7
-
.
..
-"---*--
I_-1693282
c
EL PRESENTE TRABAJO FUE REALIZADO BAJO LA DiRECCION DEL
DR. MARCO ANTONIO QUIROZ ALFAR0 EN EL LABORATORIO DE
ELECTROQUIMICA DEL DEPTO. DE QUIMICA DE LA UNIVERSIDAD
AUTONOMA METROPOLITANA-IZTAPALAPA.
EL DESARROLLO DE L A PRESENTE TESIS CONTO CON EL APOYO
ECONOMIC0 DE PRONAES (S.E.P.) POR MEDIO DEL PROYECTO
"ELECTRODEPOSICION DE METALES Y ALEACIONES"
YDEL
CONSEJO NACIONAL DE CIENCIA
Y
TECNOLOGIA A TRAVES DE
I N D I C E
INTRODUCCION CAPITULO I
.
. . .
I Fundamentos T e ó r i c o s 4
1 . 1 . C a r a c t e r i z a c i ó n s u p e r f i c i a l d e m e t a l e s p o r procesos
d e e l e c t r o s o r c i ó n d e h i d r ó g e n o y o x i g e n o
. . .
5I
.
1 . 1 . P r o c e s o s d e a d s o r c i ó n d e h i d r ó g e n o. . .
6I
. I .
2. D e t e r m i n a c i ó n de Q p o r V.
C . . . 91 . I . 3. P r o c e s o s d e e l e c t r o s o r c i ó n d e o x i g e n o
. . .
14H , S 1 . 1 . 3 . 1
.
P l a t i n o. . .
171 . 1 . 3 . 2 . P a l a d i o . . . 21
1 . 1 . 3 . 3 . Oro
. . .
251.2.' D e p ó s i t o s m e t á l i c o s a s u b p o t e n c i a l
. . .
31I .2. I . I n t r o d u c c i ó n
. . .
31I . 2 . 2. F o r m a c i ó n d e d e p ó s i t o s m e t á l i c o s a subpotencial s o b r e e l e c t r o d o s P O
I
i c r i s t a l i n o se n soluciones
a c u o s a s. . .
33I . 2 . 3. A s p e c t o s t e r m o d i n á m i c o s d e l a f o r m a c i ó n . d e d e p ó s i t o s m e t á l i c o s a s u b p o t e n c i a l
. . .
39CAPITULO I I 1 1 . 1 . C e l d a e l e c t r o q u i m i c a
. . .
1511.2. P r e p a r a c i ó n d e e l e c t r o d o s
. . .
5011.3. R e a c t i v o s u t i l i z a d o s
. . .
5111.4. M o n t a j e e l e c t r ó n i c o
. . .
52<. , . -.
-.
.
...--
___.*_cI_-..,. ... .,.
...
,._I ... --~-.*'.rr>.^il'ru~'~ru^rar---.~..
093282
CAPITULO
I11
I 1 1
.
Resultados
ydiscusión
. . .
581 1 1 . 1 .
Procesos de electrosorción de hidrógeno
y
oxigeno
. . . .
581 1 1 . 1 . 1 .
Platino
. . .
581 1 1
.
1.
1.a. E s t i m a c i ó n del área superficial expuesta
.
a p a r t
ir de la adsorción de hidrógeno
s o b r e Pt
. . .
64I l l
.
1.
1.b.
E s t i m a c i o n del área superficial expuesta
a p a r t
ir de
l adesorción de oxígeno sobre
P t .
. . .
67I l l
.
1.2.Paladio
. . .
68111.1.3.
Oro
. . . 7 3I
I 1
.
2
.
Depósito de adát o m o s de Cu a subpotencial.,
. . .
79111.2.1.
Efecto
UPD
d eCu sobre
Pt
. . .
79111.2.2.
Efecto UPD de Cu sobre
Pd
. . .
90I I
I
.2.
3 .
Efecto UPD
d e
Cu
sobre
Au . . . 981 1 1 . 3 .
Estimacion de
A r ,
de
Pd
y
Auutilizando el valor de
f obtenido por depósito de
Cua subpotencial
. . .
106
111.3.
1Paladio
. . .
106111.3.2.
Oro. . .
108
CONCLU
CI ONES
. . .
BI
B L I O G R A F
IA
. . .
110
I N T R O D U C C I O N
Los estudios cinéticos de reacciones electroquímicas,
asi
como ladeterminación de propiedades electrocataliticas de electrodos de
metáles nobles dependen, prioritariamenre, de
las
caracteristicas dela superficie expuesta, en particular de la magnitud de su área y SU
configuración cristalográfica. Es lógico, por tanto, que una adecuada
caracterización superficial
no
puedeser
subestimada.A este respecto,
se
realizan esfuerzos cadavez
más importantesa
f i n de desarrollar métodos más precisos, destacando significativamente
aquellos que se aplican "ex-situ" al sistema 187-901. Aunque estos
rnetodos dan valores muy precisos. no indican la morfología real de la
superficie. ' y a que los resultados
son
obtenidos. bajo condicionesdiferentes al medio ambiente normal del electrodo, y puesto que la
mayoria de ellos son de carácter destructivo, esto los hace poco
atractivos e imprácticos para determinaciones rutinarias.
Por
estarazón, es prácticamente
una
necesidaden
la
investigaci6neiectroquimica efectuar tal caracterización mediante técnicas
sn-situ", de manera que
se
puedan entender la naturaleza. y origen delas propiedades que dependen de manera directa o indirecta de
la
estructura que presenta la superficie expuesta. En
las
últimasdécadas, l o s métodos electroquimicos han permitido desarrollar
tecnicas de caracterización superficial cada vez mbs adecuadas, en
particular destacan aquellas empkadas en electrodos Constituidos de
metales nobles I2.31.
Es
sabido queeste
tipo de electrodos metálicosmuestran gran
afinidad hacia los procesos de electrosorción de hidrógeno y oxígeno
en soluciones ácidas, lo que ha permitido su caracterización por estas
técnicas electroquímicas [1,31. Sin embargo, metales
tales
como Pd yAu no han podido
ser
adecuadamente caracterizados poresta
vía. En Pd,el proceso de adsorción de hidrógeno es acompañado siempre por
una
absorción significativa del
mismo,
10 que impidedeterminar
lacantidad real adsorbida y
su
extención. Para Au basta decir quepresenta una adsorción casi nula de dicho eiemento, aproximadamente 2%
(37,381. Por otro lado, si bien ambos metales, Pd y Au. presentan
una
adecuada electrosorción de oxígeno, la relación O/M a nivel de la
monocapa, aún no ha sido bién definida para los electrodos
constituidos de estos metales nobles.
En los últimos años, en el ámbito electroquimico se h a despertado
un enorme interés en el estudio del depósito de metales a subpotencial
(UPD : por Under Potential Deposition) sobre sustratos metálicos
ajenos. Este interés reside en ei conocimiento de la profunda
influencia que puede tener la
monocapa
UPD(como
una fraccióno
completa),
sobre los electrodossustrato
usadospara
electrocatálisisde reacciones electrosintéticas y celdas de combustibles, refinamiento
de superficies, estudios sobre crecimiento electroquímico de
cristales, etc. Se ha comprobado que
el
proceso UPD presentauna
oportunidad única para estudiar el acoplamiento de
una
amplia variedadde fenómenos superficiales, tales como la adsorción, transferencia de
carga, difusión superficial, formación de películas metálicas
bidimencionales. etc. Sobre estas bases, estudios muy recientes
utilizado para pr6positos de caracterización superficial. La
importancia del método UPD descrito, es particularmente ref lejada para
aquellas superficies metálicas cuya caracterización por las técnicas
tradicionales de electrosorción de hidrógeno y oxigeno es inoperante.
Por lo tanto, los objetivos de este trabajo van encaminados a la
aplicación del efecto UPD sobre electrodos metálicos de Pd y Au con
los siguientes propósitos:
1. Determinar las caracteristicas del depósito formado como una
funcidn del potencial aplicado al electrodo de trabajo.
2. Establecer las condiciones experimentales que permitan la
formación de una monocapa metálica sobre la superficie expuesta.
3. Determinar las caracteristicas de la superficie expuesta, en
particular la determinación de factores de rugosidad.
J.
Analizar los procesos de electrosorción de oxigeno. a l a luzde los. resultados obtenidos por aplicación del efecto UPD.
5. Establecer las relaciones O/Pt, O/Pd y OiAu correctas, cuando
una monocapa de oxígeno es formada sobre la superficie de estos
CAPITULO I
1. FUNDAMENTOS TEORICOS
;\I estudiar la cinetica de reacciones electroquimicas, es
necesario conocer la densidad de corriente que fluye a través del
electrodo donde estas ocurren. La densidad de corriente la podemos
evaluar si conocemos de antemano el valor del área real expuesta del
electrodo.
El área real del electrodo la definiremos como el área activa de la
superficie expuesta, la cual, a su
vez.
está relacionadae?.
formadirecta al número de sitios activos accesibles en la superficie del
metal. entendiendo por sitio activo, desde el punto de vista puramente
electroquímico, .como aquel sitio capáz de efectuar una transferencia
d e carea.
Para llevar a cabo la estimación del área real dei electrodo. ya
sea mediante los procesos de electrosorción de hidrógeno u oxigeno ! / o
por
el depósito de metales a subpotencial (UPDI. es necesario establecer el comportamiento electroquímico de la capa adsobida, asicomo su estequimetria con los sitios metálicos superficiales [121.
Particularmente. el efecto UPD de metales ajenos sobre sustratos de
metales nobles, parece ser
el
que proporciona informacionmás
detallada y valiosa de
las
características superficiales de los1.1 CARACTERIZACION SUPERFICIAL D E
METALES
NOBLES PORPROCESOS
DE
ELECTROSORCION DE HIDROGENO Y OXIGENO.Los procesos de electrosorción de hidrógeno y oxígeno dependen
ampliamente' de la naturaleza del metal del electrodo. Las
características electroquímicas de dichos procesos, estan definidos y
limitados por el intervalo de electroactividad del metal en el
electrolito soporte utilizado. Así,
tanto
la adsorción de hidrógenocomo la formación de especies oxigenadas adsorbidas, constituyen
la
erapa- intermediaria para los procesos de evolución de hidrógeno y
oxigeno moleculares respectivamente. Una de las aplicaciones prácticas
que proporciona el conocimiento de las características de los
procesos
de electrosorción, es la posibilidad de realizar una adecuada
estimación del area real del electrodo para la mayoria de
los
electrodos constituidos de metales nobles
[ill.
Los procesos de electrosorción pueden
ser
estudiados de maneradirecta mediante el uso de técnicas transitorias, siendo las más
adecuadas las siguientes:
a ) aquel que utiliza como perturbación
una
señal de corrientealterna de amplitud pequeña, particularmente útil
en
el estudio deadsorción de hidrógeno. Esta técnica permite estudiar la
pseudocapacitancia originada por la transferencia de ca.rga
if51.
b) la voltamperometría cíclica
[131,
que consiste en aplicar unbarrido triangular repetitivo de potencial que varía de forma lineal
con el tiempo, dando como respuesta un flujo de corriente como una
función del potencial aplicado. El registro generalmente se denomina
"perfil potenciodinárnico o curva voltamperométrica".
se mide el potencial
en
función del tiempo. El registro transitoriose
conoce como
"curva
de carga".1.1.1. PROCESOS DE ADCORCION DE HIDROGENO.
La técnica que utiliza c.a mide
la
pseudocapacitancia comouna
función del potencial, relacionando la capacitancia diferencial con el
potencial y
la
carga mediante la relación:C = dQ/dE (1.1)
la fracción de superficie cubierta (grado de recubrimiento1 por
electroadsorción de hidrógeno se define como Q/QH.s , siendo QH.S el
recubrimiento de la superficie por hidrógeno a la monocapa, esto
es. cada atom0 del sustrato está bloqueado por un átomo de hidrógeno,
y
0 l a carga experimental obtenida asociada al proceso deelectrosorción de hidrógeno a un potencial dado. Ya que:
V dQ = Q d e ,entonces
H . S
ü = Q/Q
H . 5
sustituyendo dQ en (1.1) se obtiene,
(de/dE) (1.2)
De aqui que la isoterma electroquímica
relaciona
el grado derecubrimiento
con
el potencial, por lotanto,
la técnicacon
c.a.determinará la derivada de la isoterma en función del potencial. Un
ejemplo tipico de pseudocapacitancia en función del potencial se
muestra en la figura 1.1.1.1, en donde además se muestra la integral
de la curva, esto es, s u isoterma elecrroquimica
[I61
=
0H.SEn el caso de la
voltamperometría
cíclicaí
V.C.1,
se midela
corriente como una función del potencial. Esta corriente se relaciona
a los parametros eléctricos de la siguiente forma:
1.0
IS00
0.6
-
IO00 0.5 oE
N
.
aa 0.4
u
500 v0.2
O
o.
1 0.2 R3 0.4E (V/Eb")
Fig. 1 . 1 . 1 . 1 . Variación de la pseudocapacitancia en función del
potencial ( a ) , cuya integral es la curva ( b ) que representa la
isoterma electroquímica de electroabsorción de hidrógeno sobre Pt en
H 2 SO 4
hf
a 25 C 1161.3
dB/dE
=
0 H . S i =Cx
dE/dti
=
( Q H , s dE/dt) dWdE (1.3)Como dE/dt
=
v
(velocidad de barrido) es constante, - é s t a técnicadeberá generar una curva similar a la curva de pseudocapacitancia. La
Fig. 1.1.1.2 muestra el perfil potenciodinámico para la electrosorción
de hidrógeno sobre P t en H SO4 I
M
a 25 C. En ia misma figurase
muestra también la carga de adsorción como una función delpotencial
íisoterrna de adsorciónl. obtenida por integración del perfil
potenciodinámico para la adsorción de hidrógeno, después de r e s t a r la
Fig. 1 ; l . l . Z . Perfil potenciodinámico mostrando l a adsorcion de
hidrogeno sobre Pt en H,S04 1 bí a 25 C (curva a).
Su
integral es laciirva b (isoterma de adsorcion).
~
contribución del proceso de carga de
la
doblecapa.
En la técnica galvanostática, se aplica una corriente constante y
se mide el potencial en función del tiempo,
la
carga .eléctrica esproporcional a i tiempo, esto es:
t = Q/I t
=
O(i/dQ/dt)t = (QH,Sdt/dQ) 6 (1.4)
La curva de carga sigue el cambio de ü en función del potencial
(isoterma de adsorción), por lo que es comparable a la integral del
figuras 1.1.1.1. y 1.1.1.2. Conway 118.201 apoya
fuertemente
el uso deestas curvas, debido a que
el
procedimiento de integración dela
cargaes más fácil y
menos
complicadassus
expresiones matemáticas, aunquela curva integral es menos sensible a los detalles finos de ias curvas
pseudocapacitivas y a los voltamperogramas cíclicos.
El proceso de electrosorción de hidrógeno sobre Pt
es
reversible, por lo tanto, el
valor
de la capacitancia se puedeconsiderar constante, excepto a
altas
velocidades de barrido o afrecuencias muy elevadas. Bajo condiciones estacionarias, las
diferencias entre las características de la adsorción obtenidas por
los tres métodos previamente citados, son despreciables como se puede
apreciar
en
las figuras precedentes, y los diversos trabajospublicados 117-201. Para una determinación del número de sitios
activos de la superficie del electrodo, la voltamperornetria cíclica
(\'.Cl resulta ser la técnica más útil de las tres mencionadas, dada
su
simplicidad y versatilidad.
1.1.2. DETERMINACION DE QH.S
POR V.C.
Para llevar a cabo la determinación de QH,S
es
necesario sabersi
l a contribución debida al proceso de adsorción de hidrógeno puede ser
separada de aquellas debidas a otros procesos que ocurren
simultaneamente, como son carga de la doble capa y evolución de
hidrógeno.
La carga de la doble capa puede
ser
eliminada por extrapolacióndel voltamperograma en la zona
no faradaica
[Zl].
La exactitud de laexrrapolación se basa en el supuesto de que la capacidad de la doble
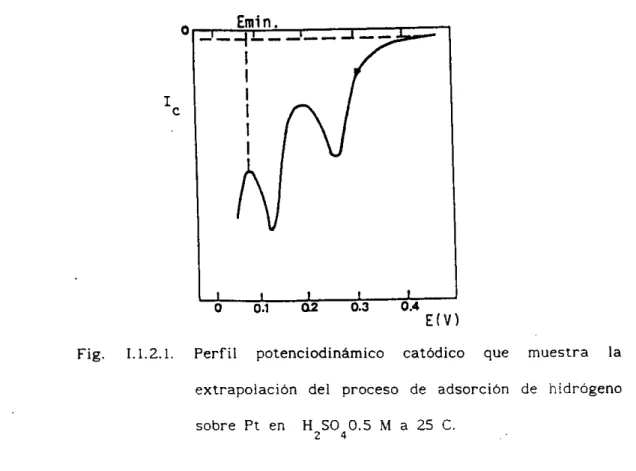
Emi
n
.
O
IC
I I 1 I I I
Fig. 1.1.2.1. Perfil potenciodinámico catódico que muestra la extrapolación del proceso de adsorción de hidrogeno
sobre Pt en
H2C040.5
M a 25 C.proceso de 'adsorción de hidrógeno, esto e s , entre 0.0 y 0.6 Y, además
de que la contribucion de los procesos que no sean los de carga de la
doble capa, son despreciables en dicha region 131. La linea horizontal
(punteada) de la Fig. i.1.2.1 muestra l a manera de efectuar esta
correccion.
El otro problema que es aún más difícil de resolver, es debido al
traslapamiento que existe entre el
proceso
de adsorción de. hidrógeno yl a evolución del
mismo
sobreel
electrodo. Por tanto, existeuna
región de potencial donde ambos procesos se dan de manera simultanea,
y no
se
puede suprimir por un simple cambio de técnica utilizada.Ambas contribuciones pueden ser estudiadas midiendo la
pseudocapacitancia con la frecuencia de la señal de a.c., mediante el
adecuado para
hacer
mediciones
rutinarias de l acantidad
de QHS, peroes interesante desarrollarlo ya que permite el estudio del fenómeno de
adsorción en ia región de potencial donde aparece la evolución de
hidrógeno. En la Fig. I.i.l.1 se puede observar la presencia de
una
capacitancia de adsorción considerable para potenciales inferiores
a
0.08 V, además de que el recubrimiento total solo
es
alcanzado hasta-0.01
v.
Los trabajos hechos sobre electrodos de disco-anillo 131
condujeron a Biegler I221 a recomendar, que la cantidad QH,S
determinada por integración de la carga pasada hasta 0.08 ir (Ernin).
segundo mínimo de la curva catódica para la adsorción de hidrógeno,
fuera dividida por el grado de recubrimiento a dicho potencial,
interpolado de la isoterma derivada de los datos capacitivos (Fig.
1.1.1.11.
Para el caso del Pt. si la carga de adsorcih de hidrógeno se
determina por integración del perfil potenciodinámico de l a Fig. 1.1.2.1. con Emin=0.08
V,
la división de esta carga por 0 . S 4 . valordeterminado por interpoiación de la isoterma mostrada en la Fig.
I.l.l.l.b, dará el
valor
de l a carga correspondiente ai total desitios inmediatamente accesibles del electroaa, esto es, OH.S.
Una vez conocido QHS, se puede estimar
ei
valor del áreareal del electrodo haciendo uso del estandar convencional determinado
para el metal. La carga integrada involucrada en ei proceso de
adsorción da cuenta del número de átomos de hidrógeno depositados
sobre el electrodo. Para el caso particular del Pt se ha determinado
que un átomo de hidrógeno se adsorbe en un átomo del metal 128,291
Pt
+
H+
+
le--
>
PtíHIad (1.5)esto permite relacionar el número de átomos adsorbidos con
una
cantidad definida de electricidad. La carga por cm de área real
asociada
con
la adsorción deuna
monocapa de hidrógeno sobre losplanos de bajo índice del P t son: 208 pC para el plano (1001, 241 pC
para el plano (111) y 147 y 295 pC para
el
plano (1101, según si losátomos de coordinación 7.
u
ambos 7 y 11 son los definidos como átomos superficiales. Para superficies policristalinas, es costumbre general1301 'asumir que la superficie consite de una distribución igual de los
tres
planos de bajo índice, aunque también se ha supuesto que el plano (1001 es el predominante 1311. Biegler [221 ha sugerido que se tomen210 pC/cm2 como estandar convencional para el Pt. considerando el
plano (1001 como predominante y para el que corresponde una población
ae 1 . 3 ~ 1 0 ~ ~ átomosícm
.
2
2
El área real expuesta para un electrodo policristalino de Pt se
puede determinar, vía electrosorción de hidrógeno, mediante i a simple
expresión:
A r = Q /210 pic cm-* 11.6)
QH,s =
(Q/ept)
pC (1.7)Para electrodos constituidos de Rh, Gilman I231 ha sugerido que
l a estimación de
OHS
para
este metal, puede ser obtenida -a partir delperfil catódico. seleccionando adecuadamente
el
valor de E m i n para laintegración de la carga de adsorción. Dividiendo este resultado por el
grado de recubrimiento
(e
=
0.59) se obtiene la carga correspondientea l total de sitios activos sobre la superficie del metal [241. El
cálculo del área real es inmediato (321 H.8
Ar
=
Q
/221 pCcm-'
(1.8)Para el Ir la determinaci6n es algo
más
complicada, aunquelos
datos capacitivos muestran que la adsorción de hidrógeno se completa
al mismo potencial que para Pt i25.261. El grado de recubrimiento
encontrado para este metal fue de 0.65, al potencial de 0.06
V.
Dividiendo la carga integrada,del perfil potenciodinámico,
se
encuentra el valor de QH.S para este metal. El
valor
del áreareal
se obtiene fácilmente a partir de la siguiente expresión [3,251:
A r = QH,S/218 pC cm-* (1.91
'En el caso de electrodos constituidos de Ru, Bagotzky y Col. [331
han puesto de manifiesto que el proceso de adsorción de hidrogeno
sobre electrodos lisos, ocurre
en
el intervalo de 0.0a
0.4 V, y sobreRu electrodepositado dicho proceso de adcorción ocurre entre 0.0 y 0.2
V.
Además, estos mismos investigadores han demostrado que el procesode adsorción de hidrógeno procede, de manera simultanea, con una
absorción significativa del
mismo,
favorecida porla
pronta evoluciónde hidrogeno molecular sobre
el
electrodo. Estudios recientes 135.351realizados por depósitos metálicos a subpotencial han conducido a
una
adecuada caracterización de éste metal. Sin embargo, Woods y Col. I361
han reportado que se puede
hacer
el cálculo del áreareal
mediante laintegración de
la
carga de adsorción de hidrógeno tomando como Emin0.03 i
con
r,un grado de recubrimiento de 0.56 y un valor. de estandar convencional de 251 pC/cm 2.
Para el Pd, el proceso de adsorción viene acompañado por el de
absorción de hidrógeno, 0.69 átomos de hidrógeno por átomo de Pd
13,241. Cuando la cantidad absorbida es pequeña, el metal conserva su
estructura. Cuando alcanza una relación atómica de H/Pd de 0.05, la fase original a es convertida a
una
fasep
considerablementeexpandida.
Esta
transformación, en el que ambasfases coexisten,
s ecompleta cuando l a relación H/Pd
es
0.6.
L a f a s e homogenea6
puedeabsorber más hidrógeno, y la cantidad absorbida se incrementa
linealmente con el potencial del electrodo L271 y por lo t a n t o con el
Log P i271.
H2
La estimación del grado de recubrimiento de hidrógeno sobre é s t e
metal, ha sido uno de los problemas más difíciles de resolver, debido
funaamentaimente a la imposibilidad de separar contribuciones
ajenas
ala corriente de adsorción de hidrógeno como consecuencia propia de la
naturaleza del metal, lo que hace más especulativa la adsorción de
hidrogeno sobre Pd que p a r a los otros metaies nobles.
Para ei caso del Au. en contraste con los metaies del grupo del
Pt. solamente adsorbe una pequeña cantidad de hidrógeno a potenciales
cercanos a la región de evolución de hidrógeno molecular. El grado de
recubrimiento de hidrógeno al potencial reversible de hidrógeno ha
sido estimado entre 2-4 % de una monocapa [37,381.
1.1.3. PROCESOS DE ELECTROSORCION DE OXIGENO
Ei estudio de los procesos de electrosorción de oxígeno sobre
metales nobies, también puede realizarse por las técnicas ya descritas
en este capitulo. Aquí también emplearemos la voltamperometria cíclica
dada su simplicidad y versatilidad p a r a el estudio de t a l e s procesos
para ei oxígeno.
Los
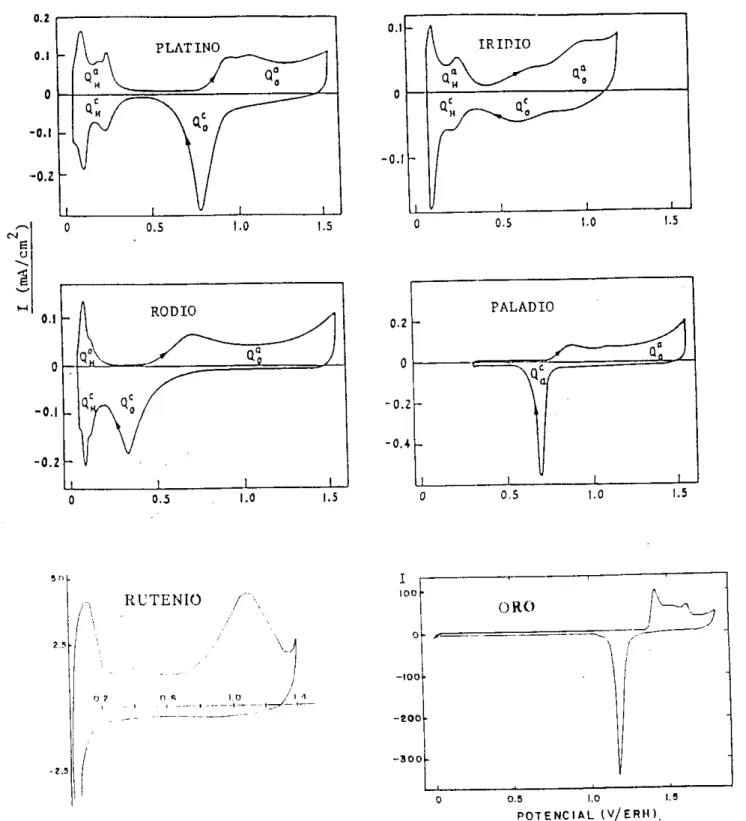
procesos de electrosorción de oxígeno sobre electrodos de metales nobles ocurren a potenciales más positivos, que loscorrespondientes a los procesos de electrosorción de hidrógeno, como
0.2 I I
I I
O 0.5 1.0 1.5
r--
PALADIOFig. 1.1.3.1. Perfiles potenciodinámicos para electrodos de
La capa de oxígeno que
se
forma sobre los electrodos de metalesnobles ha sido descrita en terminos de: a) quimisorción 1391, b)
incorporación bajo la superficie formando una capa dermasorbida
o
aleación metal-oxígeno y c) la formación de una fase de oxido metálico
[42,431. La quimisorción se caracteriza por un incremento lineal o
casi lineal del recubrimiento con el potencial, hasta su valor limite,
mientras que la fase de oxido se forma cuando hay un incremento agudo
en el' recubrimiento, el cual frecuentemente es irreproducible y se
acompaña de cambios significativos
en
la rugosidad de la superficie.En los metales nobles, las etapas iniciales de oxidación se pueden dar desde una pequeiia fracción de monocapa hasta la formación
de una monocapa completa, incluso hasta la formación de varias capas.
En la Fig. 1.1.3.1 se muestran los voltamperogramas
correspondientes a Pt. Rh. Ir, Ru, Pd y Au. resaltando la region donde
se
llevan a cabo los procesos de electrosorción de las especiesoxigenadas. Los procesos de electrosorción, aunque inician a
potenciales diferentes para cada caso, se extienden hacia potenciales
positivos,
enuna
amplia zona, hastaalcanzar la
región de evoluciónde oxigeno molecular.
Para P t , Rh. Ir, Ru y Pd
la
región de evolución. de oxígenomolecular se alcanza en el intervalo de potencial cercano a 1.5 V,
mientras que para el Au
la
evolución de oxígeno se alcanza hastapotenciales próximos a 2.0
V.
Los procesos correspondientes a laelectrodesorción de especies oxigenadas está caracterizado por un pico
de corriente catódica, situado a potenciales que son característicos
* 4c*-i---r<
-
I_-Ru, exhiben
características
electroquimicas quese alejan
deeste
comportamiento (para el proceso catódico).
Para
losmetales
objeto deeste estudio se hace un análisis más detallado de los procesos de
electrosorción de las especies oxigenadas, incluso para el Pt por ser
el sistema de .referencia utilizado.
1.1.3.1. PLATINO
las características de electroadsorci6n de oxigeno bajo
condiciones transitorias son muy diferentes a
las
exhibidas por elhidrógeno, pués hay una considerable asimetría entre las curvas
transitorias de adsorción-desorción de oxígeno, indicativo de que ei
proceso de adsorción (de oxígeno) es irreversible, mientras que el
hidrógeno es adsorbido reversiblemente. La irreversibilidad del
proceso de adsorción depende de la cantidad de especies oxigenadas
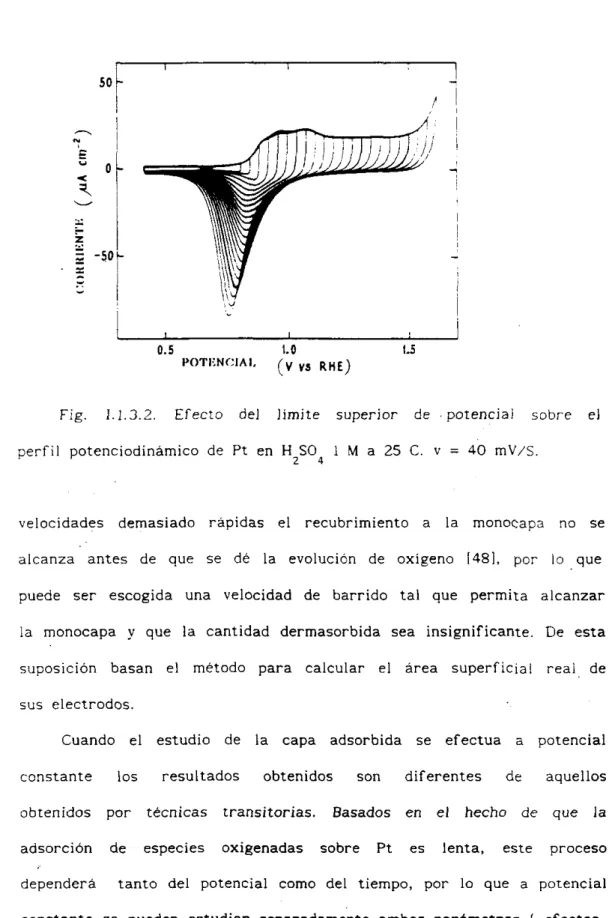
adsorbidas. lo que puede ser observado en la Fig. 1.1.3.2, en donde
para limites superiores de potencial (EL,S) <0.9 V .el comportamiento
del proceso de adsorcion es el correspondiente a un proceso reversible
l l a s curvas san aproximadamente simetricas alrededor dei eje de
potencial).
Conforme se
incrementa el
limite superior de potencial hay ~un
incremento continuo del recubrimiento. La carga que pasa antes del
comienzo de la evolución de oxigeno es aproximadamente 2 e por atom0
superficial de Pt (44-461. Estas observaciones conducen al concepto de
una monocapa de oxígeno adsorbido o PtO formado, antes del
desprendimiento de oxígeno molecular.
-
ad
Schuldiner y Co1.145,47,481 han sostenido que
en
un barridorápido, se forma una monocapa de átomos de oxigeno adsorbidos, pero
50
c
! !Fig. 1.1.3.2. Efecto del limite superior de potencial sobre el
perfil potenciodinámico de P t en
H2S04
1M
a 25 C. v = 40 mV/C.velocidades demasiado rápidas el recubrimiento a la monocapa no se
alcanza antes de que se dé la evolución de oxigeno 1481, por lo que
puede ser escogida una velocidad de barrido tal que permita alcanzar
la monocapa y que
la
cantidad dermasorbida sea insignificante. Deesta
suposición basan el método para calcular el área superficial real de
sus electrodos.
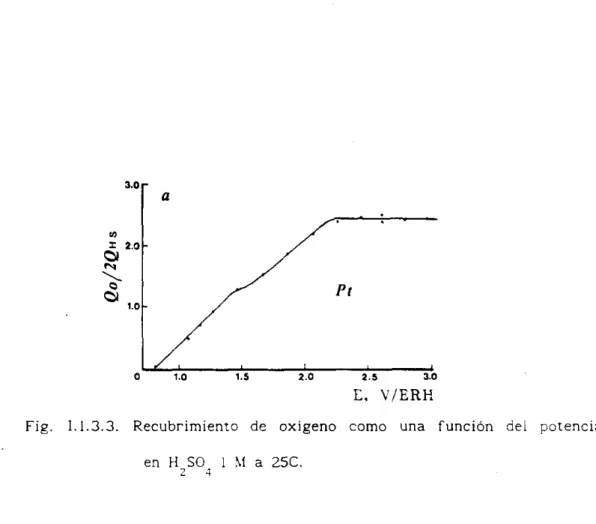
Cuando el estudio de la capa adsorbida se efectua a potencial
constante los resultados obtenidos son diferentes de aquellos
obtenidos por técnicas transitorias. Basados en el
hecho
de que laadsorción de especies oxigenadas sobre Pt es lenta, este proceso
dependerá tanto del potencial como del tiempo, por lo que a potencial
P t
.A
o 1.0 1.5 2.0 2.5 3.0
E.
VIER€?
Fig. 1.1.3.3. Recubrimiento de oxigeno como una función aei potencial
en HZSOI 1 !if
a
2SCdel potencial en funcion del tiempo).
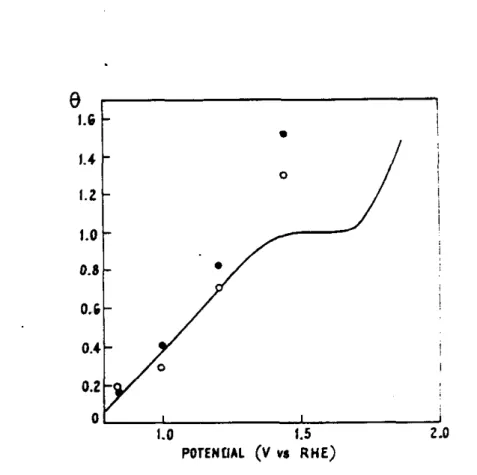
L a figura 1.1.3.3. muestra la dependencia de la cantidad de especies oxigenadas adsoroidas en función del potencial a tun tiempo
f i j o
í -
1000 SI.El recubrimiento de oxigeno alcanza un valor constante a
potenciales superiores a 2.2
V.
Biegler y Woods [491 concluyeron quee s t e recubrimiento corresponde a una monocapa que contiene aos atomos
de oxigeno por átomo superficial de Pt. En la Fig. 1.1.3.3. se observa
que hay
un
escalón a la mitad del recubrimiento, el cual debecorresponder a un átomo de oxigeno por átomo superficial de P t .
E x i s t e una extensa literatura dirigida
a
dilucidar los mecanismosde formación reducción de l a s especies oxigenadas sobre el P t
de los resultados. ya que los diversos autores
han
descritola
naturaleza de la capa de oxígeno en términos de diferentes etapas dela formación del óxido.
Estos estudios conducen
a
que el proceso más probable puedeser
representado
a
través de las siguientes reacciones:2
Q:,
Fc/cm-
4Pt + H 2 0
c--
P t 4 0 H +H+
+ e 55 1I.lOa)P t 4 0 H + H 2 0
c--
I 2PtZOH +H
+
+
e-
55 í1.1Ob). 2 ~ t
on
+ 2 n 2 0 4PtOH +ZH+
+2e- 110 (1.10c)2
La reacción global para estas etapas es la suma de ( a ) . (bl y (cl
4Pt + 4H20
-
-
4PtOH+
4H+ + 4e- 220Se ha mostrado (521 que estas etapas sucesivas de ocupación de
gxigeno sobre Pt son las responsables de los picos anódicos observados
al inicio de la adsorción de especies oxigenadas sobre e l P t (Fig.
I . 1.3.1').
Antes de. liegar a la región de evolución de oxigeno molecular
ocrre otra etapa, que es intermedia entre estos pasos sucesivos .y la
evolución de O2 (región
ancha
del perfil anódico).-
+
PtüH -IPtO
+
H
+e
220 1I.IOd)La carga total del proceso desde ( a ) hasta ( d ) es 440 pC/cm
.
'Este esquema de reacción se ajusta excelentemente tanto al perfil
experimental como al cálculo de la cantidad de electricidad
involucrada por
cm
para la fomración de una monocapa de oxigenoelectroadsorbido en
una
estequiometría PtO de 1:1, así como a losresultados reportados por Bockris y Col. 1551 en donde se cita que a
0 = 0.27 un átomo de oxígeno comparte 2e con aproximadamente cuatro
átomos de Pt.
2
2
--
---u
-Así
el área real de electrodos constituidos de Ptse
puededeterminar
en
base a la estequiometria PtO de ]:I, es decir:(1.11)
2
Ar = Qa pC / (2x210 pC/cm
1
O
donde:
Qt?
pC
es
la carga experimental integrada del perfil potenciodinámico anódico entre los intervalos de potencial apropiados.210 pC/cm2 correspnde al estandar convencional sugerido [301, para
un
electrodo de Pt policristalino, bajo el supuesto de que lasuperpicie consiste de una distribución de los tres planos cristalinos
de bajo índice de Miller, IIOO). (110) y (111) con el plano (100) como predominante (221.
1.1.3.2. PALADIO
La electroadsorción de especies oxigenadas sobre Pd comienza
aproximadamente
ai
mismo potencial que sobre Pt. y se observa e l mismocomportamiento, es decir, se incrementa la irreversibilidad a medida
que aumenta el valor del 'potencial límite superior de inversión de
barrido, tal como se muestra
en
la Fig. 1.1.3.4La cantidad de especies oxigenadas adsorbidas
en
un barrido depotencial aumenta en forma aproximadamente lineal con el potencial
[38,57-591.
Rand y Woods i601 investigaron la adsorción de especies
oxigenadas sobre Pd a potencial constante. En la Fig. 1.1.3.5. se
muestra la dependencia del recubrimiento del potencial aplicado
a
diferentes tiempos de anodización. Se puede
ver
que elrecubrimiento aumenta linealmente con el potencial hasta alcanzar una
I
: I
' 1I
i
1.0 51.
,vo<P,iN#:lAl< , v "I R H L ,
0.5
Fig. 1.1.3.4. Efecto del límite superior de potencial sobre el
perfil potenciodinámico de Pd en
H
SO 0.5hi
a 25 Cv = 40 mV/S [31.
2 4
tiempo de anodización, cambiando a potenciales mas bajos a medida que
se incrementa el tiempo. .A aproximadamente 2.0 \' el recubrimiento se
incrementa abruptamente y
no
se aproxima a un valor límite. La capade óxidos producido a estos potenciales elevados se reducen en una
serie de picos anchos, en vez de un solo pico bien definido del tipo
mostrado en la Fig. 1.1.3.4., que resulta de la desorción de las
especies oxigenadas cuando el recubrimiento es menor o igual que aquel
de la meseta. La reducción de la capa de altos potenciales viene acompañado de una considerable rugosididad de la superficie
del electrodo. Este comportamiento se debe a la formación de una fase
oxida, la cual se vuelve una capa gris oscura visible bajo condiciones
POTENCIAL
(v
vs RHE)Fig.
1.1.3.5. Recubrimiento de oxígeno sobre Pd como una funcion delpotencial en
H2SO4
IM
a 25 C. Electrodos anodizados por(1) 10 S , (21 100 S , (31 1000 S al potencial deseado.
La carga asociada con el recubrimiento a
la
meseta para unelectrodo que ha sido tratado térmicamente por calentamiento bajo
vatic para dar
una
superficie lisa fue 690 pC/crn* 1601, un valorsienificativamente menor que aquel anticipado para una relaciori 2:1 de
O/Pd, pero cercano al esperado para una capa 1:l sobre una superficie
pulida. Se concluyó, por tanto, que la meseta correspondió al escalón
observado para el
Pt,
es decir un recubrimiento I : ] . Esta conclusiónse confirmó por las medidas de recubrimiento de oxigeno de Burshtein y
Col. [S71, quienes estudiaron electrodos dispersos de área BET
(Brunauer, Emmet y Teller) conocida. En la Fig. 1.1.3.6. se muestran
los
resultados obtenidos por ambos grupos de investigación.O
:.f/
0.2
I I I
1.0 1.5 2
.o
POTENOAL (V vs RHE)
Fig. 1.1.3.6. Comparación de los valores del grado de recubrimiento
de oxígeno para Pd reportado por Rand y Woods 1601 y
Burshtein y Col. [571.
-
, curva 3 de la Fig.1.1.3.5.; y o, datos obtenidos de 1571 en HZS04
0 . 5
M.
El trazado de la Fig. 1.1.3.6. se hace asumiendo que la meseta
corresponde a una estequiometría 1:l. La discrepancia. entre los
recubrimientos a 1.45 V puede deberse a los diferentes estados
físicos de las superficies examinadas.
.
La meseta
mostrada
sobre la curva de recubrimiento de oxigenoen
función del potencial de anodización (Fig. 1.1.3.5.) presenta un
método para la determinación del área real de electrodos de Pd. Rand y
de oxígeno
en
función del potencialse
requieren para obtener lameseta. Además, recomiendan
tener
cuidadode de evitar condiciones bajolas cuales se desarrolla la fase óxido, ya que esto puede conducir
a
cambios serios en la rugosidad de la superficie.
De
la Fig. 1.1.3.6 se puede concluir que para superficiesaltamente dispersas este método no puede ser aplicado, ya que
la
meseta desaparece para este caso, además
en
la práctica es difíciltener
un
electrodo bien pulidocomo
el que utilizaron Rand y Woods ensus investigaciones.
L
Fue propuesto I601 que un cm de un plano (1001 se acepte como estandar convencional. de acuerdo con Io propuesto para los otros
metales nobles.
En
este caso,un
cm real es equivalente a un cambiode 424 pC para un proceso que intercambia 2e-, es decir, el a r e a real
es:
2
(1.12) 2
A r
=
QCpC
/ (424 pC/cm 1O
1.1.3.3.
OR@
El oxígeno comienza
a
ser adsorbido sobre Au a aproximadamente1.35 V en medio ácido 137,38,60-691 tal como se muestra en la Fig. I. 1.3.7. No obstante. varios investigadores 170-741 han reportado corrientes faradaicas a potenciales anódicas inferiores y han
sostenido que la carga que fluye
se
debe a la formación de especiesoxigenadas de Au. Bonewitz y Cchmid 1631 encontraron que el Pt cuando
es usado
como
contraelectrodo se disuelve y deposita sobre elelectrodo de trabajo, originando con ello la adsorción de especies
I 1
1100
-
0 -
-100
-
-too.
-800
-
I
O 0.6 1.0 1.3
POTENCIAL (V/ERH)
Fig. 1.1.3.7. Efecto del limite anódico de barrido de potencial
sobre el perfil potenciodinámico de Au en
H
SO IM
a 25 C. v
=
40 mV/S.2 .
electrodo de trabajo como una aleación Pt/Au.
AI analizar el barrido anódico del Au en H SO 1 M íFip. 1.1.3.71
se observan dos picos de corriente claramente definidos a 1.47 y 1.63
\',
separados por un tercero casi imperceptible a 1.52V.
.A 1.7V
sedetecta un mínimo en la corriente que marca el comienzo de l a
evolución de oxígeno. Las investigaciones hechas sobre monocristales
de Au
I751
exponiendo los planos (1001, (110) ó (111) han mosrrado quela forma del perfil potenciodinárnico depende de la orientación
cristalográf ica de la superficie del electrodo.
2 4
La desorción de especies oxigenadas la caracteriza un pico
barrido lentas, tal
como se
muestraen la
figura precedente,mientras
que a velocidades más rápidas se
alcanzan
a apreciar dos picosen
elproceso catódico 1691.
La histérisis entre las curvas anódica y catódicas sobre el
voltamperograma es similar a áquel esperado para la formación de la
fase óxido [31. con solamente corrientes anódicas fluyendo a potenciales
>
1.35 V y corrientes catódicas debajo de este potencial.Por lo tanto, el potencial metal/óxido metálico sería entonces 1.35 V;
que es un valor aigo menor a los calculados a partir de datos
termodinámicos para los pares Au/AuíOHI
v
Au/Au O que son de 1.46 y1.51 V respectivamente [761. El pico de desorción cambia a potenciales
más catódicos a medida que el potencial limite superior de inversión
del barido se incrementa. lo cual es un comportamiento analog0 ai
observado para los otros metales del grupo del P t . Este cambio sugiere
una variacidn continua en la constitución de las especies oxigenadasde
la superficie en vez de una fase óxido simple.
3 - 2 3
La duda que surge es si la capa de óxido sobre Au puede ser
considerada como oxigeno quimisorbido o identificado como una fase de
óxido definida. Se ha encontrado que la formación de
la
pelicula siguela cinética de Elovich 1651 (aplicable a quimisorciónl. o que sigue
una ecuación de velocidad basado
en
un mecanismo de crecimiento deóxido [771.
Rand y Woods 1601 midieron el recubrimiento de óxigeno a
potencial constante para diferentes tiempos de anodización (Fig.
1.1.3.8). Se encontró que el recubrimiento aumenta de manera continua con el potencial, y no fue observada ninguna región donde el
-
0.4"4
I 1 I I1.0 1.5 2.0
POTEMOAL ( V v s RHE)
Fig. 1.1.3.8. Recubrimiento de oxigeno sobre Au como una funcion del potencial en HZCO, 1 M a 25 C. Electrodos analizados por ( o ) 10 C, ( A ) 100 C y ( 0 ) 1000 C al
potencial deseado.
los otros metales nobles. Sin embargo,
la
velocidad deincremento
delrecubrimiento con el potencial se hace
menos
marcado a medida queaumenta el potencial. El proceso alcanza un mínimo a aproximadamente
2.0 V antes de registrar
un
incremento repentino del recubrimiento, similar al reportado para Pd. Bajo condiciones extremas de anodizaciónuna capa visible de color naranja intenso fue observado, que en
la
reducción
se
vuelve negro y causa severa rugosidad enla
superficiedel Au.
En
la Fig. 1.1.3.8 se observa que a potenciales<
2 . 0 ifelque o igual
a
100 C.Este
mismo comportamientofue
observado porLaitinen y Chao i561.
Brummer y Makrides 1651 consideraron que la gráfica consiste de
dos secciones lineales y que la intersección a
1.45
V marca el llenadode
una
monocapa de oxígeno. Rand y Woods I601 señalaronla
similaridadentre el comportamiento del Au y los metales del grupo del Pt e
interpretaron la curva de recubrimiento en términos de una capa incial
de átomos de oxlgeno quimisorbido que se aproxima a
un
recubrimiento ala monocapa alrededor de 2.0 V antes de que la nucleación y
crecimiento de la fase oxido cause una repentina elevación en l a carga
de oxígeno. La carga asociada con la capa inicial
justo
antes de laformación de la fase óxido, fue 500 pC/cm , 'valor que concuerda, con
el concepto de un átomo de oxígeno por átomo de Au superficial
sobre una superficie pulida.
2
Un rapido incremento en la carga que pasa
en la
formación dela
capa de oxígeno cuando se desarrolla la fase de óxido fue reportado
por Laitinen y Chao 1561 y por Ogura y
Col.
1731, pero l a cargacorrespondiente a la saturación de la capa inicial fue mucho mayor que
la
reportada en la Fig.1.1.3.8, y que fueron 1.05 mC/cm para la capa 2inicial y 1.40 mC/cm para las fase óxido.
2
Los valores reportados para la carga asociada
con
adsorcion deoxígeno sobre Au bajo condiciones similares difieren
considerablemente.
Por
ejemplo, el recubrimiento a 1.6V
se sitúaentre 3 0 0 pC/cmZ después de 20
C
I621 y 900 pC/cm2 sobre un barrido de108 mV/C 1671. El último valor fue para un electrodo pulido a espejo.
La película puede ser detectada por técnicas de reflectancia
parámetros ópticos ocurren cuando la carga fluye
a
travCs
delsistema
en el proceso de adsorción. Vinnikov y Col.
I851
consideran
quese
forma una monocapa a 1.40 V y que éste evoluciona para dar una
monocapa de una composición diferente, que se completa a 1.80
V.
Arriba de 2.0 V se forma la fase óxido, que tiene propiedades Ópticas
diferentes de la película inicial, implicando una composición
diferente.
Las características de la capa inicial de oxígeno son totalmente
distinguibles de aquéllas de la fase óxida. En este sentido el Au se
comporta de manera similar a los metales del grupo del P t . El Au
difiere en que el oxígeno es adsorbido irreversiblemente aún a bajos
recubrimientos, sugiriendo una etapa de reconstrucción que sigue
inmediatamente a la adsorción. La adsorción pudiera procedei- via
especies OHad y Oad y que aparentemente alcanza un recubrimiento a la
monocapa de átomos de oxígeno antes de la nucleación y crecimiento de
l a fase óxido sobre la superficie. Rand y Woods [60, 861 se apoyan
en esia suposición para determinar el area de los electrodos
constituidos de Au. Estos investigadores sugieren que
el
recubrimientode oxígeno sobre un electrodo sometido a 1.80 V por 100 S en
H , S 0 4
1M
puede ser considerado próximo a una monocapa. El recubrimiento puede
ser obtenido desorbiendo la capa de óxido mediante un . barrido de
potencial catódico e integrando la carga del perfil. El plano (1001
fue escogido como el estandar convencional de área real y la carga
correspondiente a la adsorción de un átomo de oxígeno por sitio
superficial sobre dicho plano
es
386 pC/cm , por l o tanto, e l áreareal esta dada por la siguiente expresión: 2
Ar
=
Qc pC/í386 pC/cm21 (1.13)I. 2. DEPOCITOC METALICOS A SUBPOTENCIAL
c93282
1.2.1. INTRODUCCION
El efecto de depósito a subpotencial (UPD). es decir, el depósito
de una submonocapa de un metal,
M,
sobre un sustrato metálico ajeno,hl:
a potenciales positivos repecto al potencial reversible de Nernst,se conoce desde hace mucho tiempo. Uno de los pioneros a este respecto
fue Hevesy 1931, quién reportó en 1912 una desviación de l a ley de
Nernsi cuando depositó trazas de metales radiactivos sobre
cobre.
Suscurvas, grado de recubrimiento contra potencial ( ü vs E), mostraron un
residuo asimétrico a potenciales positivos, indicando que la adsorción
del metal se lleva a cabo varias decenas de mV más positivo que el
potencial reversible de Nernst. Luego de este suceso Herzfeld í941
ofreció una explicación formal a esta aparente violación a l a ley de
Nernst. al. asumir que la actividad de la fase sólida es una funcion del
grado de recubrimiento,ü, mientras el depósito no cubriera la
totalidad de la superficie del sustrato.
'4
finales de la década de 1940 varios gruposinvestigaron el comportamiento del depósito y oxidación de pequeñas
cantidades de depósitos metálicos sobre electrodos de metales
nobles. Rogers y Col. 195,961 estudiaron en detalle el .deposito de
trazas de Ag y Cu radiactivos sobre diferentes sustratos metálicos
tales como Pt, Au, Pd. Rh y W en función de la concentración de iones
metálicos, electrolito soporte y agentes cornplejantes. entre otros. Su
interés principal, como el de otros grupos, fue con propósitos
analíticos.
electrolftica de la primera capa at6mica de
un
metal sobre unsustrato
metalico
extraño
y sus propiedades especiales fueron Mills y Willis1971, quienes estudiaron la formación a la monocapa de Pb, TI, Sb y Ni
sobre Au y Pb sobre Ag.
A pesar de sus diferentes enfoques, todas
estas
investigacionescondujeron a un número remarcable de conclusiones que fueron probadas
como completamente razonables en la década pasada. Se vi0 claramente
que la monocapa depositada a subpotencial resulta de una fuerte
interacción entre los átomos de la monocapa y el sustrato. que puede
ser descrita de una manera formal por una actividad menor que la
unidad para el depósito en la región de submonocapa. Varios resultados
experimentales conducen a la conclusión que la monocapa se distribuye
uniformemente y no crece sobre sitios activos del sustrato 197. 9S1, y
su estructura es predominantemente determinada por la del sustrato.
Se asume que la etapa inicial del electrodepósito corresponde
a
la formación de la monocapa. la que a su vez, es de gran importancia
en el desarrollo de capas posteriores y por lo tanto, de las
propiedades físicas del acabado superficial. Se ha encontrado que
la
segunda capa no se
inicia
hasta que la monocapa se completa, la cual2
requiere de una cantidad de electricidad de aproximadamente 250 pC/cm
para un ión metálico monovalente. También fue demostrado.1971. que la
isoterma de adsorción de la monocapa puede tener una forma complicada.
Finalmente se ha señalado que existe una correlación entre el deposito
a subpotencial y el grado de desigualdad de los parámetros de red
entre sustrato y materiales depositados (961. Esto ha permitido apoyar la idea de un desarrollo epitaxial de la monocapa, sobre la superficie
1.2.2. FORMACION DE DEPOSITOS METALICOS A SUBPOTENCIAL
SOBRE ELECTRODOS POLICRISTALINOS EN SOLUCIONES ACUOSAS.
Numerosos pares metálicos han sido investigados por métodos
electroquimicos para obtener información sobre la formación de
monocapas a subpotencial. Los sistemas que han recibido mayor interés
se enruentran en esta cita bibliográfica [61.
Algunos aspectos característicos del depósito a subpotencial
(L'PD) pueden ser muy claramente observados en las curvas
voltamperométricas, como la mostrada para el sistema AgíTI en la Fig.
1.1.7.1.
+
Cuando se comienza el barrido de potencial en l a dirección
catodi.ca desde un valor anódico (0.0 V/ESC), donde la superficie del
sustrato está limpia. se genera un pico de corriente alrededor de
-0.g \ / E X debido al depósito de TI sobre el sustrato. que es oxidado
al mismo potencial al
invertir
el sentido del barrido.
L a velocidadde barrido (dE/dtl deberá
ser
suficientemente lenta, con el propósitode evitar que se cause una polarización por concentración. Cuando
el
barrido de potencial es interrupido en la región de subpotencial la
corriente decrece inmediatamente hasta cero, indicando un proceso de
adsorción reversible dependiente del potencial. Barriendo el potencial
en la región negativa del potencial reversible de Nernst, Er. el TI
será depositado en forma masiva a una velocidad limitada por la
difusión de TI del seno de la solución al electrodo. Durante el
barrido anódico, el depósito masivo será oxidado cerca del valor
LO
E.
II
30
O
-10
I
J
O
-20
L
-1,o -0.5
E (V/ESC)
Fig. 1.2.2.1. Curvas cíclicas I-E para un electrodo de Ag P T
Na2COd 0.5 M (pH 3 ) + TINO3 ZXIO-’~M. v = ?ú rnV/S.
termodinámico,
Er,
en
un intervalo de potencial pequeño ydefinido por la velocidad de barrido de potencial; por tanto, la
cantidad de
T1
depositado dependerá del tiempo de depósito y delpotencial.
El pico de oxidación más positivo. alrededor de -0.55 V/ECC,
es
llamado generalmente como “pico de la monocapa”. Se ha observado que
el pico de la monocapa solo representa una monocapa incompleta (usualmente
<
50% de una monocapa compacta), ya que una considerabletermodinámico. La cantidad
máxima
deTI
depositadoa
subpotencial esconstante y proporcional
ai área
superficial delsustrato,
esto es,aproximadamente Z X ~ O - ~ mol/cm
161.
quees
io esperado para unrecubrimiento a la monocapa, puesto que corresponde aproximadamente al
número de átomos superficiales por cm para
un
metal. Casi todos lossistemas reportados en la literatura presentan alrededor del
mismo valor. Además, se ha encontrado que la carga requerida para
, remover ( o depositar) la cantidad máxima depositada a subpotencial es
2
2
2
del orden Zx200 pC/cm , lo cual significa que la carga transferida es
cercana al valor esperado para una descarga total de acuerdo a la
siguiente reacción:
M
'
+
+ Ze--
-
M
(1.14)Puesto que la cantidad máxima depositada a subpotencial
corresponde
en
muchos casos a valores muy próximos a un recubrimientoa la .monocapa, esto sugiere muy fuertemente la .formación de una
monocapa. Una prueba directa ha sido obtenida para sustratos de
P t al medir la supresión de la adsorción de hidrógeno debido a la
deposición metálica en la región de subpotencial 1100-1041. Se sabe que el hidrógeno se adsorbe sobre P t , antes de la evolución de
hidrógeno
molecular,
en dos regiones distintas de potencial, quecorresponde a átomos fuerte y débilmente enlazados 11051. .La cantidad
total de hidrógeno adsorbido sobre Pt corresponde a una carga
equivalente de 210 pC/cm , aunque este valor difiere para las
diferentes caras cristalinas del P t [1061. Esto se considera como un
recubrimiento a la monocapa que corresponde a una relación I : 1 de
Pt/Hads.
Por
otro lado, se sabe que el hidrógeno no se adsorbe sobreaquellos metales que
son
usualmente
depositadosen
estos
estudios.Por
2L0
F
I I I I IE
(V/ESC)Fig 1.2.2.2. Curvas
I-E
de electroxidación de Cu sobre Pt enHZS04 0.2 M + CUCO 5xlO-'M, con diferentes
cantidades de Cu depositado entre cero (a) y
recubrimiento a la monocapa íhl, depositadas
a -0.25 V/ESC. v = 200 mV/C 11021. 4
ejemplo, cuando el cobre es depositado en pequeñas cantidades
a
subpotencial la adsorción de hidrógeno es suprimida en cierto grado
(Fig. 1.2.2.2). Se observa que para pequeños recubrimientos de Cu el
hidrógeno débilmente ligado es más afectado por los átomos de Cu que
ei fuertemente enlazado, como debe esperarse por simples
consideraciones termodinámicas. Cuando una monocapa de Cu se deposita
(es decir, cuando aproximadamente la máxima cantidad de átomos de Cu
p i adsorbido a subpotencial) l a adsorción de hidrógeno es totalmente
sobre el Pt y no apilado
en
algunos sitios (debidoa
deposiciónpreferencial y nucleación en sitios activos), dejando algunos sitios
de la superficie de Pt
sin
recubrir.Generalmente existen dos formas de definir una monocapa. El
primero, principalmente usado en física de superficie, refiere el
recubrimiento a la monocapa como el número de átomos adsorbidos.
siendo iguales al número de átomos superficiales del sustrato. El
segundo, frecuentemente usado en electroquímica, define un
recubrimiento a la monocapa como la cantidad depositada por unidad de
área superficial sobre el sustrato antes del crecimiento de la segunda
capa. Aqui el término monocapa señala un cambio importante en el
enlace del adsorbato debido a que cambia de un enlace
deposito-sustrato a un enlace puro depósito-depósito. Nos referiremos
usualmente a esta última definición cuando hablemos acerca de una
monocapa. Cuando impliquemos
la
primera definición, será establecidacomo tal.
En la Fig. 1.2.2.3, está representada la cantidad de metal
depositado contra la cantidad de
H
aún adsorbido enuna
superficiede P t para Cu IiOZl, Ag 11021, Bi I1041 y
TI
IiOil. Existe unarelación lineal simple entre ambas cantidades, comprobando nuevamente
de manera cuantitativa la formación de una monocapa uniforme. Para
los
átomos metálicos más pequeños tales como Cu y Ag la pendiente es uno
correspondiendo a
una
relación 1:l. esto es, un átomo de hidrógeno es desorbido por cada átomo metálico depositado. mientras que para átomosmetálicos más grandes tales como Bi y TI la eficiencia de despl+zamiento de Hads se incrementa, siendo aproximadamente 2: 1 para
ads
I
N
- I
?I
Fig. 1.2.2.3. Cantidad total de hidrógeno que puede ser adsorbido
sobre P t , como una función de la concentración
superficial de átomos metálicos depositados para
Cu, Ag, Bi y TI I102,104,1071.
menor, como necesaria, para formar
una
monocapa compacta. Bowles[I071
na demostrado que para Cu. Cd, TI. Cn y Bi la inhibición de l a
adsorción de hidrógeno expresada por I a relación rM rHads es
directamente proporcional al cuadrado de la relación d e los radios atomicos de Pt y del metal adcorbido.
Una
observación interesanteha
sido reportada respecto al hecho de que átomos grandes tales como Bi,
Pb y Au inhiben la adsorción del hidrógeno débil y fuertemente
enlazados en igual magnitud a cualquier grado de recubrimiento,
mientras que los átomos más pequeños tales como Cu y Ag muestran una
supresión
preferencial
del hidrógeno débilmente adsorbido a bajos-" -7
recubrimientos del adátomo.
1.2.3. ASPECTOS TERMODINAMICOS DE LA FORMACION DE DEPOCITOS
METALICOS A SUBPOTENCIAL.
El depósito metálico a potenciales
positivos
respecto alpotencial reversible de
Nernst
ha sido descrito termodinámicamente poruna actividad menor que la unidad
para
la fase depositada. En base alconcepto de Herzfed [941, Roger y Stehney
[lo81
derivaron una ecuaciónde
Nernst
quefue
usada en todoslos
trabajosen los
siguientes 20años
El potencial de equilibrio para una reacción reversible esta dada
por la siguiente expresión:
(1.15)
O
E
=
E + íRT/ZF)Ln(Aox/Ared)r
rdonde:
.lox y Ared son las actividades de las especies oxidada
::
reducidarespectivamente;
para
una reacción de deposición metálica, donde esinvolucrada
una fase
sólida, laactividad
del depósitose
asume quees
constante e igual a la unidad. Sin embargo, cuando la superficie del
electrodo se recubre parcialmente por trazas de un depósito extraño,
ia actividad del depósito será menor que la unidad y variara con el
recubrimiento de la superficie [941. '
A partir de 1970, se ha venido desarrollando la descripción
termodinámica del fenómeno de formación de sub y monocapas por
depósito de metales a subpotencial a través del modelo