Aplicación de técnicas paralelas utilizando CUDA, al proceso de simulación de poblaciones en secuencias genéticas
70
0
0
Texto completo
(2) Dictamen Hago constar que el presente trabajo fue realizado en la Universidad Central “Marta Abreu” de Las Villas como parte de la culminación de los estudios de la especialidad de Ciencias de la Computación, autorizando a que el mismo sea utilizado por la institución, para los fines que estime conveniente, tanto de forma parcial como total y que además no podrá ser presentado en eventos ni publicado sin la autorización de la Universidad.. Firma del autor. Los abajo firmantes, certificamos que el presente trabajo ha sido realizado según acuerdos de la dirección de nuestro centro y el mismo cumple con los requisitos que debe tener un trabajo de esta envergadura referido a la temática señalada.. Firma del tutor. Firma del Jefe del Seminario. i.
(3) Agradecimientos Quiero expresar mis más sinceros agradecimientos: A mi tutor Leonardo Flavio del Toro Melgarejo por guiarme durante todo el proceso de investigación que dío como resultado este trabajo. A todos los amigos que de una forma u otra me ayudaron durante los cinco años de vida universitaria, y compartieron conmigo tanto los buenos momentos como los malos. A mi hermana, mi mamá y mi papá por el inmenso apoyo que siempre me brindaron, incluso en momentos en los cuales nos separaba una gran lejanía. Especialmente a Eimy por su gran ayuda en todo momento , por dedicarme incondicionalmente su atención y amor, por apoyarme y animarme a rebasar las circunstancias más difíciles. Finalmente, a todos los que me han ayudado de cualquier manera a forjarme como un profesional. Muchas gracias a todos!!!. ii.
(4) Resumen Una de las áreas de la Bioinformática que ha experimentado mayor expansión en los últimos años es la investigación de procesos evolutivos mediante la aplicación de técnicas de la Biología Molecular combinadas con los métodos matemáticos. En este trabajo se exponen los resultados obtenidos a partir de la aplicación de técnicas paralelas utilizando CUDA a la simulación de la evolución de poblaciones virales. Con el propósito de evaluar las tendencias evolutivas de las poblaciones virales, en ausencia y bajo la presión selectiva del sistema inmune, fueron implementadas dos variantes paralelas de un algoritmo de generación de secuencias mutantes en el lenguaje de programación C++. En el trabajo se implementan en paralelo las fases más costosas de los algoritmos para la obtención de nuevas variantes mutacionales y la clasificación de las nuevas secuencias a partir de su correspondiente proteína, estas fueron incorporadas en una aplicación que realiza todo el proceso. Los resultados obtenidos fueron validados utilizando una aplicación secuencial que se desarrolla y se perfecciona a partir de una existente, con la cual también se comparan los tiempos de ejecución, lográndose mejoras sustanciales de estos con el uso de CUDA.. iii.
(5) Abstract One of the fields that have experienced major breakthroughs in the last years in Bioinformatics is the research of evolutionary processes by means of the implementation of techniques used in Molecular Biology which are combined with mathematical methods. This papers aims at displaying the results obtained from the application of parallel techniques using CUDA to the simulation of the evolution of viral populations. In order to assess the evolutionary tendencies of the viral populations, in absence and under the selective pressure of the immune system, two parallel versions of an algorithm of mutant sequences were implemented in the programming language of C++. At the time of the investigation the most expensive phases of the algorithms are implemented in parallel in order to obtain new mutational variants and the classification of new sequences from their corresponding protein; these sequences were incorporated in an application that does the whole process. The obtained results were validated using a sequential application that performs intention, with which the times of execution are also compared, achieving significant progress of these with the use of CUDA.. iv.
(6) Tabla de Contenidos. Introducción. 1. 1 Fundamentos Biológicos, Matemáticos y Computacionales 1.1 Introducción a los elementos biológicos . . . . . . . . . . . . . . . . . . . . . 1.1.1 Las moléculas básicas de la vida . . . . . . . . . . . . . . . . . . . . . 1.1.2 ADN y ARN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.1.3 Los aminoácidos y el código genético . . . . . . . . . . . . . . . . . . 1.2 Procedimiento evolutivo de las poblaciones virales . . . . . . . . . . . . . . . 1.3 Simulación de la evolución molecular . . . . . . . . . . . . . . . . . . . . . . 1.3.1 Cadenas de Markov . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.3.2 Matriz de probabilidades y probabilidad de transición en n pasos . . 1.3.3 Técnica Markov Chain Monte Carlo . . . . . . . . . . . . . . . . . . . 1.3.4 Modelos Evolutivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.3.5 Modelo TN93 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.4 Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.4.1 Construcción del modelo de regresión lineal mediante la obtención de los estimadores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.4.2 Los residuos en la estadística . . . . . . . . . . . . . . . . . . . . . . . 1.5 El modelo de programación en CUDA . . . . . . . . . . . . . . . . . . . . . . 1.6 Estructura de datos Trie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.6.1 Definición formal . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. v. 5 5 5 6 7 8 9 10 11 12 14 15 16 16 20 22 24 24.
(7) 1.7 1.8. 1.6.2 Ventajas de un Trie . . . . . . . . . . . . . . . . . . . . . . . . . . . . Las bibliotecas GSL y CURAND . . . . . . . . . . . . . . . . . . . . . . . . Conclusiones parciales del capítulo . . . . . . . . . . . . . . . . . . . . . . .. 2 Implementación de los algoritmos para la simulación de las poblaciones virales 2.1 Aplicación de la técnica MCMC a la simulación de poblaciones virales. . . . 2.1.1 Cálculo de la matriz P . . . . . . . . . . . . . . . . . . . . . . . . . . 2.2 Descripción del proceso de evolución genética de las poblaciones virales y de los principales algoritmos utilizados para desarrollar el mismo . . . . . . . . 2.2.1 Generación de mutaciones sin restricciones . . . . . . . . . . . . . . . 2.2.2 Generación de mutaciones con restricciones . . . . . . . . . . . . . . . 2.3 Implementación secuencial en C++ de la simulación de poblaciones virales . 2.3.1 Diseño del diagrama de clases y relaciones entre las mismas para simular la evolución de las secuencias sin selección . . . . . . . . . . . . . . . 2.3.2 Diseño del diagrama de clases y relaciones entre las mismas para simular la evolución de las secuencias con selección . . . . . . . . . . . . . . . 2.4 Implementación paralela utilizando CUDA de la simulación de las poblaciones virales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.4.1 Estrategias de solución paralelas . . . . . . . . . . . . . . . . . . . . . 2.4.2 Presión selectiva y paralelismo . . . . . . . . . . . . . . . . . . . . . . 2.5 Conclusiones parciales del capítulo . . . . . . . . . . . . . . . . . . . . . . . 3 Resultados y Discusión 3.1 Características de software y hardware . . 3.2 Descripción de las bases de datos . . . . . 3.3 Discusión de los resultados experimentales 3.4 Conclusiones parciales del capítulo . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. . . . .. 25 26 26. 27 27 29 31 32 34 38 40 42 44 44 50 51 52 52 53 53 56. Conclusiones. 58. Recomendaciones. 59. Referencias Bibliográficas. 61. vi.
(8) Lista de Figuras. 1.1 1.2. 1.3 1.4 1.5 1.6 1.7 2.1 2.2 2.3 2.4 2.5. La doble hélice del ADN. . . . . . . . . . . . . . . . . . . . . . . . . . . . . Código genético estándar. Los aminoácidos están escritos con el símbolo de tres letras. El codón utilizado con mayor frecuencia como codón de inicio de la transcripción corresponde al aminoácido Metionina: AUG. Los codones UAA, UAG y UGA son marcadores del final de los genes. . . . . . . . . . . . . . . Relación de dependencia lineal exacta entre las variables X y Y . . . . . . . Representación de una dependencia estocástica entre X y Y . . . . . . . . . Lotes de hilos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Espacios en memoria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Representación gráfica de un trie . . . . . . . . . . . . . . . . . . . . . . . .. 8 17 18 22 24 25. Esquema general del método propuesto para la simulación de la evolución de poblaciones virales. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Diagrama de clases sin presión selectiva. . . . . . . . . . . . . . . . . . . . . Diagrama de clases con presión selectiva. . . . . . . . . . . . . . . . . . . . . Esquema general para la paralelización de la evolución de poblaciones virales. Proceso de mutación paralelizado por sitio . . . . . . . . . . . . . . . . . . .. 39 40 43 45 46. vii. 6.
(9) Lista de Algoritmos. 2.1 2.2 2.3 2.4 2.5 2.6 2.7. Algoritmo MCMC para la generación de nuevas mutaciones . . . . . . . . . . Algoritmo para encontrar los codones de parada . . . . . . . . . . . . . . . . Algoritmo para clasificar una secuencia a partir de una vacuna . . . . . . . . Algoritmo para hallar los residuos estándares de un modelo de regresión lineal Algoritmo para estudentizar un conjunto de residuos estandarizados y realizar la clasificación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Algoritmo paralelo para realizar las mutaciones . . . . . . . . . . . . . . . . Algoritmo para clasificar las mutaciones . . . . . . . . . . . . . . . . . . . .. viii. 31 34 36 37 38 47 49.
(10) Introducción. Una de las áreas de la Bioinformática que ha experimentado mayor expansión en los últimos años es la investigación de procesos evolutivos mediante la aplicación de técnicas de la Biología Molecular combinadas con los métodos matemáticos. Los resultados obtenidos en las investigaciones científicas han demostrado que la simulación es un método muy útil para validar una teoría o una implementación de un programa cuando el análisis del problema es complejo. Cuando el modelo es analíticamente intratable, la simulación provee una manera potente para estudiarlo. La simulación de Monte Carlo, y en particular, el método de Monte Carlo basado en Cadenas de Markov (Markov-Chain Monte Carlo, MCMC), es el fundamento para formar una familia de algoritmos de generación de muestras aleatorias en un tiempo continuo y razonable. De hecho, debido a que el proceso de evolución molecular puede ser tratado como un proceso estocástico, la aplicación de MCMC resulta apropiada para resolver el problema de simulación de la evolución molecular de poblaciones de virus patógenos, un fenómeno confrontado diariamente por el sistema inmune del hombre. En particular, en el área de la medicina es de vital importancia la introducción de tratamientos a los pacientes con nuevos antivirales contra virus tales como el de la influenza A/H1N1 y el VIH, los cuales poseen una velocidad de evolución muy alta. Hasta el presente la compresión de los procesos de evolución molecular es uno de los retos más encumbrados de la biología molecular evolutiva. Desde el punto de vista computacional, las Unidades de Procesamiento Gráfico (GPU por sus siglas en ingles) se han popularizado y masificado en los últimos tiempos, en principio por el auge de juegos en las computadoras y la utilización en estos de cada vez más recursos gráficos que requieren de una alta capacidad de procesamiento. Los programadores utilizan un modelo de programación de la GPU en un lenguaje de alto nivel y una arquitectura de. 1.
(11) Introducción programación paralela; estos dos elementos conforman lo que NVIDIA denomina Compute Unified Device Architecture (CUDA). Considerando los antecedentes anteriores, se propone la idea de investigar el proceso de evolución de determinadas poblaciones virales de la influenza A mediante la simulación de Monte Carlo. Para este fin se propone el uso de un modelo estocástico evolutivo que permita simular matemáticamente la aparición de nuevas variantes mutacionales en las poblaciones virales a partir de la introducción aleatoria de mutaciones en las secuencias tomadas como ancestros. La implementación del modelo evolutivo se realizará sobre una GPU NVIDIA, utilizando las facilidades que nos brinda la API CUDA.. Antecedentes Actualmente existen varias aplicaciones que se emplean para el proceso de simulación de poblaciones genéticas basadas en el algoritmo Markov Chain Monte Carlo (MCMC), constituyendo este una herramienta poderosa para la computación bayesiana. Una descripción de este método puede ser consultada en el texto “Computational Molecular Evolution” (Yang, 2006). Algunas de estas aplicaciones han sido concebidas para entornos MatLab, empleando colecciones de herramientas específicas (J.Cai et al., 2006, p.179). Otras herramientas como Evolver (Cai et al., 2006) constituyen una colección de programas diseñados para simular la evolución de la secuencia de nucleótidos de un genoma entero. Evolver es una herramienta potente y compleja, requiriendo de días o semanas de procesamiento en un clúster de computadoras para procesar grandes genomas en distancias evolutivas interesantes. En el Centro de Estudios Informáticos (CEI), el grupo de Bioinformática aplica y desarrolla diferentes modelos para el análisis del proceso evolutivo en secuencias genéticas de virus. Para ello se emplean varios modelos evolutivos (Tamura and Nei, 1993; Sánchez and Grau, 2009) y el método de Simulación de Monte Carlo. El empleo de estos modelos, y la estimación de los parámetros del mismo partiendo de los datos disponibles, hace posible el empleo de la simulación de Monte Carlo en la predicción de posibles variantes mutacionales de genes con el propósito de evaluar las tendencias evolutivas de las poblaciones virales, en ausencia y bajo la presión selectiva del sistema inmune (Sánchez et al., 2011). Algunos trabajos del grupo de Bioinformática han experimentado con la implementación en paralelo de algunos modelos evolutivos sobre un clúster de computadoras (Moreira et al., 2011) usando la biblioteca MPI. Estos procesos pueden tener un alto costo computacional si el número de secuencias a procesar constituye un gran volumen de datos por lo que las técnicas de programación paralela contribuyen a tratar este tipo de problemas. La popularidad de las unidades de procesamiento gráfico (GPU) en los computadores modernos brinda un vasto potencial de paralelismo. La tecnología CUDA (Compute Unified Device Architecture) de NIVIDIA se ha convertido en una poderosa herramienta para el desarrollo de diversas aplicaciones paralelas sobre las GPU, constituyendo actualmente un campo en. 2.
(12) Introducción. desarrollo e investigación. El empleo de esta tecnología ya se ha aplicado exitosamente en trabajos recientes que emplean el algoritmo MCMC en procesos de optimización multi-objetivo (Minh, 2010) y también para el análisis filogenético estadístico (Suchard and Rambaut, 2009). Por otro lado, el grupo de Programación e Ingeniería del Software ha empleado la tecnología CUDA (NVIDIA CUDA C Programming Guide 5.0, 2012) para explotar sus beneficios en trabajos previos aplicados a la bioinformática (Alejo, 2012; Alba, 2013).. Planteamiento del problema El proceso de simulación, usando técnicas estadísticas y el algoritmo MCMC, para la predicción de posibles variantes mutacionales en determinado gen al evaluar las tendencias evolutivas de las poblaciones virales, puede resultar en un problema con una alta complejidad computacional, que se incrementa con el número de secuencias a procesar. En la actualidad no se ha desarrollado ningún algoritmo paralelo para tratar esta problemática cuya implementación contribuya a disminuir la complejidad inherente del algoritmo y acelere el tiempo de ejecución de la misma. A partir de la problemática descrita se plantean las siguientes preguntas investigación: 1. ¿Cuáles son los segmentos con mayor costo computacional en el algoritmo general empleado para la simulación de poblaciones virales mutantes?. 2. ¿Cómo implementar de manera paralela los segmentos anteriores? Para conducir el proceso investigativo expuesto previamente se plantean los siguientes objetivos.. Objetivo general Implementar algoritmos paralelos en diferentes etapas del proceso para la simulación de variantes mutacionales usando el modelo evolutivo TN93 y el algoritmo MCMC, para evaluar tendencias evolutivas en poblaciones virales, utilizando la tecnología CUDA.. Objetivos específicos • Describir el proceso de simulación de variantes mutacionales considerando, o descartando la presión del sistema inmune. • Caracterizar los rasgos del modelo de programación en CUDA y su aplicación en la bioinformática. • Desarrollar algoritmos paralelos para: – La simulación de nuevas secuencias genéticas mutantes, partiendo de secuencias ancestrales. – Clasificación de las nuevas secuencias a partir de las proteínas sintetizadas por las mismas.. 3.
(13) Introducción. • Obtener una aplicación que implemente en CUDA estos algoritmos. • Analizar los resultados obtenidos por los algoritmos implementados, comparando sus tiempos de ejecución con el de sus versiones secuenciales.. Justificación del trabajo La aplicación que se pretende desarrollar, para ejecutarse sobre la GPU, sería de gran importancia para el grupo de investigación Bioinformática de la UCLV, como parte de un proyecto nacional del Ministerio de Ciencia, Tecnología y Medio Ambiente (CITMA), que lleva a cabo investigaciones sobre la predicción de mutaciones y la resistencia antiviral de virus como el VIH, el virus H5N1, H1N1 y otros. La aplicación de técnicas paralelas a los problemas de la Bio-Informática y Biología Computacional, como una tendencia a reducir los costos en la solución de problemas reales, es importante en Cuba, tanto científica como económicamente, considerado en el proyecto 01700032 del Programa Nacional Científico Técnico de Tecnologías de la Información.. 4.
(14) Capítulo 1. Fundamentos Biológicos, Matemáticos y Computacionales. En este capítulo se realiza una descripción de las principales bases teóricas del trabajo, pues dado el hecho de que las aplicaciones procesarán secuencias de ADN genómico y que, además, a lo largo de este trabajo se aplican una gran variedad de herramientas matemáticas y computacionales incluyendo el uso de la tecnología CUDA, se hace necesario realizar un despliegue acertado de estos conocimientos antes de hacer uso de los mismos.. 1.1. Introducción a los elementos biológicos. Se presentan en esta sección algunas ideas básicas de biología molecular y algunos de los resultados anteriores relacionados con este trabajo. Ellos son imprescindibles para la comprensión de las hipótesis de investigación y los capítulos siguientes.. 1.1.1. Las moléculas básicas de la vida. Los principales actores de la Biología Molecular son las moléculas del ADN, ARN y las proteínas. Estas moléculas son simultáneamente los principales actores de un poderoso 5.
(15) 1.1. Introducción a los elementos biológicos. sistema de comunicación, el sistema de información genética, entrelazados por el código de comunicación conocido como Código Genético. La expresión de la información almacenada en el ácido desoxirribonucleico (ADN) se produce a través de la trascripción lineal de la secuencia de nucleótidos en la secuencia de nucleótidos del ácido ribonucleico mensajero (ARNm). La secuencia de nucleótidos del ARNm es seguidamente traducida en una secuencia lineal de aminoácidos y a partir de estos se forman las proteínas, de manera que el flujo de información es ADN → ARNm → Proteína (Minh, 2010).. 1.1.2. ADN y ARN. Desde el punto de vista químico, el ADN es un polímero de nucleótidos, es decir, un polinucleótido. Un polímero es un compuesto formado por muchas unidades simples conectadas entre sí. En el ADN, cada unidad simple es denominada nucleótido, donde cada nucleótido está formado por un azúcar (la desoxirribosa), una base nitrogenada (que puede ser adenina→A, timina→T, citosina→C o guanina→G) y un grupo fosfato que actúa como enlace de cada nucleótido con el siguiente. Lo que distingue a un nucleótido de otro es, entonces, la base nitrogenada, y por ello la secuencia del ADN se especifica nombrando solo la secuencia de sus bases. La disposición secuencial de estas cuatro bases a lo largo de la cadena es la que codifica la información genética: por ejemplo, una secuencia de ADN puede ser ATGCTAGATCGC...(Alba, 2013). En la Fig. 1.1 se observa la estructura del ADN.. Figura 1.1: La doble hélice del ADN.. 6.
(16) 1.1. Introducción a los elementos biológicos. Las bases nitrogenadas se aparean en la doble hélice formando enlaces por puentes de hidrógeno de acuerdo a las siguientes reglas: G≡C, A = T, donde cada – simboliza un enlace por puente de hidrógeno y cada par contiene una purina (A o G) y una pirimidina (C o T). El ARN al igual que el ADN está formado por una cadena de nucleótidos unidos por enlaces fosfodiester. En el proceso de transformación de ADN a ARN tenemos que la base nitrogenada T es sustituida por la base uracilo (U) y se mantienen las otras bases nucleotídicas: A, C y G como en el ADN. La función del ARN es transportar hacia el citoplasma la información genética que se encuentra almacenada en el núcleo de la célula en la secuencia de nucleótidos del ADN. Cuando se traten secuencias de ADN codificantes para proteínas, estas se pueden escribir en la práctica utilizando las bases T o U indistintamente. Existen varios tipos de ARN, cada uno con una función distinta. A los que forman parte de las subunidades de los ribosomas se les denomina ARN ribosomal (rARN); los ARN que tienen la función de transportar los aminoácidos activados, desde el citosol hasta el lugar de síntesis de proteínas en los ribosomas, se les conoce por ARN de transferencia (tARN) y los ARN que son portadores de la información genética y la transportan del genoma (molécula de ADN en el cromosoma) a los ribosomas son llamados ARN mensajero (mARN) (Minh, 2010). De los tipos de ARN mencionados anteriormente al que más se hace referencia en este trabajo es al ARNm debido a que son los portadores de la información genética.. 1.1.3. Los aminoácidos y el código genético. Los aminoácidos son los pilares estructurales de las proteínas. Solo veinte aminoácidos se encuentran corrientemente presentes en las proteínas, aunque muchos otros aminoácidos desempeñan múltiples funciones en las células. La codificación de cada aminoácido se realiza a través de un grupo de tres nucleótidos comúnmente conocidos como codones. Si combinamos tres bases (tripletes) para formar un aminoácido, obtenemos un total de 64 combinaciones (43 = 64). Los científicos demostraron que hay 61 tripletes -o codones- que codifican aminoácidos, muchos de los cuales son codificados por más de un codón, por lo que se dice que el código está degenerado. Los distintos aminoácidos son codificados por un número diferente de codones (algunos por 1, otros por 2, o por 3), e incluso existen tres tripletes que no codifican para ningún aminoácido. Encontramos aminoácidos como Leucina, Serina y Arginina, cada uno de los cuales posee 6 codones que codifican para el mismo, mientras que otros como el Triptófano y la Metionina solo poseen un solo codón Fig. 1.2. El codón de los aminoácidos Metionina es usualmente utilizado en la mayoría de los seres vivos como codón de iniciación de la cadena polipeptídica. En resumen: de los 64 codones, 61 codifican aminoácidos y los tres restantes no son codificantes sino que son utilizados como señales de terminación y son llamados codones de parada (stop codons). De la 7.
(17) 1.2. Procedimiento evolutivo de las poblaciones virales. Figura 1.2: Código genético estándar. Los aminoácidos están escritos con el símbolo de tres letras. El codón utilizado con mayor frecuencia como codón de inicio de la transcripción corresponde al aminoácido Metionina: AUG. Los codones UAA, UAG y UGA son marcadores del final de los genes. observación del Código genético se destaca que la degeneración del código implica solamente a la tercera posición del codón en la mayoría de los casos (son excepciones la Arginina, la Leucina y la Serina), de esta forma resulta que las dos primeras bases de cada codón son las determinantes principales de su especificidad. La posición tercera, esto es, el nucleótido situado en el extremo 3 del codón, tiene menor importancia y no encaja con tanta precisión (Minh, 2010).. 1.2. Procedimiento evolutivo de las poblaciones virales. El estudio científico de la evolución empezó desde un artículo publicado por Darwin y Wallace en 1858. Primero, ellos postularon que la evolución ha ocurrido principalmente como resultado de la selección natural (Nei, 1975). Esta última es efectiva solo cuando existe la variación genética, y esta variedad genética es provista primeramente por la mutación de la información genética llevada en las moléculas de ADN, las cuales son transmitidas de generación a generación. En algunos virus, la información genética es llevada en las moléculas ARN, pero la característica esencial de la herencia es la misma. En el proceso de desarrollo, la información genética contenida en la secuencia de nucleótidos ADN, es 8.
(18) 1.3. Simulación de la evolución molecular. transferida primeramente a la secuencia de nucleótidos de ARNm por un proceso simple de transcripción, uno a uno, de los nucleótidos en la molécula ADN. Por el mismo proceso, el tARN y el rARN son producidos. La información transferida al ARNm determina la secuencia de aminoácidos de la proteína que será sintetizada. Los nucleótidos de ARNm son leídos secuencialmente 3 cada vez. Cada triplete o codón es traducido en un aminoácido en la cadena de proteína mediante el código genético. Cualquiera de las mutaciones que son reconocidas como cambios morfológicos o fisiológicos tiene que ser debido a algún cambio de las moléculas ADN. Existen cuatro tipos básicos de cambios en las moléculas ADN: reemplazo de un nucleótido por el otro, eliminación de nucleótidos, adición de nucleótidos, e inversión de nucleótidos. La adición, la eliminación y la inversión pueden ocurrir con uno o más nucleótidos como una unidad. Los reemplazos de los nucleótidos pueden ser divididos en dos clases diferentes: la transición y la transversión. Transición es el reemplazo de una purina (adenina A o guanina G) por otra purina, o de una pirimidina (timina T o citosina C) por otra pirimidina. Los otros tipos de sustituciones de nucleótidos donde el cambio ocurre entre purinas y pirimidinas son llamados transversiones (Nei, 1975). Los genes o segmentos de las moléculas de ADN que actúan como las plantillas del mARN son llamados genes estructurales. Puesto que la secuencia de aminoácidos es determinada por la secuencia de nucleótidos de un gen estructural, cualquier cambio en las secuencias de aminoácidos es causado por la mutación ocurrida en la molécula ADN. Sin embargo, en el sentido opuesto, un cambio de mutación en la molécula ADN no es necesariamente reflejado en el cambio de la secuencia de aminoácidos. Esto es porque hay degeneración en el código genético (Nei, 1975).. 1.3. Simulación de la evolución molecular. La simulación de Monte Carlo ha sido utilizada comúnmente en los estudios filogenéticos para probar diferentes métodos de reconstrucción de árboles filogenéticos y, consecuentemente, sus aplicaciones para probar los modelos evolutivos pueden ser consideradas como una extensión natural de este uso. La simulación repetitiva de un proceso evolutivo dado, bajo las restricciones impuestas por el modelo que se prueba, permite estimar las distribuciones de probabilidad para los parámetros deseados. A continuación, el árbol filogenético se puede reconstruir de nuevo sin las restricciones del modelo, y los parámetros de interés derivados de este árbol pueden ser comparados con la distribución de probabilidad correspondiente derivada del árbol restringido y simulado (Yang, 2006).. 9.
(19) 1.3. Simulación de la evolución molecular. 1.3.1. Cadenas de Markov. El método utilizado en este trabajo para llevar a cabo la simulación de las secuencias de ADN se basa en las cadenas de Markov (CM). Según (Bertsekas and Tsitsiklis, 2000) estas cadenas definen un tipo especial de proceso estocástico discreto o continuo en el que la probabilidad de que ocurra un evento depende del evento inmediatamente anterior. En efecto, las cadenas de este tipo tienen memoria, “recuerdan" el último evento y esto condiciona las posibilidades de los eventos futuros. Primeramente debemos decir que en todo momento las CM poseen un estado que denotaremos por xn , el cual indica el estado actual en el que se encuentra la cadena en el momento n y el mismo pertenece a un conjunto de posibles estados S que se denomina espacio de estados:. S = {1, 2, . . . , m} | m ∈ Z, m > 0. (1.1). Las CM se describen en términos de sus probabilidades de transición pij las cuales indican la probabilidad que existe de ir del estado xi al estado xj . Como se ha dicho anteriormente que la memoria de una cadena solo se refiere al último evento que ha ocurrido en la misma, entonces las probabilidades de transición matemáticamente nos quedan definidas de la siguiente forma:. P (xn+1 = j | xn = in , . . . , x0 = i0 ) = pij = P (xn+1 = j | xn = i). (1.2). ∀n | n ≥ 0, ∀i, j ∈ S También existen autores que se refieren a las cadenas de Markov como una cadena de variables aleatorias {x0 , x1 , . . . , xn } que pueden tomar valores del conjunto de estados S y a su vez satisfacen la probabilidad condicional anterior. El presente trabajo se desarrolla completamente con modelos donde el efecto del pasado en el futuro se resume a un estado, el cual cambia en el tiempo de acuerdo con probabilidades dadas. Nuestros modelos pueden tomar un número finito de estados y los cambios de estados se producen en instantes de tiempo discretos. La suma de todas las probabilidades de transición del estado i hacia todos los demás deben sumar 1, por tanto: m X. pij = 1, ∀i ∈ S. (1.3). i=1. 10.
(20) 1.3. Simulación de la evolución molecular. es importante también destacar los vectores de probabilidad asociados a una cadena, donde tenemos que πj (t) denota la probabilidad de que la cadena esté en el estado j en el instante de tiempo t, por tanto:. πj (t) = P (xt = j). (1.4). y π(t) denota un vector fila de probabilidades del espacio de estados en el tiempo t, que tiene en su i-esima componente la probabilidad de que en el instante de tiempo t nos encontremos en el estado i del conjunto de estados S. El vector π(0) está asociado con el inicio de la cadena y todas sus componentes serán iguales a 0 excepto la componente relativa al estado inicial de la cadena cuyo valor será igual a 1.. 1.3.2. Matriz de probabilidades y probabilidad de transición en n pasos. Todos los elementos de una CM pueden definirse a través de una matriz de transición de m dimensiones y el elemento de su i-esima fila y su j-esima columna es pij . Esta matriz también es posible representarla mediante un grafo de transición de probabilidad donde los nodos son los estados y las aristas son las probabilidades de transición existentes entre dichos estados. Varios problemas de las CM requieren calcular la ley de probabilidad de un estado j en un tiempo futuro teniendo como condición de que nos encontramos en el estado i; es decir, se desea conocer la probabilidad de encontrarnos en j después de recorrer n estados y teniendo a i como punto de partida. Esta ley de probabilidad se define como:. rij = P (xn = j | x0 = i). (1.5). este valor de rij se puede calcular por la siguiente recursión conocida como ecuación de Chapman-Kolmogorov:. rij (n) =. m X. rik (n − 1)pkj. . n > 1, ∀i, j ∈ S . k=1. rij (1) = pij. (1.6). . de las ecuaciones anteriores es posible demostrar que el cálculo de las probabilidades de transición en n pasos se puede ejecutar en función de la matriz de probabilidades a partir de. 11.
(21) 1.3. Simulación de la evolución molecular. la expresión siguiente:. π(t + 1) = π(t)M. (1.7). generalizando la ecuación anterior obtenemos que:. π(t) = π(0)M t. (1.8). siendo M la matriz de probabilidades de transición. Algunas CM poseen una distribución estacionaria constituida por un vector fila de probabilidades denotado por π ∗ tal que:. π∗ = π∗M. (1.9). es decir, cuando se alcanza esta distribución no importa cuántas veces multipliquemos la matriz de transición al vector estacionario, lo cual es equivalente a avanzar un instante en la cadena. Las probabilidades de estancia en cada uno de los estados no se modifican en el tiempo y se mantienen constantes. Las condiciones para que una cadena posea una distribución estacionaria es que la misma sea irreducible y aperiódica. (Bertsekas and Tsitsiklis, 2000). 1.3.3. Técnica Markov Chain Monte Carlo. Los métodos de Markov Chain Monte Carlo, o simplemente métodos MCMC, frecuentemente se aplican para resolver los problemas de integración y optimización en los espacios de dimensión grande. En la estadística computacional estos métodos son muy útiles para generar las variables de una distribución objetivo, basándose en las cadenas de Markov, cuya distribución estacionaria es la distribución de probabilidad de interés. La razón central por la cual se usan ampliamente los métodos MCMC es que estos métodos son extremadamente generales y pueden ser utilizados para generar distribuciones univariables y multivariables cuando otros métodos han fallado o son difíciles de implementar (Gentle et al., 2004). El algoritmo utilizado en este trabajo para construir las CM obtenidas a partir de las mutaciones es una variante del método Metropolis el cual constituye un método MCMC. Mediante Metropolis, podemos generar muestras aleatorias de forma tal que estas se ajusten a una distribución de probabilidad deseada p(x) teniendo como restricción que es necesario conocer. 12.
(22) 1.3. Simulación de la evolución molecular. una función f (x) proporcional a la función de densidad de p, es decir:. p(x) =. f (x). (1.10). k. donde k será un factor de normalización y no es necesario conocerlo para aplicar el algoritmo, lo cual es muy conveniente debido al hecho de que generalmente el cálculo del mismo es muy complejo. Estas muestras se producen iterativamente de forma tal que en cada paso el nuevo valor solo dependerá del valor actual, por lo cual nos encontramos con un proceso estocástico de Markov. Luego que tenemos un posible nuevo candidato se verifica si se toma el mismo como nuevo estado. La probabilidad de aceptación de este se calcula realizando una comparación entre los ajustes del estado actual y el nuevo estado a la distribución p(x) que se quiere estimar, la cual se denomina distribución objetivo. Para calcular cuánto se ajusta un valor determinado a esta distribución se hace uso de la distribución f . A continuación se describen formalmente los pasos en los que consiste el algoritmo: 1. Comenzamos con un valor inicial θ0 tal que satisfaga f (θ0 ) > 0. 2. Utilizamos el valor actual de θi para muestrear un nuevo candidato θi+1 , luego a partir de una distribución q(θi , θi+1 ) obtenemos la probabilidad de obtener θi+1 teniendo a θi como valor previo. En la literatura q es comúnmente referida como distribución propuesta o de generación de candidatos. La única restricción que debe cumplir q es que sea simétrica, por tanto q(θi , θi+1 ) = q(θi+1 , θi ) . 3. Teniendo la nueva muestra propuesta θi+1 calculamos la proporción α existente entre la densidad de esta y del estado actual θi tomando como distribución a p, entonces:. α=. p(θi+1 ) p(θi ). =. f (θi+1 ) f (θi ). (1.11). notemos que en la fórmula 1.11 podemos hacer uso de la función de densidad f debido a que estamos hallando la proporción entre 2 valores y por tanto la constante de normalización k se cancela durante el procedimiento. 4. Si la transición propuesta incrementa la densidad (α>1) entonces aceptamos el valor candidato y volvemos al paso 2. En caso de que la densidad disminuya (α < 1) aceptamos a θi+1 con probabilidad α; es decir, generamos un número aleatorio a partir de U (0, 1) y si es menor que α se acepta el nuevo estado. Si ocurre un rechazo entonces θi+1 = θi y luego retornamos al paso 2. A menudo en algunas exposiciones del algoritmo anterior se resume el cálculo de α mediante. 13.
(23) 1.3. Simulación de la evolución molecular. la siguiente ecuación: (. α = min. f (θi+1 ) f (θi ). ). ,1. (1.12). y luego se acepta el candidato con α (probabilidad de que ocurra el movimiento). De esta forma se genera una cadena de Markov θ0 , θ1 , . . . , θn , la que después de un número finito de iteraciones alcanza su distribución estacionaria y los valores del vector θk+1 , θk+2 , . . . , θk+n son muestras de p(x) (Walsh, 2004). Específicamente en este trabajo se utiliza una variante realizada por Hastings (1970) del método expuesto anteriormente conocida como Metropolis-Hastings. Lo que se plantea en esta modificación es que el algoritmo utiliza una función de probabilidad de transición q arbitraria no simétrica y la probabilidad de cambio se halla aplicando la siguiente ecuación: (. α = min. 1.3.4. f (θi+1 )q(θi+1 , θi ) f (θi )q(θi , θi+1 ). ). ,1. (1.13). Modelos Evolutivos. Los modelos evolutivos en filogenias moleculares describen el modo y la probabilidad de que una secuencia de nucleótidos cambie a otra secuencia de nucleótidos homóloga a lo largo del tiempo. Es decir, estos modelos describen para cada uno de los sitios de la matriz la probabilidad de que se produzca el cambio de un nucleótido a otro a lo largo de las ramas de un árbol filogenético dado. Los modelos de evolución de nucleótidos se definen matemáticamente mediante dos clases de parámetros que determinan el cambio: 1. Frecuencia de cada nucleótido. Parámetro que mide la frecuencia de cada nucleótido en la matriz de datos y puede tomar los valores siguientes: (a) En los modelos evolutivos más sencillos se tiene una misma frecuencia para los cuatro nucleótidos sin tener en cuenta la frecuencia de aparición de los mismos en la matriz de datos. (πA = πC = πG = πT = 0.25). (b) Los modelos más complejos asumen que las frecuencias asociadas a cada uno de los nucleótidos son diferentes, y son calculadas a partir de los datos. (πA 6= πC 6= πG 6= πT ).. 14.
(24) 1.3. Simulación de la evolución molecular. 2. Tipos de sustituciones y sus correspondientes tasas de sustitución (rate parameters) las cuales se representan a partir de una matriz de tasas de sustitución. Las tasas de sustitución se representan con las tasas relativas de cambio de un nucleótido a otro para una posición de un tiempo t0 a un tiempo t1 . Así, cada posición de la matriz tendrá una probabilidad asociada de cambio para cada unidad de tiempo (unidad de distancia evolutiva). Los modelos más sencillos asumen una misma tasa relativa para todas las sustituciones posibles, mientras que los más complicados asumen una tasa relativa diferente para cada tipo de sustitución. A partir de estas tasas relativas se calcula la tasa media de sustitución (µ).. 1.3.5. Modelo TN93. El modelo evolutivo utilizado en este trabajo fue propuesto por Tamura y Nei (1993) y es comúnmente conocido por TN93, formando parte de la familia de modelos GTR (General Time-Reversible). Este contiene varios parámetros para lograr una simulación más realista del comportamiento de una secuencia de nucleótidos, ausmiendo que las tasas de sustitución difieren entre los nucleótidos y las frecuencias de las bases se toman como diferentes. Además, los intercambios entre 2 pirimidinas y 2 purinas se presentan en este modelo como formas de sustitución distintas; por tanto, contamos con un parámetro adicional para cada uno de estos cambios con el objetivo de lograr cierta distinción entre uno y otro; estos parámetros son α1 y α2 , los cuales son valores reales entre 0 y 1. La matriz Q de tasas de sustitución propuesta por este modelo es la siguiente:. . . −(α1 πC + α1 πT Q= βπT . βπT. βπR ). α1 πC. βπA. βπG. −(α1 πT + βπR ). βπA. βπG. βπC. −(α2 πG + βπY ). α2 πG. βπC. α2 πA. . −(α2 πA + βπY ) (1.14). Aquí tenemos que la columna 1 representa un cambio partiendo de una adenina a cualquiera de las otras bases y así sucesivamente. Esta matriz es utilizada para calcular la matriz de probabilidades de transición P utilizada por la técnica MCMC, esto se abordará con mayor profundidad en el capítulo 2 (Yang, 2006).. 15.
(25) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. Con frecuencia, nos encontramos en economía con modelos en los que el comportamiento de una variable, Y , se puede explicar a través de una variable X; lo que representamos mediante:. Y = f (X). (1.15). Si consideramos que la relación f , que liga Y con X, es lineal, entonces 1.15 se puede escribir así:. Yt = β 1 + β 2 X t. (1.16). Las relaciones del tipo anterior raramente son exactas, sino que más bien son aproximaciones en las que se han omitido muchas variables de importancia secundaria. Debemos incluir un término de perturbación aleatoria, ut , que refleja todos los factores – distintos de X – que influyen sobre la variable endógena, pero que ninguno de ellos es relevante individualmente. Con ello, la relación quedaría de la siguiente forma:. Yt = β 1 + β 2 X t + u t. (1.17). La expresión anterior refleja una relación lineal que nos permite explicar el comportamiento de una variable Y denominada variable explicada (o dependiente), a partir de otra variable X que llamaremos variable explicativa (o independiente), recibiendo el nombre de regresión lineal simple (Spiegel et al., 2009).. 1.4.1. Construcción del modelo de regresión lineal mediante la obtención de los estimadores. Supongamos ahora que disponemos de T observaciones de la variable Y (Y1 , Y2 , . . . , YT ) y de las correspondientes observaciones de X(X1 , X2 , . . . , XT ). Si hacemos extensiva 1.17 a. 16.
(26) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. la relación entre observaciones, tendremos el siguiente conjunto de T ecuaciones: . Y1 = β1 + β2 X1 + u1 Y2 = β1 + β2 X2 +. u2 . (1.18). . . . . . . . . . . . . . . . . . . . YT = β1 + β2 XT + uT El sistema de ecuaciones 1.18 se puede escribir abreviadamente de la forma siguiente:. Yt = β 1 + β 2 X t + u t. t = 1, 2, . . . , T. (1.19). El objetivo principal de la regresión es la determinación o estimación de β1 y β2 a partir de la información contenida en las observaciones de dos variables X e Y que disponemos sobre una muestra de individuos. El primer paso en un análisis de regresión es representar estos datos sobre unos ejes coordenados x − y. Esta representación es el llamado diagrama de dispersión (Spiegel et al., 2009). Nos puede ayudar mucho en la búsqueda de un modelo que describa la relación entre las dos variables. Si la relación lineal de dependencia entre Y y X fuera exacta, las observaciones se situarían a lo largo de una recta Fig. 1.3. Y. Y = β2 X + β1. X Figura 1.3: Relación de dependencia lineal exacta entre las variables X y Y Pero si la dependencia entre Y y X es estocástica, entonces, en general, las observaciones no se alinearán a lo largo de una recta, sino que formarán una nube de puntos Fig. 1.4. Si 0 0 designamos mediante β1 y β2 las estimaciones de β1 y β2 respectivamente, la ordenada de. 17.
(27) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. Y. Y = β2 X + β1. X Figura 1.4: Representación de una dependencia estocástica entre X y Y la recta para el valor Xt vendrá dada por: 0. 0. Yt = β 1 + β 2 X t. (1.20). 0. 0. El problema ahora es hallar unos estimadores β1 y β2 tales que la recta que pasa por los 0 puntos (Xt , Yt ) se ajuste lo mejor posible a los puntos (Xt , Yt ). Se denomina error o residuo a la diferencia entre el valor observado de la variable dependiente y el valor ajustado, es decir:. 0. 0. 0. 0. ut = Yt − Yt = Yt − β1 − β2. (1.21). En el modelo de regresión lineal simple hay tres parámetros que se deben estimar: los coeficientes de la recta de regresión, β1 y β2 , y la varianza de la distribución normal, σ 2 . El cálculo de estimadores para estos parámetros puede hacerse por diferentes métodos, siendo el más utilizado el método de mínimos cuadrados. 0. 0. Este método consiste en buscar los valores de los parámetros β1 y β2 de manera que la suma de los cuadrados de los residuos (S) sea mínima. Esta recta es la recta de regresión por mínimos cuadrados. Siendo la suma de los cuadrados la expresión:. S=. T X. 0. 0. (Yt − β1 − β2 Xt )2. (1.22). t=1. 18.
(28) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos 0. 0. Para minimizar S, derivamos parcialmente respecto a β1 y β2 y obtenemos: ∂S. = −2. 0. ∂β1 ∂S 0. ∂β2. T X. 0. 0. (Yt − β1 − β2 Xt ). (1.23). t=1. = −2. T X. 0. 0. (Yt − β1 − β2 Xt )Xt. (1.24). t=1. ahora igualamos a cero las ecuaciones anteriores y realizando el trabajo algebraico correspondiente obtenemos el siguiente sistema de ecuaciones, conocido como sistema de ecuaciones normales de la recta de regresión: T X. 0. 0. (Yt − β1 − β2 Xt ) =. t=1 T X. 0. 0. (Yt − β1 − β2 Xt )Xt =. t=1. 0 . (1.25). 0 0. resoloviendo el sistema planteado anteriormente obtenemos que el estimador de β2 es : T X 0. β2 =. (Yt − Y )(Xt − X). t=1 T X. (1.26) 2. (Xt − X). t=1. y. 0. 0. β1 = Y − β2 X. (1.27). dividiendo el numerador y el denominador de 1.26 por T obtenemos que: PT. t=1 (Yt. T. 0. β2 =. − Y )(Xt − X). T X. (Xt − X). = 2. cov(X, Y ) var(X). (1.28). t=1 T 0. dado que la varianza de X no puede ser negativa, el signo de β2 será el mismo que el de la covarianza muestral de X y Y . La recta de regresión ahora se puede escribir de la manera. 19.
(29) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. siguiente:. 0. 0. 0. Y = β1 + β2 X. (1.29). los residuos de la recta de regresión los denotaremos por ei y se calculan a partir de: 0. ei = Yi − Yi. (1.30). 0. donde Yi es el valor estimado por la recta de regresión.. 1.4.2. Los residuos en la estadística. En la regresión lineal, los residuos o residuales son utilizados para verificar la adecuación del modelo. Los residuos expresan la diferencia entre una observación y su valor ajustado. Estos pueden ser usados para evaluar la adecuación del ajuste de un modelo, con respecto a la elección de la función de varianza, la función enlace y en términos del predictor lineal (Spiegel 0 et al., 2009). Los residuos ei = Yi − Yi de un modelo de regresión lineal son variables aleatorias con distribución ei ∼ N (0, σ 2 (1 − hii )), i = 1, 2, ..., n siendo n el tamaño de la muestra de los datos. Existen las 3 variantes de residuos siguientes: 1. Residuos crudos: Es la diferencia entre el valor observado y el valor predictivo, tal y como se muestra en la ecuación (1.30). 2. Residuos estandarizados: Son aquellos que se obtienen dividiendo los residuos crudos por la estimación del error estándar y se definen como: ri =. ei √ SR 1 − vii. i = 1, 2, . . . n. (1.31). 3. Residuos estudentizados: son los que resultan de dividir los residuos raw por la estimación de su desviación estándar y se definen como : ti =. ei √ SR(i) 1 − vii. i = 1, 2, . . . n. (1.32). Para chequear el modelo de regresión se deben utilizar los residuos estandarizados o los estudentizados. Típicamente, las desviaciones estándares de los residuos en una muestra varían considerablemente de un punto de datos al otro aunque los errores todos tienen igual la desviación estándar, particularmente en el análisis de regresión. Por tanto, no es conveniente comparar los residuos en diferentes puntos de datos sin primero estudentizarlos. Esta técnica es muy importante para la detección de los outliers, o datos atípicos, los cuales se definen. 20.
(30) 1.4. Modelo de regresión lineal simple, herramienta estadística para el análisis relacional de datos. como las observaciones con residuos estandarizados grandes (|ri > 2|). Nos interesa ahora cómo se estudentizan los residuos de un modelo de regresión lineal simple. Para ello, partimos de una matriz del tamaño (nx2) siguiente: . X=. . 1 2 . . . . n. x1 . x2 . . . . xn. . donde xi (i = 1, ..., n) son los datos de la muestra. La matriz “hat” H es la proyección ortogonal sobre el espacio columna de la matriz X y se obtiene por:. H = X(X T X)−1 X T. (1.33). var(êi ) = σ 2 (1 − hii ). (1.34). la varianza del i-esimo residuo es:. siendo hii el i-esimo elemento de la diagonal principal de la matriz H; luego el residuo estudentizado correspondiente se calcula por:. ti =. √. êi. (1.35). σ̂ 1 − hii. donde σ̂ es la estimación apropiada de σ que viene calculado de la siguiente forma:. 2. σ̂ =. 1. n X. n − m j=1. σ̂j 2. (1.36). Sabiendo que m es el número de los parámetros en el modelo (m es igual a 2 para el modelo simple). Sin embargo, es deseado excluir la i-esima observación del proceso de estimación de las varianzas cuando se considera que el i-esimo caso pudiera ser un outlier. Consecuentemente, σ̂ se puede estimar con la forma:. σ̂i2 =. 1. n X. n − m − 1 j=1. σ̂j 2. (1.37). j6=i. 21.
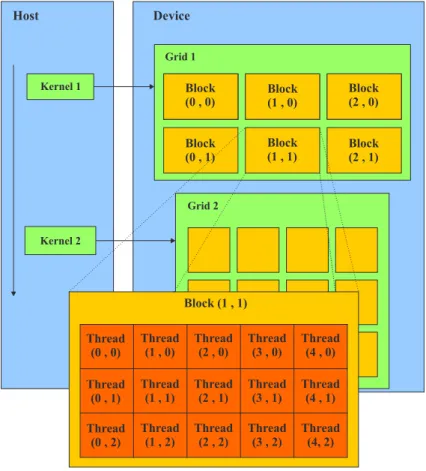
(31) 1.5. El modelo de programación en CUDA. que se basa en todos los casos, excepto el i-esimo. Si esta última estimación es utilizada, excluyendo un caso atípico, entonces el residuo se dice que es estudentizado externamente; de lo contrario, si usamos la forma (1.36) para estimar la varianza σ estamos estudentizando el residuo internamente.. 1.5. El modelo de programación en CUDA. El diseño del modelo de programación en CUDA está pensado de forma tal que las aplicaciones creadas pueden, de forma transparente, escalar su paralelismo para así incrementar el número de núcleos computacionales (del Toro Melgarejo et al., 2012). La programación en CUDA está basada en tres abstracciones básicas; una jerarquía de grupos de hilos, de tipos de memoria, y barreras de sincronización. La estructura que conforma la jerarquía de hilos en este modelo está formada por tres unidades básicas: mallas, bloques e hilos. Múltiples bloques conforman las mallas; un bloque está formado por un grupo de hilos, la unidad constituyente más elemental. Una malla puede ser definida como un grupo de bloques que ejecuta cierta función llamada kernel. La Fig. 1.5 muestra la organización de esta jerarquía. Device. Host. Grid 1 Kernel 1. Block (0 , 0). Block (1 , 0). Block (2 , 0). Block (0 , 1). Block (1 , 1). Block (2 , 1). Grid 2 Kernel 2. Block (1 , 1) Thread (0 , 0). Thread (1 , 0). Thread (2 , 0). Thread (3 , 0). Thread (4 , 0). Thread (0 , 1). Thread (1 , 1). Thread (2 , 1). Thread (3 , 1). Thread (4 , 1). Thread (0 , 2). Thread (1 , 2). Thread (2 , 2). Thread (3 , 2). Thread (4, 2). Figura 1.5: Lotes de hilos. 22.
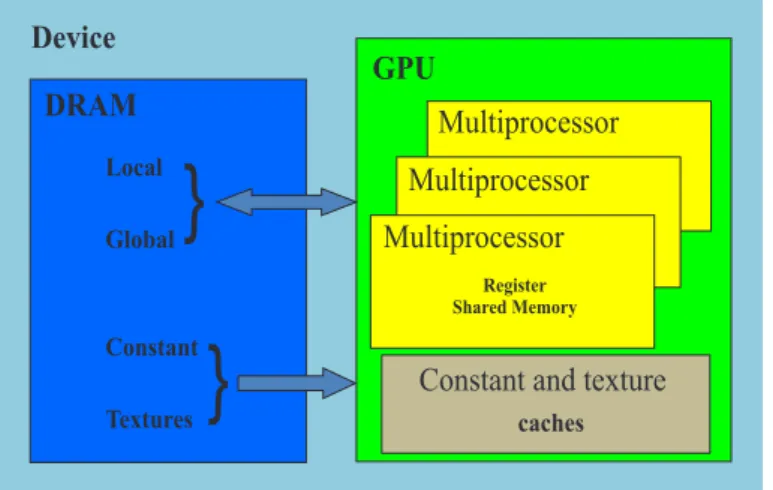
(32) 1.5. El modelo de programación en CUDA. Los hilos dentro de un bloque son ejecutados concurrentemente en un Multiprocesador de flujo (SM1 ). A su vez los bloques dentro de las mallas son distribuidos entre los diferentes multiprocesadores, lo que establece otro grado de paralelismo. Los SM son las unidades de cálculo por la cuales está constituido una moderna GPGPU2 típica, a las cuales le son asignados bloques de hilos para su ejecución paralela. Cada SM tiene una o más unidades de recolección de instrucciones, múltiples unidades aritmético lógicas (ALU), llamadas también núcleos CUDA para la ejecución paralela, cierta cantidad de memoria paralela accesible por todos los hilos del bloque, y un grupo de registros compartidos entre los hilos. Como las diferentes ALU comparten una misma unidad de instrucciones, los hilos asignados a estas ejecutan la misma instrucción utilizando una arquitectura llamada SIMT3 (Alba, 2013). Las funciones que se ejecutarán en la GPU paralelamente por varios hilos son llamadas kernel. Mediante una de las extensiones al lenguaje C creadas por NVIDIA, los kernels, se definen usando la declaración específica __global__. Uno de los rasgos principales que define a un kernel es la “configuración de ejecución”, toda llamada a una función __global__ debe especificarla. Esta configuración define la dimensión de la malla de bloques y la cantidad de hilos por bloques con que será ejecutado el kernel en la GPU. Los hilos pertenecientes a un mismo bloque pueden trabajar en colectivo compartiendo información a través una cantidad limitada de memoria compartida, accesible solo por los hilos del bloque. El trabajo de estos hilos puede ser sincronizado mediante directivas de bloqueo lo que posibilita la coordinación entre estos; sin embargo, hilos agrupados en diferentes bloques no pueden comunicarse entre sí. Este modelo permite a los kernels ejecutarse de manera eficiente en varios dispositivos sin recompilación y con diferentes capacidades paralelas en todos los bloques de una malla de forma secuencial si se tiene muy poca capacidad de paralelismo, o en paralelo si se tiene mucha capacidad de paralelismo, aunque por lo general se usa una combinación de ambos (del Toro Melgarejo et al., 2012). Los hilos que se ejecutan en un dispositivo CUDA tienen acceso a múltiples espacios de memoria: global, local, compartida, textura y registros, tal como se muestra en la Fig. 1.6. Cada hilo tiene un espacio de memoria local privado; a su vez, cada bloque de hilos posee memoria compartida visible solo a todos los hilos del bloque y con la misma duración de vida que el bloque. Además, todos los hilos ejecutando cierto kernel tienen acceso a la misma memoria global. Los espacios de memoria global, constante y de textura, son persistentes a través de los diferentes lanzamientos de kernels en la misma aplicación (Alba, 2013). 1. Del inglés Streaming Multiprocessor Del inglés General-Purpose Computing on Graphics Processin Units 3 Del inglés Single Instructions Multiple Threads 2. 23.
(33) 1.6. Estructura de datos Trie. Device GPU DRAM Local Global. Multiprocessor Multiprocessor Multiprocessor Register Shared Memory. Constant. Constant and texture Textures. caches. Figura 1.6: Espacios en memoria. 1.6. Estructura de datos Trie. Esta sección contiene los elementos teóricos básicos necesarios para lograr una correcta comprensión del funcionamiento de la estructura de datos Trie. Se hace aquí un análisis sobre la complejidad computacional que tienen algunas de las posibles operaciones que se pueden realizar sobre la estructura, además se compara de forma concisa la misma con una tabla hash siendo esta última el otro tipo de dato abstracto que se pudo haber utilizado en el desarrollo de la aplicación.. 1.6.1. Definición formal. El trie es una estructura de datos de tipo árbol que permite la recuperación de información4 . La información almacenada en un trie es un conjunto de claves, donde una clave es una secuencia de símbolos pertenecientes a un alfabeto. Las claves son almacenadas en las hojas del árbol y los nodos internos son pasarelas para guiar la búsqueda. El árbol se estructura de forma que cada letra de la clave se sitúa en un nodo de manera que los hijos de un nodo representan las distintas posibilidades de símbolos diferentes que pueden continuar al símbolo representado por el nodo padre. Por tanto, la búsqueda en un trie se hace de forma similar a como se hacen las búsquedas en un diccionario. Por eficiencia se suelen eliminar los nodos intermedios que solo tienen un hijo; es decir, si un nodo intermedio tiene solo un hijo con cierto caracter entonces el nodo hijo será el nodo hoja que contiene directamente la clave completa (Crochemore et al., 2001). En la Fig. 1.7 se presenta un trie en el que se encuentran almacenadas las palabras a, to, tea, ted, ten, in, and y inn; en la misma podemos apreciar la agrupación de caracteres que se 4. Su nombre proviene de la palabra en inglés retrieval. 24.
(34) 1.6. Estructura de datos Trie. ξ t. a. i a. t. i. o n. e to. te a tea. in n. d ted. n. inn ten. Figura 1.7: Representación gráfica de un trie realiza en los nodos con un solo hijo. Según (Crochemore et al., 2001) podemos decir que matemáticamente un trie es un caso especial de autómata finito determinista (S, Σ, T, s, A), que sirve para almacenar un conjunto de cadenas E en el que: • Σ es el alfabeto5 sobre el cual están definidas las cadenas. • S es un conjunto de estados, cada uno de los cuales representa un prefijo de E. • La función de transición T : S ×Σ → S está definida como sigue: T (x, σ) = xσ si x, xσ ∈ S, e indefinida en otro caso. • El estado inicial s corresponde a la cadena vacía ξ. • El conjunto de estado de aceptación A ⊆ S es igual a E.. 1.6.2. Ventajas de un Trie. En un trie las búsquedas de una clave de longitud m tendrán en el peor de los casos un costo lineal O(m). Esta estructura requiere muy poco espacio para almacenar gran cantidad de cadenas pequeñas, puesto que las claves no se almacenan explícitamente. Como sustitución de la estructura de datos tabla hash podemos decir que una búsqueda en implementaciones imperfectas de estas últimas tienen un coste del orden de O(n) mientras que en el trie tiene un coste de O(l) siendo n el total de claves almacenadas y l la longitud de la clave que se busca, esto se debe a las colisiones de claves, las cuales solo se producen en una tabla hash. En un trie no es necesario definir una función hash, además nos puede proporcionar un ordenamiento alfabético de las entradas por clave. También los contenedores que almacenan distintos valores asociados a una única clave solo son necesarios si tenemos más de un valor asociado a 5. En este trabajo dicho alfabeto es A,C,G,T. 25.
(35) 1.7. Las bibliotecas GSL y CURAND. una única clave mientras que en una tabla hash siempre se necesitan estos contenedores para las colisiones de clave.. 1.7. Las bibliotecas GSL y CURAND. La GNU Scientific Library(GSL) es una biblioteca escrita en el lenguaje C, destinada a cálculos numéricos necesarios en matemáticas y en la ciencia de modo general, la misma se distribuye bajo la licencia GNU GPL. Esta biblioteca puede ser utilizada junto con las clases de C++, pero sin utilizar punteros a funciones miembros(Galassi et al., 2013). De las herramientas proporcionadas por la biblioteca se han utilizado en el trabajo las destinadas al trabajo con vectores, matrices, permutaciones y álgebra lineal. La biblioteca CURAND forma parte del kit de herramientas de CUDA, su documentación es también incluida en este kit. Esta biblioteca brinda una variedad de facilidades para la generación de números pseudoaleatorios. La biblioteca consta de dos partes fundamentales, una diseñada para brindar estos servicios en la CPU, y otra en la GPU. Las funciones implementadas del lado de la GPU permiten generar números pseudoaleatorios en esta, y usarlos directamente desde la GPU sin necesidad de escribirlos a memoria global para después leerlos. Las funciones que permiten la generación de estos números lo hacen de forma concurrente, aprovechando el poder de paralelismo que brindan los GPU de NVIDIA (Alba, 2013).. 1.8. Conclusiones parciales del capítulo. Hemos expuesto los conceptos básicos de la evolución molecular y el mecanismo de aplicar la técnica de simulación para la evolución de una población viral en el cual el método de Markov es usado como el núcleo para implementar nuestro algoritmo en el capitulo siguiente. Las secuencias generadas por este método son considerados como las mutaciones de las secuencias de la población inicial teniendo en cuenta que el tamaño de las poblaciones nuevas creadas por el algoritmo son diferentes dependiendo de la configuración empleada al desarrollar los diseños experimentales. Se ha abordado también la técnica del análisis de regresión lineal como una herramienta estadística muy útil para estudiar la relación entre los datos que se usa en la implementación del algoritmo propuesto en las secciones siguientes. Finalmente, se expone el modelo de programación en CUDA a través de sus principales características, así como la estructura de datos trie, las bibliotecas CURAND y GSL, que se emplearán en el desarrollo de la aplicación. 26.
(36) Capítulo 2. Implementación de los algoritmos para la simulación de las poblaciones virales. Este capítulo tiene como objetivo exponer los principales componentes que constituyen la aplicación, además, se realiza con detalle la descripción algorítmica de las estrategias secuenciales y paralelas desarrolladas para lograr la simulación de las poblaciones virales. El desarrollo del mismo se realiza buscando la mayor correspondencia posible entre sus contenidos y los presentados en el capítulo precedente.. 2.1. Aplicación de la técnica MCMC a la simulación de poblaciones virales.. Anteriormente se presentó a MCMC como una técnica para generar aleatoriamente muestras de una distribución objetivo, haciendo uso de las cadenas de Markov para explorar el espacio de estados. Para utilizar este algoritmo en la mutación de secuencias de ADN primeramente debemos establecer los estados posibles que conformarán la cadena de Markov. En el presente trabajo las secuencias de ADN están conformadas por 4 bases nitrogenadas y estas 27.
(37) 2.1. Aplicación de la técnica MCMC a la simulación de poblaciones virales.. se representan por los enteros 0, 1, 2, 3 en lugar de adenina(A), guanina(G), citosina(C) y timina(T) respectivamente, es decir, un gen o individuo se define como un arreglo de enteros que en cada posición tendrá uno de los valores previamente mencionados; es decir, las secuencias de ADN obtenidas de esta forma se pueden considerar vectores de (Z4 )n ; por lo tanto nuestro espacio de estados S para las mutaciones es:. S = {0, 1, 2, 3} Durante el procedimiento de mutación se asume el mismo proceso de sustitución en todos los sitios, además a cada uno de ellos le corresponde una cadena de Markov cumpliéndose la condición de que dichas cadenas son independientes. En la actualidad existen varios métodos para realizar las mutaciones a partir de MCMC, uno de ellos se denomina método de desarrollar las secuencias a lo largo del árbol filogenético, conocido como un método fácil y muy usado en la simulación computacional (Yang, 2006). En este método primero se genera una secuencia para la raíz del árbol, de forma tal que los nucleótidos que constituyen la misma se crean de acuerdo a sus distribuciones equivalentes bajo el modelo de frecuencias πA , πC , πG , πT , la secuencia obtenida se denomina ancestro, y permite desarrollar y producir descendientes en su nodos hijos.1 Este proceso se repite para cada rama del árbol, de modo que se va generando una secuencia en un nodo, después que la secuencia del nodo padre ya se ha generado. Para simular la evolución de una secuencia a lo largo de una rama de longitud t, es necesario calcular la matriz de probabilidades de transición siguiente : . P (t) =. . pAA (t) pCA (t) pGA (t) . pAC (t) pAG (t) pAT (t) . pCC (t) pCG (t) pCT (t) pGC (t) pGG (t). (2.1). pGT (t) . pT A (t) pT C (t) pT G (t) pT T (t). donde pij (t) denota la probabilidad de que ocurra un cambio a j después de t instantes de tiempo, partiendo desde i.2 Este proceso se repite hasta que se generan todos los sitios de la secuencia objetivo. La simulación de la evolución de los sitios en una secuencia se manifiesta en el evento de la mutación, en la cual el nucleótido mutado es determinado aleatoriamente (E. Gultepe, 2005). Estas mutaciones estocásticas se obtienen a partir del método MCMC, aplicándose este de la forma descrita en el capítulo anterior. 1 En la implementación de la aplicación las secuencias no son generadas, estas son leídas como datos y a partir de las mismas se calculan cada una de las frecuencias, siendo esto análogo a lo que plantea el método. 2 El valor de t lo podemos interpretar como el número de mutaciones que han ocurrido desde el ancestro hasta el nodo hijo.. 28.
(38) 2.1. Aplicación de la técnica MCMC a la simulación de poblaciones virales.. 2.1.1. Cálculo de la matriz P. Las mutaciones en cada sitio que se produzcan durante la simulación dependen directamente de la matriz de probabilidades de transición (P ), pues la misma posee las probabilidades de cambio que se emplearán para formar las cadenas de Markov. Esta matriz está estrechamente relacionada con el modelo evolutivo, debido a que según sea el modelo que estemos empleando obtendremos la matriz de tasas de sustitución Q y a partir de la misma podemos calcular a P mediante la fórmula:. P (t) = eQt. (2.2). en este trabajo se utiliza el modelo TN93 por lo que Q posee la estructura definida en 1.14. Según (Yang, 2006) si Q es diagonalizable se cumple que:. Q = U DU −1. (2.3). donde U es una matriz no singular de dimensión 4x4, U −1 es su inversa y D = diag{λ1 , λ2 , λ3 , λ4 } es una matriz diagonal creada a partir del vector de valores propios λi de Q, la ecuación (2.3) se denomina descomposición espectral de Q. De esta ecuación obtenemos que:. Q2 = (U DU −1 )(U DU −1 ) = U D 2 U −1 = U (diag{(λ1 )2 , (λ2 )2 , (λ3 )2 , (λ4 )2 })U −1 (2.4) de forma análoga obtenemos Qm = U (diag{(λ1 )m , (λ2 )m , (λ3 )m , (λ4 )m })U −1 para cualquier entero m. En general cualquier función algebraica f aplicada a la matriz Q se puede calcular como f (Q) = U (diag{f (λ1 ), f (λ2 ), f (λ3 ), f (λ4 )})U −1 siempre y cuando f (Q) exista; por tanto utilizando la ecuación 2.4 podemos escribir a 2.2 de la siguiente forma:. P (t) = eQt = U (diag{eλ1 t , eλ2 t , eλ3 t , eλ4 t })U −1. (2.5). los λi (i = 1, 2, 3, 4) son los valores propios de la matriz Q. Las columnas de U y las filas de U −1 son los vectores propios derechos e izquierdos correspondientes de Q respectivamente. Para el modelo TN93 se determinan analíticamente los valores propios de Q; tenemos que los mismos son:. 29.
Figure


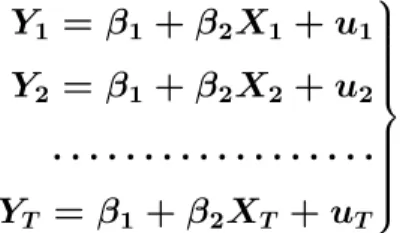

+7




Documento similar