Electronic aspects of formation and properties of local structures
around Mn in Cdi
x
Mn
x
Tei
y
Se
y
Ivana Radisavljevic , Nikola Novakovic , Nebojsa Romcevic , Miodrag Mitric ,
Bojana Kuzmanovic , Slobodan Bojanic , Nenad Ivanovic
H I G H L I G H T S
> Local structural/electronic properties around Mn in Cdi_xMnxTei_ySey are
determined.
> Influence of different types of Mn—Te bonds on electronic structure is established.
> Mn and Te excess charges are found to be distinctly different from their valences.
> The spin-up states are spatially much more extended than the spin-down states.
> Small magnetic dipole moment forms at the Mn—Te bond critical points.
G R A P H I C A L A B S T R A C T
A B S T R A C T
Local electronic and structural features around Mn in Cdi _xMnxTeo.97Seo.o3 (x = 0.02; 0.05; 0.1; y = 0.03)
were studied by means of X-ray Absorption Fine Structure (XAFS) techniques. Manganese ions with an average valence 2+, are found to be well incorporated into the host CdTe lattice, with clear preference for Te atoms as the first neighbors. However, Mn and Te are found to form two essentially different types of bonds, one short, strong and directional (cubic MnTe-alike bond), and three much longer, predominantly ionic in nature (hexagonal MnTe-alike bonds), thereby distorting the tetrahedral coordination around Mn. The origin of peculiar Mn—Te bonds distribution and details of their nature and strength are further elaborated by employing the first principle electronic structure calculations. That way a thorough insight in impact of the Mn—Te bond length variation on the electronic structure of the compound is obtained. The relations established between the local structures and electronic properties offer a reliable procedure for detailed analysis of the structural and electronic consequences of the 3d-transition metals (TM) incorporation in II—VI semiconductor host. Clear distinction between various influences makes the procedure easily adoptable also to the studies of TM impurities in other semiconductors.
1. Introduction
II—VI diluted magnetic semiconductors (DMS) [1,2]. Strong coupling (exchange interaction) between the localized Mn d-states and charge carriers (s- and p-states of the host CdTe) induces numerous important magneto-optical/transport phenomena, such as extremely fast giant Faraday rotation [3,4], bound magnetic polarons formation [5,6], magnetic field induced metal-insulator transition [7], etc. Although the antiferromagnetic (AFM) exchange coupling often prevails [8], other types of magnetic ordering have been observed as well (e.g. spin-glass [9,10], ferromagnetic ordering at low temperatures [11 J), depending primarily on the Mn concentration (x). Versatile properties of Cdi_xMnxTe DMSs
resul-ted in their numerous applications not only in the field of photonics and (opto)electronics (Faraday rotators, IR- and magnetic field de-tectors, visible and near-IR lasers, solar cells, magnetic diodes, spin injectors, ...) [12—16], but also in a new field of the spintronics of antiferromagnets (high-speed electronic devices operating in ter-ahertz range) [17—19]. The tunable energy gap [20], high resistivity
[21] and good electron transport properties [22], make Cdi_xMnxTe
prospective material also for nuclear radiation detection [23]. Ad-vances in the epitaxy of semiconductor compounds which enabled fabrication of small crystallites (nanostructures) and hetero-structures, further extended the possibilities of the material application in spintronics of ferromagnets and quantum informa-tion processing [24—28]. Another way to improve the material characteristics and to bring in new physical properties is to use two anion-system as a host [29—33]. The additional degree of freedom introduced that way, represents an important aspect for designing devices based on multi-anion layered systems [34—36]. In partic-ular, Se addition into CdTe-based solid solutions substantially re-duces the concentration of sub-grain boundaries and destroys their network, thereby ensuring compositional uniformity and effective solid solutions hardening [37,38]. Owing to the persistent photo-conductivity (which enables to continuously vary the concentra-tion of shallow donors by means of optical excitaconcentra-tions), the metal-insulator transition in Cdi_xMnxTei_ySey can be controlled by both
magnetic field and the donor density [1[. Furthermore, by increasing the interaction between localized moments and band electrons (the s—d interaction), Se could lead to enhancement of Faraday rotation in Cdi_xMnxTei_ySey. Besides, variation of the Se
content could make it possible to regulate the type of the con-ductivity and free charge carrier concentration [39]. Main requirement imposed to Cdi_xMnxTei_ySey system studied in this
work, is to possess well defined crystal structure free from pre-cipitates and secondary impurity phases. While Cdi_xMnxTe
crys-tallizes in zinc-blende (ZB) type structure for concentrations x < 0.77 [40], Cdi_xMnxTei_ySey, has several single and multiple
phase areas in its phase diagram, and the ZB-phase exists only in the limited range of concentrations x and y [41]. The Cdi_xMnxTe
lattice constant decreases with x, but its energy gap (Eg) increases
from 1.6 eV (x = 0) to the value 2.2 eV (x ~ 0.7) [20,42[ which is larger than in binary end-point NiAs-type MnTe compound (Eg = 1.3 eV), but smaller than that reported for (metastable) ZB-type MnTe (Eg = 3.2 eV) [43]. For constant y, energy gap of Cdi_xMnxTei_ySey follows similar dependence with x as in
Cd!_xMnxTe [30] (e.g. for x = 0.1 and y = 0.03, Eg ~ 1.6 eV [41]).
Atypical behavior of Eg(x) in Mn doped CdTe-based systems is
commonly related to the features of the Mn-3d states, although some details concerning their energy position and hybridization with the band states of the host crystal (pd-hybridization) are still under debate [42-50]. In Cd!_xMnxTe, the Mn-3d states form two
bands split by the exchange interaction, a completely occupied spin-up band (with reported position ranging from -0.8 eV [45]
to - 6 eV [46] below the valence band (VB) top Ev), and an empty
spin-down band (with reported position ranging from 2.9 eV to 4.5 eV above Ev [47—50]). In what manner the pd-hybridization
affects the position of the Mn-3d bands is also unclear. According to
[44], pd-hybridization causes shift of the occupied Mn-3d band deeper into VB. According to [47] the position of the occupied Mn-3d band is constant, while the position of the empty Mn-3d band de-pends on Mn concentration. The band structure of the two-anion Cdi_xMnxTei_ySey system is even less known. The reported
pref-erences for certain ions pairing [32,33,51] suggest that position of the Mn-3d band in Cdi_xMnxTei_ySey could be even more affected
by peculiarities of local structures and the pd-hybridization fea-tures, than in Cdi_xMnxTe. The local structures and their variations
which affect the electronic and magnetic correlations, play an important role in promoting ferromagnetism in novel Mn doped II—II—V DMS [52], and are strongly linked also with recently discovered superconductivity in FexTei_ySey [53].
Despite several decades of extensive research of Mn doped CdTe-based systems, there still exist some arguable issues con-cerning the position of the Mn-3d energy levels and their hybrid-ization with the band states of the host crystal (which affect the Eg,
as well as the stability and properties of the systems). To resolve some of these fundamental issues, we performed detailed analysis of the Mn K-edge X-ray Absorption Near Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) of Cdi_xMnxTeo.97Seo.o3 systems with different Mn concentration
(x = 0.02, 0.05, 0.1). In attempt to elucidate details of the nature, strength and distribution of Mn—Te bonds in the first coordination around Mn and the impact of the bond length variation on the electronic structure of the compound, first principle electronic structure calculations and simulation of the XANES were per-formed. The firm relations established between the local structural and electronic properties enable (i) atomic level understanding of the investigated compounds; (ii) offer the possibility to make modifications on the atomic-scale and (iii) to track these modifi-cations by observing the characteristic features appearing in the XANES spectra.
2. Experimental measurements
X-ray Absorption Fine Structure (XAFS) measurements were performed on Cdi_xMnxTeo.97Seo.o3 (x = 0.02; 0.05; 0.1) single
crystals grown by the Bridgman method [30]. XRD measurements were performed on the powdered Cdo.9sMno.02Teo.97Seo.03 sample using PHILIPS 1050 diffractometer (40 kV and 20 mA) with Ni filtered Cu Kai2 radiation in Bragg—Brentano geometry, in the
range of angles 20°<26 < 100° and step 0.05°, with exposure time 9 s/step. Manganese K-edge XAFS data were collected at HASYLAB at Deutsches Elektronen-Synchrotron DESY (Hamburg, Germany) bending-magnet Al beamline with a double Si-crystal mono-chromator. The synchrotron radiation source was operating at electron beam energy of 4.45 GeV and maximum stored current of 120 mA. Samples were oriented at 45° to the incident beam di-rection and the 7-cell silicon drift detector was used to collect the spectra in fluorescence mode at T = 77 K. XAFS data processing and analysis were performed by IFEFFIT [54], as implemented in ATHENA and ARTEMIS software packages [55]. Interatomic dis-tances (r), the mean-square variation of disdis-tances (a2), the number
and kind of the first neighbors (n), and the edge shift correction (Eo) were treated as free parameters. The passive electron reduction factor S02 was set to 0.63, the value estimated from the a2(S02)
dependence of the ky(k), k2y(k) and k3y(k) functions [56].
3. Theoretical calculations
performed using full potential linearized augmented plane waves method with addition of localized orbitals (FP (L)APW+(LO)lo), as implemented in the WIEN2k code [57]. In this method, the unit cell space is separated into non-overlapping muffin-tin (MT) atomic spheres with basis functions in the form of linear combination of the atomic-like functions and their energy derivatives. In the interstitital space between the MT spheres basis functions are standard plane waves. Core states were treated fully relativistic, while valence states were treated within scalar relativistic approximation. The parameter Riwrkmax. which controls the size and completeness of the basis set, was set to 7.0 in all calculations presented here. MT radii were chosen to be 1.27 Á for Cd and Te, and 1.06 Á for Mn. The unit cell parameters obtained for pure ZB—CdTe after full relaxation of atomic forces and the unit cell volume, were used as a starting point for all subsequent calculations. The Cd3MnTe4 system is constructed from the CdTe unit cell by replacing one out of four Cd atoms with Mn. The Cdi5MnTei6 sys-tem is constructed by replacing one out of 16 Cd atoms with Mn in the supercell made of 2 x 2 x 2 unit cells stacking. For the k-point sampling we used 600 k points in Brillouin zone (BZ) (29 k points in the irreducible BZ) in the case of CdTe and Cd3MnTe4, and 100 k points in BZ (6 k points in the irreducible BZ) in the case of Cdi5MnTei6. Atomic positions of both Mn-doped systems were fully
relaxed. To account for the exchange-correlation effects, standard GGA (Generalized Gradient Approximation) parameterization of Perdew, Burke and Ernzerhoff [58] is used. For more accurate description of localized Mn-3d states and for better estimation of Eg, GGA calculations were followed by GGA + U (U-Hubbard repulsion term) calculations [59]. Simulation of Mn K-edge X ray absorption spectra were therefore performed by GGA + U method. The effect of core-hole is found to be negligible. The Bader analysis method of the topological properties of the electron charge density
[60] as implemented in the CRITIC2 code [61] is used to elucidate details of the nature, strength and distribution of bonds in the first coordination around Mn.
4. Results and discussion
4.1. XANES spectra
The XRD pattern of the powdered Cdo.9sMno.02Teo.97Seo.03 sample is shown in Fig. 1. It is characteristic for the ZB-type
(1
-•
LI)
V—+- )
(220)
C d0 . , 8M n0 . 0 2T e0 . 9 7S e0 . 0 3
(3
K~J
11)
(331) (422) (440)
(400) (511) (531)
I... . 1 , A.. JLJ__
1_JLJL_
20 30 40 50 60 70 80 90
20
[°)
Fig. 1. XRD spectrum of Cdo.9sMno.02Teo.97Seo.03.
structure CdTe compound, with no indication of the existence of secondary impurity phases. The experimental Mn K-edge XANES spectra of Cd1_xMnxTeo.97Seo.03 samples (x = 0.02, 0.05, 0.1) are shown in Fig. 2a.
Nearly identical spectral shapes indicate that the local structure around Mn is similar in all investigated samples. The structure appearing in the region below the main absorption edge (pre-edge region) results mainly from the electronic transitions onto the states with pd-character, created via hybridization between local-ized Mn 3d-states and band states of the host crystal (pd-hybridi-zation). After the background subtraction, the pre-edge region was fitted with Lorentzian function. Energy positions and integrated intensities of the normalized pre-edge peaks are shown in Fig. 2b. Pre-edge peak for x = 0.02 has the energy position closest to 6540.5 eV, which is expected for 2+ valent Mn [33,62]. With in-crease of Mn concentration, the position of the pre-edge peak shifts to somewhat lower energies, probably as a consequence of slight decrease of Mn valence. Integrated intensity of the pre-edge peak decreases with x, implying that the number of unoccupied Mn states with pd-character decreases with x, and that the average distance between Mn and the atoms in its first coordination be-comes longer.
4.2. EXAFS spectra
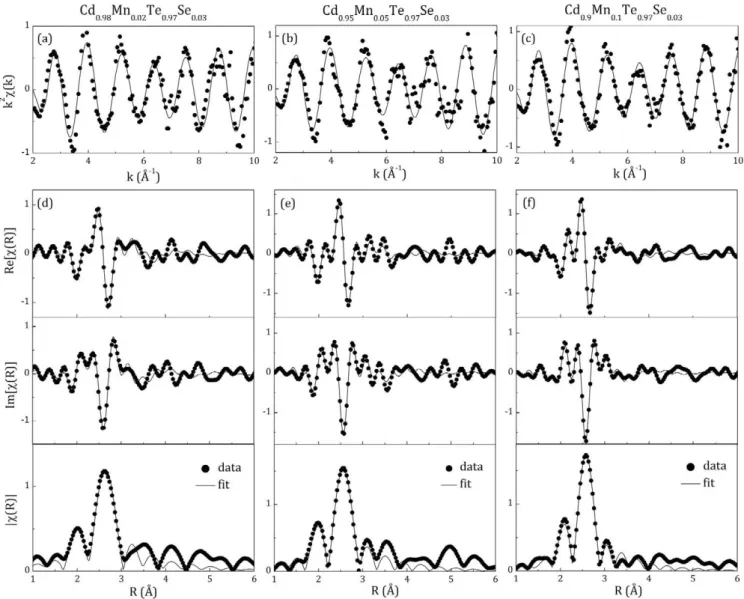
The Mn K-edge EXAFS spectra k2x(k) of the three investigated
samples Cd1_xMnxTeo.97Seo.03 (x = 0.02, 0.05, 0.1) are presented in
Fig. 3a—c. Real parts (Re[x(R)]), imaginary parts (Im[x(R)]) and magnitudes (IxWI) °f t n e corresponding Fourier transforms are
presented in Fig. 3d—f (the best fits to the experimental data are included). Results of the EXAFS data analysis are presented in
Table 1.
In all investigated Cdi_xMnxTeo.97Seo.03 (x = 0.02, 0.05, 0.1) samples, Mn is surrounded predominantly with Te atoms. Selenium atoms were not detected in the first coordination around Mn, and thus the preference for Mn—Se pairing reported in [32] cannot be confirmed. Note that the employed experimental methodology has proven to be sensitive enough to detect preferential pairing of minority elements even at concentrations as low as in the samples investigated in this work [63]. According to the structural model with Te atoms in regular tetrahedral coordination around Mn (single shell model), the Mn—Te distances slightly increase with x
(Table 1 — first entry), and are somewhat larger than those reported for Cdi_xMnxTe (2.76-2.74 A) [51,64,65] and Cdi_xMnxTeo.9Seo.i
(2.80-2.79 Á) [32]. According to the structural model with dis-torted tetrahedral coordination around Mn, Te atoms can be found at two different Mn—Te distances, both being nearly independent on x (Table 1 — second entry). The mean value of the two Mn—Te distances (Table 1 — third entry) is close to the distance obtained in the model with regular tetrahedral coordination (see Table 1 — first entry) and in the preliminary analysis of Cdo.98Mno.02Teo.97Seo.03
[33]. Longer Mn—Te distance (2.88—2.90 Á, see Table 1 — second entry) obtained in the distorted tetrahedron model is close to the length of ionic Mn—Te bond (di0n = 2.87 Á [66]), which is
charac-teristic for hexagonal NiAs-type MnTe. Shorter Mn—Te distance (2.60—2.64 Á, see Table 1 — second entry) compares to the length of covalent Mn—Te bond (dcov = 2.71 Á [67,68]) expected in cubic
•
•
•
D
• •
D
6540.5
5540.4
M 6 5 4 0 . 3 ^
6540.2
0.24
0.16 gJ
6540 6550 6560
EfeV)
6570 0.02 0.04 0.06
X
0.08 0.10
Fig. 2 . (a) Experimental Mn K-edge XANES spectra of Cdi _xMnxTeo.97Seo.o3 (x = 0 . 0 2 , 0 . 0 5 , 0 . 1 ) w i t h t h e fit of t h e p r e - e d g e region s h o w n in inset, (b) Integrated p r e - e d g e p e a k areas a n d t h e i r e n e r g y positions.
Cd Mn Te Se
0.98 0.02 0.97 0.03
Cd Mn Te Se
0.95 0.05 0.97 0.03
Cd Mn Te Se
0.9 0.1 0.97 0.03
R(A)
3 4
R(A]
Fig. 3 . The k2- w e i g h t e d Mn K-edge EXAFS d a t a of Cdo.9sMno.02Teo.97Seo.03 (a), Cdo.95Mno.05Teo.97Seo.03 (b) a n d Cdo.9Mno.1Teo.97Seo.03 (c). Real parts, imaginary parts a n d m a g n i t u d e s of t h e c o r r e s p o n d i n g Fourier transforms (d—f). Data are r e p r e s e n t e d w i t h full circles a n d t h e b e s t fits w i t h lines.
Table 1
Structural parameters obtained from the Mn K-edge EXAFS data analysis: r — the Mn—Te distance, a1 • the mean-square distance variation, E0 — the edge shift correction.
Cdo.9sMno.02Teo.97Seo.03 Cdo.95Mno.05Teo.97Seo.03 Cdo.9Mno.iTeo.97Seo.o
Hvln-Te (A)
<r2 (A2)
Eo (eV)
2.866(4) [33] *2.894(4) <2.82>
0.0037(3) [33] *0.002(1)
4 [33[
*2.8(2)
0.9(4) [33] * - l . l ( 6 )
*2.64(2)
*0.002(1)
"1.2
-1.1(6)
2.867(4) *2.88(2) <2.80> 2.80(5) [31] 0.0037(3) *0.002(2) 0.006(2) [31] 4
*2.9(5) 3.2 (5) [31] 0.5(5)
*2.60(6)
*0.002(2)
"1.1
-1(1)
2.880(7) *2.90(1) <2.85> 2.79(2) [31] 0.0034(6) *0.002(3) 0.005(1) [31] 4
*3.2(6) 3.2 (3) [31] 0.7(5) * - l ( 2 )
*2.63(8)
*0.002(3)
"0.8
-1(2)
*Model with two sub-shells in the first coordination around Mn, with the mean values 0 corresponding to the weighted sum of the two distances.
OMn-Te reported in Table 1 are found to be quite low for this kind of systems [31 ] and virtually independent on Mn concentration in the range 0.02 < x < 0.1 (at low measurement temperature they reflect primarily structural disorder). The fluctuations of the center of gravity of the Mn bonding charge, as the most probable cause of structural disorder, can trigger Mn off-centering from the regular lattice position and initiate formation of one very short and strong Mn—Te bond, as observed experimentally.
4.3. Theoretical calculations
To further investigate nature, strength and distribution of the Mn—Te bonds in the first coordination around Mn, the impact of the bond length variation on the electronic structure of the compound and their influence on the characteristic features appearing in the Mn K-edge XANES spectra, we performed detailed theoretical cal-culations. Following the results of EXAFS measurements, according to which the first coordination around Mn consists predominantly of Te atoms, calculations were performed on the optimized Cdi5MnTei6 and Cd3MnTe4 structures. These systems were then used to construct several specific types of deformation of the local structure around central Mn atom: expansion of the first coordi-nation tetrahedron with all the Mn—Te distances set to 2.9 Á (configuration labeled 4/[email protected]); contraction of the first coordina-tion tetrahedron with all the Mn—Te distances set to 2.5 Á (configuration labeled 4/[email protected]); displacement of the central Mn atom from the regular lattice site toward one, and between the two neighboring Te atoms with the shorter Mn—Te distance(s) set to 2.5 Á (configurations labeled l/[email protected] and 2/[email protected]).
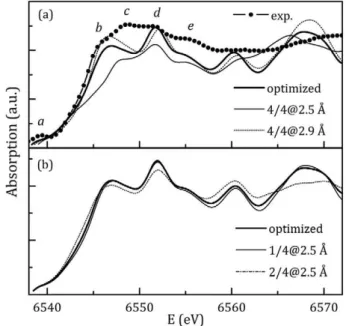
Experimental Mn K-edge XANES spectrum of Cdo.98Mno.02Teo.97Seo.03 and theoretical XANES spectra of the optimized Cd3MnTe4 system with regular and distorted first coor-dination around Mn are presented in Fig. 4. XANES spectrum of the Cdi5MnTei6 is very similar in appearance and therefore it is not shown. Characteristic spectral features denoted by letters a—e in
Fig. 4a, enable to track the influence of local structural deformations on the XANES spectrum.
Contraction and expansion of the first coordination tetrahedron (see Fig. 4a) have much more pronounced impact on the XANES spectrum than Mn-displacement from the regular lattice site (see
Fig. 4b). Position, intensity and shape of feature a are almost insensitive to the analyzed deformations, in accordance with deli-cate changes of the pre-edge region observed in the experimental XANES spectra. The feature b can be related to the number of long Mn—Te bonds, since its intensity is largest in configuration 4/[email protected]. As the number of short Mn—Te bonds increases, XANES spectrum markedly changes shape, feature b shifts towards higher energy positions, and ultimately alters the feature c in 4/[email protected]
3
re
e
a s. o - Q
<
optimized
J 1 l _
6540 6550 6560
E(eV)
6570
Fig. 4. Experimental Mn K-edge XANES spectrum of Cdo.9Mno.1Teo.97Seo.03 and theo-retical XANES spectra of (a) optimized Cd3MnTe4 system and Cd3MnTe4 system with
contracted/expanded tetrahedral coordination around Mn; (b) Cd3MnTe4 system with
local distortion resulting from two characteristic displacements of Mn inside its first coordination tetrahedron (see text for more details). Characteristic features of the experimental XANES spectrum are denoted by letters a—e.
configuration. Features d and e are visible in all theoretical XANES spectra, though their relative intensity is closest to the experi-mental in configuration 4/[email protected].
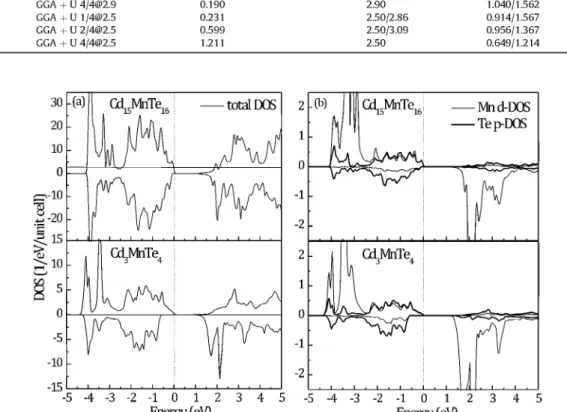
Main results of the electronic structure calculations of Cd3MnTe4 and Cdl5MnTei6 systems are presented in Table 2. Total and
selected 1-projected densities of states (DOS) of the two systems are presented in Fig. 5a and b, respectively.
Among all the deformed structures analyzed, the most stable configuration is 4/[email protected] in Cdi5MnTei6, while in both Mn-doped systems configuration 4/[email protected] is the least stable. The configuration l/[email protected] in Cd3MnTe4 is closer in energy to the optimized structure than the configuration 2/[email protected]. Calculated nearest neighbor Mn—Te distances slightly increase with Mn-concentration, in agreement with EXAFS results (see Table 2). As can be seen from
Fig. 5a, with the increase of Mn concentration, the asymmetry between spin-up and spin-down DOS gets more pronounced and the negative exchange splitting (spin-up Eg < spin-down Eg)
be-comes stronger. The calculated spin-up Eg decreases with x, in
Table 2
Calculated parameters of Cd3MnTe4 and Cd15MnTe16 systems: total energy relative to the optimized structure, nearest neighbor distances (rMn_Te), energy gap (Eg) for spin-up/ spin-down states and spin-down conduction band bottom (Ec) relative to the Fermi level.
System Relative energy (eV) rMn-Te (A) Eg (eV) up/down Ec (eV)
Cd15MnTe16
Cd3MnTe4
GGA
GGA + U optimized GGA + U 4/[email protected] GGA + U 4/[email protected] GGA
GGA + U optimized GGA + U 4/[email protected] GGA + U l/[email protected] GGA + U 2/[email protected] GGA + U 4/[email protected]
-0.776
--0.449 0.952 -0.680
-0.190 0.231 0.599 1.211
2.73 2.76 2.90 2.50 2.74 2.77 2.90 2.50/2.86 2.50/3.09 2.50
1.142/1.289 1.193/1.304 1.197/1.300 1.046/1.249 0.889/1.650 0.977/1.625 1.040/1.562 0.914/1.567 0.956/1.367 0.649/1.214
0.980 1.065 1.068 0.816 0.713 0.905 0.989 0.720 0.759 0.356
Mnd-DOS Tep-DOS
- 5 - 4 - 3 - 2 - 1 0 1 2 3 4 5 Energy (eV)
5 -4 -3 - 2 - 1 0 1 Energy (eV)
Fig. 5. (a) Total DOS of optimized Cd15MnTe16 (upper part) and Cd3MnTe4 (lower part); (b) Mn d- and Te p-projected DOS of optimized Cd15MnTe16 (upper part) and Cd3MnTe4
(lower part). Spin-up and spin-down states are presented with the opposite orientation. Vertical lines denote the position of EF.
in conduction band (CB) decreases with x, which is expected to shift the CB bottom (Ec) upwards in energy (positive shift). Manganese 3d spin-up states are spread over the entire VB (between - 5 and 0 eV), while empty Mn-3d spin-down states dominate the Ec. Strong hybridization with Te-5p states causes downward energy shift (negative shift) of the two most prominent Mn-3d spin-up peaks at the VB bottom (see Fig. 5b). Despite negative shift of the two main spin-down Mn-3d peaks at the Ec, an even larger nega-tive shift of the spin-down states (predominantly Te-5p in char-acter) close to the Ev causes the spin-down Eg to increase (see
Fig. 5b). Note that Eg estimation is a weak point of the DFT
calcu-lations, and at present we are not able to provide more reliable conclusion on the fine details of the states involved. The second coordination around Mn is mainly composed of Cd atoms. While Cd-5s states are located around - 4 eV, majority of Cd-4d states reside deeper in the VB (not shown). Their influence on the band states of the host crystal, although indirect, probably is not negli-gible, and is yet to be determined.
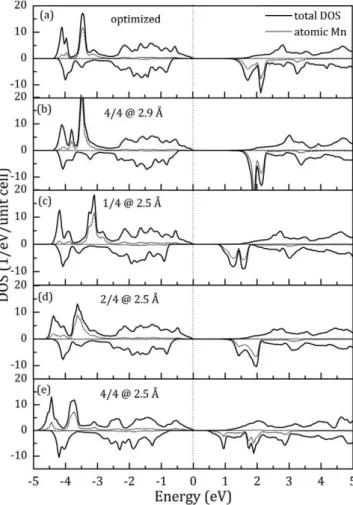
Total and atomic Mn DOSs of the optimized Cd3MnTe4 system with regular and distorted tetrahedral coordination around Mn, are presented in Fig. 6. Variations of Mn—Te bond length affect pri-marily the DOS regions dominated by Mn-3d states. The main differences are reflected in the distribution of Mn states, VB width, Eg and the position of Ec relative to EF.
Contraction of the first coordination tetrahedron (configuration 4/[email protected]) has the largest impact on the electronic structure of the compound. The most prominent Mn-3d spin-up peaks are shifted deeper into VB, making it wider. The spin-up states in CB are more spatially extended, which results in narrower spin-up Eg.
Appear-ance of additional peaks in the spin-down DOS close to Ec causes also spin-down Eg to shrink.
Expansion of the first coordination tetrahedron (configuration 4/[email protected]) leads to less prominent changes in the Cd3MnTe4 elec-tronic structure. The position of the most intense spin-up DOS peak at around -3.5 eV (predominantly Mn-3d in character) coincides with the corresponding peak in the optimized structure. The VB width is comparable to the optimized structure, but the spin-up Eg
is slightly wider. Although the spin-down Mn-3d states at the Ec are more localized and shifted upwards in energy, the spin-down Eg is
narrower than in the optimized structure due to larger positive shift of the spin-down states at the Ev.
Intermediate configurations (l/[email protected] and 2/[email protected]) enable to track modifications in Mn-3d DOS between the two end configu-rations, in particular influence of the Mn—Te bond length on Eg and
J I I I I I I I I I I I I I I I I I L
- 5 - 4 - 3 - 2 - 1 0 1 2 3 4 5
Energy (eV)
Fig. 6. Total and atomic Mn DOSs of optimized Cd3MnTe4 system with (a) regular tetrahedral coordination around Mn; (b) expanded tetrahedral coordination with all four Mn—Te distances set to 2.9 A (configuration 4/402.9); (c) distorted tetrahedral coordination resulting from Mn displacement toward Te atom (configuration l/[email protected]); (d) distorted tetrahedral coordination resulting from Mn displacement between the two Te atoms (configuration 2/[email protected]); (e) contracted tetrahedral coordination with all four Mn—Te distances set to 2.5 A (configuration 4/402.5).
most prominent spin-up peaks at the VB bottom, which interest-ingly, does not affect much the VB width. Also, when compared to 2/[email protected], configuration l/[email protected] is characterized with larger nega-tive shift of the spin-down states (at both the Ev and the Ec), which is an indicator of stronger pd-hybridization. In this way, the spin-down Eg follows the trend (it is larger in l/[email protected] than in 2/[email protected]), but the spin-up Eg does not (it is smaller in l/[email protected] than in 2/
[email protected]), implying that the exchange interaction is also stronger in configuration l/[email protected]. The fact that the electronic features specific for the optimized structure DOS can be recognized in configura-tions l/[email protected] and 4/[email protected] ensures the "electronic" arguments for existence of short and long Mn-Te bonds experimentally identified in the investigated compounds.
The results of Bader's charge density topology analysis of Cd3MnTe4 are summarized in Tables 3 and 4. The results obtained for Cdi5MnTei6 follow similar trends, and therefore they are not given explicitly. The attractor basin volumes are much larger than the volumes of the corresponding MT-spheres (see Table 3) and allow for more reliable estimation of the ions' excess charge and the spin polarization. Manganese and Te excess charges are smallest in the most compact 4/[email protected] configuration, and increase both with Mn concentration x and the number of long Mn—Te bonds (the only exception is the excess charge on Te in the single short bond of 1/ [email protected] configuration). The overall excess charge on Mn does not
exceed 0.945 e (donor) and the overall excess charge on Te is around -0.6 e (acceptor). Both values are considerably smaller than the estimated ions' valences (Mn2+, Te2~) supporting recent
statements [70] regarding essential difference between the charge state and the valence state, the two properties frequently misused as synonyms. Spin magnetic moment of Mn ranges from 4.87 (iB (in
configuration 4/[email protected]) to 5.19 (iB (in configuration 4/[email protected]).
Mag-netic moment induced on neighboring Te atoms is small and has opposite sign. The Mn spin-up attractor basin is not only much more populated, but it is also considerably larger than the spin-down attractor basin, which indicates pronounced delocahzation of the Mn spin-down states. The trend is opposite in case of Te, but with much smaller differences between the two basins (see
Table 3). The situation described above could be considered a typical electron charge distribution portrait of the exchange inter-action in DMS systems composed of tetrahedral local structures around 3d-TM ion.
The presented results indicate that contraction and expansion of the first coordination tetrahedron around Mn both have significant impact on the nature and properties of the Mn—Te bonds. When compared to the optimized Cd3MnTe4 structure, the complete contraction of the tetrahedron (configuration 4/[email protected]) causes in-crease, while its complete elongation (configuration 4/[email protected]) leads to decrease of the charge density (p) and its gradient (Vp) in bond critical points (CPs). Behavior of the Laplacian p (Ap) in the bond CPs has opposite trend — it decreases for contracted and increases for elongated Mn—Te bonds (see Table 4). Along with the excess charges of Mn and Te atomic basins, this implies that in the sym-metrical tetrahedral configurations (optimized, 4/[email protected] and 4/ [email protected]) Mn—Te bonds are partly ionic in nature (the ionicity in-creases with the bond length, in agreement with XAFS results). As oppose, in case when the tetrahedral configurations are distorted (l/[email protected] and 2/[email protected]), the contracted Mn-Te bonds' CPs have the largest p (see Table 4) and Ap changes sign (becomes negative), which indicates predominantly covalent nature of these bonds. A similar situation, perceived in our earlier studies of the nature of the Mn—Te bonds in Mn doped PbTe [69], suggests that the observed effects could be common for rather different DMSs, pro-vided that the TM-local coordination is composed of the same el-ements). Strikingly different spatial distributions of the Mn spin-up and spin-down states result in a slight mismatch between the positions of the spin-up and spin-down bond CPs (see Table 4),
which leads to ultimate formation of a small "bond critical point magnetic dipole" between them.
5. Conclusion
In conclusion, X-ray Absorption Fine Structure (XAFS) mea-surements, simulation of XAFS spectra, the electronic structure calculations and charge distribution topology analysis are per-formed to reveal local structural features and electronic properties produced by Mn impurity in Cdi_xMnxTeo.97Seo.o3 (x = 0.02, 0.05,
0.1). It has been established that Mn ions (in the given concentra-tion range) substitute for Cd in the host CdTe lattice (ZB-type structure) and that they are predominantly coordinated with Te atoms. With the increase of the concentration from x = 0.02 to x = 0.1, Mn valence slightly decreases from the initial 2+. The decrease in Mn valence is most likely accompanied with the decrease in the degree of hybridization between Mn-3d and Te-5p states.
Table 3
The results of Cd3MnTe4 electron charge density topology analysis. Attractor basin volumes and charges are given for spin-up and spin-down states, as indicated in the first row (Excess charge = Atomic - Total charge; Spin polarization = spin-up - spin-down charge).
System Atom Attractor basin volume (A3; Total attractor charge (e) Excess charge (e) Attractor basin charge (e) Spin polarization (jiB)
Cd3MnTe4
GGA
Cd3MnTe4
GGA + U optimized
Cd3MnTe4
GGA + U 4/[email protected]
Cd3MnTe4
GGA + U l/[email protected]
Cd3MnTe4
GGA + U 2/[email protected]
Cd3MnTe4
GGA + U 4/[email protected] Mn nnTe Mn nnTe Mn nnTe Mn nnTe short nnTe long Mn nnTe short nnTe long Mn nnTe 25.873 10.009 44.578 48.293 25.742 9.817 44.614 48.489 28.060 10.985 44.795 48.838 26.032 9.842 42.614 47.303 45.352 48.930 26.111 9.933 42.545 47.512 46.732 49.665 21.562 8.224 43.922 47.531 24.153 52.596 24.088 52.612 24.054 52.616 24.100 52.593 52.614 24.120 52.597 52.611 24.245 52.572 0.847 0.596 0.912 0.612 0.945 0.616 0.900 0.593 0.614 0.880 0.597 0.611 0.755 0.572 14.579 9.574 26.282 26.314 14.613 9.476 26.277 26.335 14.623 9.431 26.277 26.339 14.610 9.490 26.250 26.343 26.286 26.328 14.604 9.516 26.254 26.343 26.303 26.308 14.556 9.689 26.271 26.301 5.006 -0.032 5.137 -0.059 5.192 -0.061 5.120 -0.093 -0.042 5.088 -0.089 -0.005 4.867 -0.030 Table 4
Properties of the Mn bond critical points (CPs) in the Cd3MnTe4 systems with regular (optimized) and distorted tetrahedral coordination around Mn. p — electron charge
density in bond CP; rTe(Mn) — distance from Te(Mn) atom to the corresponding bond CP; | r - d / 2 | — deviation of the bond CP position from the half of the Mn—Te distance (d).
System Mn bond CPs P (e/A3 Vp-10-15(e/A4) Ap-lO-^e/A5) rTe (A) rMn (A) |r-d/2| (A)
Cd3MnTe4
GGA
Cd3MnTe4
GGA + U optimized Cd3MnTe4
GGA + U 4/[email protected] Cd3MnTe4
GGA + U 1/4S2.5
Cd3MnTe4
GGA + U 2/[email protected]
Cd3MnTe4
GGA + U 4/[email protected]
Up Down Total Up Down Total Up Down Total 1 short 2.5 A 3 long 2.86 A 2 short 2.5 A 2 long 3.09 A Up Down Total Up Down Total Up Down Total Up Down Total Up Down Total 0.178 0.146 0.336 0.173 0.137 0.323 0.142 0.112 0.265 0.261 0.212 0.487 0.147 0.116 0.275 0.260 0.211 0.485 0.101 0.078 0.188 0.256 0.209 0.480 2.60 1.62 2.48 2.06 1.48 4.85 0.78 0.55 3.18 1.09 0.14 10.9 1.88 0.42 2.52 4.86 0.47 8.77 1.05 0.40 2.07 2.26 0.94 2.53 7.168 6.639 6.304 7.078 1.167 6.893 6.331 1.275 6.848 4.827 1.074 0.975 6.809 0.478 6.948 4.640 1.033 0.853 5.694 1.058 6.915 6.052 9.298 2.213 1.470 1.667 1.541 1.483 1.693 1.559 1.564 1.780 1.638 1.326 1.466 1.423 1.542 1.763 1.616 1.330 1.466 1.424 1.678 1.904 1.749 1.325 1.458 1.409 1.275 1.077 1.203 1.283 1.073 1.206 1.336 1.120 1.261 1.173 1.033 1.076 1.322 1.102 1.249 1.172 1.037 1.079 1.413 1.187 1.343 1.175 1.042 1.090 0.097 0.295 0.169 0.099 0.310 0.176 0.114 0.329 0.188 0.076 0.216 0.173 0.110 0.330 0.184 0.079 0.215 0.173 0.132 0.358 0.203 0.075 0.207 0.160
are much longer and more ionic in nature (hexagonal MnTe-alike bonds).
This distinctive local atomic arrangement, caused by Mn off-centering from the regular lattice position, is accompanied by the appearance of specific electronic features. The electronic structure analysis of the Cd3MnTe4 system with the first coordination tetra-hedron around Mn distorted in several characteristic ways, revealed that the optimized structure DOS shares certain common characteristics with distorted configurations l/[email protected] (Mn
Uniqueness of the experimentally detected first coordination around Mn is also reflected in the fact that the features of its electronic structure deviate from the trends linking the two end configurations (4/[email protected] and 4/[email protected]). Larger accumulation of p into contracted Mn—Te bond CPs in distorted tetrahedral configu-rations (l/[email protected] and 2/[email protected]) and the change of Ap sign, indicate their pronounced covalent character. As oppose, long Mn—Te bonds are found to be predominantly ionic (the iconicity slightly rises with the bond length). The excess charges on Mn and Te ions are considerably different from their nominal and XAFS-determined valences, implying that the two concepts (charge state and valence state) should not be thought of as being equivalent.
In all configurations analyzed, the attractor basins of the Mn-3d spin-up states are much larger than the spin-down attractor basins, implying that the spin-up states are considerably more spatially extended than the spin-down states. As a consequence, the spin-up and the spin-down CPs corresponding to the same Mn—Te bond are slightly displaced from each other and generate a small magnetic dipole at the bond CP. If this new kind of magnetic order and interaction receives an experimental confirmation, some novel possibilities to manipulate DMS characteristics by external means would be opened.
Presented results have enabled to determine electronic prop-erties and to establish positive relations between details of the first coordination structures around Mn and characteristic features of the theoretical and experimental XAFS spectra, and to make clear distinctions between various influences. That way, a consistent and adoptable procedure for methodical analysis of structural and electronic consequences of transition metal impurity incorporation in II—VI (and presumably other semiconductor hosts) has been established.
Acknowledgments
The authors gratefully acknowledge HASYLAB @ DESY for providing the beamtime and the partial financial support (Project No. 1-20100379 EC). Al beamline scientist Dr. E. Welter is acknowledged for assistance during XAFS measurements. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/ 2007—2013) under the Grant agreement No 226716 and is sup-ported by Serbian Ministry of Education, Science and Technological Development under the Grant III 45003.
References
[1] J. Kossut, J.A. Gaj (Eds.), Introduction to the Physics of Diluted Magnetic Semiconductors, Springer—Verlag, Berlin Heidelberg, 2010.
[2] T. Dietl, A ten-year perspective on dilute magnetic semiconductors and oxides, Nat. Mater. 9 (2010) 965-974.
[3] W. Zaets, K Ando, Magneto-optical mode conversion in Cdi xMnxTe wave-guide on GaAs substrate, Appl. Phys. Lett. 77 (2000) 1593-1595.
[4] Y. Hwang, S. Chung, Y. Um, Giant Faraday rotation in Cdi xMnxTe (0 < x < 0.82) crystals, Phys. Status Solidi C 4 (2007) 4453-4456.
[5] M. Nogaku, R. Pittini, T. Sato, J. Shen, Y. Oka, Formation dynamics of free excitonic magnetic polarons in Cdi xMnxTe, J. Appl. Phys. 89 (2001) 7287-7289.
[6] P. Wojnar, J. Suffczyriski, K. Kowalik, A. Golnik, M. Aleszkiewicz, G. Karczewski, J. Kossut, Size-dependent magneto-optical effects in CdMnTe diluted magnetic quantum dots, Nanotechnology 19 (2008) 235403-235405.
[7] C. Leighton, I. Terry, P. Becla, Metal-insulator transition in the persistent photoconductor Cdi xMnxTe:In, Europhys. Lett. 42 (1998) 6 7 - 7 2 .
[8] S. Geschwind, A.T. Ogielski, G. Devlin, J. Hegarty, P. Bridenbaugh, Activated dynamic scaling and magnetic ordering in Cdi xMnxTe: spin glass or random
antlferromagnet? J. Appl. Phys. 63 (1988) 3291-3296.
[9] M.A. Novak, O.G. Symko, D.J. Zheng, S. Oseroff, Spin glass behavior of Cdi xMnxTe below the nearest-neighbor percolation limit, J. Appl. Phys. 57 (1985) 3418-3420.
[10] R.R. Galazka, S. Nagata, P.H. Keesom, Paramagnetic-spin-glass-antiferromag-netic phase transitions in Cdi xMnxTe from specific heat and magnetic
susceptibility measurements, Phys. Rev. B 22 (1980) 3344-3355.
G.-L. Tan, M. Wang, Ferromagnetism of ternary Cdi xMnxTe nanocrystals, IEEE Trans. Nanotechnol. 11 (2012) 236-238.
A. Mycielski, L. Kowalczyk, R.R. Galazka, R. Sobolewski, D. Wang, A. Burger, M. Sowiñska, M. Groza, P. Siffert, A. Szadkowski, B. Witkowska, W. Kaliszek, Applications of II—VI semimagnetic semiconductors, J. Alloy. Compd. 423 (2006) 163-168.
A. Rohatgi, S.A. Ringel, J. Welch, E. Meeks, K. Pollard, A. Erbil, C.J. Summers, P.V. Meyers, C.H. Liu, Growth and characterization of CdMnTe and CdZnTe polycrystalline thin films for solar cells, Sol. Cells 24 (1988) 185-194. I.S. Yahia, F. Yakuphanoglu, S. Chusnutdinow, T. Wojtowicz, G. Karczewski, Photovoltaic characterization of n—CdTe/p—CdMnTe/GaAs diluted magnetic diode, Curr. Appl. Phys. 13 (2013) 537-543.
L.A. Kosyachenko, R. Yatskiv, N.S. Yurtsenyuk, O.L Maslyanchuk, J. Grym, Graphite/CdMnTe Schottky diodes and their electrical characteristics, Semi-cond. Sci. Technol. 29 (2014) 015006-015010.
C. Gould, G. Schmidt, G. Richter, R. Fiederling, P. Grabs, L.W. Molenkamp, Spin injection into semiconductors using dilute magnetic semiconductors, Appl. Surf. Sci. 190 (2002) 395-402.
E.V. Gomonay, V.M. Loktev, Spintronics of antiferromagnetic systems, Low. Temp. Phys. 40 (2014) 1 7 - 3 5 .
H. MacDonald, M. Tsoi, Antiferromagnetic metal spintronics, Philos. Trans. R. Soc. A 369 (2011) 3098-3114.
R. Rungsawang, F. Perez, D. Oustinov, J. Gómez, V. Kolkovsky, G. Karczewski, T. Wojtowicz, J. Madéo, N. Jukam, S. Dhillon, J. Tignon, Terahertz radiation from magnetic excitations in diluted magnetic semiconductors, Phys. Rev. Lett. 110 (2013) 177203-177205.
K.H. Kim, A.E. Bolotnikov, G.S. Camarda, G. Yang, A. Hossain, Y. Cui, R.B. James, J. Hong, S.U. Kim, Energy-gap dependence on the Mn mole fraction and temperature in CdMnTe crystal, J. Appl. Phys. 106 (2009) 023706-23713. D. Kochanowska, et al., Growth and characterization of (Cd, Mn)Te, IEEE Trans. Nucl. Sci. 60 (2013) 3805-3814.
K. Strzalkowski, F. Firszt, A. Marasek, Thermal diffusivity, effusivity, and conductivity of CdMnTe mixed crystals, Int. J. Thermophys. 35 (2014) 2140-2149.
J. Zhang, L Wang, J. Huang, K. Tang, Z. Yuan, Y. Xia, Properties of CdMnTe single crystal used in nuclear radiation detectors, Adv. Mat. Res. 311—313 (2011) 1209-1212.
H. Boukari, P. Kossacki, M. Bertolini, D. Ferrand, J. Cibert, S. Tatarenko, A. Wasiela, J.A. Gaj, T. Dietl, Light and electric field control of ferromagnetism in magnetic quantum structures, Phys. Rev. Lett. 88 (2002), 207204—4. J. Fernández-Rossier, R. Aguado, Mn-doped II—VI quantum dots: artificial molecular magnets, Phys. Status Solidi C 3 (2006) 3734-3739.
L Villegas-Lelovsky, F. Qu, LO. Massa, V. Lopez-Richard, G.E. Marques, Hole-mediated ferromagnetism in coupled semimagnetic quantum dots, Phys. Rev. B 84 (2011) 07531-75412.
C. Rice, L.C. Smith, J.J. Davies, D. Wolverson, M. Wiater, G. Karczewski, T. Wojtowicz, Exchange interactions in Cdi xMnxTe wide quantum wells, Phys. Rev. B 86 (2012) 155318-155326.
Z. Ben Cheikh, S. Cronenberger, M. Vladimirova, D. Scalbert, F. Perez, T. Wojtowicz, Electron spin dephasing in Mn-based II—VI diluted magnetic semiconductors, Phys. Rev. B 88 (2013) 201306-201314.
S. Chehab, G. Lamarche, A. Manoogian, J.C. Woolley, Magnetic properties of Cdi zMnJei ySey alloys, J. Magn. Magn. Mater. 59 (1986) 105-114. N. Romcevic, M. Romcevic, A. Golubovic, L Van Khoi, A. Mycielski, D. Jovanovic, D. Stojanovic, S. Nikolic, S. Duric, Far-infrared and Raman spectroscopy of Cdi xMnxTei ySey: phonon properties, J. Alloy. Compd. 397
(2005) 5 2 - 5 7 .
B. Pukowska, J. Jaglarz, B. Such, T. Wagner, A. Kisiel, A. Mycielski, Optical in-vestigations of the CdTeSe and CdMeTeSe (Me = Mn, Fe) semiconductors, J. Alloy. Compd. 335 (2002) 3 5 - 4 2 .
B.V. Robouch, A. Marcelli, M. Cestelli Guidi, Statistical model analysis of local structure of quaternary sphalerite crystals, Low. Temp. Phys. 33 (2007) 214-225.
I. Radisavljevic, N. Novakovic, N. Romcevic, N. Ivanovic, Local and electronic structure around manganese in Cdo.98Mno.02Teo.97Seo.03 studied by XAFS, J. Phys. Conf. Ser. 430 (2013) 012083-012084.
M. O'Toole, D. Diamond, Absorbance based light emitting diode optical sen-sors and sensing devices, Sensen-sors 8 (2008) 2453—2479.
A. Klein, Energy band alignment in chalcogenide thin film solar cells from photoelectron spectroscopy, J. Phys. Condens. Matter 27 (2015) 134201-134224.
J. Jack Li, J.M. Tsay, X. Michalet, S. Weissa, Wavefunction engineering: from quantum wells to near-infrared type-II colloidal quantum dots synthesized by layer-by-layer colloidal epitaxy, Chem. Phys. 318 (2005) 8 2 - 9 0 .
K. Kim, J. Hong, S. Kim, Electrical properties of semi-insulating CdTe0.9Se0.i :C1 crystal and its surface preparation, J. Cryst. Growth 310 (2008) 91—95. U.N. Roy, A.E. Bolotnikov, G.S. Camarda, Y. Cui, A. Hossain, K. Lee, G. Yang, R.B. James, Growth and characterization of CdTeSe for room-temperature radiation detector applications, J. Cryst. Growth 389 (2014) 99-102. V.A. KulbachinskiT, LA. Churilov, P.D. Maryanchuk, R.A. Lunin, Effect of sele-nium on the galvanomagnetic properties of the diluted magnetic semi-conductor Hgi xMnxTei ySey, Semiconductors 32 (1998) 4 9 - 5 1 .
inverse-photoemission spectroscopies, Phys. Rev. B 61 (2000) 10622—10627. [41] S.F. Chehab, J.C. Woolley, Energy gap and valence band structure of
Cdi zMnzTei ySey alloys, Phys. Status Solidi B 139 (1987) 213-222. [42] S.-H. Wei, A. Zunger, Total-energy and band-structure calculations for the
semimagnetic Cdi xMnxTe semiconductor alloy and its binary constituents, Phys. Rev. B 35 (1987) 2340-2365.
[43] S.M. Durbin, J. Han, O. Sungki, M. Kobayashi, D.R. Menke, R.L Gunshor, Q_. Fu, N. Pelekanos, A.V. Nurmikko, D. Li, J. Gonsalves, N. Otsuka, Zinc-blende MnTe: epilayers and quantum well structures, Appl. Phys. Lett. 55 (1989) 2087-2089.
[44] J. Oleszkiewicz, M. Podgórny, A. Kisiel, E. Burattini, Theoretical and experi-mental analysis of the near-edge x-ray absorption structure in MnTe and Cdt xMnxTe alloys, Phys. Rev. B 60 (1999) 4920-4927.
[45] M.P. Vecchi, W. Giriat, L Videla, Photoluminescence studies of the Mn2 +
d-levels in Cdi xMnxTe, Appl. Phys. Lett. 38 (1981) 9 9 - 1 0 1 .
[46] B.A. Orlowski, Photoemission evidence of the Mn(3d5) band in Cdo.4Mno.6Te
crystals, Phys. Status Solidi B 95 (1979) K31-K35.
[47] M. Taniguchi, K. Mimura, H. Sato, J. Harada, K. Miyazaki, H. Namatame, Y. Ueda, Ultraviolet inverse-photoemission and photoemission spectroscopy studies of diluted magnetic semiconductors Cdi xMnxTe (0 < x < 0.7), Phys. Rev. B 51 (1995) 6932-6939.
[48] A.E. Merad, M.B. Kanoun, S. Goumri-Said, Ab initio study of electronic struc-tures and magnetism in ZnMnTe and CdMnTe diluted magnetic semi-conductors, J. Magn. Magn. Mater. 302 (2006) 536-542.
[49] C. Webb, M. Kaminska, M. Lichtensteiger, J. Lagowski, Valence band states of semi-magnetic semiconductors: Cdi xMnxTe, Solid State Commun. 40 (1981) 6 0 9 - 6 1 1 .
[50] N. Happo, H. Sato, K. Mimura, S. Hosokawa, M. Taniguchi, Y. Ueda, M. Koyama, d4 identification of the satellite in the Mn 3d photoemission spectra of Cdi xMnxTe alloys, Phys. Rev. B 50 (1994) 12211-12214.
[51 ] A. Balzarotti, N. Motta, A. Kisiel, M. Zimnal-Starnawska, M.T. Czyzyk, M. Podgórny, Model of the local structure of random ternary alloys: experi-ment versus theory, Phys. Rev. B 31 (1985) 7526-7539.
[52] J.K Glasbrenner, I. Zutic, LI. Mazin, Theory of Mn-doped II—II—V semi-conductors, Phys. Rev. B 90 (2014) 140403-140405.
[53] T. Mousavi, C.R.M. Grovenor, S.C. Speller, Structural parameters affecting su-perconductivity in iron chalcogenides: review, Mater. Sci. Tech. 30 (2014) 1929-1943.
[54] M. Newville, IFEFFIT: interactive XAFS analysis and FEFF fitting, J. Synchrotron Radiat. 8 (2001) 322-324.
[55] B. Ravel, M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat. 12 (2005)
537-541.
[56] B. Ravel, EXAFS Analysis with FEFF and FEFFIT, http://cars9.uchicago.edu/ -ravel/course/.
[57] P. Blaha, K. Schwarz, J. Luitz, WIEN 2k, Vienna University of Technology, Vienna, 1997. Improved and updated UNIX version of the original copyrighted WIEN code, published by P. Blaha, K Schwarz, P. Sorantin, S.B. Trickey, Full-potential, linearized augmented plane wave programs for crystalline sys-tems, Comput. Phys. Commun. 59 (1990) 399-415.
[58] J.P. Perdew, K. Burke, M. Ernzerhoff, Generalized gradient approximation made simple, Phys. Rev. Lett. 77 (1996) 3865-3868.
[59] F. Tran, P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential, Phys. Rev. Lett. 102 (2009) 226401-226404.
[60] R. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, USA, 1994.
[61] A. Otero-de-la-Roza, M.A. Blanco, A. Martín Pendas, V. Luana, Critic: a new program for the topological analysis of solid-state electron densities, Comput. Phys. Commun. 180 (2009) 157-166.
[62] I. Radisavljevic, N. Novakovic, H.-E. Mahnke, N. Romcevic, M. Medic, B. Paskas Mamula, N. Ivanovic, X-ray absorption near edge structure studies of Pbi xMnxTe(In, Ga) systems, Int. J. Mater. Res. 104 (2013) 319-325. [63] I. Radisavljevic, N. Novakovic, N. Romcevic, M. Manasijevic, H.-E. Mahnke, N.
Ivanovic, Quaternary Fe-based Wide-gap Diluted Magnetic Semiconductors: Structural Investigations, HASYLAB Annual Report 2010, http:// photon—science.desy.de/annual_report/files/2010/20101410.pdf.
[64] B.V. Robouch, A. Kisiel, J. Konior, Statistical model for atomic distances and site occupation in zinc-blende diluted magnetic semiconductors (DMSs), J. Alloy. Compd. 340 (2002) 13-26.
[65] N.-E. Sung, H.-Y. Park, M.-S. Jang, EXAFS analysis of the local structure of Cd(i x)MxTe (M = Cr, Fe, Ni), AIP Conf. Proc. 882 (2007) 550-552.
[66] R.D. Shannon, Revised effective ionic radii and systematic studies of inter-atomic distances in halides and chalcogenides, Acta Cryst. A 32 (1976) 751-767.
[67] R.J. Iwanowski, K. Lawniczak-Jablonska, Z. Golacki, A. Traverse, Tetrahedral covalent radii of Mn, Fe, Co and Ni estimated from extended X-ray absorption fine structure studies, Chem. Phys. Lett. 283 (1998) 313-318.
[68] J.A. Van Vechten, J.C. Phillips, New set of tetrahedral covalent radii, Phys. Rev. B 2 (1970) 2160-2167.
[69] I. Radisavljevic, N. Ivanovic, N. Novakovic, N. Romcevic, M. Mitric, V. Andric, H.-E. Mahnke, Structural aspects of changes induced in PbTe by doping with Mn, In and Ga, J. Mater. Sci. 48 (2013) 8084-8100.