ellas se ejecutaron los métodos implementados. Más adelante en este capítulo aparece un resumen de los resultados que se obtuvieron. Para generar los conglomerados verdaderos se utilizó una distribución de Bernoulli (0.2) en el primer y último cuarto de la población, representando esto un 50% del total mientras que para el porcentaje restante se utilizó Bernoulli (0.8). Ejemplo de verdaderos conglomerados: 0 1 0 0 0 0 1 1 1 1 1 1 1 1 1 1 1 1 0 0 1 0 0 1 Para generar los conglomerados falsos se utilizó una distribución de Bernoulli (0.2) en el total de los casos. Ejemplo de falsos conglomerados: 0 1 0 0 0 0 0 0 1 0 0 0 0 1 0 0 0 0 1 0. 37.
(47) 3.2 Resultados y discusión con datos simulados Los métodos de detección de conglomerados, entre los que se encuentran las técnicas Scan, no pueden aplicarse sin tener idea de los valores adecuados para sus parámetros: •. ancho de la ventana móvil,. •. paso con el que se moverá dicha ventana,. •. parte borrosa de la ventana. Este último parámetro es válido sólo en las variantes borrosas de los métodos Scan. En los problemas de Bioinformática generalmente el conocimiento es escaso. No resulta obvio entonces que un “especialista” pueda identificar con precisión valores “adecuados” para resolver un problema de detección de aglomeraciones inusuales de una subsecuencia dentro de un genoma completo o dentro de una porción de este. Por estudios de simulación previos, para casos particulares en los que el paso (segundo parámetro) se considera igual a la unidad, se conoce el comportamiento de las técnicas Scan. (Rodríguez, Casas et al. 2008) Ante secuencias en las que se sabe que existe a menos un cluster, (verdaderos conglomerados) las técnicas Scan responden de forma adecuada en dependencia del tamaño de la ventana que se utilice. La figura 3.1 muestra estos resultados. Obsérvese que el método borroso (línea negra) resuelve el problema mucho mejor que el clásico (línea azul) en el caso de las ventanas pequeñas.. Figura 3.1 Resultados de los métodos Scan ante conglomerados verdaderos Scan clásico (línea azul), Scan borroso (línea negra) Ante secuencias en las que no existen clusters (falsos conglomerados), ambas técnicas Scan responden de la manera adecuada, es decir no detectan aglomeraciones con independencia del tamaño de la ventana utilizada, (Rodríguez, Casas et al. 2009). 38.
(48) Es por esta razón que decide aplicarse el PSO, como método de optimización. Su función objetivo es el cálculo de la significación asociada al método Scan correspondiente. Si el valor de la significación final es superior a 0.05, entonces puede afirmarse que no existe un ancho de ventana con el que se detecten conglomerados. Para corroborar la idea, puede aplicarse un método de Monte Carlo creando secuencias similares y aplicando sobre ellas el PSO, como se explicó en el capítulo 1. Si el valor de la significación mínimo es aún mayor que 0.05, quedará “demostrado” la no existencia de conglomerados sobre esa secuencia. Si producto de la aplicación el PSO, se devuelve un valor de p significativo, entonces los valores de los parámetros hallados, podrán utilizarse para detectar el conglomerado. A continuación se mostrarán los resultados obtenidos al ejecutar Optimus sobre varias secuencias de verdaderos y falsos conglomerados.. 3.2.1 Análisis con verdaderos conglomerados El sistema Optimus se ejecutó con cinco secuencias de verdaderos conglomerados, generadas como se explicó en el epígrafe 3.1.1. La tabla 3.1 muestra los resultados obtenidos para el método Scan Lineal Clásico: Fichero. Tamaño de la ventana. Paso. Significación. V1.gen. 71. 32. 0.0000. V2.gen. 47. 24. 0.0000. V3.gen. 60. 47. 0.0000. V4.gen. 36. 36. 0.0000. V5.gen. 52. 18. 0.0000. Tabla 3.1 Resultados obtenidos con el PSO para el método Scan Lineal Clásico sobre verdaderos conglomerados Obsérvese que todos los resultados son significativos, luego puede afirmarse que, con esa ventana y ese paso, se encuentra al menos un conglomerado.. 39.
(49) La tabla 3.2 muestra los resultados para el método Scan Lineal Borroso. Fichero. Tamaño de la ventana. Paso. Parte borrosa Significación. V1.gen. 36. 29. 6. 0.0000. V2.gen. 42. 27. 6. 0.0000. V3.gen. 49. 26. 7. 0.0000. V4.gen. 44. 35. 6. 0.0000. V5.gen. 49. 46. 7. 0.0000. Tabla 3.2 Resultados obtenidos con el PSO para el método Scan Lineal Borroso sobre verdaderos conglomerados Igualmente puede observarse que los resultados son significativos, luego puede afirmarse que, con esos parámetros, se encuentra al menos un conglomerado. La combinación de métodos matemáticos propuesta en la tesis detectó correctamente la presencia de conglomerados.. 3.2.2 Análisis con falsos conglomerados El sistema Optimus se ejecutó con cinco secuencias de falsos conglomerados, generadas como se explicó en el epígrafe 3.1.1. La tabla 3.3 muestra los resultados obtenidos: Fichero. Tamaño de la ventana. Paso. Significación Significación mínima máxima. F1.gen. 266. 1. 0.5213. 0.5734. F2.gen. 300. 183. 0.5462. 0.5604. F3.gen. 300. 96. 0.5243. 0.5533. F4.gen. 150. 50. 0.4467. 0.5081. F5.gen. 291. 132. 0.4848. 0.589. Tabla 3.3 Resultados obtenidos con el PSO para el método Scan Lineal Clásico sobre falsos conglomerados Para todos estos casos, se ejecutó también la técnica de Monte Carlo, generando 20 cada vez secuencias similares al 5% con respecto a la original. Los valores de la significación mínima y máxima halladas se muestran en la tabla 3.3. Puede observarse que en ninguno de los casos los resultados fueron significativos, por tanto los valores que se muestran del ancho de la ventana y del paso no son útiles.. 40.
(50) Los valores no significativos demuestran que estamos en presencia de secuencias que no tienen conglomerados. La tabla 3.4 muestra resultados análogos para el caso del método Scan Lineal Borroso Fichero. Tamaño de la ventana. Paso. Parte borrosa. Significación Significación mínima máxima. F1.gen. 168. 28. 12. 0.3106. 0.3597. F2.gen. 196. 82. 14. 0.2155. 0.4371. F3.gen. 212. 66. 14. 0.2621. 0.3332. F4.gen. 95. 90. 9. 0.1110. 0.2573. F5.gen. 202. 43. 14. 0.2461. 0.3816. Tabla 3.4 Resultados obtenidos con el PSO para el método Scan Lineal Borroso sobre falsos conglomerados Como puede apreciarse, los resultados usando como base el método Scan Lineal Borroso coinciden con los resultados mostrados anteriormente. Por tanto se corrobora que la combinación de métodos matemáticos propuesta en la tesis dictaminó correctamente la ausencia de conglomerados.. 3.3 Estudio del genoma de la Escherichia Coli Recientemente se han publicado numerosos estudios relacionados con el ADN de la E. Coli. En este trabajo se pretende analizar el genoma completo de la E. Coli con el objetivo de demostrar la existencia de conglomerados de sitios Dam. Los sitios Dam son subsecuencias pequeñas (GACT) dentro del genoma competo que tienen una importancia especial desde el punto de vista bioquímico, (Karlin and Brendel 1992; Hénaut, Rouxel et al. 1996; Cardellá and Hérnandez 1999; Glaz and Balakrishnan 1999). Debe mencionarse que el genoma de la E. Coli tiene una longitud aproximada de 4,7 millones de pares de bases. El ADN de la E. Coli es circular. Por ese motivo se aplicarán las variantes circulares del método Scan Clásico y del Borroso. 3.3.1 Análisis de la E. Coli usando los métodos Scan. Debido a estudios bioquímicos previos, se seleccionó como ancho de la ventana el valor 245. Como que no se tenía información acerca de los valores posibles de los demás 41.
(51) parámetros, se decidió tomar el paso igual a la unidad y la parte borrosa de la ventana (en el caso de los métodos borrosos), como los valores 2 y 4. La tabla 3.5 muestra los resultados obtenidos: Escherichia Coli IAI1, GenBank, NC_011741, 4.7Mb Ancho de la ventana móvil: 245bp Método. # ‘GACT’. Significación. - Classic Circular Scan. 13. 0.0000. - Fuzzy Circular Scan (g=2). 13. 0.0000. - Fuzzy Circular Scan (g=4). 13. 0.0000. Tabla 3.5 Resultados obtenidos con los métodos Scan circulares para la E. Coli Los valores de la significación demuestran la existencia de conglomerados de sitios Dam dentro del genoma de la E. Coli. 3.3.2 Análisis de la E. Coli usando los métodos Scan y el PSO. Supongamos ahora que no se conoce un valor adecuado para los parámetros de de los métodos Scan y que se desea de la misma forma, determinar la existencia de conglomerados de sitios Dam dentro del genoma de la E. Coli. Debe aclararse que esta es la secuencia más larga que se ha ejecutado con nuestro sistema Optimus. Los resultados aparecen recogidos en la tabla 3.6: Escherichia Coli IAI1, GenBank, NC_011741, 4.7Mb 10 partículas, 10 iteraciones, posición de las partículas 1-300. 10 iteraciones y 5 mutaciones. PSO Método. Fuz. Vent Paso. PSO+ Monte Carlo. # ‘GACT’. Sig. Fuz. Vent Paso. # ‘GACT’. Sig. - Classic Circular Scan. 0. 43. 30. 12. 0.000. 0. 40. 38. 14. 0.000. - Fuzzy Circular Scan. 1. 44. 34. 12. 0.000. 10. 36. 36. 13.18. 0.000. Consideraciones finales del capítulo En este capítulo se ha mostrado, de manera breve, los resultados obtenidos con la combinación de métodos matemáticos propuestos en la tesis, al presentarle secuencias generadas con verdaderos y falsos conglomerados. 42.
(52) Al presentar secuencias de verdaderos conglomerados, se hallaron parámetros adecuados que, incorporados a los métodos Scan, detectan conglomerados. Al presentar secuencias de falsos conglomerados, no se hallaron valores significativos ni siquiera en secuencia similares a la original. Tales secuencia se obtuvieron por el método de Monte Carlo, lo que se considera novedoso. Se detectaron conglomerados de sitios Dam en el genoma completo de la E. Coli.. 43.
(53) Conclusiones En el presente trabajo se combinan varios métodos matemáticos para dar solución al problema de la detección eficiente de conglomerados en secuencias binarias: 1. Se utilizan los métodos Scan Clásico y Borroso, Lineal y Circular en dependencia de la aplicación, para detectar conglomerados en secuencias binarias. 2. Se utiliza un algoritmo bioinspirado de optimización: el PSO para determinar valores óptimos de los parámetros de los métodos Scan. 3. Se utiliza el método de simulación por Monte Carlo para generar secuencias similares a la original y sobre cada una de ellas ejecutar el PSO. De esta manera disminuye la probabilidad de error al aplicar el PSO. Los métodos están implementados en el software Optimus, que es sencillo y ofrece un ambiente amigable. Los métodos se validaron con secuencias simuladas de verdaderos y falsos conglomerados. En todos los casos se obtuvieron resultados correctos. Para finalizar se detectaron conglomerados de sitios Dam en el genoma completo de la E. Coli, siendo esta una aplicación bioinformática real.. 44.
Figure



+7





Documento similar