METODOLOGÍA SINTÉTICA
APLICADA A LA SÍNTESIS DE
FÁRMACOS
Tema 3
Enfermedades del sistema
nervioso central: síntesis de
antidepresivos, antiepilépticos y
anti-Parkinson
Tema 3. Enfermedades del sistema nervioso central
3.1. Neurotransmisores 1
3.1.1. Tipos de neurotransmisores 3
3.2. Inhibidores Selectivos de la Recaptación de Serotonina (ISRS) 6
3.2.1. Liberación de la serotonina 8
3.2.2. Receptores se seronotina 9
3.3. Otros inhibidores selectivos de recaptación de serotonina 11
3.4. Fármacos Inhibidores Selectivos de la Recaptación de Serotonina 11
3.5. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina 15
3.5.1. Síntesis de escitalopram 15
3.5.1.a. Análisis retrosintético 15
3.5.1.b. Síntesis 15
3.5.1.c. Cuestiones 17
3.5.2. Síntesis de dapoxetina 17
3.5.2.1a. Análisis retrosintético 17
3.5.2.1b. Síntesis 16
3.5.2.2b. Síntesis de dapoxetina empleando (R)-fenilglicina como
material quiral de partida 18
3.5.2.2c. Cuestiones 19
3.5.3. Síntesis de fluoxetina (Prozac) 19
3.5.3.a. Análisis retrosintético 19
3.5.3.b. Síntesis 19
3.5.3.c. Cuestiones 20
3.5.4. Síntesis de sertralina 21
3.5.4.a. Análisis retrosintético 21
3.5.4.b. Síntesis 22
3.5.4.c. Cuestiones 23
3.5.5. Síntesis de paroxetina 26
3.5.5.1a. Análisis retrosintético 26
3.5.5.1b. Síntesis 27
3.5.5.1c. Cuestiones 28
3.5.5.2a. Análisis retrosintético de paroxetina mediante la estrategia de
Adición de Quiralidad (AQ) 28
3.5.5.2b. Síntesis de (+)-paroxetina mediante el empleo de un fragmento
quiral (Adición de Quiralidad) 29
3.5.5.2c. Cuestiones 30
3.5.5.3a. Análisis retrosintético de paroxetina mediante una estrategia
3.5.5.3b. Síntesis de paroxetina mediante desimetrización enantioselectiva
de un diéster simétrico 32
3.5.5.3c. Cuestiones 33
3.6. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina y
Norepinefrina (ISRSN) 35
3.6.1. Síntesis de venlafaxina 35
3.6.1.a. Análisis retrosintético 35
3.6.1.b. Síntesis 35
3.6.1.c. Cuestiones 36
3.6.2. Síntesis desvenlafaxina 36
3.6.2.a. Análisis retrosintético 36
3.6.2.b. Síntesis 37
3.6.3. Síntesis de milnacipran 37
3.6.3.a. Análisis retrosintético 37
3.6.3.b. Síntesis 38
3.6.3.c. Cuestiones 39
3.6.4. Sïntesis de duloxetina 39
3.6.4.1a. Análisis retrosintético 39
3.6.4.1b. Síntesis 40
3.6.4.1c. Cuestiones 41
3.6.4.2a. Análisis retrosintético de duloxetina mediante reducción
enantioselectiva 41
3.6.4.2b. Síntesis de duloxetina mediante reducción enantioselectiva 42
3.6.4.2c. Cuestiones 42
3.6.4.3a. Análisis retrosintético de duloxetina mediante esterificación
enzimática enantioselectiva 44
3.6.4.3b. Síntesis de duloxetina mediante mediante esterificación
enzimática enantioselectiva 44
3.6.4.3c. Cuestiones 45
3.7. Síntesis de Inhibidores Selectivos de la Recaptación de Noradrenalina (ISRN) 47
3.7.1. Síntesis de atomoxetina 47
3.7.1.1a. Análisis retrosintetico 47
3.7.1.1b. Síntesis 48
3.7.c.1c. Cuestiones 49
3.7.1.2a. Análisis retrosintetico de atomoxetina mediante reacción SNAr 49
3.7.1.2b. Síntesis de atomoxetina mediante reacción SNAr 50
3.7.1.2c. Cuestiones 51
3.8.1. Síntesis de amitriptilina 52
3.8.1.a. Análisis retrosintético 52
3.8.1.b. Síntesis 53
3.8.1.c. Cuestiones 53
3.8.2. Síntesis de imipramina 53
3.8.2.a. Análisis retrosintético 54
3.8.2.b. Síntesis 54
3.8.2.c. Cuestiones 54
3.9. Epilepsia 55
3.10. Fármacos antiepilépticos 55
3.10.1. GABA: un neurotransmisor inhibitorio cerebral 58
3.10.1.a. Receptores de GABA 60
3.10.2. Fármacos antiepilépticos: gabapentina y pregabalina 64
3.10.3. Modo de acción de la pregabalina 66
3.11. Sintesis de fármacos antiepilépticos 68
3.11.1. Síntesis de gabapentina 68
3.11.1.1a. Análisis retrosintético 68
3.11.1.1b. Síntesis 68
3.11.1.1c. Cuestiones 69
3.11.1.2a. Análisis retrosintético 69
3.11.1.2b. Síntesis 69
3.11.2. Sintesis de pregabalina 70
3.11.2.1a. Análisis retrosintético 70
3.11.2.1b. Síntesis 70
3.11.2.1c. Cuestiones 71
3.11.2.2a. Análisis retrosintético de (S)-pregabalina mediante el empleo
de un auxiliar quiral 71
3.11.2.2b. Síntesis de (S)-pregabalina mediante el empleo de una
oxazolidinona quiral de Evans 72
3.11.2.2c. Cuestiones 73
3.11.2.3a. Análisis retrosintético de (S)-pregabalina mediante el empleo
del pool quiral 75
3.11.2.3b. Síntesis de (S)-pregabalina a partir de L-leucina 76
3.11.2.3c. Cuestiones 77
3.11.3. Síntesis de rufinamida 78
3.11.3.a. Análisis retrosintético 79
3.11.3.c. Cuestiones 80
3.11.4. Síntesis de lacosamida 80
3.11.4.a. Análisis retrosintético 70
3.11.4.b. Síntesis 81
3.11.5. Síntesis de perampanel 82
3.11.5.a. Análisis retrosintético 83
3.11.5.b. Síntesis 84
3.11.5.c. Cuestiones 86
3.12. Enfermedad de Parkinson 88
3.12.1. Causas de la enfermedad de Parkinson 88
3.13. Fármacos antiParkinson 89
3.13.1. Levodopa 90
3.13.2. Agonistas de dopamina 91
3.13.3. Inhibidores de la monoaminooxidasa B: selegilina y rasagilina 92
3.13.4. Liberadores presinápticos de dopamina: amantadina 93
3.13.5. Antagonistas del receptor muscarínico de la acetilcolina: benztropina 94
3.14. Síntesis de fármacos antiParkinson 94
3.14.1. Síntesis de pramiprexol 94
3.14.1.a. Análisis retrosintético 94
3.14.1.b. Síntesis 95
3.14.1.c. Cuestiones 95
3.14.2. Síntesis de ropinirol 95
3.14.2.a. Análisis retrosintético 96
3.14.2.b. Síntesis 97
3.14.2.c. Cuestiones 98
3.14.2.2b. Síntesis a partir de isocromano 98
3.14.2.2c. Cuestiones 99
3.14.3. Síntesis de selegilina 100
3.13.3.a. Análisis retrosintético 100
3.13.3.b. Síntesis 100
3.13.3.c. Cuestiones 102
3.14.4. Síntesis de mesilato de rasagilina 103
3.13.4.a. Análisis retrosintético 103
3.1. Neurotransmisores
Los neurotransmisores (NT) son los compuestos encargados de transmitir el impulso nervioso entre neuronas. Estos metabolitos son sintetizados por enzimas existentes en el cuerpo neuronal y son almacenados en vesículas de las células presinápticas.
El mecanismo de comunicación interneuronal se denomina sinapsis y se inicia con una descarga química que origina una corriente eléctrica en la membrana de la célula presináptica (célula emisora). Cuando el impulso nervioso alcanza el extremo del axón (la conexión con la otra célula), la neurona presináptica segrega el neurotransmisor que se une a los receptores ubicados en la célula postsináptica (figura 3.1).
Figura 3.1. Representación esquemática del proceso de sinapsis
Los receptores de los NT pueden ser canales iónicos abiertos por ligando (receptor en color amarillo de la figura 3.2) o receptores acoplados a proteínas G.
Dominio de unión del ligando
Poro de entrada de iones
Bicapa lipídica
Canal iónico controlado por ligando
Proteína G Citoplasma Exterior celular Dominio de unión
N-terminal Receptor acoplado a proteína G
Los receptores acoplados a proteína G están constituidos por una larga cadena de proteína que serpentea dentro y fuera de la célula (receptor en color naranja de la figura 3.2, véase el tema anterior). La interacción NT-receptor debe concluir de forma inmediata para que el mismo receptor pueda ser activado repetidamente. Para ello, el NT es captado rápidamente por la terminación presináptica mediante un proceso activo (recaptación) introduciéndolo de nuevo en las vesículas presinápticas (figura 3.3).
Célula presináptica
Reabsorción del neurotransmisor
Célula postsináptica Neurotransmisor
Figura 3.3. Proceso de liberación y recaptación del neurotransmisor
Algunos neurotransmisores como la acetilcolina (ACh), la glicina, el glutamato, el aspartato y el ácido γ-aminobutírico (GABA), tienen una actividad biológica directa, aumentando la conductancia a ciertos iones por adherencia a canales iónicos activados en la membrana postsináptica (parte a de la figura 3.4).
Otros neurotransmisores, como la noradrenalina (NA), la dopamina (DA) y la serotonina (5-HT), no tienen actividad directa, pero provocan la respuesta postsináptica actuando indirectamente en sistemas que implican adenosín-monofosfato-cíclico (cAMP, véase la parte b de la figura 3.4), guanidín-monofosfato-cíclico (GMPc), inositol trifosfato (ITP), diacilglicerol (DAG), prostaglandinas (Pgs), leucotrienos, epóxidos y Ca2+.
Los receptores acoplados a proteínas G (GPCRs) son el grupo más grande de receptores. Se han identificado unos 700 genes en el genoma humano que sirven para la producción de GPCRs. El acoplamiento del NT al receptor provoca la unión de éste a la proteína heterotrimérica G (paso 1 de la figura 3.4). La proteína G está anclada a la membrana celular y está constituida por tres subunidades diferentes denominadas alfa, beta y gamma. En su estado inactivo, la subunidad alfa contiene un grupo de guanosina difosfato (GDP).
La unión de la proteína G estimula la adenililato ciclasa, lo que conduce al aumento de la concentración intracelular del adenosin monofosfato cíclico (cAMP, paso 4 de la figura 3.4). La producción de cAMP activa los procesos indicados en la figura 3.4.
Figura 3.4. Modos de acción de los neurotransmisores
La cantidad de neurotransmisor en las terminaciones se mantiene relativamente constante e independiente de la actividad nerviosa mediante una regulación estrecha de su biosíntesis. Este control varía de unas neuronas a otras y depende de la capacidad de recaptación del neurotransmisor y de la actividad enzimática encargada de su formación y catabolismo. La estimulación o el bloqueo de los receptores postsinápticos también pueden aumentar, o disminuir, la síntesis presináptica del neurotransmisor.
Las alteraciones en la síntesis, almacenamiento, liberación, degradación o recaptación de los NT, o el cambio en el número y/o actividad de los receptores, afectan a la neurotransmisión y pueden producir trastornos mentales.
3.1.1. Tipos de neurotransmisores
Los principales neurotransmisores pueden clasificarse según su tamaño en:
a) Neurotransmisores de pequeño tamaño de tipo aminoácido: glicina, ácido aspártico, ácido glutámico:
Figura 3.5. Aminoácidos con actividad neurotransmisora
Los aminoácidos glutamato y aspartato son los principales NT excitatorios del sistema nervioso central. Están presentes en la corteza cerebral, el cerebelo y la médula espinal. b) Neurotransmisores de pequeño tamaño derivados de aminoácidos: GABA, histamina, serotonina, norepinefrina y dopamina:
Figura 3.6. Neurotransmisores derivados de aminoácidos
El ácido -aminobutírico (GABA) es el principal NT inhibitorio cerebral. Deriva del ácido glutámico mediante la descarboxilación provocada por la enzima glutamato-descarboxilasa. Tras la interacción con los receptores específicos, el GABA es recaptado y metabolizado.
La histamina es un NT del sistema nervioso central. También interviene decisivamente en las reacciones de hipersensibilidad inmediata y alérgica. La histamina se forma por descarboxilación del aminoácido histidina catalizada por el enzima L-histidina-descarboxilasa.
La serotonina (5-hidroxitriptamina) (5-HT) participa en el control de los estados de sueño y de vigilia. Interviene regulando los estados de ánimo y las emociones y es decisiva en el desencadenamiento de algunos tipos de depresión. También interviene en el control de la temperatura del cuerpo, de la conducta sexual y de ciertos estados alucinatorios inducidos por drogas. La serotonina se origina en el núcleo del rafe (estructuras del encéfalo) y en las neuronas de la línea media de la protuberancia y del meséncefalo.
La norepinefrina (noradrenalina) es el NT de la mayor parte de las fibras simpáticas postganglionares y de muchas neuronas centrales, por ejemplo del locus ceruleus y del hipotálamo. El precursor de la noradrenalina es la tirosina, que se convierte en dopamina, que a su vez es hidroxilada por la dopamina β-hidroxilasa a noradrenalina. Cuando se libera la noradrenalina interactúa con los receptores adrenérgicos (véase el tema 2), proceso que finaliza con su recaptación por las neuronas presinápticas y su degradación por la monoaminoxidasa (MAO) y por la catecol-O-metiltransferasa (COMT). La tirosina-hidroxilasa y la MAO regulan los niveles intraneuronales de noradrenalina.
c) Neuropéptidos: metabolitos compuestos por más de 3 aminoácidos como la somatostatina, la vasopresina y la oxitocina. Muchos de estos neuropéptidos actúan también como hormonas, denominándose en estos casos neurohormonas.
d) Otros neurotransmisores: acetilcolina
Figura 3.7. Estructura de la acetilcolina
La acetilcolina es el NT fundamental de las neuronas motoras bulbo-espinales, las fibras preganglionares autónomas, las fibras colinérgicas postganglionares (parasimpáticas) y muchos grupos neuronales del SNC, como los de los ganglios basales y de la corteza motora. Se biosintetiza a partir de la colina y de la acetil-coenzima A mitocondrial mediante acción de la enzima colinacetiltransferasa. Al ser liberada, la acetilcolina estimula receptores colinérgicos específicos y su interacción finaliza rápidamente por hidrólisis local a colina y acetato mediante la acción de la acetilcolinesterasa. Los niveles de acetilcolina están regulados por la acetilcolintransferasa y por el grado de recaptación de colina.
Los neurotransmisores también se pueden clasificar en función de su estructura química del siguiente modo:
3.2. Inhibidores Selectivos de la Recaptación de Serotonina (ISRS)
La depresión severa (Trastorno Depresivo Mayor, en inglés Major Depressive Disorder MDD) se manifiesta por una combinación de síntomas como tristeza patológica, apatía, ansiedad, etc, que interfieren en la capacidad para trabajar, estudiar, dormir, comer y disfrutar de actividades que antes le eran placenteras al enfermo que sufre la MDD.
Se acepta en general que la depresión está relacionada con la reducción de la trasmisión del impulso nervioso en zonas específicas del sistema nervioso central provocada por un déficit de neurotransmisores en la sinapsis. De hecho, todos los antidepresivos actúan aumentando la concentración de aminas neurotransmisoras en la sinapsis.
Una vez producido el impulso nervioso, el 95% de aminas liberadas son vueltas a recaptar por la neurona presináptica en preparación del siguiente impulso. El 5% no recaptado es destruido por la enzima monoaminooxidasa (MAO). Las pérdidas de neurotransmisores son repuestas a partir de precursores metabólicos.
La serotonina se clasifica dentro del grupo de los neurotransmisores adrenérgicos, que son aquéllos que se unen a receptores acoplados a proteína G.
Célula presináptica
Célula postsináptica Serotonina
Receptor de serotonina
Figura 3.9. Unión de la setononina al receptor postsináptico
La serotonina, también denominada 5-hidroxitriptamina (5-HT) se genera en el organismo mediante hidroxilación del L-triptófano catalizada por la enzima triptófano-hidroxilasa (esquema 3.1). La hidroxilación del triptófano produce el 5-hidroxi-L-triptófano, el cual se transforma en serotonina por descarboxilación catalizada por la enzima 5-hidroxi-L-triptófano descarboxilasa.
Esquema 3.1. Biosíntesis de la serotonina
Los niveles de serotonina están regulados por la disponibilidad de L-triptófano y por la acción de la monoaminooxidasa (MAO). La biodegradación de la serotonina, se lleva a cabo tanto a nivel intracelular como en la hendidura sináptica por la acción enzimática de la MAO, que la convierte en 5-hidroxi-indolacetaldehído, que es su principal metabolito inactivo. Este metabolito es oxidado por la enzima aldehído-deshidrogenasa y transformado en ácido 5-hidroxi-indolacético (esquema 3.2).
Esquema 3.2. Degradación enzimática de la serotonina
En humanos existen dos tipos de MAO: MAO-A y MAO-B. La MAO-A es particularmente importante en el catabolismo de monoaminas ingeridas con el alimento. Ambas MAOs juegan un papel clave en la inactivación de los neurotransmisores monoaminérgicos. Así, la serotonina, norepinefrina (noradrenalina), y epinefrina (adrenalina) son degradadas en su mayoría por la MAO-A. La fenetilamina es degradada por la MAO-B, mientras que la dopamina es degradada por ambas MAO.
Célula presináptica
Célula postsináptica Serotonina
Receptor de serotonina Destrucción por
monoamino-oxidasa
5-HTP
Serotonina Triptófano
Recaptación
Liberación de serotonina
Figura 3.10. Liberación y recaptación de serotonina
Los inhibidores selectivos de la recaptación de la serotonina (ISRS) son una clase de antidepresivos utilizados en el tratamiento de la depresión, trastorno por ansiedad y algunos trastornos de la personalidad. Actúan aumentando los niveles extracelulares del neurotransmisor serotonina, inhibiendo su recaptación por la neurona presináptica e incrementando de esta forma el nivel de serotonina disponible para unirse con el receptor postsináptico.
Aunque existe serotonina en todo el cuerpo ésta es incapaz de atravesar la barrera hematoencefálica, por lo que el cerebro produce su propia serotonina. La biosíntesis de serotonina cerebral depende del aporte del aminoácido L-triptófano. Este es un aminoácido esencial y por tanto el organismo no lo puede biosintetizar. El L-triptófano sólo puede provenir de la dieta, por lo que sus niveles cerebrales dependen, en parte, de los alimentos ingeridos. El L-triptófano abunda en los huevos, la leche, los cereales integrales, el chocolate, la avena, los dátiles, las semillas de sésamo, los garbanzos, las pipas de girasol, las pipas de calabaza y los cacahuetes. Las personas que no ingieren estos alimentos tienen mayor riesgo de deficiencia de triptófano, así como aquellas personas sometidas a altos niveles de estrés. Para un buen metabolismo del triptófano se requieren niveles adecuados de vitamina B6 y de magnesio.
3.2.1. Liberación de la serotonina
Representación esquemática de un canal iónico
1= Dominio del canal iónico. 2=Vestíbulo externo 3= Filtro de selección. 4=Diámetro del filtro de selección
5=Sitio de fosforilación. 6=Membrana celular
Figura 3.11. Estructura de un canal inónico
Los canales iónicos de calcio son proteínas oligoméricas constituidos por una subunidad principal α1, que sirve como poro y sensor del cambio de potencial, y diversas
subunidades reguladoras o auxiliares tales como la subunidad β, las subunidades α2σ
(unidas por puentes disulfuro) y, dependiendo del tejido, una quinta subunidad (véase la figura 3.12).
Figura 3.12. Vista superior de un canal de calcio
Cuando llega un impulso nervioso a la neurona presináptica ésta abre los canales de Ca2+ y los iones entran en la neurona, lo que activa el proceso de exocitosis provocándose
el traslado de las vesículas a los lugares de su liberación con la ayuda de proteínas de membrana plasmática y de la membrana vesicular. Este proceso desemboca en la liberación del neurotransmisor en el espacio sináptico.
3.2.2. Receptores de serotonina
Los principales receptores de serotonina son el 5-HT1, el 5-HT2 y el 5-HT3. Éstos, a su
vez, se subdividen en cuatro subtipos del 5-HT1 (de la A a la D), dos del 5-HT2 (A y B) y uno
del 5-HT3. De todos ellos, la mayoría son postsinápticos, pero al menos dos de ellos (el
neurotransmisor. Los receptores de serotonina se localizan en la membrana celular de las células nerviosas. Con la excepción del receptor 5-HT3, que es un canal iónico asociado a
ligando, los demás receptores son receptores acoplados a proteínas G, que son también conocidos como receptores 7TM (transmembrana) o "en serpentina", debido a la región incluida en la membrana, que asoma siete veces. En la figura 3.13 se representa esquemáticamente la estructura de un receptor GPCR de serotonina (5-HT).
Figura 3.13. Representación esquemática de un receptor GPCR de serotonina (5-HT)
En la figura inferior 3.14 se ilustra una representación del receptor visto desde la cara extracelular. La flecha señala la zona de interacción con el neurotransmisor.
NT
Figura 3.14. Vista superior extracelular de un receptor GPCR
Los receptores 5-HT1A están asociados a la apertura de canales de K+,
presumiblemente de forma directa a través de una proteína G. En las áreas del campo terminal como el hipocampo, los receptores 5-HT1A están también asociados, mediante
proteína G, a la inhibición de la actividad de la adenilciclasa.
Los receptores 5-HT1B y 5-HT1D también están asociados a la inhibición de
adenilciclasa a través de la proteína G.
Los receptores 5-HT1C y 5-HT2 están asociados a través de la proteína G a la
El receptor 5-HT3 es de tipo canal iónico, por lo que su activación no es mediada por
segundo mensajero o a través de proteínas G.
El receptor 5-HT4 está asociado a la estimulación de la actividad de la adenilciclasa y a la inhibición de canales de K+. Se ha demostrado que la inhibición de canales de K+ en
neuronas del colículo implica la producción de adenosin monofosfato cíclico (AMPc) y la activación de proteína-quinasa A dependiente de AMPc.
Figura 3.15. Acción de los receptores en el proceso de liberación y recaptación de serotonina
3.3. Otros inhibidores selectivos de recaptación de serotonina
Los ISRS pertenecen a una subclase de inhibidores de la recaptación de serotonina que incluye también a otros inhibidores no selectivos como:
a) Los inhibidores de la recaptación de serotonina-noradrenalina-dopamina. b) Los inhibidores de la recaptación de serotonina-norepinefrina.
c) Los estimulantes selectivos de la recaptación de serotonina.
3.4. Fármacos inhibidores selectivos de la recaptación de neutrotransmisores
Las primeras moléculas empleadas en el tratamiento del Trastorno Depresivo Mayor fueron los denominados antidepresivos tricíclicos, como la imipramina, que se introdujeron en el mercado en la década de 1950. En la década de l960 se introdujeron los inhibidores de monoaminooxidasa (IMAO), de entre los cuales cabe destacar a la isocarboxazida.
Los inhibidores de MAO (IMAO) ejercen su acción antidepresiva aumentando los niveles de monaminas, como la serotonina, la norepinefrina o la dopamina. Desafortunadamente los inhibidores de MAO tienen importantes efectos secundarios, entre los que destaca la supresión de la reabsorción de tiramina, por lo que se debe evitar la administración de inhibidores de MAO con la ingesta de alimentos que contengan una alta concentración de tiramina, tales como alimentos fermentados, arenques, o hígado de pollo, ya que la combinación de tiramina con IMAO puede provocar hemorragias cerebrales debido a aumentos bruscos de la presión arterial.
Figura 3.17. Estructura de la tiramina
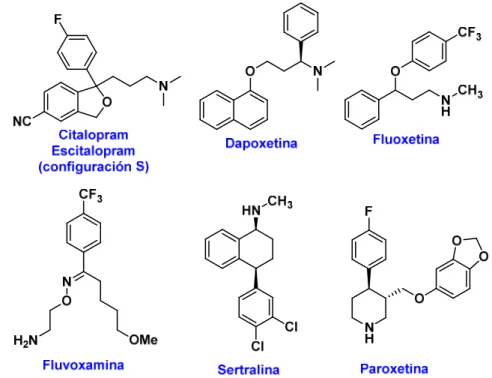
Las estructuras de fármacos inhibidores selectivos de la recaptación de la serotonina (ISRS) se indican en la figura 3.18.
Figura 3.18. Estructuras de fármacos ISRS
Figura 3.19. Liberación e inhibición de la recaptación de serotonina por ISRS
En la década de 1990 se introdujo una nueva generación de fármacos denominados inhibidores selectivos de la recaptación de serotonina y norepinefrina (ISRSN, en inglés SSNRI Selective Serotonin Norepinephrine Reuptake Inhibitors). Estos nuevos fármacos, como la venlafaxina, el milnacipran o la duloxetina (figura 3.20), son capaces de reducir más eficientemente que los ISRS los síntomas de la depresión debido a su acción dual sobre vías neuronales diferentes.
Figura 3.20. Estructuras de fármacos ISRSN
Otros fármacos empleados en el tratamiento de la depresión y desórdenes de tipo nervioso son inhibidores selectivos de la recaptación de noradrenalina (ISRN). Las estructuras de algunos de estos fármacos se indican en la figura 3.21.
Figura 3.21. Estructuras de fármacos ISRN
indica, resaltada en azul y en trazo más grueso, esta parte de 3-ariloxipropilamina que contienen algunos de los fármacos empleados en el tratamiento de la depresión y otras enfermedades relacionadas.
3.5. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina
3.5.1. Síntesis de escitalopram
El citalopram es un inhibidor selectivo de la recaptación de serotonina utilizado en el tratamiento de los síntomas de depresión. También se prescribe para el tratamiento de la fobia social, trastorno de pánico y el trastorno obsesivo compulsivo. El enantiómero S del racemato citalopram se denomina escitalopram. El desarrollo de este fármaco se inició conjuntamente en 1997 por los laboratorios Lundbeck y Forest. En 2001 la FDA aprobó su comercialización en Estados Unidos.
NC O F N Me Me NC O F N Me Me Citalopram Escitalopram Figura 3.23. Estructuras del citalopram y del escitalopram
3.5.1.a. Análisis retrosintético
El análisis retrosintético del escitalopram se inicia con la escisión del anillo tetrahidrofuránico que se construirá mediante una reacción SNi sobre el alcohol
funcionalizado 3.1 (X=grupo saliente, esquema 3.3).
O NC O F Escitalopram C-O NC OH F X NC O F N Me Me X NC F Met C-C C-C Met OH + + NC O O Br O O H2N
O O
N
Me Me MeN Me
3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 IGF IGF Esquema 3.3
La desconexión de la cadena de N,N-dimetil propilo en el compuesto 3.1 genera la cetona 3.2 y el compuesto organometálico 3.3. La desconexión del grupo p-fluorofenilo en la cetona 3.2 conduce al sintón catiónico 3.4 y al reactivo organometálico 3.5. El equivalente sintético del sintón catiónico 3.5 es la 5-cianoftalida 3.6 que por interconversiones de grupo funcional se convierte primero en la 5-bromoftalida 3.7 y luego en la 5-aminoftalida 3.8.
3.5.1.b. Síntesis
por hidrogenación se convierte en la 5-aminoftalimida 3.11.1 La reducción del anillo de
ftalimida con zinc en presencia de sulfato cúprico e hidróxido sódico, en agua a reflujo, proporciona directamente la aminoftalida 3.8, que se convierte en la correspondiente sal de arildiazonio, con NaNO2 y HBr, y luego en la bromoftalida 3.7 por reacción de Sandmeyer
con CuBr. La reacción de 3.7 con cianuro de zinc en presencia de Pd(PPh3)4 conduce a la
5-cianoftalida 3.6.2
Esquema 3.4
Cuando la ftalida 3.6 se trata con bromuro de 4-fluorofenilmagnesio se obtiene la hidroxicetona 3.2, que por reacción con el bromuro de (3-dimetilaminopropil)magnesio se transforma en el aminodiol 3.12. La resolución de este compuesto se consigue con el ácido (+)-O,O´-di-p-toluiltartárico, lo que permite la obtención del (S)-3.12. Cuando este compuesto se trata con cloruro de mesilo se genera el mesilato 3.1 que se convierte en escitalopram por reacción SNi.3
1 T. W. Bell, J. I. Cline, C. R. Cremo, S. L. Gillett, J. H. Frederick, John H. Patent:US 2011/77394A1,
2011.
2H. Lundbeck A/S Patent: US198391A1, 2002.
3 Para patentes relacionados con la síntesis del citalopram y del escitalopram véase: (a) K. P. Boegesoe, J. Perregaard, Patente: US4943590 1990. (b) K. P. Boegesoe, A. S. Toft, Patente: US4136193 1979. (c) H. Ahmadian, H. Petersen, Patente: WO03051861 2003. (d) K. P. Boegesoe,
3.5.1.c. Cuestiones
1) Proponga un mecanismo para la reducción de 3.11 con Zn ¿Por qué la reducción de 3.11 para dar 3.8 es regioselectiva?
2) Explique mecanísticamente la conversión de 3.7 en 3.6.
3) ¿Por qué en el último paso de la síntesis del escitalopram, en la que se crea el anillo furánico mediante tratamiento con MsCl, no se produce inversión de la configuración en el estereocentro?
3.5.2. Síntesis de dapoxetina
La dapoxetina es un inhibidor selectivo de la recaptación de serotonina de corta duración de acción. Su poca eficacia como antidepresivo llevó a investigar otras aplicaciones terapéuticas como el tratamiento de la eyaculación precoz.
3.5.2.1a. Análisis retrosintético
En el esquema 3.5 de indica un análisis retrosintético de la dapoxetina que se inicia con la desconexión del sistema naftalénico basado en una reacción SNAr. La operacicón
retrosintética conduce al aminoalcohol 3.13 (el nucleófilo de la reacción SNAr) y el
-halonaftaleno 3.14. La operación de intercambio de grupo funcional en el aminoalcohol 3.13 genera el -aminoéster 3.15 que se sintetizará mediante adición conjugada Michael de la dimetilanina al éster conjugado 3.16.
Esquema 3.5
3.5.2.1b. Síntesis
La síntesis de la dapoxetina se inicia con la adición conjugada de la dimetilamina al cinamato de etilo 3.16 lo que proporciona el β-aminoéster 3.15 (esquema 3.6).4 Este
compuesto, por reducción con LiAlH4 se convierte en el aminoalcohol 3.13. Cuando este
compuesto se calienta a 100ºC con α-fluoronaftaleno 3.14, en N,N-dimetilacetamida (DMA) en prencia de NaOH, se obtiene la dapoxetina racémica. La resolución con ácido (R,R)-tartárico proporciona la (S)-dapoxetina.
Esquema 3.6
3.5.2.2b. Síntesis de dapoxetina empleando (R)-fenilglicina como material quiral de partida
En el esquema 3.7 se indica una síntesis de (S)-dapoxetina llevada a cabo por los mismos autores que efectuaron la síntesis anterior. En este caso se elige como compuesto de partida el aminoácido no natural (R)-fenilglicina 3.17 que se convierte en el aminoácido
3.18 N-Boc protegido.
Esquema 3.7
La reducción de 3.18 con borano proporciona el alcohol 3.19 el cual, mediante mesilación y reacción con cianuro sódico, se convierte en el nitrilo 3.20. La hidrólisis de este compuesto conduce al aminoácido 3.21 que por reducción con borano forma el aminoalcohol 3.21. La metilación de Eschweiler-Clarke de 3.21 lo convierte en el N,N-dimetilaminoalcohol (S)-3.13. La (S)-dapoxetina se obtiene mediante reacción SNAr de
3.5.2.2c. Cuestiones
1) Explique mecanísticamente la reacción de metilación de Eschweiler-Clarke que permite la obtención de (S)-3.13 a partir de 3.22.
3.5.3. Síntesis de fluoxetina (Prozac)
La fluoxetina se comercializa en forma de racemato, puesto que ambos enantiómeros presentan similar actividad in vitro. Sin embargo, el enantiómero con mayor poder terapéutico es la (S)-fluoxetina debido a que es eliminada más lentamente que el enantiómero (R). Investigaciones recientes han apuntado la posibilidad de que la duración prolongada del enantiómero (S) sea la causante de las contraindicaciones del fármaco.
3.5.3.a. Análisis retrosintético
La fluoxetina (Prozac) se comercializó por primera vez en 1986. A pesar de que ya lleva en el mercado 25 años, sigue siendo uno de los fármacos antidepresivos más recetados.
En el esquema 3.8 se indica un análisis retrosintético para la fluoxetina. El proceso de desconexión comienza con la escisión del enlace C-O. La escisión del enlace C-O conduce al p-trifluorometilfenol 3.23 y la amina funcionalizada 3.24 (X=grupo saliente), cuyo precursor será el aminoalcohol 3.25. El aumento del estado de oxidación de la función hidroxilo genera la -aminocetona 3.26, cuya desconexión, basada en una reacción de tipo Mannich, conduce a la acetofenona 3.27, el formaldehído 3.28 y la amina 3.29.
Esquema 3.8
3.5.3.b. Síntesis
La síntesis de la fluoxetina se describe en el esquema 3.9 y comienza con la reacción de Mannich entre la acetofenona 3.27, el formaldehído 3.28 y la dimetilamina 3.30.5 La
reacción de Mannich proporciona la -aminocetona 3.31, que se transforma en el aminoalcohol 3.32 mediante reducción de la función cetónica. La transformación del alcohol
3.32 en el cloruro 3.33, seguida de reacción SN2 con p-triflurometilfenóxido, generado in situ
a partir del p-trifluorometilfenol, conduce al compuesto 3.34. Para conseguir la conversión
5 (a) B. B. Molloy, K. K. Schmiegel, US Patent 1982, 4,314,081. (b) J. Saunders Top Drugs. Top
de 3.34 en fluoxetina se debe llevar a cabo la N-desmetilación reductiva del grupo N,N-dimetilamino. Esta operación se consigue en una secuencia de dos pasos. En primer lugar el N,N-dimetilamino, compuesto 3.34, se convierte en la N-metil-N-cianoamina 3.35 por reacción con bromuro de cianógeno (BrCN). A continuación, el compuesto 3.35 se transforma en fluoxetina por descianación reductiva con KOH acuoso en etilenglicol.
Esquema 3.9
La fluoxetina preparada según la síntesis que se describe en el esquema 3.9 se obtiene en forma racémica. Conviene indicar que ambos enantiómeros presentan una actividad similar in vitro. Así, la constante de inhibición Ki (concentración de fármaco necesaria para inhibir la mitad de la actividad enzimática in vitro) para la serotonina, en cortex de rata, es de 21 nM para el enantiómero (S)-(+) y de 33 nM para el enantiómero (R)-(-). El enantiómero S es el que lleva a cabo la mayor parte del efecto terapéutico, ya que este enantiómero es eliminado más lentamente que el enantiómero R.
3.5.3.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 3.31 mediante la reacción de Mannich ¿Por qué se utiliza dimetilamina en lugar de metilamina en esta reacción?
2) ¿Cuál es el mecanismo que explica la conversión del compuesto 3.34 en la N-cianoamina
3.35?
3.5.4. Síntesis de sertralina
La sertralina, también conocida por las marcas comerciales Zoloft®, Altruline®, Sertex® o Besitrán®, es un antidepresivo perteneciente al grupo de los ISRS (Inhibidores Selectivos de la Recaptación de Serotonina). Actúa inhibiendo la recaptación de la serotonina en el espacio intersináptico por parte de la neurona emisora, lo cual aumenta la disponibilidad de la misma. La sertralina no tiene afinidad sobre el bloqueo de la recaptación de la noradrenalina (norepinefrina) y dopamina.6
La sertralina se utiliza principalmente en el tratamiento de la depresión, esté o no asociada con estados de ansiedad, en el tratamiento del trastorno por estrés postraumático, en el trastorno obsesivo compulsivo, en los ataques de pánico, en el trastorno esquizoide de la personalidad y en la fobia social.
3.5.4.a. Análisis retrosintético
La sertralina se desarrolló en los laboratorios Pfizer en los cuales se había descubierto que una serie de trans-1-amino-4-ariltetralinas presentaban potente actividad biológica en la reabsorción de norepinefrina. La actividad era altamente específica para el trans-(1R,4S) (figura 3.22). El enantiómero trans-(1S,4R) era mucho menos activo y el racemato cis era inactivo en el bloqueo de la reabsorción de norepinefrina. Posteriormente a estos descubrimientos se encontró que los isómeros cis eran potentes inhibidores de la reabsorción de serotonina.
MeHN Ar 1 4 trans-(1R,4S) MeHN Ar 1 4 trans-(1S,4R) MeHN 1 4 MeHN Ar 1 4 cis-(1R,4R) Sertralina Cl Cl Figura 3.22
El compuesto (1S,4S)-cis-1-amino-4-ariltetralina se denominó sertralina. Este enantiómero dextrogiro es mucho más potente en la inhibición de la reabsorción de serotonina que el enantiómero levogiro (1R,4R). Por tanto, y en contraposición a la fluoxetina, la sertralina se comercializa únicamente en su forma de enantiómero (1S,4S).
En el esquema 3.10 se indica un análisis retrosintético para la sertralina que se inicia con una operación de intercambio del grupo funcional metilamina por cetona. La dihidronaftalenona 3.36 genera en la operación IGF se convierte en el ácido 4-arilfenilbutanoico 3.37 mediante una operación retrosintética basada en una reacción SEAr
intramolecular. El análisis se continúa con la adición de un doble enlace en el punto de ramificación de la estructura 3.37. Esta operación genera la olefina 3.38 que se desconecta en el doble enlace a la cetona 3.39 y al sintón nucleofílico 3.40. Este sintón no tiene existencia real y su equivalente sintético es el anión 3.41, derivado del succinato de dialquilo.
Esquema 3.10
3.5.4.b. Síntesis
La síntesis de sertralina se inicia con la condensación de Stobbe entre la diarilcetona
3.39 y el succinato de dietilo 3.42 (esquema 3.11).7 La condensación de Stobbe se lleva a
cabo en presencia de t-butóxido de potasio como base y proporciona el acidoéster insaturado 3.43. El tratamiento del compuesto 3.243 con HBr en ácido acético provoca la hidrólisis de la función éster y la subsiguiente descarboxilación. El resultado final es la formación del ácido insaturado 3.38, que por hidrogenación se convierte en el ácido 4-arilfenilbutanoico 3.37. La construcción del anillo de dihidronaftalenona se consigue mediante reacción SEAr intramolecular del cloruro de ácido derivado de 3.37. El producto de
la ciclación, compuesto 3.36, se convierte en la N-metilimina 3.44 por condensación con metilamina en presencia de TiCl4. La hidrogenación de 3.44, en presencia de Pd/C al 10%,
proporciona una mezcla racémica de cis- y trans-aminas diastereoisoméricas (+/-)-3.45 y
(+/-)-3.46, en relación 70:30 respectivamente. La cis-amina racémica (+/-)-3.45 se separa de
la mezcla mediante cristalización fraccionada en forma de clorhidrato. Finalmente, la sertralina se obtiene en forma ópticamente activa por resolución óptica de la mezcla racémica (+/-)-3.45 con ácido D-(-)-mandélico (ácido (R)-α-hidroxifenilacético).
Esquema 3.11
3.5.4.c. Cuestiones
1) Explique mecanísticamente la formación del ácidoéster 3.43 mediante la reacción de condensación de Stobbe.
2) Explique mecanísticamente la conversión del ácidoéster 3.43 en el ácido 3.38.
3) ¿Por qué la reacción SEAr intramolecular sobre el cloruro de ácido derivado de 3.37 se
produce sobre el anillo de fenilo y no sobre el anillo de 3,4-diclorofenilo?
4) Una síntesis más eficiente de la tetralona 3.36 se indica en el esquema 3.12.8 Todas las
reacciones de formación de enlace C-C de esta secuencia sintética se llevan a cabo mediante reacciones SEAr. Explique mecanísticamente la formación del compuesto 3.36 a
partir de 3.50.
Esquema 3.12
5) En el esquema 3.13 se describe una síntesis enantioselectiva de la tetralona (S)-3.36. En esta síntesis se parte del cetoácido 3.49 que se convierte en el t-butiléster 3.51.9 El paso
clave en la secuencia sintética es la reducción enantioselectiva del carbonilo cetónico de
3.51 que se lleva a cabo con BH3 y una oxazaborolidina quiral derivada de prolina. La
reacción proporciona el hidroxiéster 3.52 con un rendimiento químico del 100% y con un exceso enantioselectivo del 90%. La mesilación del hidroxilo forma el mesilato 3.53 que experimenta una reacción de tipo SN2, con inversión de la configuración, por reacción con el
difenilcianocuprato de dilitio (Ph2Cu(CN)Li2).10 Esta reacción conduce al diariléster 3.54 que
se convierte en la tetralona quiral (S)-3.36 mediante reacción SEAr intramolecular promovida
por ácido trifluoroacético. La tetralona (S)-3.36 se transforma en sertralina mediante la secuencia de reacciones indicada en el esquema 3.11.
Esquema 3.13
El método de reducción enantioselectiva de cetonas, aplicado en la síntesis del cetoéster 3.51 se debe a E. J. Corey, R. K. Bakshi y S. Shibata y se conoce como método
9 G. J. Quallich, T. M. Woodall. Tetrahedron, 1992, 48, 10239-10248.
CBS, por las iniciales de estos tres autores.11 Este método permite la reducción
enantioselectiva de determinado tipo de cetonas por reacción con BH3, en presencia de
oxazaborolidinas quirales, que se preparan a partir de L-prolina. En el esquema 3.14 se describe la preparación de la oxazaborolidina quiral (S)-3.55 a partir de L-prolina.
Esquema 3.14
La oxazaborolidina enantiomérica (R)-3.55 se obtiene mediante un proceso similar que implica una etapa de resolución (esquema 3.15).
Esquema 3.15
En el esquema 3.16 se indica el ciclo catalítico de la reducción enantioselectiva con el método CBS. En el primer paso se produce la coordinación del borano al átomo de nitrógeno de la oxazaborolidina (S)-3.55. Esta coordinación activa el BH3 como dador de
hidruro y aumenta la acidez de Lewis del boro endocíclico del catalizador. A continuación, el átomo de boro del catalizador se coordina a la cetona 3.51 por el par de electrones no enlazantes estéricamente más accesibles, que es el par electrónico en sin con respecto del sustituyente estéricamente menos impedido. Esta coordinación disminuye las interacciones estéricas entre la cetona y el catalizador, puesto que el sustituyente más voluminoso de la cetona está orientado en trans, y por tanto alejado del grupo metilo del catalizador. En el esquema 3.16 se dibuja la conformación del estado de transición (estructura III), en la que se observa cómo el carbonilo cetónico y el borano adquieren un orientación que permite la transferencia favorable de hidruro desde la cara Si de la cetona mediante la intervención de un estado de transición de seis eslabones. La transferencia de hidruro produce el alcoxiborano 3.34, que por hidrólisis ácida proporciona el alcohol 3.30.
BH3
N BO H PhPh
Me
H3B
Estado de transición favorecido ataque a la cara Si del grupo carbonilo
(S)-3.33 II III IV N O B Me O B H H H H N O B Me O B H H H H
N BO
H PhPh
Me Cl Cl O O OtBu 3.29 Cl Cl COOtBu COOtBu Cl Cl Cl Cl O
OBH2
BuOt 3.34
H
Esquema 3.16
¿Por qué en la secuencia de reacciones del esquema 3.13 no se ha sintetizado la tetralona (S)-3.36 mediante una reacción SEAr intramolecular, de modo similar a lo
efectuado en el esquema 3.12, en el cual se obtiene la tetralona 3.36 mediante reacción SEAr inducida por ácido sulfúrico?
3.5.5. Síntesis de paroxetina
La paroxetina es un inhibidor selectivo de la recaptación de serotonina con efecto ansiolitico (tranquilizante). La compañía farmacéutica GlaxoSmithKline lo lanzó al mercado en 1992 y, desde entonces, es uno de los antidepresivos más prescritos del mercado debido a su eficacia en el tratamiento de la depresión. También se prescribe en el tratamiento del trastorno obsesivo-compulsivo, la ansiedad, el trastorno del pánico y el trastorno por ansiedad social, también conocido como fobia social.
3.5.5.1a. Análisis retrosintético
El análisis retrosintético de la paroxetina comienza con la escisión del enlace C-O (esquema 3.17). Esta operación, que se basa en una reacción SN2, conduce al sustrato
Esquema 3.17
3.5.5.1b. Síntesis
Para la síntesis de la paroxetina se elige como material de partida la arecolina 3.62 (esquema 3.18), un alcaloide de origen natural que se obtiene de la nuez de areca, una palmera originaria de Indonesia. La adición conjugada a la arecolina del p-flurorofenilcuprato de litio, generado in situ a partir del bromuro de p-flurorofenilmagnesio 3.50, proporciona una mezcla de los diastereoisómeros cis y trans, ambos obtenidos como racematos (compuestos (+/-)-3.63 y (+/-)-3.64).12
La separación de la mezcla diastereoisomérica permite obtener el diastereoisómero trans puro (+/-)-3.64 el cual, por hidrólisis de la función éster y reacción con cloruro de tionilo, se transforma en el cloruro de ácido (+/-)-3.65. En este punto se lleva a cabo el proceso de separación de enantiómeros. Para ello, la mezcla racémica (+/-)-3.65 se esterifica con (-)-mentol (3.66) y la mezcla de diastereoisómeros formada por los ésteres
3.67 y 3.68 se separa por destilación fraccionada, lo que permite la obtención del
compuesto 3.67 puro. La reducción de 3.67 con LiAlH4 conduce al alcohol 3.69, que se
activa frente al proceso SN2 mediante conversión en el cloruro 3.70. La reacción SN2 de
3.70 con sesamol 3.58 proporciona la N-metilparoxetina 3.71. La paroxetina se obtiene en
dos pasos a partir de 3.71: en el primero de ellos se produce la transformación de 3.71 en el vinilcarbamato 3.73 y en el segundo la metanolisis ácida del carbamato.
Esquema 3.18
3.5.5.1c. Cuestiones
1) Explique mecanísticamente la reacción de adición conjugada Michael a 3.62. 2) Explique mecanísticamente la conversión de 3.73 en paroxetina.
3.5.5.2a. Análisis retrosintético de (+)-paroxetina mediante la estrategia de Adición de Quiralidad (AQ)
En el esquema 3.19 se indica un análisis retrosintético de la (+)-paroxetina conceptualmente diferente al indicado en el esquema 3.17. Aunque la primera desconexión es idéntica a la aplicada en el esquema 3.17 y, por tanto, origina el sustrato electrofílico
ent-3.57 (X=grupo saliente) y el nucléofilo fenólico 3.58, la diferencia se establece en la
(AQ), simbolizada con la unión al átomo de nitrógeno de P*, que representa un fragmento quiral. En el sentido sintético la estructura 3.74, que surge de la doble operación IGF/AQ, se obtendrá estereoselectivamente en la adición conjugada Michael del compuesto organometálico 3.60 a la lactama conjugada 3.75, que contiene un fragmento quiral unido al átomo de nitrógeno, y cuya misión será inducir un elevado grado de estereocontrol en la adición del reactivo organometálico al sistema aceptor Michael.
Esquema 3.19
3.5.5.2b. Síntesis de (+)-paroxetina mediante el empleo de un fragmento quiral (Adición de Quiralidad)
El compuesto quiral empleado en la síntesis de la paroxetina fue el (R)-fenilglicinol 3.54. La instalación de este fragmento quiral se consigue mediante reacción de ciclocondensación con el 5-oxopentanoato de metilo 3.53 (esquema 3.14).13 Esta reacción conduce a la mezcla
de oxazolidinas diastereoisoméricas 3.55/3.56 en relación 85:15. La equilibración de la mezcla por tratamiento con ácido trifluoroacético, seguida de separación cromatográfica, permite la obtención del diastereoisómero 3.56 puro. La enolización de 3.56 con la base hexametildisililamiduro de litio (LiHMDS), seguida de reacción del correspondiente enolato lítico con cloroformiato de metilo y bromuro de feniselenilo (PhSeBr), proporciona la selenolactama 3.57. Cuando este compuesto se oxida con ozono se obtiene directamente la lactama insaturada 3.58. Sobre este compuesto quiral se lleva a cabo la adición conjugada Michael. En este caso se emplea el p-fluorofenilcianocuprato de dilitio 3.59. La reacción da lugar a una mezcla de diastereoisómeros 3.61/3.60 en relación 97:3. El tratamiento de 3.61 con LiAlH4 en presencia de AlCl3 provoca las reducciones de las funciones éster y lactama,
y también la ruptura reductiva del enlace C-O el anillo de oxazolidina, y proporciona el aminodiol 3.62. La hidrogenolisis de este compuesto, en presencia de dicarbonato de di-t-butilo (Boc2O), conduce directamente al aminoalcohol N-Boc protegido 3.63. La mesilación
del hidroxilo seguida de reacción de desplazamiento nucleofílico SN2 con sesamol, en
presencia de hidruro sódico como base, proporciona la paroxetina N-Boc protegida 3.65. La paroxetina se obtiene mediante eliminación del grupo Boc con ácido trifluoroacético.
Esquema 3.14
3.5.5.2c. Cuestiones
1) Explique mecanísticamente la formación de las oxazolidinas 3.55 y 3.56.
2) Explique mecanísticamente la transformación del selenoderivado 3.57 en el compuesto
3.58.
3) ¿Qué ventajas puede tener el empleo del dicarbonato de di-t-butilo en lugar del carbonato de t-butilo en la preparación de N-Boc aminas? Explique mecanísticamente la reacción de eliminación del grupo Boc mediante tratamiento con ácido trifluoroacético.
Esquema 3.15
En una primera etapa el auxiliar quiral (Xq* en el esquema anterior) se une de forma covalente al sustrato aquiral, compuesto 3.66 del esquema anterior. Esta reacción proporciona el compuesto 3.67, que ya es quiral debido a que ha incorporado en su estructura el auxiliar quiral. Si, por ejemplo, lo que se pretende es llevar a cabo una reacción de alquilación asimétrica, el sustrato quiral 3.67 se enoliza y el correspondiente enolato se trata con el agente alquilante (R´X en el esquema 3.15). Como el enolato es quiral provocará inducción asimétrica en la reacción de alquilación y el resultado será la formación diastereoselectiva del compuesto alquilado 3.68. En una etapa posterior el auxiliar quiral se elimina del sustrato, obteniéndose el producto de alquilación 3.69 de forma enantioselectiva.
Las condiciones que debe cumplir un auxiliar quiral son las siguientes:
a) Se debe poder instalar en el sustrato a enolizar con alto rendimiento y pureza óptica. b) Debe ser estable a las condiciones de enolización y alquilación.
c) Debe inducir una elevada selectividad diastereofacial.
d) Debe ser fácilmente recuperado mediante desinstalación del sustrato en condiciones que no provoquen pérdida de pureza óptica.
A tenor de todo lo explicado anteriormente ¿se puede calificar el empleo del (R)-fenilglicinol en la síntesis de la paroxetina como un ejemplo de aplicación de la estrategia de auxiliar quiral?
3.5.5.3a. Análisis retrosintético de paroxetina mediante una estrategia de desimetrización
En el esquema 3.16 se indica un análisis retrosintético de paroxetina que emplea el concepto de desimetrización. La primera desconexión es similar a la efectuada en los dos análisis retrosintéticos precedentes y, por tanto, origina el sustrato electrofílico 3.35 (X=grupo saliente) y el nucléofilo fenólico 3.36. En el segundo paso de la retrosíntesis se llevan a cabo simultáneamente dos operaciones retrosintéticas. Una de ellas es una adición del grupo funcional carbonilo (AGF) y convierte la amina en lactama. La otra es una operación de interconversión de grupo funcional en la cual se transforma la parte del grupo saliente en un grupo funcional éster. El resultado es la generación de la estructura 3.70 la cual, por escisión del grupo alcoxicarbonilo, conduce a la lactama 3.71. La desconexión del enlace lactámico en 3.71 forma el aminoéster 3.72 que derivará del éster funcionalizado
3.73 (X=grupo saliente) que es un compuesto quiral. Una operación IGF en este último
Esquema 3.16
3.5.5.3b. Síntesis de paroxetina mediante desimetrización enantioselectiva de un diéster simétrico
Para la preparación del diéster simétrico 3.74 (R=Me) se emplean como compuestos de partida el p-fluorobenzaldehído 3.75 y el acetilacetonato de etilo 3.76 (esquema 3.17). La síntesis de 3.74 se lleva a cabo en tres pasos. En primer lugar se prepara el dicetodiéster
3.77 por condensación entre el p-fluorobenzaldehído 3.75 y el acetilacetonato de metilo 3.76
en presencia de piperidina. Cuando el dicetoéster 3.77 se somete a reacción con metóxido sódico en metanol acuoso se obtiene el diácido simétrico 3.78.14 La esterificación del diácido
proporciona el diéster simétrico 3.74. La desimetrización enantioselectiva del diéster 3.74 se consigue mediante hidrólisis enzimática con PLE (Pig Liver Esterase, Esterasa de Hígado de Cerdo) en acetona acuosa a pH=7.15 En las condiciones de hidrólisis enzimática
enantioselectiva se obtiene el acidoéster quiral 3.79 con un rendimiento químico del 86% y con un 95% de exceso enantiomérico. La reducción quimioselectiva de la función éster en el acidoéster 3.79 se consigue mediante adición de hidruro de litio en THF, calentamiento a reflujo durante 1 hora, luego adición de LiBH4 y calentamiento a reflujo durante 10 horas. El
hidroxiácido resultante del proceso de reducción se esterifica con sulfato de dimetilo y proporciona el hidroxiéster 3.80 que por mesilación se convierte en el mesilato 3.73. La reacción de éste con bencilamina conduce directamente a la lactama 3.81. La metoxicarbonilación de este compuesto en condiciones de control termodinámico proporciona el trans-amidoéster 3.82, el cual se convierte en aminoalcohol 3.83 mediante reducción con BH3·SMe2. La mesilación del hidroxilo, seguida de desplazamiento
nucleofílico del mesilato con sesamol 3.36 en presencia de hidruro sódico, proporciona la
14 J. Ritter, T. Kaniecki. J. Org. Chem. 1962, 27, 622-623.
bencilparoxetina 3.85. La paroxetina se obtiene mediante N-desbencilación hidrogenolítica de 3.85.
Esquema 3.17
3.5.5.3c. Cuestiones
CHO F OMe O O piperidina, 20ºC MeOOC COOMe F O CH3 O H3C 3.75 3.77 3.76 + 2
+ H2O
Esquema 3.18
2) Explique mecanísticamente la formación del compuesto 3.78 a partir de 3.77 (esquema 3.19). MeOOC COOMe F O CH3 O
H3C
NaOMe, MeOH H2O, EtOH, 90ºC
MeOOC COOMe F
3.77 3.78
HCl, H2O
HOOC COOH F
Esquema 3.19
3) En la conversión de 3.77 en 3.78 se forma como subproducto el compuesto 3.86.16
Proponga una explicación mecanística para la formación de 3.86 a partir de 3.77. 4) ¿Por qué se añade LiH en la reducción de la función éster en el acidoéster 3.79?
5) En la reacción de metoxicarbonilación de 3.81 se forma el isómero trans 3.82 ¿Por qué no sea forma el isómero cis?
3.6. Síntesis de Inhibidores Selectivos de la Recaptación de Serotonina y Norepinefrina (ISRSN)
3.6.1. Síntesis de venlafaxina
La venlafaxina es un potente inhibidor de la recaptación de aminas en la neurona presináptica y, a diferencia de la sertralina y de la paroxetina, es un fármaco que es capaz de inhibir selectivamente la recaptación de serotonina y de norepinefrina (fármaco ISRSN). La venlafaxina logra controlar los síntomas depresivos en lapsos de tiempo más cortos que los que se necesitan en el tratamiento con fluoxetina.
3.6.1.a. Análisis retrosintético
En el esquema 3.20 se indica un análisis retrosintético para la venlafaxina que se inicia con la desconexión de los grupos metilo de la parte de dimetilamina. Este proceso conduce a la hidroxiamina 3.101 que por interconversión del grupo amino en grupo ciano se convierte en el hidroxinitrilo 3.102. Este compuesto se obtendrá mediante adición a la ciclohexanona
3.103 de la base conjugada derivada del 4-metoxifenilacetonitrilo 3.102.
OH MeO N Venlafaxina OH MeO
H2N
N-metilación IGF OH MeO C N MeO C N O + 3.100 3.101 3.103 3.102 Esquema 3.20 3.6.1.b. Síntesis
La venlafaxina se comercializa en forma de racemato, por tanto no es necesario llevar a cabo una síntesis enantioselectiva de este fármaco. La síntesis de la venlafaxina comienza con la adición de la base conjugada derivada del 4-metoxifenilacetonitrilo 3.102 a la ciclohexanona 3.103 (esquema 3.21).17 La adición se lleva a cabo en presencia de NaOH y
Bu4NBr en metanol-agua y proporciona el hidroxinitrilo 3.101 con un 96% de rendimiento. La
hidrogenación del nitrilo con hidrógeno molecular a 10 atmósferas de presión, en una mezcla de MeOH/NH3 y en presencia de Ni-Raney como catalizador, genera el
aminoalcohol 3.100. Después de acabada la hidrogenación se añade formaldehído acuoso a la mezcla de reacción, se agita durante 3 horas, se filtra el catalizador, se evapora el metanol y se añade hexano. Este procedimiento experimental proporciona la oxacina 3.104, que es un compuesto sólido estable que se obtiene sin necesidad de ninguna separación
cromatográfica. La transformación de la oxazina 3.104 en la venlafaxina se consigue mediante reacción de N-metilación con formaldehído acuoso en presencia de ácido fórmico como reductor. La venlafaxina obtenida en la reacción anterior se convierte en el correspondiente clorhidrato por reacción con HCl en isopropanol.
Esquema 3.21
3.6.1.c. Cuestiones
1) Explique mecanísticamente la formación de la venlafaxina a partir de la oxazina 3.104.
3.6.2. Síntesis de desvenlafaxina
Uno de los principales metabolitos de la venlafaxina es el derivado O-desmetilado (desvenlafaxina) que presenta mayor eficacia y perfil de seguridad que aquélla. La desvenlafaxina fue aprobada por la FDA en 2008 para el tratamiento del Trastorno Depresivo Mayor.
3.6.2.a. Análisis retrosintético
La conversión de la función amina de la desvenlafaxina en amida conduce al compuesto
3.105 (esquema 3.22). Este compuesto se puede preparar por adición del reactivo
OH HO N Desvenlafaxina OH PO N O C-C O PO N O 3.105 3.106 + 3.103 IGF Esquema 3.22 3.6.2.b. Síntesis
El material de partida para la síntesis de la desvenlafaxina es el ácido 4-benciloxifenilacético 3.107.18 Este compuesto se convierte en la N,N-dimetilamida 3.108 la
cual, por ionización con LiHMDS y reacción con ciclohexanona, se transforma en la hidroxiamida 3.105. Finalmente, la desvenlafaxina se obtiene por reducción de la amida
3.105 a la amina 3.109 seguida de hidrogenolisis de la parte de benciléter.
OH BnO N O 3.105 BnO COOH 3.107
1) SOCl2, DMF
(90% 2 pasos) 2) Me2NH·HCl, Et3N
BnO
N O
3.108
LiHMDS, THF. -70ºC luego ciclohexanona
(82%)
BH3·THF, THF
(66%) OH
BnO N
H2, Pd/C
EtOH (87%) 3.109 OH HO N Desvenlafaxina Esquema 3.23
3.6.3. Síntesis de milnacipran
El antidepresivo milnacipran inhibe la recaptación de serotonina y de norepinefrina (fármaco ISRSN) en una relación aproximada de 1:3. Su uso se aprobó por primera vez en Francia (nombre comercial Ixel®) en diciembre de 1996. En enero de 2009 la FDA aprobó el empleo de este fármaco para el tratamiento de la fibromialgia.
3.6.3.a. Análisis retrosintético
La primera desconexión en el análisis retrosintético del milnacipran es la escisión del enlace C-N (esquema 3.24). Esta operación genera amoniaco y el fragmento electrofílico
3.110 (X=grupo saliente). La siguiente operación retrosintética no es evidente y para seguir
el proceso de desconexión de enlaces se ha indicado el mismo mediante flechas. Así, la ruptura intramolecular del anillo ciclopropánico, por ataque nucleofílico del grupo X origina el anión heterociclopropánico 3.111. En el sentido sintético este anión, generado a partir de la
amida 3.110, formará el compuesto ciclopropánico 3.111 mediante apertura nucleofílica intramolecular del anillo heterociclopropánico. El análisis retrosintético se continua con la desconexión del fragmento metilenheterociclopropánico en la estructura 3.112. Esta operación genera el compuesto 3.113 y el anión 3.114. Un equivalente sintético de este anión puede ser el anión derivado de fenilacetonitrilo 3.115, fácilmente generable a partir del propio fenilacetonitrilo 3.116.
Esquema 3.24
3.6.3.b. Síntesis
El milnacipran se comercializa en forma de racemato, por tanto no es necesario llevar a cabo una síntesis enantioselectiva de este fármaco. La síntesis se inicia con la reacción entre la base conjugada del fenilacetonitrilo 3.116 y el clorometiloxirano 3.117 (esquema 3.25).19 Este proceso proporciona una mezcla cis/trans de los compuestos
hidroxinitrilo-ciclopropánicos 3.118. La hidrólisis del grupo nitrilo conduce a la mezcla cis/trans de los hidroxiácidos ciclopropánicos 3.119. La convergencia de la mezcla de diastereoisómeros
3.119 en la lactona ciclopropánica 3.120 se consigue mediante calentamiento de aquélla a
150ºC. En el siguiente paso sintético se emplea la ftalimida potásica 3.121 como equivalente sintético de amoniaco (síntesis de Gabriel). Así, la reacción de la ftalimida potásica 3.121 con la lactona ciclopropánica 3.120 proporciona el derivado ftalimidociclopropánico 3.122. La función dietilamida se instala por conversión del ácido carboxílico en cloruro de ácido y reacción subsiguiente con dietilamina. Esta secuencia de dos pasos conduce a la amida 3.123 que por aminólisis de la parte de ftalimida con metilamina se convierte en el milnacipran neutro. La adición de HCl proporciona el clorhidrato de milnacipran.