NFAT5 Is Protective Against Ischemic Acute Kidney Injury
Texto completo
Figure
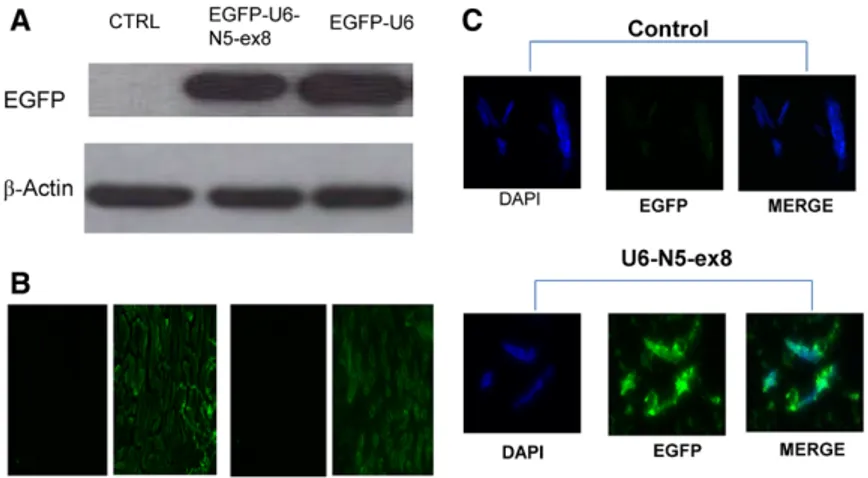


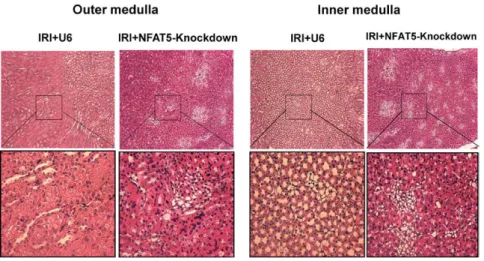
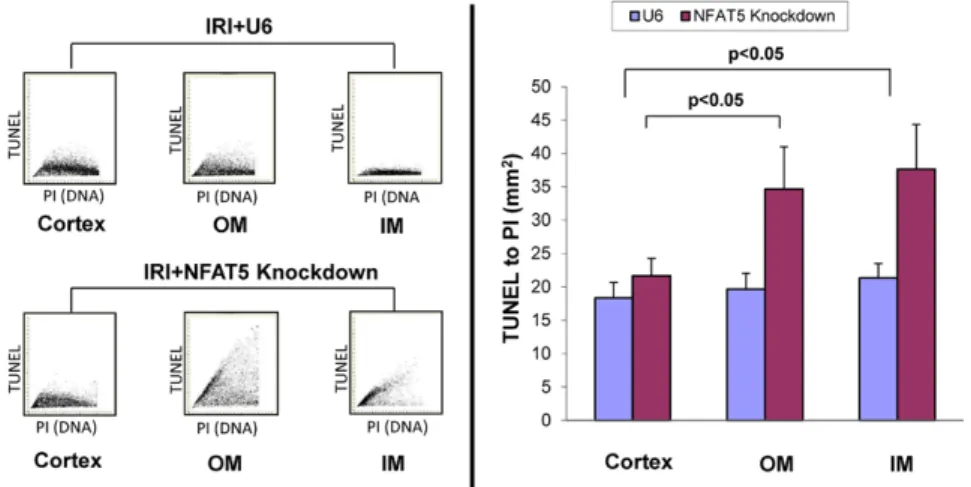
Documento similar
In addition, a signif- icant improvement of tubulointerstitial damage was indicated by reduced gene expression of kidney injury molecule-1 28 in diabetic gapoE compared with apoE
Nrf2 activation and ATF-1 phosphorylation also increase in isolated perfused hearts subjected to IR or ischemic preconditioning (IPC), from both UCP3-KO and
Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy (CREDENCE), Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients with Chronic Kidney
In vivo administration of active NBD peptide improved albuminuria (>40% reduction on average) and kidney damage, decreased podocyte loss and basement membrane thickness,
AKI depends on the duration and severity of the insult [9]. When acute renal damage occurs, there is a first phase of tubular death, followed by a phase of cell regeneration
On average, increased urinary levels for RBP4 (A) and decreased urinary levels for KNG1 (B) in response to AKI was confirmed: graphs show average data for all patients
The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB2 and adenosine receptors.. Carrier EJ,
St-PGA-CL-BDMC protected cultured renal tubular cells at lower concentrations that non-conjugated free cur- cuminoids and protected from AKI through the inhibition of NF-κB
