Síntesis de precursores y monómeros con potencialidad estructural para la preparación de polímeros con microporosidad intrínseca
77
0
0
Texto completo
(2) AGRADECIMIENTOS. Me gustaría agradecer a mis padres, hermanos y familia cercana por estar conmigo durante toda la trayectoria de esta carrera, por su apoyo incondicional que me brindaron y por la paciencia que me tuvieron.. A mis compañeros y amigos que me acompañaron en este proceso y que siempre tuvieron algún consejo que otorgarme. A mi pareja que me acompañó desde el comienzo de este proyecto, que estuvo a mi lado y que nunca dudo de mí.. Al profesor Alain Tundidor por su confianza, cariño, apoyo, paciencia y sabiduría en la realización de este proyecto, y a mis compañeros de laboratorio por todo lo que me enseñaron.. Por último, también agradezco al proyecto FONDECYT Nº 1190772 del profesor Alain Tundidor por su financiamiento.. II.
(3) LISTA DE ABREVIACIONES T.f.. Temperatura de fusión. IR. Infrarrojo. RMN. Resonancia Magnética Nuclear. DMSO-d6. Dimetilsulfóxido deuterado. Ac2O. Anhídrido acético. DMAc. N,N-Dimetilacetamida. DCM. Diclorometano. DMF. N,N-Dimetilformamida. Abreviatura y nomenclatura de precursores y monómeros. P1:. Anhídrido 3,4,5,6-tetrafluoroftálico. P2:. 4,5,6,7-Tetrafuoro-2-fenilisoindolina-1,3-diona. P3:. 4,5,6,7-Tetrafluoro-2-(4-metoxifenil)isoindolina-1,3-diona. P4:. 4,5,6,7-Tetrafluoro-2-(3,4,5-trimetoxifenil)isoindolina-1,3-diona. P5:. 2-(4-(Tert-butil)fenil)-4,5,6,7-tetrafluoroisoindolina-1,3-diona. P6:. 2-(2,6-Diisopropilfenil)-4,5,6,7-tetrafluoroisoindolina-1,3-diona. P7:. 2-(Adamantil)-4,5,6,7-tetrafluoroisoindolina-1,3-diona. P8:. 3-Bromo-4-metilcatecol. M1:. 2,11-Dibromo-3,12-dimetil-7-fenil-6H-benzo[5,6][1,4]dioxina[2,3-e] benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona. M2:. 2,11-Dibromo-7-(4-metoxifenil)-3,12-dimetil-6H-benzo[5,6][1,4] dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona. M3:. 2,11-Dibromo-3,12-dimetil-7-(3,4,5-trimetoxifenil)-6H-benzo[5,6][1,4] dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona. M4:. 2,11-Dibromo-7-(4-(tert-butil)fenil)-3,12-dimetil-6H-benzo[5,6][1,4] dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona. M5:. 2,11-Dibromo-7-(2,6-diisopropilfenil)-3,12-dimetil-6H-benzo[5,6][1,4]. III.
(4) dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona M6:. 7-(1-Adamantil)-2,11-dibromo-3,12-dimetil-6H-benzo[5,6][1,4] dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)-diona. M7:. 2,9-Dibromo-3,10-dimetilbenzo[5,6][1,4]dioxino[2,3-b] dibenzo [b,e][1,4]dioxina-6,13-dicarbonitrila. IV.
(5) ÍNDICE GENERAL AGRADECIMIENTOS .................................................................................... II LISTA DE ABREVIACIONES ........................................................................ III ÍNDICE GENERAL ......................................................................................... V ÍNDICE DE FIGURAS ................................................................................... VII ÍNDICE DE TABLAS ................................................................................... VIII ÍNDICE DE ESQUEMAS ............................................................................. VIII RESUMEN ..................................................................................................... IX ABSTRACT .................................................................................................... X 1.. INTRODUCCIÓN ................................................................................. 1. 2.. HIPÓTESIS .......................................................................................... 7. 3.. OBJETIVOS ......................................................................................... 7. 3.1.. Objetivo general ............................................................................... 7. 3.2.. Objetivos específicos ........................................................................ 7. 4.. PARTE EXPERIMENTAL..................................................................... 8. 4.1.. Material y equipos ............................................................................ 8. 4.2.. Síntesis de Precursores.................................................................... 8. 4.1.1.. Síntesis de P1 ............................................................................... 8. 4.1.2.. Síntesis de P2 ............................................................................... 9. 4.1.3.. Síntesis de P3 ............................................................................. 10. 4.1.4.. Síntesis de P4 ............................................................................. 11. 4.1.5.. Síntesis de P5 ............................................................................. 12. 4.1.6.. Síntesis de P6 ............................................................................. 13. 4.1.7.. Síntesis de P7 ............................................................................. 14. 4.1.8.. Síntesis de P8 ............................................................................. 16. 4.3. 4.3.1.. Síntesis de monómeros .................................................................. 17 Síntesis de M1 ............................................................................ 17. V.
(6) 4.3.2.. Síntesis de M2 ............................................................................ 18. 4.3.3.. Síntesis de M3 ............................................................................ 19. 4.3.4.. Síntesis de M4 ............................................................................ 20. 4.3.5.. Síntesis de M5 ............................................................................ 21. 4.3.6.. Síntesis de M6 ............................................................................ 22. 4.3.7.. Síntesis de M7 ............................................................................ 22. 5.. RESULTADOS Y DISCUSIÓN ........................................................... 24. 5.1.. Síntesis y caracterización de precursores ...................................... 24. 5.1.1.. Síntesis y caracterización de P1 ................................................. 24. 5.1.2.. Síntesis y caracterización de P2 a P7 ......................................... 27. 5.1.3.. Síntesis y caracterización de P8 ................................................. 38. 5.2.. Síntesis y caracterización de monómeros ...................................... 42. 6.. CONCLUSIONES .................................................................................. 54. 7.. BIBLIOGRAFÍA ...................................................................................... 55. Anexos .......................................................................................................... 59. VI.
(7) ÍNDICE DE FIGURAS. Figura 1. Representaciones de PIM-1............................................................ 3 Figura 2. Formación de dioxanos. .................................................................. 4 Figura 3. Ejemplo de polimerización vía CANAL............................................ 5 Figura 4. Ejemplos de monómeros tetrafluorados………………………………5 Figura 5. Polímeros de microporosidad intrínseca usando monómeros tetrafluorados con grupos voluminosos. .................................................. 6 Figura 6. Espectro de IR de P1 .................................................................... 26 Figura 7. Espectros de 1H-RMN de los precursores P2, P3, P4 y P5. ......... 31 Figura 8. Espectros de 1H-RMN de los precursores P6 y P7. ...................... 32 Figura 9. Visualización de los protones del precursor P2.. .......................... 33 Figura 10. Diferentes representaciones de P7. ............................................ 35 Figura 11. Espectros de IR de los precursores P2 a P7 .............................. 37 Figura 12. Productos de la bromación de 4-metilcatecol.. ........................... 38 Figura 13. Espectro de 1H-RMN del precursor P8 ....................................... 39 Figura 14. Espectro HMBC del precursor P8 ............................................... 40 Figura 15. Efecto de la recristalización desde n-hexano del precursor P8... 41 Figura 16. Espectro de IR del precursor P8 ................................................. 42 Figura 17. Espectro de 19F-RMN del monómero M5. ................................... 44 Figura 18. Isómeros estructurales posibles para los monómeros M1-M6.. .. 46 Figura 19. Asignación de algunos de los protones en los isómeros obtenidos. .............................................................................................. 47 Figura 20. Espectro de 1H-RMN del monómero M2 ..................................... 48 Figura 21. Espectro de 1H-RMN del monómero M5 ..................................... 50 Figura 22. Espectro de 1H-RMN del monómero M7 ..................................... 51 Figura 23. Espectros de IR de los monómeros M1-M7 ................................ 53. VII.
(8) ÍNDICE DE TABLAS. Tabla 1. Solubilidad de los monómeros en diferentes solventes. ................. 45. ÍNDICE DE ESQUEMAS Esquema 1. Síntesis de P1 ............................................................................ 8 Esquema 2. Síntesis de P2 ............................................................................ 9 Esquema 3. Síntesis de P3 .......................................................................... 10 Esquema 4. Síntesis de P4 .......................................................................... 11 Esquema 5. Síntesis de P5 .......................................................................... 12 Esquema 6. Síntesis de P6 .......................................................................... 13 Esquema 7. Síntesis de P7 .......................................................................... 14 Esquema 8. Síntesis de P8 .......................................................................... 16 Esquema 9. Síntesis de M1 ......................................................................... 17 Esquema 10. Síntesis de M2 ....................................................................... 18 Esquema 11. Síntesis de M3 ....................................................................... 19 Esquema 12. Síntesis de M4 ....................................................................... 20 Esquema 13. Síntesis de M5 ....................................................................... 21 Esquema 14. Síntesis de M6 ....................................................................... 22 Esquema 15. Síntesis de M7 ....................................................................... 22 Esquema 16. Mecanismo para la síntesis de P1 ......................................... 24 Esquema 17. Mecanismo de síntesis de imidas por condensación directa. 27 Esquema 18. Mecanismo de ciclodeshidratación empleando anhídrido acético y acetato de sodio. .................................................................... 28 Esquema 19. Mecanismo general para la síntesis de los monómeros. ....... 42. VIII.
(9) RESUMEN En este trabajo, se realizó la síntesis de siete nuevos monómeros dibromados con anillos dioxano fusionados. La estructura fundamental de estos monómeros se basa en anillos fusionados planares que poseen estructuras retorcidas que impiden un empaquetamiento eficiente de los polímeros que se obtendrán a futuro. Derivado de esto, se tendrá una pérdida de libertad de rotación de las cadenas poliméricas, permitiendo así tener una estructura molecular de alta rigidez. Debido a esta característica estructural, estos monómeros son la base para la futura síntesis de polímeros con microporosidad intrínseca. El trabajo experimental comenzó con la síntesis del precursor anhídrido tetrafluoroftálico mediante una reacción de ciclodeshidratación entre el precursor ácido tetrafluoroftálico y cloruro de tionilo. Posteriormente, dicha materia prima se hizo reaccionar con seis aminas comerciales para obtener los precursores imidas tetrafluorados. Finalmente, la síntesis de los monómeros se basó en la reacción entre cada precursor imida y un metilcatecol bromado a través de una sustitución nucleofílica aromática doble. El precursor 3-bromo-4-metilcatecol se obtuvo mediante una reacción de sustitución electrofílica aromática empleando bromo molecular como reactivo. Los rendimientos obtenidos para los precursores estuvieron entre 67 % y 96 %, mientras que para los monómeros fue entre 50 % y 88 %. Los compuestos fueron caracterizados mediante análisis espectroscópico de 1H-RMN,. 13C-. RMN, 19F-RMN, DEPT y FT-IR, sin embargo, algunos resultaron insolubles en los solventes deuterados disponibles, por lo que en estos casos no fue posible una caracterización en disolución.. IX.
(10) ABSTRACT In this work the synthesis of seven new dibrominated monomers with fused dioxane rings was performed. The structure of these monomers is based on planar fused rings having twisted structures that prevent efficient packing of the polymers that will be obtained in the future. Thus, the polymeric chains lose free rotation, which increase their molecular rigidity. All of these characteristics make those monomers excellent candidates for the future synthesis of polymers of intrinsic microporosity. The experimental work began with the synthesis of the tetrafluorophthalic anhydride precursor by means of a cyclodehydration reaction between the tetrafluorophthalic acid precursor and thionyl chloride. This raw material was then reacted with six commercial amines to obtain the tetrafluorinated imide precursors. Finally, the synthesis of the monomers was based on the reaction between each imide precursor and a brominated methylcatechol through a double aromatic nucleophilic substitution. The 3-bromo-4-methylcatechol precursor was obtained by an aromatic electrophilic substitution reaction using molecular bromine as a reagent. The performance obtained by these precursors were in between 67 %– 96 %, while the performance for the monomers were between a 50 % – 88 %. Each new compound was characterized by spectroscopic analysis (1H-NMR, NMR,. 19F-NMR,. 13C-. DEPT and FT-IR). However, some monomers were insoluble. in the available deuterated solvents, in these cases the characterization in solution was not possible.. X.
(11) 1. Introducción El potencial de las membranas poliméricas para separar mezclas de gases es conocido mucho antes de la década de los 80, pero la tecnología para fabricar membranas de alto rendimiento es algo que continúa desarrollándose. En las últimas décadas, se han observado avances significativos en la búsqueda de membranas para la separación de gases, demostrándose que se puede reducir en gran cantidad el consumo de energía para la separación de éstos en comparación con los procesos de separación convencionales (ej: destilación criogénica). El mercado de tecnología de membranas para la separación de gases se ha multiplicado por cinco desde el 2000 hasta la actualidad [1-2]. Algunos de los gases que causan el calentamiento global y que son parte de la contaminación del aire en general son el dióxido de carbono (CO2) y el metano (CH4). La clave del éxito de las membranas poliméricas que se utilizan para la separación de gases es poder aislar selectivamente un gas cuando se encuentra mezclado con otro(s). En este contexto, las membranas poliméricas juegan un rol importante para la separación de gases. Hay que tener presente que sólo ocho o nueve materiales poliméricos se utilizan para fabricar el 90 % de las membranas de separación de gases existentes [1-2]. En los últimos años, se ha buscado que el proceso de separación de gases sea rentable y respetuoso con el medio ambiente. La mayoría de las investigaciones se han centrado en la síntesis de nuevos materiales con características que favorezcan la separación de gases en cuanto a lograr elevada permeabilidad y selectividad para gases específicos [2-3]. Existe un gran interés en el estudio de los materiales microporosos que llevan a potenciales aplicaciones como almacenamiento de gases, membranas de separación. de. gases,. catálisis. heterogénea. y. encapsulación. de. nanopartículas. La mayoría de los materiales microporosos se basan en. 1.
(12) estructuras de red que son inherentemente insolubles. La microporosidad se genera a partir de la incapacidad de las cadenas de polímeros solubles para empaquetarse de manera eficiente en el estado sólido. Los ejemplos más conocidos como materiales microporosos son las zeolitas cristalinas y carbonos amorfos, pero éstos tienen aplicaciones limitadas, por lo que se ha buscado diseñar nuevos materiales microporosos que se preparen a partir de componentes orgánicos. [4-6] Por ejemplo, los armazones organometálicos (MOF) y los armazones orgánicos covalentes (COF) poseen un orden cristalino y una distribución estrecha del tamaño de poro, por lo tanto, son análogos orgánicos de las zeolitas. Otros ejemplos son los materiales poliméricos porosos amorfos, polímeros lineales de cadena rígida como los poliacetilenos, polímeros reorganizados térmicamente (TR), polímeros a base de ferroceno y los polinorbornenos. Estos polímeros poseen grandes áreas de superficie y amplias oportunidades sintéticas para modificaciones y, por lo tanto, son de gran interés como membranas o materiales de almacenamiento de gas. [5] Uno de los tipos de materiales microporosos que se ha estudiado ampliamente son los polímeros de microporosidad intrínseca (PIMs), los cuales han exhibido una alta eficiencia en la separación de gases. Los PIMs suelen ser solubles en disolventes orgánicos comunes como cloroformo y tetrahidrofurano, lo que permite una fácil fabricación de membranas mediante la evaporación lenta del disolvente empleado [3, 7]. Los PIMs son definidos como polímeros que contienen una red continua de espacios o “poros” intermoleculares interconectados, con tamaño menor a 2 nm que se forman como consecuencia directa de la forma y rigidez de la estructura de los monómeros que lo conforman. Esto hace que dichos materiales posean un área superficial elevada (típicamente entre 300 - 2000 m2g-1). Esta elevada área superficial hace que estos polímeros sean. 2.
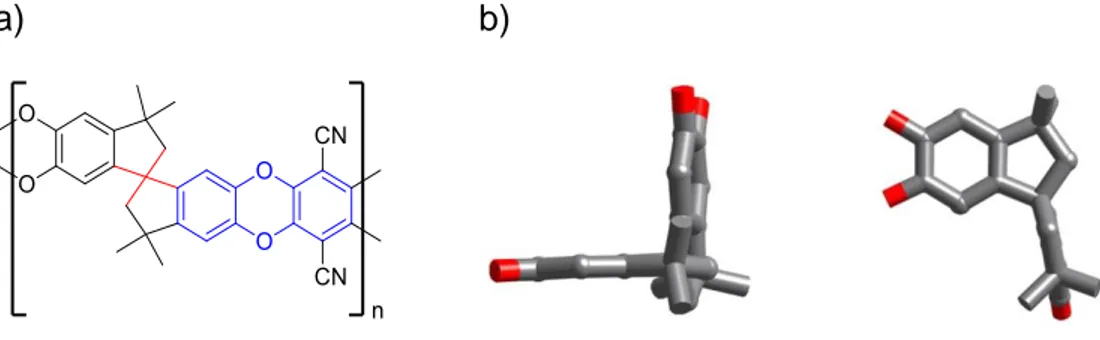
(13) excelentes candidatos en el campo de la separación de gases, y también, para otras aplicaciones como almacenamiento de gases y catálisis [3, 6-11]. Budd y McKeown sintetizaron el primer PIM (PIM-1), cuya estructura se basa en sistemas de anillos fusionados que tienen estructuras retorcidas que impiden un empaquetamiento eficiente y, por ende, las cadenas del polímero no pueden rotar libremente y tampoco pueden empaquetarse a un estado más compacto. La estructura del PIM-1 se muestra a continuación. [12] a). b). Figura 1. Representaciones de PIM-1. a) Estructura química de PIM-1. b) Representación del ángulo que se forma entre los anillos fusionados debido al carbono espiro. Es muy importante tener en cuenta que la estructura del polímero que se utilizará como membrana para la separación de gases, además de afectar las propiedades físicas, puede afectar el desempeño de la membrana tal como la separación y la adsorción de gases. [13] Los PIMs pueden ser obtenidos con estructuras lineales cuando se emplean monómeros con cuatro sitios reactivos, mientras que si se emplean monómeros con tres o seis sitios reactivos se obtienen cadenas en forma de red entrecruzadas, que suelen ser insolubles. El PIM-1, cuya síntesis es a través de cuatro sitios reactivos, forma un polidioxano lineal con una elevada área superficial, alta rigidez y una estructura molecular contorsionada que no puede llenar el espacio de manera eficiente, lo que da lugar a una mayor microporosidad, interconexión y volumen libre. [13, 14]. 3.
(14) La pérdida de libertad de rotación debido a la estructura molecular rígida y retorcida es la responsable del aumento considerable en el volumen libre y el área superficial en estado sólido de estos materiales [3, 7-10]. Para lograr este requisito estructural es necesario diseñar monómeros con estructuras rígidas y contorsionadas que impidan la libre rotación de la cadena derivada y que disminuyan el empaquetamiento eficiente. Para el diseño de estos monómeros, se han definido algunas estrategias básicas: 1) Incorporar sitios de contorsión a través de centros espiro. 2) Incorporar grupos voluminosos cercanos al sitio de crecimiento de la cadena para que impidan la rotación de ésta y 3) Incorporar estructuras cíclicas mediante la formación de anillos de dioxano o biciclos, como se puede observar en la figura 2. [15, 16]. Figura 2. Formación de dioxanos. Síntesis de PIMs mediante una reacción de polimerización. usando. una. combinación. de. monómeros. aromáticos. hidroxilados y monómeros aromáticos halogenados. [16] Otra de las reacciones que permiten obtener moléculas con estructuras rígidas, cíclicas y no planares es la anulación (anelación) catalítica entre un bromoareno y norborneno. Esta reacción se puede emplear para polimerizar si se utilizan dibromoarenos y norbornadieno, la cual se conoce como polimerización vía CANAL (Catalytic Arene-Norbornene Annulation) (Figura 3). [17-19]. 4.
(15) Figura 3. Ejemplo de polimerización vía CANAL. Teniendo en cuenta esta reacción, y usando las bondades de la química orgánica, es posible diseñar nuevos monómeros con características estructurales afines a la aplicación que se desee. Así es el caso del diseño de monómeros dibromoarenos que contienen unidades de dibenzodioxano, que otorga al futuro polímero, una estructura molecular rígida y contorsionada que no permite la libre rotación de las cadenas poliméricas y el empaquetamiento eficiente de éstas. Además, si se incorporan grupos voluminosos en el diseño del monómero se aumenta el volumen libre y, por ende, la microporosidad del material final. Un ejemplo de esto, son los precursores tetrafluroados reportados por Makhseed y colaboradores (figura 4), quienes estudiaron el efecto de sustituyentes de distinta naturaleza y tamaño en las propiedades de separación de gases de los polímeros derivados. [13-15]. Figura 4. Ejemplos de monómeros tetrafluorados con sustituyentes de distinta naturaleza y tamaño. Dichos monómeros se hicieron reaccionar con un tetraol para formar polímeros que contenían sitios de contorsión, anillos dioxano y grupos voluminosos en su estructura (figura 5).. 5.
(16) Figura 5. Polímeros de microporosidad intrínseca usando monómeros tetrafluorados con grupos voluminosos. Estos polímeros tipo escalera con grupos voluminosos fueron solubles en THF y cloroformo, y presentaron elevada permeabilidad a gases como hidrógeno y nitrógeno. [15] Teniendo en cuenta los antecedentes anteriores, la investigación propuesta se basó en el diseño y síntesis de moléculas orgánicas con un alto grado de rigidez estructural debido a la presencia de anillos fusionados y con la presencia de grupos voluminosos de distinta naturaleza. Estos nuevos compuestos serán empleados como monómeros dibromados para la futura preparación de nuevos polímeros de microporosidad intrínseca a través de una polimerización tipo CANAL.. 6.
(17) 2. Hipótesis La síntesis y el uso de precursores aromáticos tetrafluorados con grupos que retiran densidad electrónica permitirán la preparación de monómeros dibromados con anillos de dioxano fusionados a partir de una reacción de sustitución nucleofílica aromática doble. Estos monómeros de estructura rígida podrán ser usados en la posterior preparación de polímeros de microporosidad intrínseca vía polimerización tipo CANAL. 3. Objetivos 3.1. Objetivo general Sintetizar. y. caracterizar. espectroscópicamente. siete. monómeros. dibromados con anillos de dioxano fusionados los que serán empleados como monómeros en una futura polimerización tipo CANAL. 3.2. Objetivos específicos •. Sintetizar y caracterizar por técnicas espectroscópicas los precursores P1-P7.. •. Sintetizar y caracterizar por técnicas espectroscópicas el precursor P8.. •. Sintetizar y caracterizar por técnicas espectroscópicas los monómeros M1-M7.. 7.
(18) 4. PARTE EXPERIMENTAL 4.1.. Material y equipos. Los reactivos utilizados para la síntesis de los compuestos fueron de procedencia Aldrich, AKScientific y Merck, al igual que los disolventes empleados. La temperatura de fusión de los precursores y monómeros se determinó a través de un equipo SMP10 Stuart Cientific. Los espectros de FT-IR fueron obtenidos en un espectrofotómetro Shimadzu IRTracer 100, empleando como soporte pastillas de KBr. Los resultados se expresan en número de onda (cm-1). Los espectros de RMN de 1H,. 13C,. DEPT-135º,. 19F. y HMBC fueron obtenidos. en un espectrofotómetro Brucker AC de 400 MHz, como disolvente se usó DMSO-d6 y tolueno-d8. Los desplazamientos químicos (δ) están expresados en partes por millón (ppm) con relación a tetrametilsilano (TMS). Todos los materiales y equipos necesarios para la síntesis de los precursores y monómeros se obtuvieron del Laboratorio de Investigación en Polímeros Orgánicos 1 y 2 (RLOP) de la Facultad de Química y de Farmacia.. 4.2.. Síntesis de Precursores. 4.1.1. Síntesis de anhídrido 3,4,5,6-tetrafluoroftálico (P1). P1 Esquema 1. Síntesis de P1.. 8.
(19) En un balón de dos bocas de 250 mL se agrega el ácido 3,4,5,6tetrafluoroftálico (15 g, 63 mmol) y con una jeringa se agregan 24 mL de cloruro de tionilo. Una vez disuelto el diácido, se deja reaccionar 24 horas a 90 °C y constante agitación con una trampa de CaCl2 anh. Pasadas las 24 horas, se destila la mayor cantidad de cloruro de tionilo posible. Se deja enfriar a temperatura ambiente y el sólido resultante se filtra al vacío, se lava con nhexano y se seca en una estufa a 40 °C por 24 horas. P1. Rendimiento: 86 %. T.f.: 95 ºC. IR (KBr, , cm-1): 1874 y 1772 (C=O); 1627, 1520 y 1401 (C=C); 1214 (C-O-C). Esta molécula no posee hidrógenos, por lo que no se realizan análisis de 1HRMN. Tampoco se registró el espectro de RMN de. 13C. debido a que el. anhídrido puede reaccionar con el agua presente en el solvente deuterado generando trazas de ácido, lo que llevaría a una interpretación errónea del resultado. 4.1.2. Síntesis de 4,5,6,7-tetrafuoro-2-fenilisoindolina-1,3-diona (P2). P2 Esquema 2. Síntesis de P2. En un balón de dos bocas de 100 mL se disuelve anilina (1,37 mL; 6,16 mmol) en 8 mL de ácido acético glacial. Una vez disuelta, se agrega P1 (1,5 g; 6,8 mmol) y se añaden 2 mL de ácido acético glacial. La reacción se mantiene en agitación y bajo reflujo a 100 °C por 24 horas. La suspensión obtenida se vierte en una mezcla de 400 mL de agua:HCl (3:1 v/v). El sólido en suspensión se. 9.
(20) filtra al vacío, se lava con abundante agua destilada y se seca en una estufa a 80 °C por 24 horas.. P2. Rendimiento: 74 %. T.f.: 210 ºC. FT-IR (KBr, , cm-1): 3090 (C-H arom.); 1789 y 1720 (C=O); 1597, 1512 y 1411 (C=C); 1373 (C-N); 763 (arom. monosust.). 1H-RMN (DMSO-d6, δ, ppm): 7,44 (d, J= 7,2 Hz, 2H, H6); 7,49 (t, J= 7,4 Hz, 1H, H8); 7,56 (t, J= 7,5 Hz, 2H, H7). 13C-RMN (DMSO-d6, δ, ppm): 114,09 (C3); 127,37 (C6); 128,75 (C8); 129,09 (C7); 130,92 (C5); 142,66 (d, J= 257 Hz, C2); 144,13 (d, J= 266 Hz, C1); 161,82 (C4). 4.1.3. Síntesis de 4,5,6,7-tetrafluoro-2-(4-metoxifenil)isoindolina-1,3diona (P3). P3 Esquema 3. Síntesis de P3. Se empleó la misma metodología y proporciones molares informadas para la síntesis de P2, usando 4-metoxianilina en vez de anilina.. 10.
(21) P3. Rendimiento: 67 %. T.f.: 221 ºC. FT-IR (KBr, , cm-1): 3070 y 3001 (C-H arom.); 2839 (C-H alif.); 1782 y 1728 (C=O); 1612, 1512 y 1411 (C=C); 1303 (C-N); 1242 y 1087 (C-O-C); 840 y 810 (arom. para-disust.). 1H-RMN (DMSOd6, δ, ppm): 3,81 (s, 3H, H9); 7,09 (d, J= 8,9 Hz, 2H, H7); 7,34 (d, J= 8,9 Hz, 2H, H6).. 13C-RMN. (DMSO-d6, δ, ppm): 55,43 (C9); 114,04 (C3); 114,35 (C7);. 123,36 (C5); 129,74 (C6); 142,57 (d, J= 256 Hz, C2); 144,11 (d, J= 275 Hz, C1); 159,32 (C8); 162,06 (C4). 4.1.4. Síntesis de 4,5,6,7-tetrafluoro-2-(3,4,5trimetoxifenil)isoindolina1,3-diona (P4). P4 Esquema 4. Síntesis de P4. Se empleó la misma metodología y proporciones molares informadas para la síntesis de P2, usando 3,4,5-trimetoxianilina en vez de anilina.. 11.
(22) P4. Rendimiento: 88 %. T.f.: 255 ºC. FT-IR (KBr, , cm-1): 3109 (C-H arom.); 2839 y 2546 (C-H alif.); 1782 y 1705 (C=O); 1597, 1512 y 1411 (C=C); 1303 (C-N); 1242 y 1087 (C-O-C). 1H-RMN (DMSO-d6, δ, ppm): 3,72 (s, 3H, H10); 3,77 (s, 3H, H9); 6,78 (s, 2H, H6).. 13C-RMN. (DMSO-d6, δ, ppm): 56,06 (C9);. 59,69 (C10); 105,91 (C6); 113,7 (C3); 125,95 (C5); 138,34 (C8); 142,28 (d, J= 269 Hz, C2); 143,80 (d, J= 261 Hz, C1); 152,7 (C7); 161,88 (C4). 4.1.5. Síntesis de 2-(4-(tert-butil)fenil)-4,5,6,7-tetrafluoroisoindolina1,3-diona (P5). P5 Esquema 5. Síntesis de P5. Se empleó la misma metodología y proporciones molares informadas para la síntesis de P2, usando 4-terc-butilanilina en vez de anilina.. 12.
(23) P5. Rendimiento: 96 %. T.f.: 228 ºC. FT-IR (KBr, , cm-1): 3090 (C-H arom.); 2970 y 2870 (C-H alif.); 1789 y 1720 (C=O); 1604, 1512 y 1411 (C=C); 1365 (C-N); 848 y 825 (arom. para-disust.). 1H-RMN (DMSO-d6, δ, ppm): 1,33 (s, 9H, H10); 7,35 (d, J= 8,6 Hz, 2H, H6); 7,57 (d, J= 8,6 Hz, 2H, H7).. 13C-. RMN. (DMSO-d6, δ, ppm): 31,04 (C10); 34,53 (C9); 114 (C3); 125,9 (C7); 126,9 (C6); 128,28 (C5); 142,56 (d, J= 269 Hz, C2); 144,15 (d, J= 261 Hz, C1); 151,32 (C8); 161,93 (C4). 4.1.6. Síntesis de 2-(2,6-diisopropilfenil)-4,5,6,7-tetrafluoroiso indolina-1,3-diona (P6). P6 Esquema 6. Síntesis de P6. Se empleó la misma metodología y proporciones molares informadas para la síntesis de P2, usando 2,6-diisopropilanilina en vez de anilina. Sin embargo, el sólido obtenido corresponde al ácido ámico correspondiente, el cual se añade en un balón de dos bocas de 100 mL (2,4 g; 6,04 mmol), junto a acetato de sodio (0,24 g, 2,9 mmol) y Ac2O (5 mL, 52,9 mmol). La mezcla de reacción. 13.
(24) se mantiene en agitación a 100 °C por 4 horas. Posteriormente, se enfría a temperatura ambiente y se agregan 50 mL de agua destilada con agitación hasta la formación de un sólido de color blanco. Finalmente, el producto se filtra al vacío y se lava con abundante agua.. P6. Rendimiento: 89 %. T.f.: 184 ºC. FT-IR (KBr, , cm-1): FT-IR (KBr) (cm-1): 3078 (C-H arom); 2970 y 2877 (C-H alif.); 1782 y 1720 (C=O); 1589, 1519 y 1442 (C=C); 1357(C-N). 1H-RMN (DMSO-d6, δ, ppm): 1,09 (d, J= 8.6 Hz, 12H, H10); 2,74 (m, 2H, H9); 7,37 (d, J= 7,8 Hz, 2H, H7); 7,51 (t, J= 7,8 Hz, 1H, H8). 13C-RMN. (DMSO-d6, δ, ppm): 23,76 (C10); 28,43 (C9); 113,57 (C3); 124,1. (C7); 125,64 (C5); 130,57 (C8); 143,03 (d, J= 266 Hz, C2); 144,60 (d, J= 261 Hz, C1); 147,05 (C6); 162,4 (C4). 4.1.7. Síntesis de 2-(adamantil)-4,5,6,7-tetrafluoroisoindolina-1,3diona (P7). P7 Esquema 7. Síntesis de P7.. 14.
(25) En un balón de 100 mL se disuelve 1-adamantilamina (1 g; 6,6 mmol) en 50 mL de DMAc. Por otro lado, en un vaso precipitado de 50 mL se disuelve P1 (1,5 g; 6,8 mmol) en 20 mL de DMAc y se agrega a un embudo isobarico, desde el cual, por goteo se adiciona a la disolución 1-adamantilamina en DMAc que se encuentra en agitación. La reacción se mantiene a temperatura ambiente y agitación por 24 horas. Posteriormente la suspensión obtenida se vierte en una mezcla de 400 mL de agua acida (agua:HCl; 3:1 v/v). El sólido en suspensión es filtrado al vacío y lavado con abundante agua destilada. Posteriormente, se seca en una estufa a 80 °C por 24 horas. Una vez seco el sólido se añade 1,00 g (2,7 mmol) a un balón de dos bocas de 100 mL, junto con acetato de sodio (0,47 g; 5,8 mmol) y Ac2O (10 mL; 0,1 mol). La reacción se mantiene en agitación a 100 °C por 12 horas. Una vez finalizado este tiempo, se añaden 50 mL de agua al balón y se agita mecánicamente hasta la formación de un sólido. El sólido obtenido es filtrado al vacío y lavado con agua. Finalmente, el sólido se seca en una estufa a 80 ° C por 24 horas.. P7. Rendimiento: 79 %. T.f.: 179 ºC. FT-IR (KBr, , cm-1): 2908 y 2854 (C-H alif.); 1774 y 1705 (C=O); 1643, 1504 y 1411 (C=C); 1303 (C-N). 1H-RMN (DMSO-d6, δ, ppm): 1,68 (m, 6H, H8); 2,11 (m, 3H, H7); 2,4 (m, 6H, H6).. 13C-. RMN (DMSO-d6, δ, ppm): 20,11 (C7); 35,57 (C8); 39,74 (C6); 60,74 (C5); 113,61 (C3); 142,93 (d, J= 424 Hz, C2); 145,27 (d, J= 366 Hz, C1); 163,69 (C4).. 15.
(26) 4.1.8. Síntesis de 3-bromo-4-metilcatecol (P8). P8 Esquema 8. Síntesis de P8. En un balón de tres bocas de 2 L se disuelve 4-metilcatecol (10 g; 80,6 mmol) en 1,4 L de DCM, bajo atmósfera de nitrógeno y con una de las bocas conectada a una trampa para gases rellena con una solución de KOH (ac) al 5 %. Una vez disuelto el 4-metilcatecol se agrega, a través de un embudo isobárico, 4,2 mL de una disolución 0,82 M de bromo en DCM. Esta adición debe ser mediante goteo lento; aproximadamente entre 1 a 2 horas. La reacción se mantiene en agitación y a temperatura ambiente hasta que termine la adición del bromo. Posteriormente se elimina el solvente en un evaporador rotatorio y el producto crudo se recristaliza en n-hexano. El sólido obtenido se seca en una estufa a 40° C por 24 horas.. P8. Rendimiento: 76 %. T.f.: > 300 ºC. FT-IR (KBr, , cm-1): 3587 y 3402 (OH); 3150 (C-H arom.); 2916 (C-H alif.); 1658, 1597 y 1458 (C=C); 1280 (C-O). 1H-RMN. (DMSO-d6, δ, ppm): 2,14 (s, 3H, H7); 6,70 (s, 1H, H5); 6,88 (s, 1H,. H2); 9,03 (s, 1H, HB); 9,12 (s, 1H, HA).. 13C-RMN. (DMSO-d6, δ, ppm): 21,89. (C7); 111,57 (C3); 117,79 (C5); 118,63 (C2); 127,06 (C4); 144,45 (C1); 144,92 (C6).. 16.
(27) 4.3.. Síntesis de monómeros. 4.3.1. Síntesis de 2,11-dibromo-3,12-dimetil-7-fenil-6H-benzo[5,6][1,4] dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g]isoindol-6,8(7H)diona (M1). M1 Esquema 9. Síntesis de M1. En un balón de dos bocas de 100 mL se disuelve 3-bromo-4-metilcatecol (7,45 mmol) y P2 (3,38 mmol) en 65 mL de DMF. Una vez disuelto, se agrega K2CO3 (18,6 mmol). La reacción se calienta a 100 °C y se agita durante 12 horas. Posteriormente, la suspensión se vuelca sobre 250 mL de agua destilada en agitación y el sólido obtenido se filtra a vacío, se lava con abundante agua y luego con 200 mL de metanol. El producto final se seca en una estufa a 80 °C por 24 horas.. 17.
(28) M1. Rendimiento: 59 %. T.f.: > 300 ºC. FT-IR (KBr, , cm-1): 3062 (C-H arom.); 2990 (C-H alif.); 1766 y 1712 (C=O); 1589, 1496 y 1411 (C=C); 1373 (C-N); 1141 y 1087 (C-O-C); 748 (arom. mono-sust.). 1H-RMN (DMSO-d6, δ, ppm): 2,32 (s, 6H, H5 y H5´); 7,15-7,71 (m, 9H, H1, H2, H3, H4, H6, H7, H8). No se cuenta con espectros de. 13C-RMN,. debido a que la molécula es poco. soluble en los solventes deuterados disponibles en el laboratorio. 4.3.2. Síntesis. de. 2,11-dibromo-7-(4-metoxifenil)-3,12-dimetil-6H-. benzo[5,6][1,4]dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g] isoindol-6,8(7H)-diona (M2). M2 Esquema 10. Síntesis de M2. Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando P3 en vez de P2.. 18.
(29) M2. Rendimiento: 86 %. T.f.:> 300 ºC. T-IR (KBr, , cm-1): 3050 (C-H arom.) 2831 (C-H alif.); 1766 y 1720 (C=O); 1589 y 1512 (C=C); 1242 (C-O-C); 1334 (C-N); 1141 y 1080 (C-O-C); 871 y 833 (arom. para-disust.). 1H-RMN (DMSOd6, δ, ppm): 2,33 (s, 2H, H5 y H5´); 3,86 (s, 3H, H8); 7,06 (d, J = 9,08 Hz, 2H, H6); 7,13 (d, J = 8,21 Hz, 2H, H7); 7,28 - 7,32 (m, 4H, H1, H2, H3, H4). No se cuenta con espectros de. 13C-RMN,. debido a que la molécula es poco. soluble en los solventes deuterados disponibles en el laboratorio. 4.3.3. Síntesis de 2,11-dibromo-3,12-dimetil-7-(3,4,5-trimetoxifenil)-6Hbenzo[5,6][1,4]dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g] isoindol-6,8(7H)-diona (M3). M3 Esquema 11. Síntesis de M3. Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando P4 en vez de P2. M3. Rendimiento: 83 %. T.f.: > 300 ºC. FT-IR (KBr, , cm-1): 3100 (C-H arom.); 2939 y 2839 (C-H alif.); 1774 (C=O); 1589 y 1504 (C=C); 1226 (C-N); 1180 y 1080 (C-O-C). No se cuenta con espectros de RMN, debido a que la molécula es insoluble en los solventes deuterados disponibles en el laboratorio.. 19.
(30) 4.3.4. Síntesis de 2,11-dibromo-7-(4-(tert-butil)fenil)-3,12-dimetil-6Hbenzo[5,6][1,4]dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g] isoindol-6,8(7H)-diona (M4). M4 Esquema 12. Síntesis de M4. Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando P5 en vez de P2.. M4. Rendimiento: 77 %. T.f.: > 300 ºC. FT-IR (KBr, , cm-1): 3070 (C-H arom.); 2954 (C-H alif.); 1766 y 1720 (C=O); 1597 y 1535 (C=C); 1265 (C-N); 1141 y 1080 (C-O-C). 1H-RMN (DMSO-d6, δ, ppm): 1,32 - 1,36 (s, 9H, H8); 2,31 (s, 6H, H5 y H5’); 6,61 - 7,61 (m, 8H, H1, H2, H3, H4, H6, H7). No se cuenta con espectros de. 13C-RMN,. debido a que la molécula es poco. soluble en los solventes deuterados disponibles en el laboratorio.. 20.
(31) 4.3.5. Síntesis de 2,11-dibromo-7-(2,6-diisopropilfenil)-3,12-dimetil-6Hbenzo[5,6][1,4]dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g] isoindol-6,8(7H)-diona (M5). M5 Esquema 13. Síntesis de M5. Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando P6 en vez de P2.. M5. Rendimiento: 81 %. T.f.: >300 ºC. FT-IR (KBr, , cm-1): 3050 (C-H arom.); 2962 y 2870 (C-H alif.); 1766 y 1720 (C=O); 1589,1489 y 1411 (C=C); 1357 (C-N); 1141 y 1087 (C-O-C). 1H-RMN (DMSO-d6, δ, ppm): 1,15 (d, J= 6,9 Hz, 12H, H9); 2,34 (s, 6H, H5 y H5’); 2,77 (s, 2H, H8); 7,14 (d, J= 7,1 Hz, 2H, H6); 7,29 - 7,32 (m, 4H, H1, H2, H3, H4); 7,13 (t, J= 8,31 Hz, 1H, H7). No se cuenta con espectros de. 13C-RMN,. debido a que la molécula es poco. soluble en los solventes deuterados disponibles en el laboratorio.. 21.
(32) 4.3.6. Síntesis. de. 7-(1-adamantil)-2,11-dibromo-3,12-dimetil-6H-. benzo[5,6][1,4]dioxina[2,3-e]benzo[5,6][1,4]dioxina[2,3-g] isoindol-6,8(7H)-diona (M6). M6 Esquema 14. Síntesis de M6. Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando P7 en vez de P2. M6. Rendimiento: 88 %. T.f.: > 300 ºC. FT-IR (KBr, , cm-1): 3070 (C-H arom.) 2908 y 2854 (C-H alif.); 1759 y 1705 (C=O); 1653, 1589 y 1411 (C=C); 1373 (C-N); 1149 y 1033 (C-O-C). No se cuenta con espectros de RMN, debido a que la molécula es insoluble en los solventes deuterados disponibles en el laboratorio. 4.3.7. Síntesis de 2,9-dibromo-3,10-dimetilbenzo[5,6][1,4]dioxino[2,3b]dibenzo[b,e][1,4]dioxine-6,13-dicarbonitrilo. M7 Esquema 15. Síntesis de M7.. 22.
(33) Se empleó la misma metodología y proporción molar enunciadas en la síntesis de M1, utilizando 2,3,5,6-tetrafluorotereftalonitrilo en vez de P2.. M7. Rendimiento: 50 %. T.f.: >300 ºC. FT-IR (KBr, , cm-1): 3060 (C-H arom.); 2890 (C-H alif.); 2237 (CΞN); 1589,34 (C=C); 1496 y 1303 (C-H deformación); 1126 y 1018 (C-O-C). 1H-RMN (DMSO-d6, δ, ppm): 2,27 (s, 6H, H5); 7,25 (s, 2H, H2, H3), 7,45 (s, 2H, H1, H4). No se cuenta con espectros de. 13C-RMN,. debido a que la molécula es poco. soluble en los solventes deuterados disponibles en el laboratorio.. 23.
(34) 5. RESULTADOS Y DISCUSIÓN 5.1.. Síntesis y caracterización de precursores. 5.1.1. Síntesis y caracterización de P1 La síntesis del precursor anhídrido 3,4,5,6- tetrafluoroftálico (P1) a partir de ácido 3,4,5,6-tetrafluoroftálico se basa en una reacción de ciclodeshidratación con cloruro de tionilo, cuyo mecanismo de reacción se muestra en el siguiente esquema:. Esquema. 16.. Mecanismo. para. la. síntesis. de. anhídrido. 3,4,5,6-. tetrafluoroftálico (P1) El mecanismo principal es una adición-eliminación, la cual comienza con el ataque nucleofílico de uno de los pares de electrones libres del átomo de oxígeno hacia el azufre electrofílico donde se produce el desplazamiento de los electrones hacia el oxígeno enlazado al azufre, formando un intermediario tetraédrico. En un segundo paso, se regenera el grupo sulfonilo por el movimiento de los electrones del oxígeno para la expulsión del ion cloruro, por ser un buen grupo saliente, quedando con carga positiva el oxígeno, el cual se regenera por la sustracción de un hidrógeno, obteniéndose un anhídrido clorosulfínico altamente reactivo. Este anhídrido sufre una. 24.
(35) sustitución nucleofílica en el carbono carbonílico por parte de un ion cloruro, produciendo el cloruro de ácido respectivo y la eliminación de ácido clorhídrico y dióxido de azufre. Posteriormente, se produce el ataque nucleofílico de uno de los pares de electrones libres del oxígeno hacia el carbono carbonílico activado por la presencia del cloro, desplazando los electrones hacia el átomo de oxígeno, formando un intermediario tetraédrico. En el paso final, la carga del oxígeno regenera el doble enlace y elimina el ion cloruro, el cual, por medio de una reacción ácido-base con el único hidrógeno de la especie, forma el producto deseado, eliminándose ácido clorhídrico como subproducto. [20] El uso de cloruro de tionilo como agente clorante, de deshidratación o ciclante, varía dependiendo del sustrato empleado. Al utilizar ácido ftálico se obtiene un anhídrido como se mostró en el esquema 16. Este anhídrido sólo se obtendrá si el ácido ftálico posee dos grupos ácidos carboxílicos en posición orto, en cambio, si los grupos ácidos están en posición meta o para, sólo se originará un dicloruro. Por otro lado, el SOCl2 posee una temperatura de ebullición de 76 ºC, por lo cual, es fácilmente extraíble del medio de reacción mediante una destilación simple, lo cual ayuda en el proceso de aislación del producto formado. [21, 22] El precursor P1 se hidroliza en presencia de agua, generando la reacción inversa y volviendo al sustrato ácido tetrafluoroftálico. Es por esto, que se vuelve imperativo que la síntesis de P1 se realice en ausencia de agua, para lo cual, se utilizó una pipa de cloruro de calcio anhidro, para mantener el sistema seco. La eliminación de SOCl2, luego de actuar como solvente y reactivo en la síntesis, es sumamente importante por su alta peligrosidad, dado que origina productos tóxicos gaseosos, como óxidos de azufre y cloruro de hidrógeno, los cuales se forman al reaccionar con el reactivo ácido 3,4,5,6tetrafluoroftálico o agua. Estos sub-productos de reacción son tóxicos para el ser humano con un olor sofocante a acre como el dióxido de azufre, el cual. 25.
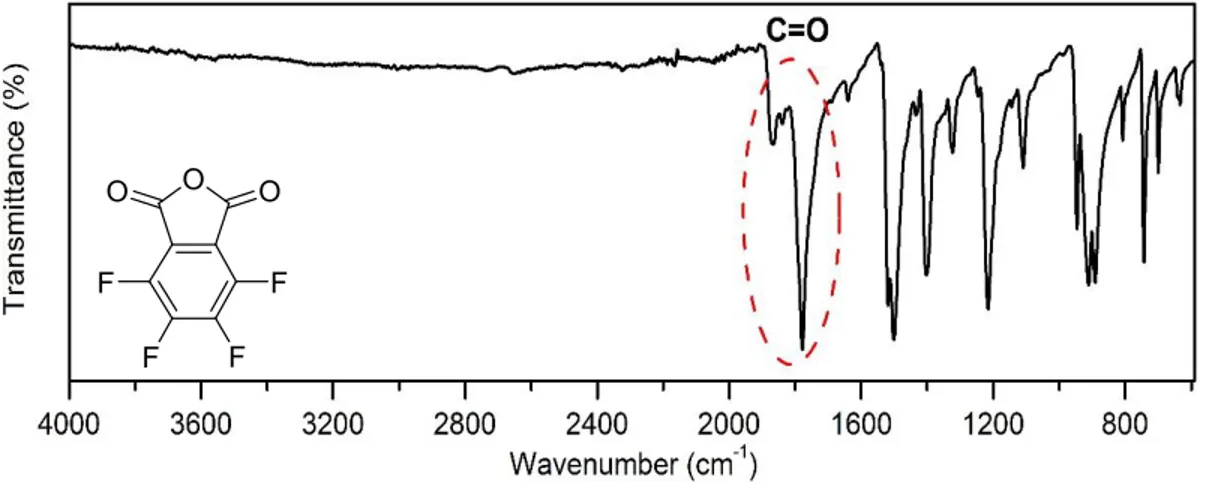
(36) causa daños al ser inhalado, ingerido y estar en contacto, produciendo irritación en los ojos, piel, mucosas y puede llegar hasta provocar quemaduras. Luego de eliminar la mayor cantidad posible de SOCl 2 remanente por destilación, el residuo obtenido se lava con abundante n-hexano, ya que éste favorece la precipitación del precursor P1 y solubiliza el SOCl2 residual. [23] Debido a que en el laboratorio no se poseían solventes deuterados libres de agua, no se pudo caracterizar P1 mediante técnicas de RMN, ya que ante la presencia de agua daría una respuesta falsa de hidrólisis. Por lo tanto, sólo se procedió a caracterizarlo mediante técnica de IR. En la figura 6 se observa el espectro de IR del precursor anhídrido P1. La presencia de señales correspondientes a los estiramientos de los carbonilos (C=O) a 1772 cm-1 y 1874 cm-1, junto a la ausencia de la señal correspondiente al estiramiento OH de ácido carboxílico, confirman el éxito de la reacción de ciclodeshidratación.. Figura 6. Espectro de IR de P1.. 26.
(37) 5.1.2. Síntesis y caracterización de P2 a P7 La obtención de los precursores imidas P2 a P5 se basa en una condensación directa, cuyo mecanismo de reacción se muestra en el siguiente esquema:. Esquema 17. Mecanismo síntesis de imidas por condensación directa. El mecanismo comienza con el ataque nucleofílico del par de electrones libres del átomo de nitrógeno de la amina al carbono electrofílico carbonílico. Producto de esto, se abre el doble enlace carbono-oxígeno, desplazando el par de electrones hacia el oxígeno cargándose este negativamente y dejando al átomo de nitrógeno con una carga parcial positiva, en donde luego sucede una transferencia de protones (T.P.). Posteriormente, se produce un segundo ataque desde el par electrónico sin compartir del nitrógeno amida hacia el carbono carbonílico del grupo ácido carboxílico, originando así una especie amida inestable doblemente cargada, produciéndose nuevamente una transferencia de protones que resulta en un producto dihidroxi-amida. Debido a la presencia de protones en el medio, se promueve la salida de agua por uno de los grupos hidroxilos, en cambio el segundo, da origen a un grupo carbonílico protonado, el cual, por medio de una reacción ácido-base intermolecular, forma el producto deseado de ftalimida. [24] El mecanismo, a través de una condensación directa, resulta efectivo para las aminas que no poseen una estructura que otorgue impedimento estérico al formarse la imida, como es el caso de las siguientes aminas: anilina, 4-. 27.
(38) metoxianilina, 3,4,5-trimetoxianilina y 4-tert-butilanilina (correspondientes a los precursores P2, P3, P4 y P5), que por sus sustituyentes metoxilo y tert-butilo en posiciones meta y para al grupo amina no otorgaron problema alguno para la condensación directa. Sin embargo, para el precursor P6 donde se utilizó 2,5-diisopropilanilina, los sustituyentes isopropilo ocupan ambas posiciones orto del anillo aromático, otorgándole un mayor impedimento estérico para que suceda la condensación directa. En el caso del precursor P7, donde la amina utilizada tiene como sustituyente un grupo alifático cíclico voluminoso (adamantilo), el impedimento estérico producido es mucho mayor que en el caso del precursor P6.. Esquema 18. Mecanismo ciclodeshidratación empleando anhídrido acético y acetato de sodio. La ciclación de P6 y P7 emplea como reactivos anhídrido acético y acetato de sodio, tal como se puede observar en el esquema 18. El anhídrido acético se usa ampliamente como agente deshidratante en presencia de acetato de sodio, ácido trifluoroacético o trietilamina. El uso de una base no nucleofílica como el acetato de sodio es indispensable para que ocurra la ciclación, dado que, por medio de una reacción ácido-base, transforma el grupo ácido carboxílico en un carboxilato de mayor nucleofilia (paso 1). En una segunda instancia, el oxianión del grupo carboxilato ataca a uno de los carbonos. 28.
(39) carbonílicos del anhídrido acético para dar lugar a un anhídrido intramolecular, donde posteriormente, el ataque del nitrógeno amídico desplaza un acetato. Finalmente, éste, por medio de una reacción ácido-base, origina el respectivo anillo ftalimida y ácido acético como sub-producto. [25, 26] Para la ciclación de P7 se hicieron pruebas de tiempo versus concentración de acetato de sodio y anhídrido acético, tomando como referencia la ciclación de P6. Como resultado de este estudio, para el uso de 1-adamantil (síntesis de P7) se utilizó el doble molar en comparación a lo empleado en la ciclación que llevó a la preparación de P6 y, además, requirió el triple de tiempo. Así, se demostró que al utilizar el doble de concentración de acetato de sodio y anhídrido acético se obtiene el producto ciclado a las 12 horas. Esto se desarrolló mediante la toma de una alícuota a las 12 y 24 horas, analizándola mediante espectroscopía de RMN. Es importante recalcar, que en el caso de la preparación de P7, al utilizar una amina alifática cíclica básica con un pKa de 10,5, no se puede recurrir al mismo método de condensación que en la síntesis de los precursores P2-P6. Estos últimos se prepararon en presencia de ácido acético glacial, que presenta un pKa de 4,8. Su uso, en la síntesis de P6, llevaría a la protonación de la amina alifática, dejándola soluble e impediría el ataque nucleofílico en la formación del enlace amida. El método utilizado se base en el uso de DMAc como disolvente. Se eligió este disolvente por ser de tipo polar con un pKa cercano a 30, lo cual asegura la ausencia de reacciones ácido-base indeseadas. [27, 28] De igual forma, se debe tener en consideración que para asegurar una mayor formación del producto deseado y garantizar que todo el anhídrido 3,4,5,6tetrafluoroftálico reaccione, se agrega un 10 % de exceso de cada amina. El remanente de éstas, es fácilmente eliminable luego de la reacción, mediante lavados sucesivos con agua ácida.. 29.
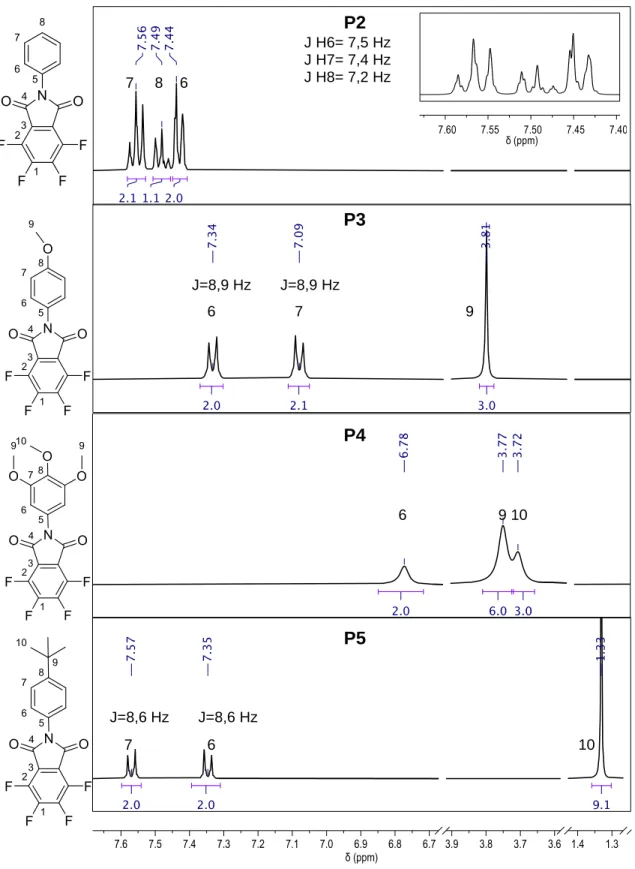
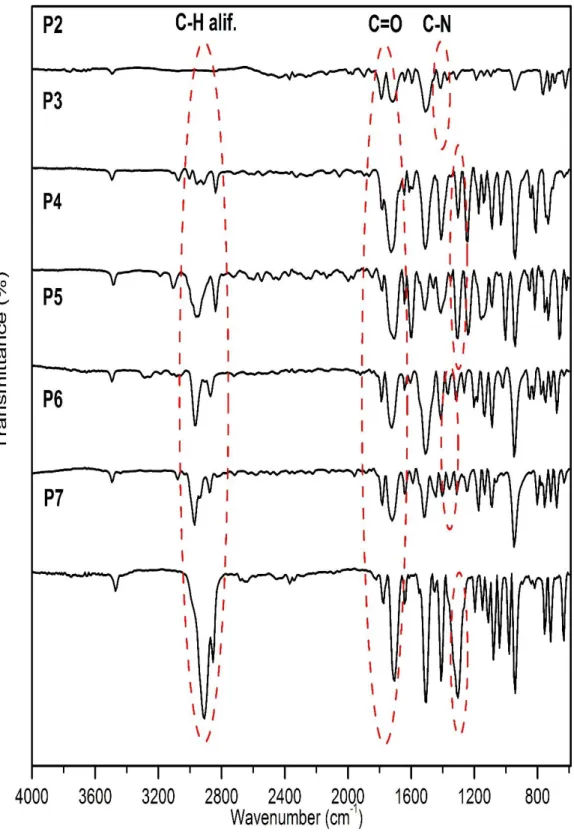
(40) En la figura 7 y figura 8 se pueden observar los espectros de 1H RMN de los precursores P2-P5 y P6-P7, respectivamente. De igual forma, se incluyen sus respectivas estructuras, asignaciones y desplazamientos químicos acotados en el rango de las señales de cada compuesto para una mayor facilidad de comprensión. Los espectros de. 13C. RMN y DEPT-135° de los precursores P2 a P7 se. encuentran disponibles en la sección de anexos.. 30.
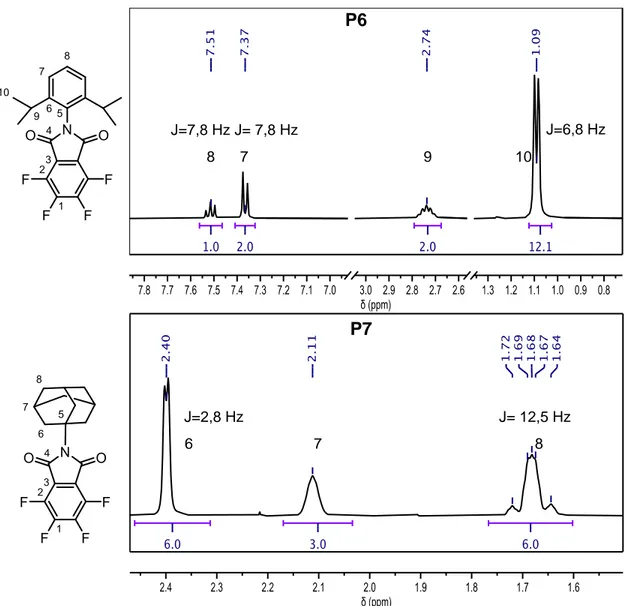
(41) P2 7. 8. J H6= 7,5 Hz J H7= 7,4 Hz J H8= 7,2 Hz. 6. P3 9 J=8,9 Hz 6. J=8,9 Hz 7. 9. 9P4 9 6. 9 10. P5. J=8,6 Hz 7. J=8,6 Hz 6. 10. 9 Figura 7. Espectros de 1H-RMN (DMSO-d6) de los precursores P2, P3, P4 y P5.. 31.
(42) P6. J=6,8 Hz. J=7,8 Hz J= 7,8 Hz 8. 7. 9. 10. 9 9 P7. J=2,8 Hz 6. J= 12,5 Hz 7. 8. 9. 9. Figura 8. Espectros de 1H-RMN (DMSO-d6) de los precursores P6 y P7. En el espectro de 1H-RMN del precursor P2 (figura 7), sólo se observan los protones. del. anillo. bencénico.. Además,. se. corrobora. que. la. ciclodeshidratación fue exitosa debido a que no se observa una señal correspondiente al hidrógeno de la amida, el cual generalmente se encuentra entre 8-11 ppm. Tampoco se observa la señal del hidrógeno del grupo ácido carboxílico, aunque esta evidencia no es concluyente por dos razones; al ser un protón activo, durante el análisis, la recuperación al estado base tras la excitación es muy rápida, lo cual provoca una señal ancha y que no siempre es visible en la región que debería observarse (entre 10-13 ppm). Por otro lado,. 32.
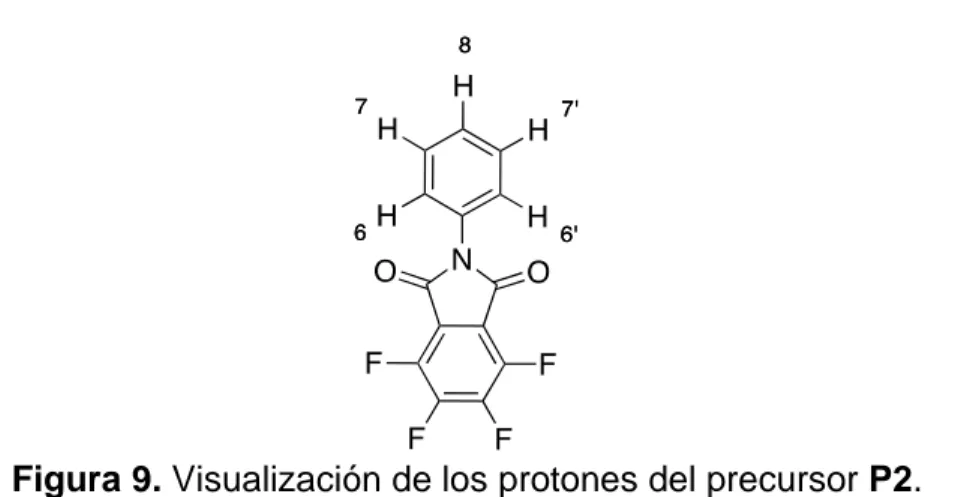
(43) el intercambio isotópico parcial o total con el deuterio del solvente empleado, minimiza o incluso anula su señal. [29]. Figura 9. Visualización de los protones del precursor P2. Los precursores P2-P7 poseen una estructura simétrica, por ende, presentan protones químicamente equivalentes. La equivalencia química es cuando los protones con entornos químicos idénticos poseen igual apantallamiento y, por ende, muestran el mismo desplazamiento químico. Los protones del precursor P2, como se observa en la figura 9, H-7 con H-7’ y H-6 con H-6’ son químicamente equivalentes, pero no magnéticamente equivalentes. La equivalencia magnética entre dos núcleos presupone la equivalencia química de dichos núcleos y, además, exige que: los acoplamientos escalares de ambos con cualquier otro núcleo magnético del sistema de espines (que no sean químicamente equivalente con ellos) tengan igual magnitud y que también acoplen entre sí. Casi todos los compuestos bencénicos poseen protones que no son equivalentes magnéticos por las constantes de acoplamientos que se generan entre sus protones. Hay tres acoplamientos en esta molécula, los que se producen entre protones en posiciones orto, meta y para. Por ejemplo: centrándose en los acoplamientos orto y para: H-6 acopla en posición orto con H-7 y en posición para con H-7´, en cambio H-6´acopla en posición orto con H-7` y en posición para con H-7, lo que da como resultado distintas constantes de acoplamiento donde J67 J6`7 y J. 76´. J. 7´6´.. Por ende,. la no equivalencia magnética genera la presencia de picos adyacentes en los. 33.
(44) dobletes y tripletes en el espectro, señales que dificultan la lectura del espectro. [29, 30] La constante de acoplamiento (J) que se conoce como la distancia entre los picos de un multiplete, es idéntica entre los dos tipos de protones que acoplan. Estos acoplamientos pueden ser dobletes, tripletes y multipletes, y pueden otorgar información estructural importante. La J se mide en Hz, y su magnitud de acoplamiento es variable en dependencia de la disposición relativa de los protones acoplados, de los grupos vecinos y de la cantidad de enlaces que separan a los protones. El efecto por el que un protón se puede desdoblar al o a los protones próximos (acoplados) tienen que tener los mismos efectos uno sobre el otro, es decir, es mutuo y de la misma magnitud. [31] El acoplamiento entre protones aromáticos vecinos (acoplamiento protones en posición orto) tiene un valor aproximado de 6 Hz a 9,5 Hz. Como se observa en los espectros de 1H-RMN de la figura 7 y figura 8, los valores obtenidos para la constante de acoplamiento son cercano a 8 Hz, lo que sugiere acoplamiento en posición orto entre los correspondientes protones. [29] En los precursores P3 y P5, también se observa un acoplamiento en orto de los protones H-6 y H-7, donde la constante obtenida para el precursor P3 fue de 8,9 Hz y para el precursor P5 de 8,6 Hz. En el espectro del precursor P6, que se muestra en la figura 8, se observa un acoplamiento entre los protones H-7 y H-8 de igual magnitud, correspondiente a 7,8 Hz, se observa el triplete de H-8 y el doblete de H-7, también se aprecia una nueva constante de acoplamiento para el H-10 de un valor de 6,8 Hz. Este protón (H-10) corresponde al CH3 del grupo sustituyente isopropilo, acopla con el protón H-9 del carbono terciario. El acoplamiento entre protones alifáticos vecinos, es un acoplamiento directo que se genera a un enlace de distancia, y generalmente presenta un valor entre 6 Hz y 8 Hz, pero puede variar según la estructura química. [29]. 34.
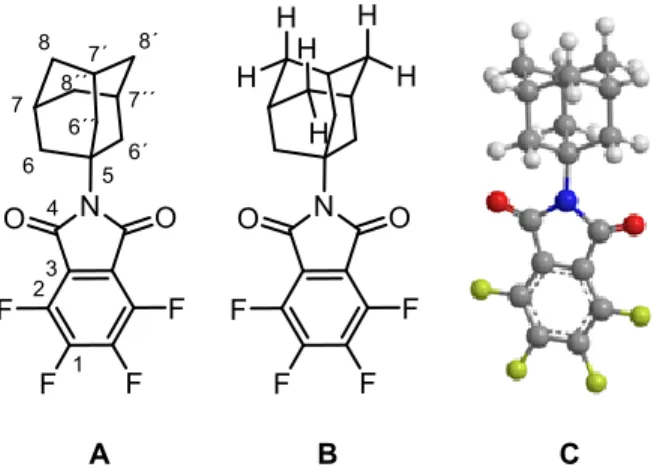
(45) Para el precursor P7, en la figura 8 se observan 3 señales, las cuales corresponden a los protones en las posiciones 6, 7 y 8, respectivamente. Como se puede observar en la figura 10A, el grupo adamantilo presenta protones que son químicamente equivalentes como los protones en posición 6, 6’ y 6’’, así como los protones 7, 7’ y 7’’. También los protones que se encuentran en posición axial y los protones que se encuentran en posición ecuatorial son químicamente equivalentes (Figura 10B). Por ende, se comprueba que la molécula es simétrica y presenta una equivalencia química, lo cual puede ver reflejado en la representación 3D del compuesto P7 (Figura 10C).. Figura 10. Diferentes representaciones de P7. El acoplamiento que se observa en el hidrógeno más desapantallado H-6, debido a su cercanía al nitrógeno de la imida, es un doblete con una J = 2,8 Hz. Los hidrógenos H-6, H-6’ y H-6’’ presentan el mismo desplazamiento químico y con multiplicidad de doblete, además, se conoce que el acoplamiento entre protones vecinos alifáticos cíclicos es muy variable ya que depende de la disposición de los núcleos. Dicho valor varía entre 2 Hz y 13 Hz, por ende, en el caso estudiado, se estaría observando un acoplamiento de tipo vecinal. [29]. 35.
(46) Para el núcleo H-7 no se evidencia claramente el acoplamiento con sus protones vecinos, sino que se observa una señal ancha, que debe corresponder a un multiplete, debido al acoplamiento con los protones en posiciones 6 y 8. No obstante, se observa dos doblete de tripletes para el hidrógeno en posición 8. Como se observa en la figura 10, se puede observar que los dos hidrógenos de C-8 se encuentran en posición axial y ecuatorial respecto al eje de simetría, y cada protón (axial o ecuatorial) acopla como triplete con los protones 7 adyacentes (también 7’ y 7’’) y como doblete con su respectivo protón geminal, dando en consecuencia dos dobletes de tripletes, con un triplete en el desplazamiento 1,68 ppm de mayor intensidad por la suma de las señales de ambos hidrógenos. El acoplamiento geminal se puede corroborar por la constante J medida desde el desplazamiento 1,72 ppm hasta 1,69 ppm, dando un valor de J = 12,5 Hz, sugiriendo, efectivamente, que se trata de un acoplamiento geminal entre los protones H-7 ecuatoriales y axiales. Esto fue corroborado por datos de bibliografía en donde se encuentra que el acoplamiento geminal presenta valores de J entre 12 Hz y 15 Hz. [29] En la figura 11 se pueden observan los espectros de IR correspondientes a los precursores P2-P7. Las señales características de estos compuestos están señalizadas y con el grupo funcional correspondiente. En los seis compuestos se observan las señales más importantes: una banda doble correspondiente al estiramiento simétrico y asimétrico del C=O del grupo imida ubicada entre 1720 cm-1 y 1760 cm-1 y la señal correspondiente a la vibración del enlace CN, la cual se ubica entre 1303 cm-1 y 1373 cm-1.. 36.
(47) Figura 11. Espectros de IR de los precursores P2 a P7. 37.
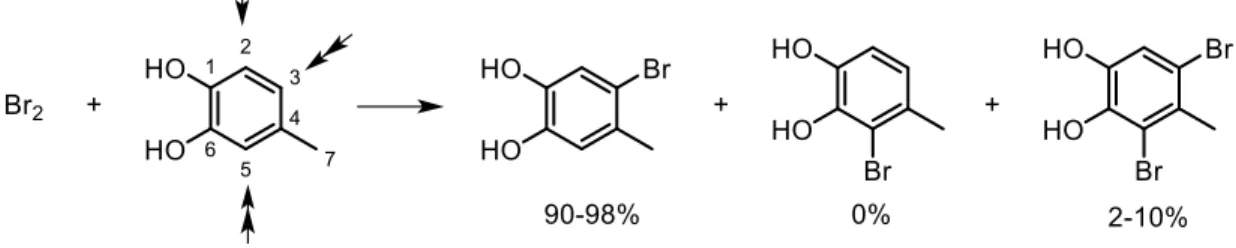
(48) 5.1.3. Síntesis y caracterización de P8 La síntesis del precursor 3-bromo-4-metilcatecol a partir de 4-metilcatecol se basa en una bromación a través de una sustitución electrofílica aromática (SEA). El electrófilo (Br+) reacciona con el anillo aromático, produciéndose un intermediario tipo carbocation estabilizado por resonancia a través de la deslocalización de la carga positiva. El bromo forma un nuevo enlace al unirse al anillo bencénico, formándose un carbono tetraédrico, provocando la pérdida momentánea de la aromaticidad del anillo, la cual es restituida eliminando un hidrógeno del carbono tetraédrico en forma de protón. [32] La halogenación por SEA convencional conlleva el uso de un catalizador: un ácido de Lewis como cloruro de aluminio. En la síntesis de P8, el anillo aromático de 4-metilcatecol está activado por dos grupos hidroxilos en posición 1 y 6, lo cual permite que el catalizador no sea necesario, bastando solamente con el uso de bromo disuelto en DCM. El uso de este disolvente es conveniente, ya que disuelve a 4-metilcatecol y al bromo molecular, y, además, es eliminable fácilmente por su baja temperatura de ebullición (40 °C). [33] Como se observa en la figura 12, la bromación podría ocurrir en tres posiciones del anillo (2, 3 y 5), siendo las posiciones 3 y 5 las más favorecidas por la orientación de los grupos sustituyentes. Sin embargo, dentro de estas dos, la posición 5 se encuentra menos favorecida debido al impedimento estérico que generan los grupos hidroxilo y metilo. Por lo tanto, la bromación ocurrió en posición 3 del anillo aromático, debido al menor impedimento estérico existente en esa región.. Figura 12. Productos de la bromación de 4-metilcatecol.. 38.
(49) Para asegurar la total bromación de 4-metilcatecol, el avance de la reacción se siguió mediante análisis de 1H-RMN tomando alícuotas del sistema cada media hora. De esta manera, si quedara sustrato sin bromarse sería muy fácil de identificar, pues al integrar las señales del sustrato y haciendo una simple regla de tres con la integral del producto, se puede calcular la cantidad de bromo faltante. Al ser una reacción rápida, una de las precauciones que hay que tener en consideración en la síntesis de este precursor es que la adición del bromo debe ser lenta. Para esto, lo ideal es ajustar la llave del embudo isobárico para que el goteo de reactivo ocurra cada 4 segundos. Si bien, la adición se hizo de forma controlada, siempre se obtuvo un pequeño porcentaje del producto dibromado en posiciones 3 y 5 (2 % al 10 %), debido a que el contenido de bromo en una gota podía incluir el exceso de éste. Además, se comprobó con el análisis del espectro de 1H-RMN que no existe mono-bromación en la posición 5. De existir una reacción en esta posición, se observarían dos dobletes correspondientes al acoplamiento entre los protones H-2 y H-3 en la región aromática (5 ppm a 9 ppm), lo cual no se evidenció. [28, 32] La caracterización del precursor P8 se realizó mediante espectroscopía de 1HRMN, 13C-RMN, DEPT-135°, HMBC e IR.. Figura 13. Espectro de 1H-RMN (DMSO-d6) del precursor P8.. 39.
(50) Como se observa en la figura 13, los hidrógenos de ambos grupos hidroxilos presentan desplazamientos químicos muy similares. En este sentido, el bromo es un desactivante del anillo aromático por efecto inductivo y orientador orto y para, por ende, la señal más desapantallada a 9,12 ppm corresponde al hidroxilo en posición A que se encuentra más cercana al éste. Para corroborar esta asignación, se procedió a realizar espectros bidimensionales, como se puede observar en el espectro de HMBC (figura 14). Se puede observar que el protón del hidroxilo en posición A corresponde al más desapantallado y que en posición 9,03 ppm se encuentra el protón del hidroxilo en posición B. El espectro 13C-RMN se encuentra disponible en los anexos. [32]. 0. 20. 40. 60. 100. δ (ppm). 80. 120. 140. 160. 180. 200. 10. 9. 8. 7. 6. 5 δ (ppm). 4. 3. 2. 1. 0. Figura 14. Espectro HMBC del precursor P8 Se realizó una recristalización del precursor P8 debido a la detección de producto dibromado en el crudo de reacción. En la figura 15 se puede observar dos espectros de protones acortados en desplazamientos químicos. Por medio del análisis de las integraciones de las señales, se observa que en el espectro de la izquierda (crudo de reacción), hay un 10 % de producto. 40.
(51) dibromado. Luego de la recristalización practicada desde n-hexano, la señal cercana a 7 ppm está ausente (espectro de la derecha), comprobando la total eliminación del producto dibromado.. Figura 15. Efecto de la recristalización desde n-hexano del precursor P8. En la figura 16, se puede observar el espectro de IR de P8. La señal más importante que se observa corresponde al estiramiento del enlace O-H de los grupos OH en 3402 cm-1 y 3587 cm-1. Estas bandas se localizan generalmente en el rango de 3500-3700 cm-1, las cuales varían si el grupo hidroxilo está asociado o no. Así, si el hidroxilo está asociado, la vibración O-H aparece en un rango de 3200 cm-1 y 3600 cm-1; por debajo del rango usual. Este efecto se debe a que al estar ambos grupos OH en posición orto del anillo, son capaces de formar dímeros y/o enlaces poliméricos entre varias moléculas por asociación de puente de hidrógenos. En este sentido, se observa la formación de puentes de hidrógenos en las señales a 3402 cm-1 y 3587 cm-1, ya que la señal es aguda y no ancha como se observaría para hidroxilos libres. Además, se observa una señal en 1658 cm-1 la cual corresponde a la flexión H-O-H. Otras señales importantes son las que dan cuenta de la presencia del estiramiento C-H del grupo metilo en 2916 cm-1 y las correspondientes al estiramiento C=C del núcleo bencénico en 1658 cm-1, 1597 cm-1 y 1458 cm-1.. 41.
(52) Figura 16. Espectro de IR del precursor P8. 5.2.. Síntesis y caracterización de monómeros. La síntesis de los monómeros dibromados se basa en una doble sustitución nucleofílica aromática (SNA), donde el reactivo que actúa como nucleófilo es el precursor P8 y como electrófilo se tienen a los halogenuros de arilos tetrafluorados, correspondientes a P2-P7 y al producto comercial 2,3,5,6tetrafluorotereftalonitrilo. Para que suceda una SNA, es indispensable que la molécula que será atacada por el nucleófilo contenga grupos que retiren densidad electrónica desde el anillo aromático. En este caso, la baja densidad electrónica del anillo radica en la presencia de cuatro átomos de flúor y dos grupos carbonilos en P2 – P7 o dos grupos nitrilos en la molécula comercial. [32]. Esquema 19. Mecanismo general para la síntesis de los monómeros.. 42.
(53) Como se puede observar en el esquema 19, una vez que los hidroxilos se encuentran desprotonados por la acción de la base, son más nucleofílicos para atacar al carbono electrofílico unido al flúor, los pares electrónicos libres del oxígeno que atacan primero al tetrafluorado son los del oxígeno más nucleofílico y corresponde al oxígeno en posición para al metilo. Al ser el metilo un grupo alquilo actúa como donor de electrones, y en este caso, activa al anillo bencénico por efecto inductivo concentrando la densidad electrónica en las posiciones orto y para, otorgándole así, mayor densidad electrónica al oxígeno en posición para a este metilo y, por ende, mayor nucleofília. Además, ambos iones fenóxidos se otorgan una mayor densidad electrónica entre sí, debido al efecto resonante tipo donor, atribuible a este tipo de grupo funcional. En conclusión, los efectos inductivos son el principal motivo por el cual el hidroxilo en posición meta al bromo, como también, posición para al metilo, posee una mayor nucleofilicidad. [32] Posteriormente, al suceder la reacción SNA, el sistema aromático se rompe por el ataque nucleófilico del oxígeno en el carbono electrofílico del anillo aromático, se forma un carbanión intermediario, donde la carga negativa se deslocaliza dentro del sistema aromático y es distribuido entre los cinco carbonos restantes del anillo. Finalmente, el sistema aromático se restaura cuando el flúor es eliminado del sistema en forma de fluoruro. En un último paso, sucede nuevamente una SNA con el ion fenóxido libre en posición meta al grupo metilo mediante el mismo mecanismo descrito. Este mecanismo se repite nuevamente en los dos carbonos electrofílicos restantes de la molécula, formándose por ambos lados el núcleo dibenzodioxano. [32] La síntesis se realiza empleando DMF como solvente por ser éste polar aprótico sin capacidad de solvatar en gran capacidad al nucleófilo, dejándolo libre para su desprotonación. El nucleófilo se prepara in situ debido a la desprotonación de los grupos hidroxilos mediante la base K 2CO3.. 43.
(54) Para la síntesis de los monómero se utilizaron dos equivalentes de 3-bromo4-metilcatecol (P8) y un equivalente del precursor tetrafluorado, para que se produjera la doble SNA formándose el anillo dibenzodioxano. Además, se utilizó un exceso de K2CO3, para asegurar la desprotonación cuantitativa de los grupos hidroxilos presentes. Para la recristalización de los monómeros se hicieron pruebas de solubilidad de todos éstos en distintos solventes orgánicos comunes. De esta forma, sólo se logró recristalizar dos monómeros, M1 y M2, donde M1 se recristalizó desde tolueno, mientras que M2 se obtuvo desde sulfolano.. Figura 17. Espectro de 19F-RMN (Tolueno-d8) del monómero M5. Se realizó un espectro de. 19F-RMN. para uno de los monómero (M5). Este. compuesto presenta una baja solubilidad en tolueno deuterado, por lo tanto, se desarrolló el análisis a fin de ratificar la ausencia de núcleos de flúor y comprobar así, el éxito de la SNA (figura 17). Para la caracterización mediante RMN de los monómeros, primero se realizaron pruebas de solubilidad a todos éstos en solventes con un homólogo deuterado disponible en el laboratorio: DMSO, cloroformo, tolueno y acetona. Como se observa en la tabla 1, los monómeros M3, M6 y M7 son insolubles en todos los solventes y condiciones de ensayo, mientras que M1, M2, M4 y M5 son solubles en DMSO a 100 ºC y a una baja concentración, lo que permitió su análisis mediante 1H-RMN.. 44.
(55) Tabla 1. Solubilidad de los monómeros en diferentes solventes. DMSO. CHCl3. Tolueno. Acetona. M1. +*. +/-. -. -. M2. +*. +/-. -. -. M3. -. -. -. -. M4. +*. -. -. -. M5. +*. -. +*. -. M6. -. -. -. -. M7. +/-. -. -. -. Aproximadamente 5 mg/mL. (+*: Soluble a aplicar calor (100 ºC); +/-: Medianamente soluble; -: Insoluble).. La baja solubilidad presentada por estos compuestos se debe a que su estructura se compone de la unión de tres anillos aromáticos a través de enlaces oxiéter (C-O-C) formando un núcleo benzodioxano por ambos lados. De esta forma, se genera una estructura plana, lo cual le otorga a la molécula la posibilidad de solapamiento con otras, por medio de interacciones − stacking y de tipo Van der Waals. Estas interacciones provocan que los monómeros se mantengan unidos y que dificulten la separación de ellos por acción del solvente. A mayor temperatura, aumenta el grado de dispersión de los monómeros y por lo tanto aumenta su solubilidad, pero no lo suficiente para realizar espectros de 13C-RMN. A excepción de M6, que presenta un sustituyente tridimensional (grupo adamantilo), los diferentes sustituyentes R enlazados al átomo de nitrógeno imídico de los restantes monómeros, también presentan una base estructural. 45.
Figure
+7
Documento similar
d) que haya «identidad de órgano» (con identidad de Sala y Sección); e) que haya alteridad, es decir, que las sentencias aportadas sean de persona distinta a la recurrente, e) que
Tejidos de origen humano o sus derivados que sean inviables o hayan sido transformados en inviables con una función accesoria.. Células de origen humano o sus derivados que
You may wish to take a note of your Organisation ID, which, in addition to the organisation name, can be used to search for an organisation you will need to affiliate with when you
Where possible, the EU IG and more specifically the data fields and associated business rules present in Chapter 2 –Data elements for the electronic submission of information
The 'On-boarding of users to Substance, Product, Organisation and Referentials (SPOR) data services' document must be considered the reference guidance, as this document includes the
In medicinal products containing more than one manufactured item (e.g., contraceptive having different strengths and fixed dose combination as part of the same medicinal
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
