Collective ionic dynamics in the liquid Na-Cs alloy: An ab initio molecular dynamics study
J. Blanco,1D. J. Gonza´lez,1,2L. E. Gonza´lez,1J. M. Lo´pez,1and M. J. Stott21
Departamento de Fı´sica Teo´rica, Facultad de Ciencias, Universidad de Valladolid, 47011 Valladolid, Spain
2Department of Physics, Queen’s University, Kingston, Ontario, Canada K7L 3N6
共Received 17 September 2002; published 21 April 2003兲
We present results for several structural and dynamical properties of the liquid Na-Cs alloy. The study has been carried out by means of the orbital-free ab initio molecular dynamics method, combined with local ionic pseudopotentials constructed within the same framework. The results show good agreement with the available experimental data, reproducing the homocoordinating tendency exhibited by this alloy.
DOI: 10.1103/PhysRevE.67.041204 PACS number共s兲: 61.20.Ja, 61.25.Mv
I. INTRODUCTION
Molecular dynamics共MD兲has become a useful technique to study the properties of liquid systems. The classical mo-lecular dynamics 共CMD兲techniques require interatomic po-tentials, whereas the ab initio molecular dynamics 共AIMD兲 methods compute the forces acting on the nuclei from elec-tronic structure calculations performed as the MD trajectory is generated. Therefore the nuclear positions evolve accord-ing to classical mechanics, while the electronic subsystem follows adiabatically.
Recently, there have been many applications of the AIMD methods based on the density functional theory共DFT兲 关1,2兴. This theory allows us to calculate the ground state electronic energy of a collection of atoms, for given nuclear positions, and yields the forces on the nuclei via the Hellmann-Feynman theorem. However, the computational demands of those AIMD methods where a Kohn-Sham 共KS兲orbital rep-resentation of DFT is used 共KS-AIMD methods兲grow very rapidly with the system size and limits the sizes of the sys-tems and the simulation times 关3– 6兴. These limitations can be partly overcome by the so-called orbital-free ab initio molecular dynamics共OF-AIMD兲method which by eliminat-ing the orbitals of the KS formulation provides a simulation method where the number of variables describing the elec-tronic state is greatly reduced, enabling the study of larger samples共several hundreds of particles兲and for longer simu-lation times共tens of picosecond兲.
The study of the dynamical properties of liquid metals has already produced much experimental and theoretical work 关7兴. Most experimental investigations have been performed by means of inelastic neutron scattering although the recent development of new synchrotron radiation has opened the possibility of using x rays too. It has also been stimulated by MD simulations because of its ability to determine several dynamical magnitudes that are not accessible to experiment, and, in this way, they have supplemented the experimental information. On the theoretical side, the development of mi-croscopic theories such as the memory function formalism or the kinetic theory has created a theoretical framework, whose application to simple liquids has lead to good qualitative results for several dynamical magnitudes 关7–9兴.
Less attention has been devoted to the dynamics of liquid binary alloys, although the last 15 years have witnessed an increasing effort. Following a CMD study of liquid Na-K
关10兴, the field was stimulated by a CMD simulation of liquid Li4Pb by Jacucci et al. 关11兴, where a new, high-frequency
mode, supported by the Li atoms only 共the so-called ‘‘fast sound’’兲 was found. Subsequently, several theoretical 关12– 14兴, computer simulations 关15–17兴 and experimental 关16,18 –20兴studies have investigated the existence and prop-erties of the collective excitations in liquid binary systems. For binary systems with disparate masses, two main branches 共low- and high-frequency ones兲 of the collective excitations have been found to contribute to the longitudinal dynamics, although its origin is still unclear. However, the transverse dynamics has been much less investigated, mainly because it is not visible in scattering experiments and only the MD simulations can provide information about the trans-verse excitations. At this stage we mention that the recent application of the generalized collective model 共GCM兲 ap-proach, which combines CMD simulations with the memory function formalism, to binary Lennard-Jones fluids and liq-uid alloys has shown the existence of transverse optic modes that arise in connection with the concentration fluctuations.
This paper reports an AIMD study on the structural and dynamical properties of the liquid Na-Cs alloy. This system exhibits significant homocoordinating tendencies, and has al-ready attracted much experimental 关21–24兴 and theoretical 关6,24 –28兴 work. The experimental works have measured several thermodynamic magnitudes 关21–23兴, as well as the static structure factor by means of x-ray-diffraction and neutron-diffraction experiments 关24兴. A marked departure from ideality has been observed in several properties; espe-cially a substantial increase in the concentration fluctuation structure factor SCC(0), at sodium concentrations xNa⬇0.8.
On the theoretical side, several studies using either semi-empirical 关25–28兴 or more fundamental 关6,24兴 approaches have calculated the static structure and/or some thermody-namic magnitudes such as the concentration dependence of
SCC(0) or the entropy of mixing which according to the
ex-periment is nearly ideal. Among the semiempirical models, we mention Refs. 关25–28兴that calculated the concentration dependence of SCC(0)关25,26,28兴and the free energy of
although strongly overestimated the oscillations of the ex-perimental total static structure factor. Recently, Costa Ca-bral and Martins 关6兴 have performed an AIMD study, with ionic pseudopotentials derived by the Troullier-Martins method关29兴, to determine the static structure and the diffu-sion coefficients 共DC’s兲 in the alloy. Their results for the total static structure factor showed a good agreement with the experiment, although for xNa⫽0.8 some discrepancies
appeared for the amplitude of the oscillations. Moreover, be-cause of the huge computational demands of their method, these authors used 108 particles and the simulation ranged for 400 configurations only, which obviously does not allow to study most of the dynamical magnitudes.
II. THEORY
A simple liquid metallic alloy AxB1⫺xcan be regarded as an assembly of NA, A type, and NB, B type, bare ions with charges ZvA and ZvB, respectively, interacting with Ne⫽NA
ZvA⫹NBZv B
valence electrons through electron-ion potentials
vA(r) andvB(r). Therefore, the total potential energy of the system can be written, within the Born-Oppenheimer ap-proximation, as the sum of the direct ion-ion interaction ergy, which we assume Coulombic, and the ground state en-ergy of the electronic system subject to the external potential created by the ions, Vext(rជ,兵Rជl其)⫽兺i⫽A,B兺l(i)vi(兩rជ⫺Rជl兩),
E共兵Rជl其兲⫽ 1
2 i, j
兺
⫽A,Bl(i)兺
⫽m( j)ZiZj
兩Rជl⫺Rជm兩
⫹Eg关g共rជ兲,Vext共rជ,兵Rជl其兲兴, 共1兲
where Rជl are the ionic positions, the sum over l(i) extends over the sites occupied by the i-type ions, and g(rជ) is the ground state electronic density. According to DFT,g(rជ) can be obtained by minimizing the energy functional
E关共rជ兲兴⫽Ts关兴⫹Eext关兴⫹EH关兴⫹Exc关兴, 共2兲
where the terms represent, respectively, the electronic kinetic energy Ts关兴 of a noninteracting system with density (rជ), the energy of interaction with the external potential due to the ions,
Eext关兴⫽
冕
drជ共ជr兲Vext共rជ兲; 共3兲the classical electrostatic energy共Hartree term兲,
EH关兴⫽1
2
冕 冕
drជdsជ共rជ兲共sជ兲
兩rជ⫺sជ兩 ; 共4兲
and the exchange-correlation energy Exc关兴 whose exact
ex-pression is unknown, although several approximations have been proposed. The simplest one is the so-called local den-sity approximation共LDA兲,
ExcLDA关兴⫽
冕
drជ共rជ兲⑀xc„共rជ兲…, 共5兲where ⑀xc() is the density of exchange-correlation energy in a uniform electron gas of density , which is known through simulations 关30兴 and further fittings 关31,32兴. Al-though the LDA is clearly inexact, however, it is remarkable that hundreds of calculations with the LDA have yielded accurate calculated properties for many systems. This suc-cess is partially attributed to the fact that it gives the correct sum rule for the exchange-correlation hole关33兴. Further im-provements have been developed, which include dependence on the gradient of the density. However, because of its sim-plicity, most AIMD calculations have used the LDA, and we have also adopted it in the present calculations.
A. The kinetic energy functional
The kinetic energy functional Ts is a basic ingredient of the energy functional. It is generally considered关34兴that the von Weizsa¨cker term
TW关共rជ兲兴⫽ 1
8
冕
drជ兩ⵜជ共rជ兲兩2/共rជ兲 共6兲
is essential for a good description of the kinetic energy. It applies in the case of rapidly varying densities, and it is exact for one- or two-electron systems. Usually other terms are added in order to correctly reproduce some exactly known limits. In the uniform density limit, the exact kinetic energy is given by the Thomas-Fermi functional
TTF关共rជ兲兴⫽
3
10
冕
drជ共rជ兲kF共rជ兲2, 共7兲
where kF(rជ)⫽(32)1/3(rជ)1/3 is the local Fermi wave vec-tor. In the limit of almost uniform density, the linear response theory共LRT兲is correct, with a response function correspond-ing to a noninteractcorrespond-ing uniform electron gas, which is given by the Lindhard functionL(q,0).
The computational advantages of the OF-AIMD method have stimulated the development of accurate kinetic energy functionals 关35–38兴 which correctly recover some known limits, such as the Thomas-Fermi, the linear response, and the quadratic response limits. However, an undesirable fea-ture of those functionals is that they are not positive definite, and therefore can lead to unphysical negative kinetic ener-gies.
Within the ‘‘averaged density’’ approach, Chaco´n and co-workers 关39,40兴 have developed some kinetic energy func-tionals that, besides recovering the uniform and LRT limits, are also positive definite. However, they are complicated to apply as they require order N more fast Fourier transforms 共FFT’s兲 than simpler functionals. Therefore we have used a simplified version关41兴,
T关兴⫽ 3
10
冕
drជ共rជ兲5/3⫺2˜k共rជ兲2, 共9兲
k
˜共rជ兲⫽共2k
F
0兲3
冕
dsជk共sជ兲w共2kF
0兩 r
ជ⫺sជ兩兲,
k共rជ兲⫽共32兲1/3共rជ兲,
where kF
0
is the Fermi wave vector corresponding to a mean electron density 0 and w(x) is a weighting function,
de-termined by requiring the correct recovery of the LRT and uniform density limits. This kinetic energy functional is posi-tive definite for any value of; moreover, in the kF0→0 limit when the mean electron density vanishes 共e.g., for a finite system兲, the von Weizsa¨cker term is recovered if ⫽4/9, whereas for other values of , the limit is TW⫹C2TTF. In
the present simulations, we have used ⫽0.51, which is close to 4/9 and for which C⫽0.046.
B. Pseudopotentials
We briefly describe the main steps in the construction of the ionic local pseudopotentials vps(r) used to describe the ion-electron interaction; further details are in Refs. 关41,42兴. The pseudopotentials are developed from first principles by fitting to a generic model of an ion immersed in a metallic medium. In the first step关42兴, for a given ion type, the elec-tron density distribution is calculated in a model system composed of an homogeneous electron gas of density 0
⫽Ne/V, in which a spherical cavity has been made and where the ion has been inserted. This is performed using the KS-DFT approach and includes core and valence electron states. The latter are used to determine the change, n␣(r) (␣⫽A,B), in the valence electron density induced in the
jellium by the presence of the␣-type ion and the cavity共the displaced valence electron density兲. The n␣(r) is pseudized so as to eliminate the core-orthogonality oscillations. In the second step关41兴, an effective local pseudopotentialvps␣(r) is constructed, so that when inserted into the jellium together with the cavity, it reproduces, within the OF-DFT procedure, the same pseudized n␣(r) as previously obtained. This is achieved by inserting each n␣(r) into the Euler equation, obtained by minimizing the energy functional of Eq. 共2兲, together with the normalization constraint兰Vdrជn␣(r)⫽Zv␣,
␦Ts关兴
␦共rជ兲 ⫹
␦Eext关兴
␦共rជ兲 ⫹ ␦EH关兴
␦共rជ兲 ⫹
␦Exc关兴
␦共rជ兲 ⫺⫽0, 共10兲
with (r)⬅␣(r)⫽0⫹n␣(r) and ⬅␣ is a Lagrange
multiplier. Moreover, due to the spherical symmetry of the system, all the magnitudes depend on兩rជ兩 ⬅ r.
C. Technical details
We have considered 600 ions in a cubic cell with periodic boundary conditions. Given the ionic positions at time t, the electronic energy functional is minimized with respect to
(rជ), yielding the ground state electronic density and energy.
In order to keep (rជ)⭓0 everywhere, the minimization is most conveniently performed in terms of an effective orbital
(rជ), defined through (rជ)⫽(rជ)2, with real (rជ). This effective orbital is expanded in plane waves compatible with the simple cubic periodic boundary conditions of the simu-lation:
共rជ兲⫽
兺
Gជ
cGជe⫺iG ជ•rជ,
cGជ⫽ 1
V
冕
Vdrជ共rជ兲eiGជ•rជ,
Gជ⫽2
L 共n1,n2,n3兲, 共11兲
where L is the superlattice parameter, V is the volume of the cell, and the expansion is truncated at wave vectors corre-sponding to a given cutoff energy ECut. Following Car and
Parrinello 关43兴, we introduce a fictitious kinetic energy T ⫽1
2Mc兺Gជ兩dcGជ/dt兩2, and determine the electronic ground state using damped Newtonian mechanics with an electronic time step ⌬tc. We have used Mc⫽3.5⫻107 hartree a.u.3 and a ⌬tc⫽1.1⫻10⫺4 ps. The forces on the ions were obtained via the Hellmann-Feynman theorem, Fជi⫽ ⫺ⵜជRជiEg关(rជ),兵Rជl其兴, (i⫽1, . . . N) and the ionic positions and velocities were updated by numerically solving New-ton’s equations, d2Rជi/dt2⫽Fជi/ Mi, using the Verlet leapfrog algorithm 关44兴 with a time step ⌬t⫽2.5⫻10⫺3 ps. The ECut⫽10 Ry and the calculation of properties was made
av-eraging over 40 ps after an equilibration time of 4 ps.
III. RESULTS
A. Structural properties
The liquid Na-Cs alloy has been simulated for three ther-modynamic states at T⫽373 K and concentrations xNa
⫽0.3, 0.5, and 0.75, with the experimental total ionic num-ber densities taken from Refs. 关25,26兴.
The simulations allow a direct evaluation of the partial pair distribution functions gi j(r), which are shown in Fig. 1, and the partial Ashcroft-Langreth 共AL兲 structure factors
Si j(q). The gNa-Na(r) changes substantially with
concentra-tion, whereas both gNa-Cs(r) and gCs-Cs(r) change little. As
the Na concentration is increased, the height of the main peak of gNa-Na(r) decreases, whereas those for gNa-Cs(r) and gCs-Cs(r) are relatively unchanged and slightly smaller,
re-spectively. Also, for xNa⫽0.30 and 0.50, the gNa-Cs(r)
re-mains smaller than the average of gCs-Cs(r) and gNa-Na(r),
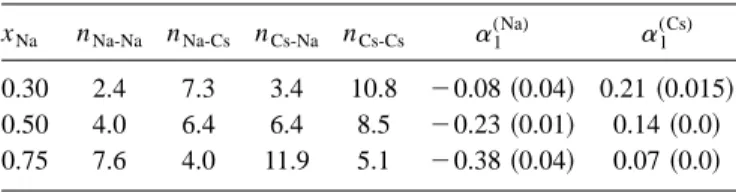
which is an indication of homocoordinating tendencies. The Warren-Cowley关45– 47兴short range order共SRO兲 pa-rameter for the first neighbor shell,␣1(i), is a simple way of displaying the short range order in the alloy. It is defined as
␣1 (i)⫽
1⫺ ni j
where ni j is the number of j-type particles around an i-type particle, within a sphere of radius Ri j. The ni j can be calcu-lated from the gi j(r) by
ni j⫽4xj
冕
0
Ri j
r2gi j共r兲dr, 共13兲
where Ri j is usually identified关48兴as the position of the first minimum of gi j(r). For a random distribution of atoms,
␣1 (i)⫽
0, whereas a positive共negative兲value for␣1(i)suggests homocoordinating共heterocoordinating兲tendency. Calculated values for ␣1(Na) and␣1(Cs) for the liquid NaxCs1⫺x alloy at
T⫽373 K are shown in Table I, but they are inconclusive since ␣1(Cs) is always positive whereas ␣1(Na) is negative.
However, the Warren-Cowley SRO parameters may be inad-equate because of the large atomic size mismatch between the pure components 共the volume ratio is around 3.0兲, and we have resorted to the local mole fractions method关49–53兴 that has also been widely used to study the homocoordinat-ing tendencies in mixtures. For a binary system, the local mole fractions xiiare defined as
xii⫽
nii
nii⫹ni j
, j⫽i⫽1,2. 共14兲
Since there has been much discussion关49,54 –56兴of the suit-able upper integration limit Ri j in Eq. 共13兲, we have evalu-ated xii as a function of Ri j. For an homogeneous phase, when both species are nearly equally distributed, xs⫺1 ⫽x11⫹x22⫺1 is around zero; whereas for a demixed state
particles of the same species dominate the distribution and
xs⫺1 grows关50,51兴. Figure 2 shows, for xNa⫽0.75, a plot
of xs⫺1 as a function of the upper integration limit Ri j in Eq. 共13兲. Notice that xs, x11, and x22 change rapidly when Ri j increases, but a plateau is observed when Ri j reaches values around the position of the first minimum of the
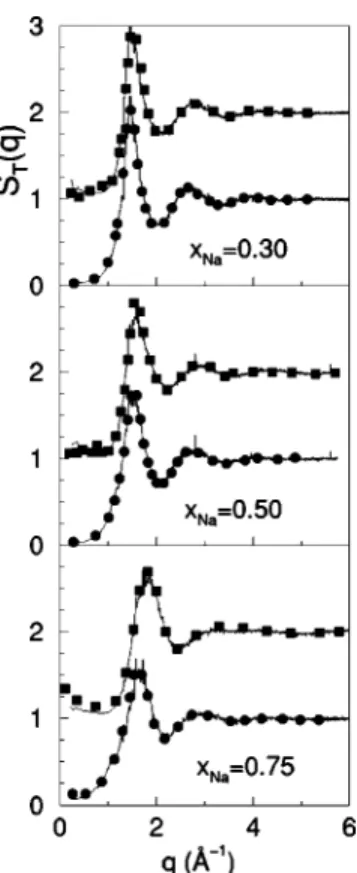
gCs-Cs(r). Taking this minimum as the upper integration limit Ri j, xs⫺1 takes small positive values, suggesting a week homocoordinating tendency. Furthermore, the corresponding Warren-Cowley SRO parameters now become positive 共see Table I兲. The total static structure factor at several concen-trations and temperatures has been measured using x-ray-diffraction and neutron-x-ray-diffraction experiments by van der Lugt and co-workers 关24兴. Several experimental x-ray and neutron weighted total static structure factors ST(q) are shown in Fig. 3 along with the OF-AIMD simulation results that reproduce rather well the basic features of the measured
ST(q), namely, the position and amplitude of the oscilla-tions, including the damping of the oscillations with increas-ing sodium concentration. From the experimental x ray and neutron ST(q)’s, and assuming SNa-Na(q)⬇1, van der Lugt
and co-workers 关24兴 derived semiempirical results for
SCs-Cs(q) and SNa-Cs(q). Their results for xNa⫽0.30 are
shown in Fig. 4 along with the OF-AIMD simulations that partly support the SNa-Na(q)⬇1 assumption, although notice-FIG. 1. Partial pair distribution functions gi j(r) for the liquid
Na-Cs alloy at T⫽373 K and three concentrations. The full, dashed, and dot-dashed lines represent the gCs-Cs(r), gNa-Na(r), and
gNa-Cs(r), respectively.
TABLE I. Calculated values of coordination numbers ni j and
the Warren-Cowley SRO parameters␣1(i)for the Na-Cs liquid alloy at T⫽373 K. The numbers in parenthesis give the values of ␣1
(i)
when ni jare evaluated by integrating up to the position of the first
minimum of gCs-Cs(r).
xNa nNa-Na nNa-Cs nCs-Na nCs-Cs ␣1
(Na) ␣ 1 (Cs)
0.30 2.4 7.3 3.4 10.8 ⫺0.08共0.04兲 0.21共0.015兲
0.50 4.0 6.4 6.4 8.5 ⫺0.23共0.01兲 0.14共0.0兲
0.75 7.6 4.0 11.9 5.1 ⫺0.38共0.04兲 0.07共0.0兲
FIG. 2. Local mole fractions xNa-Na 共dot-dashed line兲, xCs-Cs
共dashed line兲, and xs⫺1 共full line兲, as function of the upper
inte-gration limit Ri j, for the liquid Na0.75Cs0.25alloy at T⫽373 K. The
dotted lines represent the partials gNa-Na(r), gNa-Cs(r), and
able deviations appear at q⬇1.35 Å⫺1, where its first mini-mum is located. Discrepancies in SNa-Cs(q) also appear in
this region, but for other q’s the agreement between the semiempirical/simulation results for SCs-Cs(q) and SNa-Cs(q)
is satisfactory. The recent AIMD simulations 关6兴 for xNa
⫽0.60 and 0.80 also gave reasonable results for Si j(q) and
ST(q), although a detailed comparison with the experiment is not possible because the results are rather noisy共due to the small sample size and simulation time兲.
For investigating the ordering tendencies in a liquid alloy, the Bhatia-Thornton 共BT兲 partial structure factors 关57兴 are ideally suited. For a binary alloy, they are the concentration-concentration 关SCC(q)兴, the number-number 关SNN(q)兴, and the number-concentration 关SNC(q)兴 partial structure factors,
defined as
SCC共q兲⫽x1x2关x2S11共q兲⫹x1S22共q兲⫺2共x1x2兲1/2S12共q兲兴,
SNN共q兲⫽x1S11共q兲⫹x2S22共q兲⫹2共x1x2兲1/2S12共q兲,
SNC共q兲⫽S11共q兲⫺S22共q兲⫹共x2⫺x1兲/共x1x2兲1/2S12共q兲,
where Si j(q) are the AL partial structure factors. Moreover, their long-wavelength limit provides microscopic informa-tion on the liquid alloy, e.g., SCC(q→0) portrays short range
order. The experimental results 关22–24兴 for SCC(q
→0)/xNaxCs are slightly greater than unity for 0⭐xNa
⭐0.5, then increase toward a maximum value of ⬇7 at
xNa⫽0.80 and thereafter quickly decrease toward 1. These
values clearly indicate homocoordinating tendencies for all concentrations. Although this behavior is qualitatively repro-duced by the present OF-AIMD simulations, the results for
SCC(q→0)/xNaxCs remain around ⬇1.3 with a minimum
value of⬇1.0 for xNa⬇0.50.
B. Dynamic properties
1. Single-particle dynamics
The information about the transport properties in multi-component liquid systems is provided by various time corre-lation functions among the atomic velocities. However, they cannot be determined experimentally and must be obtained by other methods such as MD simulation. We present results for the relative velocity correlation functions 共VCF兲 Zi j(t) defined 关58兴 as the time correlation function of the relative velocity of the center of mass of species i with respect to that of species j:
Zi j共t兲⫽1
3xixjN
具
关uជi共t兲⫺uជj共t兲兴•关uជi共0兲⫺uជj共0兲兴典, 共15兲where N is the total number of particles and uជi(t) the mean velocity of component i, is given in terms of the velocity of the i-type particle l(i), uជl(i)(t), as
FIG. 3. Total static structure factor ST(q) for the liquid Na-Cs
alloy at T⫽373 K and three concentrations. Continuous lines are the results from the present OF-AIMD simulations, whereas the full circles and squares are the experimental x-ray-diffraction and neu-tron diffraction-data, respectively. The neuneu-tron data have been shifted upward by one unit.
FIG. 4. Partial static structure factors Si j(q) for the liquid Na-Cs
uជi共t兲⬅共xiN兲⫺1
兺
l(i)⫽1Ni
uជl(i)共t兲. 共16兲
The Zi j(t) can be separated into self-关Zi j
0(t)兴, and distinct,
关Zi jd(t)兴, contributions which reflect the self-dynamic and cross-dynamic correlations, and give information about the mobility of particles and the coupling between the velocities of distinct particles, respectively,
Zi j共t兲⫽共1⫺␦i j兲Zi j
0共
t兲⫹xixjZi j d共
t兲, 共17兲
where␦i j is Kronecker’s delta. In terms of the well-known velocity autocorrelation function Zis(t)⫽1
3
具
uជ1i(t)uជ1i(0)典
ofa tagged i-type particle in the fluid, we write Zi j0(t) ⫽xjZi
s
(t)⫹xiZj s
(t). The Zi jd(t) account for the contribution of the distinct velocity correlations. The time integrals of
Zi j(t), Zi j
0
(t), Zi j d
(t), and Zi s
(t) give the associated DC’s, namely Di j, Di j
0, D
i j d , and D
i
s, respectively, where D i sis the
usual self-diffusion coefficient. Simplifications for the binary mixtures give
D12⫽D12 0 ⫹
x1x2, D12
d⬅
D12 0共
1⫹␥12兲, 共18兲
with D120 ⫽x2D1
s⫹
x1D2
s
and␥12is the measure of the
devia-tion from an ideal mixture, vanishing when species are the same. Finally, we note that the interdiffusion coefficient is written as
Dint⫽D12⬅共1⫹␥12兲D12
0 , 共19兲
where ⫽x1x2/SCC(q→0). For a nearly ideal mixture, ⬇1, ␥12⬇0 and consequently Dint⬇D12
0
.
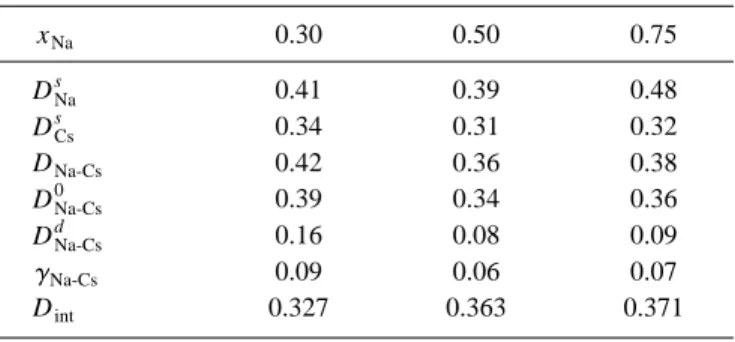
The results for the self- and relative VCF for NaxCs1⫺x liquid alloy are shown in Fig. 5, along with the correspond-ing quantities for both pure Na and Cs at T⫽373 K and number densities at their corresponding coexisting lines共see Table II兲. First, we note that ZNa
s
(t) and ZCs
s
(t) have shapes 共rate of decay and depth of first minimum兲similar to the pure components. However, due to the large mass difference be-tween the Na and Cs ions, the Na ions in the alloy will experience, during the collisions, larger relative changes in their velocities than the Cs ions. This is reflected in their VCF, with ZCss (t) exhibiting a slower decay and smaller backscattering than the ZNas (t). Moreover, as xNa increases,
the backscattering effect is slightly reduced because of fewer collisions with the Cs ions, tending to enhance the corre-sponding self-diffusion coefficient DNas , an effect that is somewhat counterbalanced by the increase in the total num-ber density.
The ZNa-Cs(t) lies between ZNas (t) and ZCss (t) suggesting that distinct correlation effects are weak; in fact the contri-bution of distinct effects to DNa-Cs is usually measured by ␥Na-CsEq.共18兲. For all concentrations, we obtain small
posi-tive values of␥Na-Cs, which as shown in Table III are smaller than 10%. Moreover, the positive values of DNa-Cs
d
indicate 关58,59兴that particles of the same species have a greater ten-dency to diffuse together than those of distinct species, an-other indicator of homocoordinating tendencies in the alloy. These tendencies are weaker at xNa⬇0.5, where the values of
both␥Na-Csand DNa-Csd reach minima.
The experimental determination of the self-diffusion co-efficients in metallic melts is rather difficult and, to our
FIG. 5. Normalized self- and relative VCF’s for the liquid Na-Cs alloy at T⫽373 K and three concentrations. The dashed, dot-dashed, and dotted lines represent the ZNas (t), ZCss (t), and
ZNa-Cs(t), respectively. The full thick lines represent the VCF’s of
pure Na共fast decay兲and Cs共slower decay兲at T⫽373 K.
TABLE II. Input data for the series Na-Cs at T⫽373 K, studied in this work, along with some simulation details.is the total ionic number density taken from Ref.关25,26兴.
x⫽xNa (Å⫺3) Cutoff共Ry兲
0.0 0.008111 10.0
0.30 0.010564 10.0
0.50 0.012897 10.0
0.75 0.017132 10.0
1.0 0.024221 10.0
TABLE III. Diffusion coefficients共in 10⫺4cm2/s), and related quantities for the Na-Cs liquid alloy at T⫽373 K.
xNa 0.30 0.50 0.75
DNa
s
0.41 0.39 0.48
DCss 0.34 0.31 0.32
DNa-Cs 0.42 0.36 0.38
DNa-Cs 0
0.39 0.34 0.36
DNa-Cs
d
0.16 0.08 0.09
␥Na-Cs 0.09 0.06 0.07
knowledge, have only been measured for the Na-K system for 32% sodium at several temperatures关60兴. As a check on the accuracy of our results, we have also calculated the self-diffusion coefficients of the pure components at 373 K. Re-sults are DNa0 ⫽0.48 and DCs0 ⫽0.39, which compare favor-ably with the corresponding experimental values关61兴of 0.42 and 0.39 (10⫺4 cm2/s units兲, respectively. The AIMD simu-lation of Costa Cabral and Martins 关6兴 gave somewhat smaller values for DCss and DNas , which may be a conse-quence of the small number of particles and short time used in their simulations.
2. Collective dynamics
The collective dynamics of density fluctuations in the al-loy is usually described through the partial intermediate scat-tering functions Fi j(qជ,t)⫽
具
i(qជ,t)•*j(qជ,0)典
, where the as-terisk denotes the complex conjugation andj共qជ,t兲⫽ 1
冑
Nj l( j)兺
⫽1 Njexp关iqជ•Rជl( j)共t兲兴 共20兲
is the density fluctuation of the jth component with wave vector qជ, in which Rជl( j)(t) is the position of the j-type par-ticle l( j ) and
具
•••典
denotes the ensemble average. The time Fourier transform of the Fi j(qជ,t) into the frequency domain leads to the partial dynamic structure factors Si j(qជ,), which are directly related to the inelastic neutron scattering data. Associated with the density fluctuations is the j th com-ponent particle currentj
ជj共qជ,t兲⫽ 1
冑
Nj兺
l⫽1 Njuជl( j)共t兲exp关iqជ•Rជl( j)共t兲兴, 共21兲
which is usually split into a longitudinal component jjL(qជ,t), parallel to qជ, and a transverse component jTj(qជ,t),
perpen-dicular to qជ. The partial longitudinal, Ci j L
(qជ,t), and trans-verse, Ci jT(qជ,t), current correlation functions are obtained from these through
Ci jL共qជ,t兲⫽
具
jiL共qជ,t兲jjL*共qជ,0兲典, 共22兲Ci jT共qជ,t兲⫽1 2
具
jiT共
q
ជ,t兲jTj*共qជ,0兲典.
The corresponding time Fourier transforms give the associ-ated spectra, Ci jL(qជ,)⫽2Si j(qជ,) and Ci j
T(qជ,),
respec-tively. Finally, we note that for isotropic systems all these correlation functions depend only on q⫽兩qជ兩.
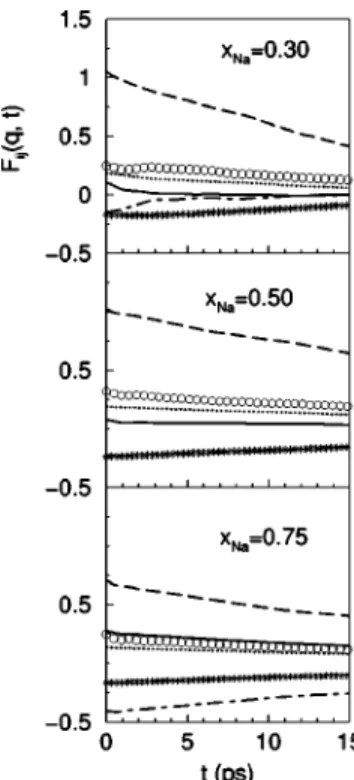
Figures 6 and 7 show, for two q values, the partial inter-mediate scattering functions FCs-Cs(q,t), FNa-Na(q,t), and
FNa-Cs(q,t), as well as the BT partial intermediate scattering
functions FNN(q,t) and FCC(q,t). FCs-Cs(q,t), FNa-Na(q,t), FNN(q,t), and FCC(q,t) are always positive, whereas FNa-Cs(q,t) can have either sign. This is consistent with SNa-Cs(q) oscillating about zero, since Fi j(q,t⫽0)⫽Si j(q). The Fi j(q,t) go monotonically to zero, showing slower
de-cays for the smaller q’s. In the hydrodynamic regime (q →0), the decay of Fi j(q,t) for binary mixtures is related to both entropy and concentration fluctuations, and the slow decay observed in Fig. 6 is mainly due to diffusion effects resulting from concentration fluctuations.
FIG. 6. Partial intermediate scattering functions Fi j(q,t) at q
⫽0.27 Å⫺1, or the liquid Na-Cs alloy at T⫽373 K and three con-centrations. The full, dashed, dot-dashed lines, circles, stars, and dotted lines represent FCs-Cs(q,t), FNa-Na(q,t), FNa-Cs(q,t),
FNN(q,t), FNC(q,t), and FCC(q,t), respectively.
The Si j(q,) also provide information concerning the mi-croscopic mechanisms of propagating longitudinal modes, e.g., the peaks in SLi-Li(q,) for the Li4Pb liquid alloy have
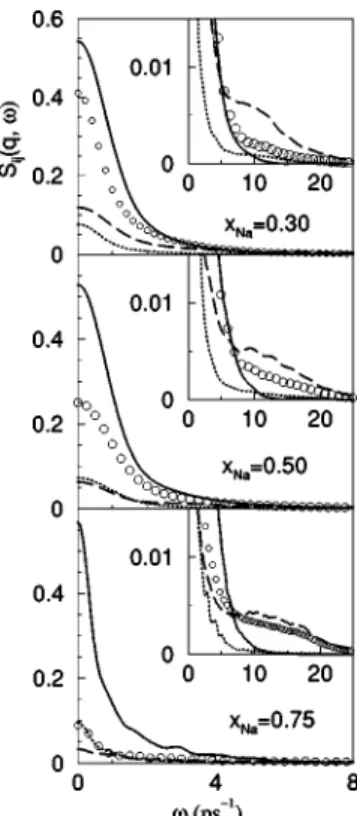
been related to propagating fast modes supported by the Li ions. Figures 8 and 9 show results for SCs-Cs(q,), SNa-Na(q,), SNN(q,), and SCC(q,). For the smaller q’s
and all concentrations, SCs-Cs(q,), SNa-Na(q,), and SNN(q,) exhibit clear side peaks that for q⭐0.20 Å⫺1 are
located at very similar frequencies; this is the typical behav-ior of Si j(q,) in the hydrodynamic regime and represents a propagating sound mode.
The range of existence of the side peaks or shoulders depends on the concentration, with the majority component exhibiting a wider range. For the Na0.3Cs0.7 alloy, both
SCs-Cs(q,) and SNN(q,) show side peaks up to q
⬇0.90 Å⫺1, at which point they shoulder and vanish for q ⭓1.0 Å⫺1. Meanwhile, SNa-Na(q,) has side peaks up to q
⬇0.40 Å⫺1, and shoulders up to q⬇0.85 Å⫺1. For the Na0.75Cs0.25alloy, both SNa-Na(q,) and SNN(q,) have side
peaks or shoulders up to q⬇1.40 Å⫺1, while in SCs-Cs(q,)
they appear only up to q⬇0.50 Å⫺1.
The SNN(q,) reflects the average behavior of the system
and in the limit of a binary mixture of identical species,
SNN(q,) reduces to the usual dynamic structure factor of a
one-component system. Therefore, it is not surprising that in the hydrodynamic regime, SNN(q,) exhibits a clear Rayleigh-Brillouin structure 关62兴, similar to that of the dy-namic structure factor in one-component liquids. Our results show Brillouin peaks共see insets of Fig. 8兲for small q values, which become remnant shoulders for larger q and eventually
disappear. These Brillouin peaks at small q’s are associated with acoustic modes propagating through the binary alloy with adiabatic velocity of propagation, cs⫽B/q, whereB is the position of the Brillouin peak. For the concentration
xNa⫽0.30, SNN(q⫽0.23,) shows a clear Brillouin peak at B⬇2.45 ps⫺1 giving cs⫽1065 m/s, which compares well with the observed value关63兴of 1070 m/s. Similarly, we ob-tain cs⫽1100 m/s for xNa⫽0.50 and cs⫽1480 m/s for xNa
⫽0.75, which are close to the experimental values 关63兴 of ⬇1160 and 1450 m/s, respectively. For the limits xNa⫽1
共pure Na兲and xNa⫽0 共pure Cs兲, we obtain cs⫽2450 m/s and
cs⫽940 m/s, respectively, which also compare well with the corresponding experimental values 关63兴 of 2520 and 960 m/s.
No clear side peaks have been observed in SCC(q,), but
shoulders appear which become weaker as q increases, and vanish for q⭓1.0 Å⫺1. This absence of concentration modes in SCC(q,) can be attributed to the dominance of diffusive
contributions in the partial intermediate scattering functions. However, such modes may be apparent in the associated par-tial longitudinal current correlation function CCCL (q,) be-cause of the 2 factor that kills the low-frequency compo-nents and enhances the higher ones. Further information about the longitudinal collective modes can be obtained from the longitudinal current correlation functions Ci jL(q,). These we have also evaluated, and Figs. 10 and 11 show results for CCs-CsL (q,), CLNa-Na(q,), CNa-CsL (q,)
CNNL (q,), and CCCL (q,). For all q’s, CCs-Cs
L
(q,), CNa-Na
L
(q,), and CNN
L (q,) have at least one peak whose frequency coincides, at very small q’s, with that of the Brillouin peak in their respective
SCs-Cs(q,), SNa-Na(q,), and SNN(q,). From those fre-FIG. 8. Partial dynamic structure factors Si j(q,), at q
⫽0.27 Å⫺1, for the liquid Na-Cs alloy at T⫽373 K and three con-centrations. The full, dashed, circles, and dotted lines represent the
SCs-Cs(q,), SNa-Na(q,), SNN(q,), and SCC(q,), respectively.
quencies, the longitudinal dispersion relations Cs-CsL (q),
Na-Na
L
(q), andNNL (q) are obtained共see Fig. 12兲. Note that
Cs-Cs
L (q) always takes markedly smaller values that those of
Na-Na
L (q), due to the large atomic mass difference. For the
three concentrations, Cs-CsL (q) has one branch and
Na-Na
L
(q) has two, with the lower one present only at small
q’s, including the hydrodynamic region, and at frequencies
close to that of Cs-CsL (q); and the higher one is present at frequencies comparable to those of the pure Na system. In the binary alloy, the heavy Cs ions have a characteristic low frequency, and the light Na ions a much higher frequency that persists up to the hydrodynamic regime, although for smaller q some Na ions start to oscillate with the Cs ions. In the hydrodynamic limit, all the particles must oscillate at the same frequency, and only the lower-frequency peak will sur-vive in CNa-Na
L
(q,). The marked differences between
Cs-Cs
L (q), and
Na-Na
L (q), for most q’s, suggest that the
mo-tions of the Na and Cs ions are largely uncorrelated. As shown in Fig. 12, at small q’s, the Cs-CsL (q) has an initial linear increase up to a maximum followed by a mini-mum at q⬇1.5 Å⫺1, which corresponds to the main peak
position of SCs-Cs(q). As xNa increases, the main peak of SCs-Cs(q) drops, widens, and maintains its position at ⬇1.5 Å⫺1; this explains that at the same time the first mini-mum ofCs-CsL (q) becomes less marked while the first maxi-mum moves toward smaller q values. A similar behavior is exhibited by the high-frequency branch ofNa-NaL (q), with a maximum and minimum at same positions as those of
SNa-Na(q). Moreover, as xNa is increased the structure of Na-Na
L (q) becomes more marked as it happens with the
maxima and minima in SNa-Na(q).
The NNL (q) dispersion curve also has two branches be-cause for some q region, the CNNL (q,) exhibits two peaks. As the CNa-CsL (q,) is substantially smaller than the other partial currents, the behavior of the CNN
L
(q,) is dominated by CCs-CsL (q,) and CNa-NaL (q,), with the latter one playing a greater role even when Na is the minoritary component.
FIG. 10. Partial longitudinal current correlation functions
Ci j L
(q,), at q⫽0.27 Å⫺1, for the liquid Na-Cs alloy at T ⫽373 K and three concentrations. The full, dashed, dot-dashed, circles, stars, and dotted lines represent CCs-Cs
L
(q,), CNa-Na
L
(q,),
CNa-Cs
L
(q,), CNN
L
(q,), CNC
L
(q,), and CCC
L
(q,), respectively.
FIG. 11. Same as Fig. 10, but for q⫽1.34 Å⫺1.
FIG. 12. Longitudinal dispersion relation of the partials,
Na-Na
L
(q) 共open and full squares兲, Cs-Cs
L
(q) 共open circles兲,
number-number NN
L
(q) 共open and full triangles兲, and
concentration-concentrationCC
L
Therefore, as q decreases, CNNL (q,) shows two peaks, in-duced by the high-frequency peak of CNa-NaL (q,) and that of
CCs-CsL (q,), respectively. At the hydrodynamic regime, the high-frequency peak disappears and only the low-frequency one remains that comes from the corresponding Brillouin peak in the SNN(q,). Therefore, the two branches of
NN
L (q) are closely connected with
Cs-Cs
L (q) and with the
high-frequency Na-NaL (q) branches, respectively. Moreover, the high-frequency NNL (q) branch disappears in the hydro-dynamic regime, whereas the low-frequency branch exhibits now a linear behavior similar to that ofCs-CsL (q).
Two branches are also exhibited by theCCL (q) dispersion curve because CCCL (q,), which is substantially small as compared with the other partial currents, shows either one or two peaks that may be connected with propagating concen-tration modes. At small q’s, including the hydrodynamic re-gime, CCCL (q,) shows only one peak, but for greater wave vectors two maxima appear in CCCL (q,): one at a similar frequency as the high-frequency branch of CNNL (q,) and another at a frequency slightly lower than that in
CCs-CsL (q,). In conclusion, the high-frequency branch of
CC
L
(q) exists for all q values, closely follows the high fre-quencyNNL (q) and goes toward a finite value when q→ 0; this is a typical trend of the kinetic modes. On the other hand, the low-frequency branch of CCL (q) appears outside the hydrodynamic regime and closely followsCs-CsL (q).
3. Transverse currents
The partial transverse current correlation functions
Ci j T
(q,) provide information about the existence of shear modes in the system. These modes are not associated directly with any measurable quantity and can only be analyzed by either theoretical models or by MD simulations. Few works have addressed the transverse currents in liquids, and most of them have focused on one-component systems where
CT(q,) evolves 关7兴, as a function of , from a Gaussian 共when q → ⬁) toward a Lorentzian curve 共when q→0). Transverse modes do not propagate in these extreme regimes but at intermediate q’s, the CT(q,) may exhibit a peak associated with propagating shear waves.
The first CMD simulations of transverse current correla-tions in binary fluids were performed on molten salts 关64兴.
CNN
T
(q,) showed a peak at intermediate q’s as in one-component liquids. Also, a peak appeared in the charge-charge transverse current correlation functions at both small and intermediate q’s, which was related to transverse optic modes with a finite value of frequency in the hydrodynamic limit. Subsequent CMD studies in binary Lennard-Jones sys-tems 关14兴 and metallic alloys 关65兴 have found opticlike modes associated with CCCT (q,).
Figures 13 and 14 show some calculated Ci jT(q,). Both
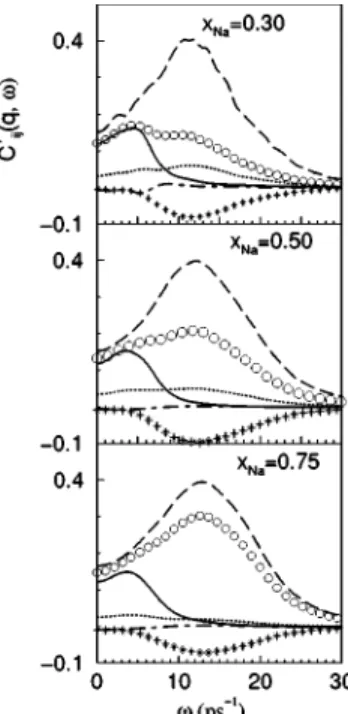
CCs-CsT (q,) and CNa-CsT (q,) show no peaks for the smallest
q’s reached in the simulations (qmin ⬇0.19, 0.17, and 0.16 Å⫺1 for xNa⫽0.75, 0.50, and 0.30, respectively兲,
how-ever, starting from a qc(qc⬇0.23 Å⫺1 for Na0.75Cs0.25 and qc⬇0.32 Å⫺1for Na0.3Cs0.7) a peak appears in both partials,
which persists for a limited range of q values. In contrast,
CNa-NaT (q,) already exhibits a high-frequency peak at qmin, which lasts up to q ⬇4 Å⫺1; moreover, a low-frequency
peak appears in CNa-NaT (q,) at qc, which persists for a lim-ited range and has a similar frequency to those in
CCs-Cs
T
(q,) and CNa-Cs
T
(q,). Notice that CNa-Cs
T
(q,) quickly diminishes as q increases 共see Figs. 13 and 14兲,
in-FIG. 13. Partial transverse current correlation functions
Ci j T
(q,), at q⫽0.43 Å⫺1, for the liquid Na-Cs alloy at T ⫽373 K and three concentrations. The full, dashed, dot-dashed lines, circles, stars, and dotted lines represent CCs-Cs
T
(q,),
CNa-Na
T
(q,), CNa-Cs
T
(q,), CNN
T
(q,), CNC
T
(q,), and CCC
T
(q,), respectively.
dicating strong decoupling of the dynamics of the two spe-cies.
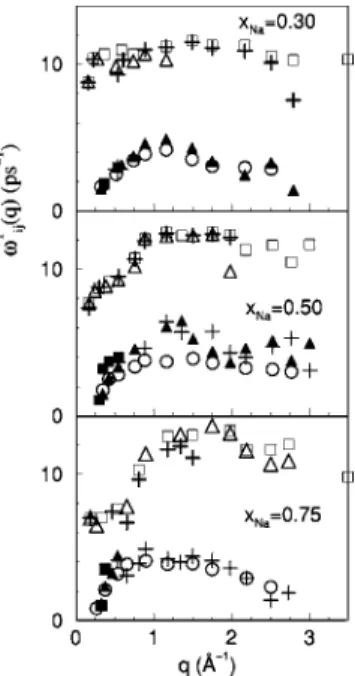
From the peaks in Ci jT(q,), the transverse dispersion relations i jT(q) are obtained 共Fig. 15兲. Cs-CsT (q) has one branch only, whereas Na-NaT (q) has two, with the lower-frequency branch staying rather close to Cs-Cs
T
(q). The ap-pearance of the two branches in the dispersion relation of the lighter species was found earlier in the longitudinal disper-sion relation, and is typical of binary systems with a signifi-cant mass ratio.
The CNNT (q,t) gives information on the average behavior of the system. At qmin, it already shows a high-frequency peak similar to that of CNa-NaT (q,), moreover, a second peak appears for a limited range and shows a behavior typical of a one-component system. Consequently, the dispersion curve,
NN
T (q), has two branches: a high-frequency branch taking a
finite value as q→0, and a low-frequency branch with a linear behavior for very small q values and going to zero as
q → qc. In this linear region the low-frequency branch is quite close toCs-CsT (q) and to the low-frequencyNa-NaT (q) branch, which suggests that the propagation of shear modes involves both species, although the appearance of the high-frequency branch ofNa-NaT (q) shows that some Na particles exhibit kinetic modes in this low q region. Similar behavior was found in the longitudinal dispersion curves in the region within and close to the hydrodynamic regime. The appear-ance of the two branches for the NNT (q) is qualitatively different from the recent CMD simulations of Anento and Padro´关65兴for liquid Li-Mg, Li-Na and Li4Pb alloys, where
the NNT (q) had one branch only. Their NNT (q) in Li-Na, and Li-Mg alloys exhibited behavior qualitatively similar to the present low-frequency NNT (q) branch, whereasNNT (q)
for the Li4Pb alloy behaved similarly to the present
high-frequency branch, remaining finite as q→0. This absence of the propagating transverse collective modes for the Li4Pb
alloy was associated关65兴with the large mass ratio (⬇30) of the components along with a majority concentration of the light Li particles. In the Na-Cs system, the mass ratio ⬇6, and we find two NNT (q) branches for the three concentra-tions considered. As the concentration of the light Na par-ticles is increased, the high frequency NNT (q) branch exists for a larger q-range, while the range of the low-frequency
NN
T (q) branch shrinks toward smaller q values.
A fitting of the low-frequencyNNT (q) branch in the linear region,NN
T
(q)⬃cT(q⫺qc), allows estimation of the veloc-ity of propagation of the shear modes in the alloy: cT⬇570, ⬇850, and ⬇1500 m/s for xNa⫽0.30, 0.50, and 0.75,
re-spectively. For comparison, we have also calculated the ve-locities of the shear modes in pure Na and Cs at the same temperature, 373 K, obtaining cT⬇1700 m/s共pure Na兲 and
cT⬇475 m/s共pure Cs兲.
CCC
T
(q,) is smaller than any of the other Ci j T
(q,), sug-gesting a weak contribution of concentration fluctuations to the collective transverse dynamics; however, clear peaks are seen in CCCT (q,). At qmin, there is a high-frequency peak for a limited q region, it takes finite values as q→0 and follows roughly the high-frequency Na-NaT (q) branch. This behavior has also been observed in molten salts, and was associated not with shear propagating modes but with trans-verse optic modes of kinetic character关14,64兴. The damping of the concentration modes estimated from peak widths does not go to zero as q→0. Bryk, Mryglod, and Kahl关14兴, in their GCM study of transverse dynamics in the liquid Kr-Ar, Mg0.7Zn0.3, Li4Pb, and He0.75Ar0.25systems, reported similar results that they attributed to opticlike nature of the trans-verse concentration fluctuations. This conclusion was cor-roborated by Anento and Padro´ 关65兴 in their CMD simula-tions of liquid Li-based alloys.
CC
T (q) also has a low-frequency branch in a limited q
range, starting at q⬇0.6 Å⫺1, outside the hydrodynamic re-gion and even beyond the linear rere-gion associated with the shear modes. Its disappearance for q⭐0.6 Å⫺1is further evi-dence of the homocoordinating tendencies of the liquid Na-Cs alloy 关14,65兴.
In the hydrodynamic limit 关7兴, the one-component trans-verse current correlation function CT(q,t) becomes
CT共q→0,t兲⫽共1/m兲exp兵⫺q2兩t兩/m其, 共23兲
where  is the inverse temperature times the Boltzmann’s constant, m is the mass of the particles, and is the shear viscosity coefficient. For finite q, using the memory function representation,
C
˜T共q,z兲⫽ 1
m
冋
z⫹ q2m˜共q,z兲
册
⫺1
, 共24兲
where the tilde denotes the Laplace transform and ˜ (q,z) is a generalized shear viscosity coefficient. The area under the normalized CT(q,t) givesmC˜T(q,z⫽0), from which
val-FIG. 15. Transverse dispersion relation of the partials,Na-Na
T
(q)
共open and full squares兲, Cs-Cs
T
(q) 共open circles兲, number-number
NN
T
(q) 共open and full triangles兲, and concentration-concentration
CC
T
ues for˜ (q,z⫽0) can be obtained and, when extrapolated to
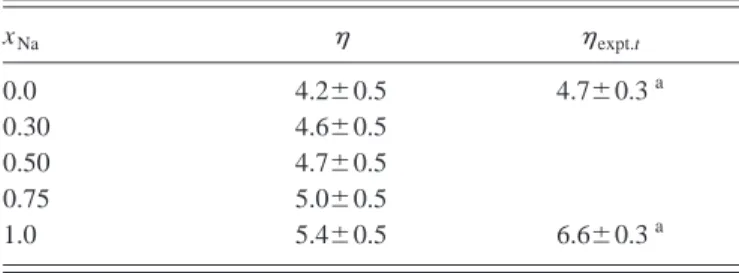
q⫽0, give the shear viscosity. The results for obtained in this way for pure Na and Cs at T⫽373 K are presented in Table IV, showing good agreement with the experiment关66兴. Extension of this scheme to binary systems is straight-forward. The equivalent correlation function is the total transverse current correlation function CttT(q,t) ⫽
具
jtT(q,t) jtT*(q,0)典
, where jtT(q,t)⫽x11/2m1j1T(q,t) ⫹x21/2m2j2T
(q,t) is the total transverse current and jiT(q,t) are defined according to Eq.共21兲. In the hydrodynamic limit,
CttT共q→0,t兲⫽共m¯ /兲exp兵⫺q2兩t兩/m¯其, 共25兲
where m¯⫽x1m1⫹x2m2 and is the alloy shear viscosity.
The results for the alloy shear viscosity calculated in this way are presented in Table IV. No experimental results are available.
The viscosity of simple liquid alloys shows either a linear or slightly concave behavior with concentration, whereas large positive deviations from linearity are exhibited by liq-uid alloys with heterocoordinating tendencies关47,67兴. When homocoordinating tendencies are present, the available ex-perimental data suggest a mixed behavior, with a marked predominance for a negative departure from linearity. More-over, some semiempirical models 关47,67兴 establish a close connection between positive 共negative兲 deviations from the linear law and a negative共positive兲enthalpy of mixing in the alloy. The positive experimental enthalpy of mixing of Na-Cs alloy 关21兴 suggests some negative deviation of the alloy shear viscosity from the linear law. The calculated val-ues obtained for the shear viscosity of the liquid Na-Cs alloy, given in Table IV, show a linear variation with concentration, which according to the previous comments may reflect the real behavior of the system in view of its weak homocoordi-nating tendencies.
IV. CONCLUSIONS
Several static and dynamic properties of the liquid Na-Cs alloy have been calculated for three concentrations. The simulations have been performed using the orbital-free ab
initio molecular dynamics method combined with first
prin-ciples local pseudopotentials derived within the same frame-work. This demonstrates the feasibility of this technique for investigating time correlation functions and the dynamical properties of binary alloys.
The results for the static structural properties show good agreement with the available experimental data, although the simulations predict somewhat weaker homocoordinating ten-dencies. We have analyzed the local arrangement of particles in the mixture and found that when there is a large size mismatch, the biggest value of the minimum of the gi j(r)’s should be used for estimating the coordination numbers.
For the dynamical properties, we have analyzed several time correlation functions, although comparison with the ex-periment could only be made at the level of some transport coefficients. The excellent agreement with the experiment of the calculated self-diffusion coefficients for the pure compo-nents suggests that the simulations of the alloys treats self-diffusion very well. The calculated longitudinal dispersion relations show the existence of kinetic concentration modes. In addition, the results for the adiabatic sound velocity in the pure components and the alloy are in very good agreement with the experiment.
An interesting feature is the appearance of the two branches in the transverse dispersion relation for NNT (q),
Na-Na
T (q), and
CC
T (q), with the high-frequency branch
rep-resenting overdamped kinetic modes. The low-frequency
NN
T
(q) andNa-NaT (q) branches, along with the Cs-CsT (q), go linearly to zero at a finite q value qcand represent propa-gating shear modes. However, the low-frequency CCT (q) branch does not exist for low q values, in agreement with the predictions of the GCM model. The presence of two branches contrasts with the recent results for other binary systems关65兴, where only one branch forNN
T
(q) was found. The shear viscosity of the alloy has been estimated by using its relationship with the hydrodynamic limit of the total transverse current correlation function. The results seem plausible, given the weak homocoordinating tendencies and the positive enthalpy of mixing of the alloy.
The main approximations in the orbital-free ab initio mo-lecular dynamics method used here are the kinetic energy functional and the local pseudopotentials describing the electron-ion interactions. Further improvements in the method will come from the development of more accurate kinetic energy functionals, which would lead to improved local pseudopotentials.
ACKNOWLEDGMENTS
This work has been supported by the Junta de Castilla y Leo´n 共Project No. VA 073/02兲 and the DGES 共Project No. PB98-0641-C02-01兲. D.J.G. acknowledges the financial sup-port of the Spanish Ministry of Education, Culture and Sports, and the University of Valladolid. M.J.S. acknowl-edges the support of the NSERC of Canada.
TABLE IV. Calculated and experimental values of the shear viscosity 共in GPa ps兲for the Na-Cs liquid alloy at T⫽373 K.
xNa expt.t
0.0 4.2⫾0.5 4.7⫾0.3a
0.30 4.6⫾0.5
0.50 4.7⫾0.5
0.75 5.0⫾0.5
1.0 5.4⫾0.5 6.6⫾0.3a
关1兴P. Hohenberg and W. Kohn, Phys. Rev. 136, 864共1964兲. 关2兴W. Kohn and L.J. Sham, Phys. Rev. 140, A1133共1965兲. 关3兴F. Shimojo, Y. Zempo, K. Hoshino, and M. Watabe, J.
Non-Cryst. Solids 205, 983共1996兲; B.J. Costa Cabral and J.L. Mar-tins, Phys. Rev. B 51, 872共1995兲.
关4兴G. Kresse, J. Non-Cryst. Solids 205, 833 共1996兲; G. Kresse and J. Hafner, Phys. Rev. B 55, 7539共1997兲.
关5兴Y. Senda, F. Shimojo, and K. Hoshino, J. Phys. Soc. Jpn. 67, 2753共1998兲.
关6兴B.J. Costa Cabral and J.L. Martins, J. Chem. Phys. 111, 5067 共1999兲.
关7兴U. Balucani and M. Zoppi, Dynamics of the Liquid State共 Clar-endon, Oxford, 1994兲; J.P. Hansen and I.R. McDonald, Theory of Simple Liquids共Academic, London, 1986兲; J.P. Boon and S. Yip, Molecular Hydrodynamics 共McGraw-Hill, New York 1980兲.
关8兴L. Sjo¨gren and A. Sjo¨lander, J. Phys. C 12, 4369 共1979兲; L. Sjo¨gren, ibid. 13, 705 共1980兲; Phys. Rev. A 22, 2866共1980兲; 22, 2883共1980兲.
关9兴J. Casas, D.J. Gonza´lez, L.E. Gonza´lez, M.M.G. Alemany, and L.J. Gallego, Phys. Rev. B 62, 12 095共2000兲.
关10兴G. Jacucci and I.R. McDonald, J. Phys. F: Met. Phys. 10, L15 共1980兲.
关11兴G. Jacucci, M. Ronchetti, and W. Schirmacher, J. Phys. Colloq. 8, 385共1984兲.
关12兴A. Campa and E.G.D. Cohen, Phys. Rev. A 41, 5451共1990兲. 关13兴P. Westerhuijs, W. Montfrooij, L.A. de Graaf, and I.M. de
Schepper, Phys. Rev. A 45, 3749共1992兲.
关14兴T. Bryk, I. Mryglod, and G. Kahl, Phys. Rev. E 56, 2903 共1997兲; T. Bryk and I. Mryglod, Phys. Lett. A 261, 349共1999兲; J. Phys.: Condens. Matter 12, 6063共2000兲.
关15兴E. Enciso, N.G. Almarza, P. Dominguez, M.A. Gonza´lez, and F.J. Bermejo, Phys. Rev. Lett. 74, 4233共1995兲.
关16兴R.M. Crevecoeur, H.E. Smorenburg, and I.M. de Schepper, J. Low Temp. Phys. 105, 149共1996兲.
关17兴R. Fernandez-Perea, M. Alvarez, F.J. Bermejo, P. Verkerk, B. Roessli, and E. Enciso, Phys. Rev. E 58, 4568共1998兲. 关18兴W. Montfrooij, P. Westerhuijs, V.O. de Haan, and I.M. de
Schepper, Phys. Rev. Lett. 63, 544共1989兲.
关19兴P.H.K. de Jong, P. Verkerk, C.F. de Vroege, L.A. de Graaf, W.S. Howells, and S.M. Bennington, J. Phys.: Condens. Matter 6, L681共1994兲.
关20兴M. Alvarez, F.J. Bermejo, P. Verkerk, and B. Roessli, Phys. Rev. Lett. 80, 2141共1998兲.
关21兴F. Yokokawa and O.J. Kleppa, J. Chem. Phys. 40, 46共1964兲. 关22兴K. Ichikawa, S.M. Grandstaff, and J.C. Thomson, J. Chem.
Phys. 61, 4059共1974兲.
关23兴F.E. Neale and N.E. Cusack, J. Phys. F: Met. Phys. 12, 2839 共1982兲.
关24兴M.J. Huijben, W. van der Lugt, W.A.M. Reimert, J.Th.M. de Hosson, and C. van Dijk, Physica B & C 97, 338共1979兲. 关25兴E.G. Visser, W. van der Lugt, and J.Th.M. de Hosson, J. Phys.
F: Met. Phys. 10, 1681共1980兲.
关26兴F.E. Neale and N.E. Cusack, Phys. Chem. Liq. 14, 115 共1984兲.
关27兴J.A. Alonso and L.J. Gallego, J. Phys. F: Met. Phys. 15, L185 共1985兲.
关28兴K. Hoshino, J. Phys.: Condens. Matter 2, 7345共1990兲. 关29兴N. Troullier and J.L. Martins, Phys. Rev. B 43, 1993共1991兲.
关30兴D.M. Ceperley and B.J. Alder, Phys. Rev. Lett. 45, 566共1980兲. 关31兴S.H. Vosko, L. Wilk, and M. Nussair, Can. J. Phys. 58, 1200
共1980兲.
关32兴J.P. Perdew and A. Zunger, Phys. Rev. B 23, 5048共1981兲. 关33兴D.C. Langreth and J.P. Perdew, Phys. Rev. B 15, 2884共1977兲. 关34兴E.S. Kryachko and E.V. Luden˜a, Energy Density Functional Theory of Many-Electron Systems共Kluwer Academic, London, 1990兲, and references therein.
关35兴F. Perrot, J. Phys.: Condens. Matter 6, 431共1994兲.
关36兴E. Smargiassi and P.A. Madden, Phys. Rev. B 49, 5220 共1994兲.
关37兴M. Foley and P.A. Madden, Phys. Rev. B 53, 10 589共1996兲. 关38兴Y.A. Wang, N. Govind, and E.A. Carter, Phys. Rev. B 58, 13
465共1998兲; 60, 16 350共1999兲.
关39兴E. Chaco´n, J.E. Alvarellos, and P. Tarazona, Phys. Rev. B 32, 7868共1985兲.
关40兴P. Garcia-Gonza´lez, J.E. Alvarellos, and E. Chaco´n, Phys. Rev. A 54, 1897 共1996兲; Phys. Rev. B 53, 9509 共1996兲; 57, 4857 共1998兲.
关41兴L.E. Gonza´lez, D.J. Gonza´lez, and J.M. Lo´pez, J. Phys.: Con-dens. Matter 13, 7801共2001兲.
关42兴L.E. Gonza´lez, A. Meyer, M.P. In˜iguez, D.J. Gonza´lez, and M. Silbert, Phys. Rev. E 47, 4120共1993兲.
关43兴R. Car and M. Parrinello, Phys. Rev. Lett. 55, 2471共1985兲. 关44兴See, for instance, M.P. Allen and D.J. Tildesley, Computer
Simulation of Liquids 共Oxford University Press, Oxford, 1987兲.
关45兴B.E. Warren, X-Ray Diffraction 共Addison-Wesley, Reading MA, 1969兲.
关46兴J.M. Cowley, Phys. Rev. 77, 667共1950兲.
关47兴R.N. Singh and F. Sommer, Rep. Prog. Phys. 60, 57共1997兲. 关48兴R.L. McGreevy, A. Baranyai, and I. Ruff, Phys. Chem. Liq. 16,
47共1986兲.
关49兴K. Nakanishi, S. Okazaki, K. Ikari, T. Iguchi, and H. Tanaka, J. Chem. Phys. 76, 629共1982兲.
关50兴M. Schoen and C. Hoheisel, Mol. Phys. 53, 1367共1984兲. 关51兴P. Borgelt, C. Hoheisel, and G. Stell, J. Chem. Phys. 92, 6161
共1990兲.
关52兴C. Rey, L.J. Gallego, L.E. Gonza´lez, and D.J. Gonza´lez, J. Chem. Phys. 97, 5121共1992兲.
关53兴M.C. Abramo, C. Caccamo, and G. Giunta, Phys. Rev. A 34, 3279共1986兲.
关54兴C. Hoheisel and F. Kohler, Fluid Phase Equilib. 16, 13共1984兲. 关55兴L.L. Lee, T.H. Chung, and K.E. Starling, Fluid Phase Equilib.
12, 105共1983兲.
关56兴R.G. Rubio, M.G. Prolongo, M. Diaz-Pen˜a, and J.A.R. Renun-cio, J. Phys. Chem. 91, 1177共1987兲.
关57兴A.B. Bhatia and D.E. Thornton, Phys. Rev. B 2, 3004共1970兲. 关58兴J. Trulla´s and J.A. Padro´, J. Chem. Phys. 99, 3983 共1993兲;
Phys. Rev. E 50, 1162共1994兲.
关59兴T. Kato, J. Phys. Chem. 89, 5750共1985兲.
关60兴A. Feinauer and G. Majer, Phys. Rev. B 64, 134302共2001兲. 关61兴M. Gerl and A. Bruson, in Handbook of Thermodynamic and
Transport Properties of Alkali Metals, edited by R.W. Oshe 共Blackwell, Oxford, 1985兲, p. 843.
关62兴N.H. March and M.P. Tosi, Atomic Dynamics in Liquids共 Do-ver Publications, New York, 1991兲.
London, Ser. A 357, 37共1977兲.
关65兴N. Anento and J.A. Padro´, Mol. Phys. 99, 275共2001兲. 关66兴E.E. Shpil’rain, K.A. Yakimovich, V.A. Fomin, S.N.
Sko-vorodjko, and A.G. Mozgovoi, Handbook of Thermodynamic
and Transport Properties of Alkali Metals, edited by R.W. Ohse共Blackwell, Oxford, 1985兲, p. 753.