Inhibition of the angiotensin converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN 2)
16
0
0
Texto completo
(2) 174. Cell Tissue Res (2013) 353:173–187. expression and activity without affecting TGF-β1 signaling. Our data provide insights into the pathogenic events in dystrophic muscle. We propose ACE as a target for developing therapies for DMD and related diseases. Keywords Angiotensin-converting enzyme . Fibrosis . Skeletal muscle . Extracellular matrix . Duchenne muscular dystrophy (DMD) . Mouse (mdx model of DMD) Abbreviations Ang-II ACE ARB AT-1 receptor CTGF DMD ECM mdx RAS TGF-β. Angiotensin II Angiotensin-converting enzyme Angiotensin II receptor type 1 blocker Angiotensin II receptor type I Connective tissue growth factor Duchenne muscular dystrophy Extracellular matrix DMD mouse model Renin angiotensin system Transforming growth factor beta. Introduction Fibrosis is a pathological condition characterized by the replacement of normal and functional tissue by connective tissue with excessive accumulation of extracellular matrix (ECM) proteins (Graham et al. 2010; Leask 2007; Moyer and Wagner 2010). Several pro-fibrotic factors are involved in the beginning and progression of fibrosis, including transforming growth factor type beta 1 (TGF−β1), connective tissue growth factor (CTGF) and angiotensin II (Ang-II; Abraham 2008; Pereira et al. 2009; Pohlers et al. 2009). In addition, Ang-II, the main component of the renin angiotensin system (RAS), together with angiotensin II receptor type I (AT-1 receptor) and the angiotensin-converting enzyme (ACE), induces increased ECM protein synthesis and increased CTGF and TGF-β1 levels (Cabello-Verrugio et al. 2011; Morales et al. 2012). The blocking of AT-1 receptors or ACE activity decreases ECM and profibrotic factor levels and the fibrosis observed in kidney, lung and cardiac diseases. These observations suggest that RAS components might be attractive therapeutic targets to prevent or improve fibrotic disease. Duchenne muscular dystrophy (DMD) is a genetic disease characterized by the absence of dystrophin, an anchor protein essential for the maintenance of sarcolemma stability and structure. One of the animal models of DMD, the dystrophin-deficient (mdx) mouse, exhibits milder damage to skeletal muscle fibers than in DMD; this effect can be increased and exacerbated to resemble human DMD pathology by exercising mdx mice (Bizario et al. 2009; Brussee et. al. 1997; De Luca et al. 2003; Okano et al. 2005; Sandri et al. 1997; Vilquin et al. 1998; Weller et al. 1990; Zeman et al. 2000). Under these conditions, damage is increased and the replacement of normal muscular tissue by fibrotic and connective tissue leads to a decrease in muscle strength. CTGF and TGF-β1, two pro-fibrotic factors that are increased in DMD (Sun et al. 2008; Zhou et al. 2006), are involved in the fibrotic phenotype of the DMD murine model with an increase in ECM protein, such as fibronectin and several types of collagen (Andreetta et al. 2006; Morales et al. 2011; Vial et al. 2008). RAS has been implicated in the skeletal muscle fibrosis associated with DMD (Cabello-Verrugio et al. 2012b; Cohn et al. 2007; Morales et al. 2011). The components of the RAS axis, such as ACE and AT-1 receptors, are expressed in skeletal muscle and are elevated in dystrophic skeletal muscle (Cabello-Verrugio et al. 2012a; Sun et al. 2009). Evidence showing the regulatory effect of the AT-1 receptor blocker (ARB) on TGF-β1 and CTGF activity suggests that the local classical RAS axis is activated in dystrophic skeletal muscle and contributes to the fibrotic process in DMD (Cabello-Verrugio et al. 2012b; Cohn et al. 2007; Morales et al. 2011). The inhibition of ACE activity has been used as a therapeutic treatment in mdx mice to improve the cardiomyopathy associated with dystrophy (Bauer et al. 2009). Interestingly, a unique report of the effect of ACE inhibition on skeletal muscle function in DMD has demonstrated an increase of muscle function in dystrophic mice by decreasing pro-inflammatory and prooxidant pathways (Cozzoli et al. 2011). However, the effect of ACE inhibition on fibrosis and CTGF expression and its pro-fibrotic activity has not been reported. The purpose of the current study has been to assess the role of ACE inhibition on the fibrosis associated with dystrophic muscle in the mild or more severe dystrophic phenotype represented by sedentary or exercised mdx mice, respectively. Here, we show that treatment with the ACE inhibitor enalapril significantly increases the net force and tetanic isometric force of the gastrocnemius muscle in sedentary and exercised dystrophic mice. Enalapril decreases the necrotic areas in the gastrocnemius muscles in both conditions of mdx mice. ACE inhibition treatment also leads to a decrease in fibrosis, as manifested by a reduction in ECM protein levels and collagen amount. Interestingly, the administration of enalapril decreases the increment of CTGF expression in the gastrocnemius muscle from sedentary and exercised mdx mice. Moreover, enalapril also decreases CTGF-dependent pro-fibrotic activity in skeletal muscle. However, ACE inhibition does not have an effect on the expression or activity of TGF-β1. Thus, we propose that ACE inhibition is a potential pharmacological strategy that can be used to decrease the fibrotic response associated with DMD..
(3) Cell Tissue Res (2013) 353:173–187. Materials and methods Animals and experimental exercise Normal or mdx (8 weeks old) male mice of the C57BL/10 ScSn strain were used. The animals were kept at room temperature with a 24-h nightday cycle and were fed with pellets and water ad libitum (Cabello-Verrugio et al. 2012b). Experimental exercise was performed by running the mice in a treadmill under the following conditions: 3 times/week, 30 min each time at 12 m/min, for 6 months (De Luca et al. 2005). During this time, three experimental groups were designated: those treated with a vehicle (water), enalapril (5 mg/kg per day), or losartan (90 mg/kg per day; Cohn et al. 2007; Cozzoli et al. 2011). The blood pressure of the mice was measured and monitored as previously described (Salas et al. 2004). At the end of the experiment, gastrocnemius muscles were dissected and removed under anesthesia and then the animals were killed. Tissues were rapidly frozen and stored at –80°C until processing or were used for electrophysiological measurements (Cabello-Verrugio et al. 2012b). All protocols were conducted in strict accordance and with the formal approval of the Animal Ethics Committee of P. Universidad Católica de Chile. Experimental skeletal muscle adenoviral infection Infection of tibialis anterior muscles was performed in 12-week-old male mice of the C57BL/10 ScSn strain by adenovirus injection of 2×1011 viral particles for the control adenovirus (containing green fluorescent protein; GFP) or 2×1011 particles of adenovirus codifing CTGF, as previously described (Cabello-Verrugio et al. 2012b; Morales et al. 2011). Mice were treated with a vehicle (water), enalapril (5 mg/kg per day), or losartan (90 mg/kg per day) for 10 days prior and 5 days post adenoviral infection (Cabello-Verrugio et al. 2012b; Cohn et al. 2007; Cozzoli et al. 2011). At the end of the experiment, animals were killed under anesthesia at day 5 post infection and the tibialis anterior muscles were dissected, removed, rapidly frozen and stored at –80°C until processing. RNA isolation, reverse transcription and quantitative realtime polymerase chain reaction Total RNA was isolated from gastrocnemius muscles by using TRIzol (Invitrogen, USA) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse-transcribed to cDNA by using random hexamers and Superscript II reverse transcriptase (Invitrogen, USA). Taqman quantitative real-time polymerase chain reactions (qPCR) were performed in triplicate on an Eco Real-Time PCR System (Illumina, USA) with predesigned primer sets for mouse CTGF, TGF-β1 and the housekeeping gene D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Taqman Assays-on-Demand, Applied Biosystems, USA). mRNA expression was quantified by. 175. using the comparative ΔCt method (2-ΔΔCT) with GAPDH as the reference gene. The mRNA levels are expressed relative to the mean expression in the normal mice (Morales et al. 2012). Immunoblot analysis For skeletal muscle extracts, gastrocnemius muscles were homogenized in TRIS-EDTA buffer with a cocktail of protease inhibitors and 1 mM phenylmethane sulfonyl-fluoride. Proteins were subjected to SDS-polyacrylamide gel electrophoresis, transferred onto polyvinylidene difluoride membranes (Millipore, USA) and probed with rabbit anti-fibronectin (1:5000; Sigma-Aldrich, USA), rabbit anti-collagen III (1:1000; Rockland, USA), goat anti-CTGF (1:500; Santa Cruz Biotech, USA), rabbit antiphospho-p38 (1:1000), rabbit anti-phospho-extracellular signal-related kinase (ERK; 1:1000), rabbit anti-total p38 (1:1000), rabbit anti-total ERK (1:1000; Cell Signaling, USA), mouse anti-tubulin (1:10,000; Sigma-Aldrich) and mouse anti-GAPDH (1:10,000; Chemicon, USA). All immunoreactions were visualized by enhanced chemiluminescence (Thermo Scientific, USA). Immunofluorescence microscopy For immunofluorescence analysis, transverse sections of gastrocnemius muscle were fixed, blocked and incubated with the rabbit anti-fibronectin (1:100), rabbit anti-collagen type III (1:100), rabbit anticollagen type I (1:100) (Chemicon, USA), or rabbit antiphospho-Smad3 (Ser423/425) antibodies (Cell Signaling, USA). As a secondary antibody, flourescein- or rhodamine-conjugated goat-anti rabbit IgG was used. The coverslips were mounted by using Fluoromont (Dako, USA) and viewed under a Nikon Diaphot inverted microscope equipped for epifluorescence. Skeletal muscle histology and Sirius red staining Architecture and histology were detected by hematoxylin and eosin (H&E) staining of transverse sections of gastrocnemius skeletal muscle. Total collagen content was detected by staining with Sirius red and quantification was performed by ImageJ software (Cabello-Verrugio et al. 2012b) in a blinded fashion. Centered nuclei and fibrotic area quantification Myofibers with or without centered nuclei were quantified manually in cross-sections of gastrocnemius muscle stained with H&E. Quantification was carried out on 10 micrographs of the cross-sections of the gastrocnemius muscle of each animal. The percent of fibrotic area was measured in 10 randomly selected fields in each animal by using Image J software (NIH, USA) in an analysis of the percentage of the total area accounted for by immunostaining for fibronectin versus the total area. All scoring were performed in a blinded fashion on coded slides..
(4) 176. Cell Tissue Res (2013) 353:173–187. Contractile properties After treatment, mice were anesthetized and then killed. The gastrocnemius muscles were removed and the muscle contractile properties were measured as previously described (Cabello-Verrugio et al. 2012b; Gregorevic et al. 2002). Maximum isometric tetanic force was determined from the plateau of the frequencyforce relationship after successive stimulations at 1–100 Hz for 450 ms, with 2-min rests between stimuli. After determination of isometric contractile properties, muscles were subjected to a triple repeated tetanic stimulation protocol. Muscles at loading were maximally stimulated for 450 ms once every 5 s. After functional testing, muscles were removed from the bath, trimmed of their tendons and of any adhering non-muscle tissue, blotted once on filter paper and weighed. Muscle mass and loading were used to calculate the specific net force (force normalized per total muscle fiber cross-sectional area [CSA], mN/mm2). All the contractile measures were performed in a blind fashion. Running test Mice were subjected to a running test for 15 min at 15 m/min on a treadmill. The number of times that mice were detained on the first 1/3 of the moving platform was counted. All scoring were performed in a blinded fashion. Statistics Statistical analysis was carried out by using a oneway analysis of variance (ANOVA) with a post-hoc Bonferroni multiple-comparison test (Sigma Stat). A difference was considered statistically significant at a P-value of <0.05.. Results ACE inhibition is able to improve skeletal muscle strength in sedentary and exercised dystrophic mice. Fig. 1 Treatment with an angiotensin-converting enzyme (ACE) inhibitor produces an improvement in skeletal muscle strength in dystrophic mice. Gastrocnemius muscles of normal, sedentary (a) and exercised (b) mdx mice treated with a vehicle, enalapril, or losartan for 6 months were compared with respect to the specific isometric force (mN/mm2)–stimulation frequency (Hz) relationship. Values correspond to means ± SD from three independent experiments; n=5 mice/group. *P<0.05. c Running test (number of detention) of normal and sedentary mdx mice treated with a vehicle, enalapril (Enal) and losartan (Los). *P<0.05 relative to mdx treated with a vehicle, #P<0.05 relative to normal mice. We decided to evaluate the effects of enalapril treatment on muscle function in sedentary and exercised mdx mice by analyzing force-generating capacities. The mdx mice at rest or exercised on a treadmill for 6 months were treated systemically with enalapril and the gastrocnemius was measured for maximum isometric force. Figure 1a, b shows the curves of net force generated by the gastrocnemius muscles of sedentary and exercised mdx mice, respectively, treated with enalapril and stimulated with frequencies ranging from 1 to 100 Hz. Under sedentary conditions, the dystrophic muscles produced a minor net force, about 1/4 of the force generated by the gastrocnemius muscles of normal mice, in all the ranges of stimulatory frequency evaluated (Fig. 1a). Under chronic exercise, dystrophic muscles produced only about 1/10th of the force generated by the muscles of normal mice (Fig. 1b). The gastrocnemius muscles of the sedentary and exercised mdx mice treated with enalapril.
(5) Cell Tissue Res (2013) 353:173–187. 177. generated significantly more net force (almost twice) than those treated with a vehicle (Fig. 1a, b). In Fig. 1a, b, the force generated by the gastrocnemius of mdx mice treated with ARB losartan is shown as the control of strength recovery as previously demonstrated (Cohn et al. 2007). The maximal isometric tetanic force was also evaluated (Fig. S1a). Under these conditions, treatment with enalapril significantly increased the maximal isometric tetanic force in the gastrocnemius muscles of sedentary mdx mice. Similar results were obtained for exercised dystrophic mice (Fig. S1b). In addition, we investigated whether treatment of the mdx mice with losartan or enalapril had a beneficial impact on muscle function. In a running test on a treadmill, sedentary mdx mice treated with enalapril showed a significant decrease in the number of detention times compared with the exercised mdx mice treated with a vehicle (Fig. 1c). Together, these results strongly suggest that ACE inhibition by enalapril treatment improves skeletal muscle contractile properties in sedentary and exercised dystrophic mice. Skeletal muscle damage in mdx mice is reduced by ACE inhibition We (in this report) and others have showed that enalapril treatment improves muscle strength in dystrophic mice (Cozzoli et al. 2011). However, its relationship with skeletal muscle damage has not been demonstrated. Thus, we decided to evaluate the effect of enalapril treatment on muscle damage in sedentary and exercised dystrophic skeletal muscle. Exercise (Fig. 2e) did not alter muscle histology in sedentary normal mice (Fig. 2a) but an increase in damage of the gastrocnemius muscles was seen in exercised mdx mice (Fig. 2f) compared with sedentary mdx mice (Fig. 2b) as evaluated by H&E staining. The administration of enalapril to sedentary (Fig. 2c) and exercised (Fig. 2g) mdx mice clearly prevented any increase of damaged areas. A decrease of muscle damage occurred after losartan treatment (Fig. 2d, h), as previously demonstrated (Cohn et al. 2007). Interestingly, we did not observe differences in the numbers of central nuclei in the fibers of mdx muscles treated with enalapril compared with mdx mice treated with a vehicle (Fig. S2a) suggesting similar regenerative ability. Together, these results indicate that ACE inhibition is able to decrease damage in sedentary mdx mice and to prevent further impairment of the tissue produced by exercise in dystrophic skeletal muscles. Enalapril administration decreases ECM proteins in skeletal muscle from sedentary and exercised mdx mice We and other researchers have shown that the development of fibrosis in dystrophic muscle is characterized by an increase in ECM proteins, such as fibronectin and several. Fig. 2 Treatment of mdx mice with an ACE inhibitor partially prevents damage in skeletal muscle. C57BL/10J (normal) and mdx mice were placed under sedentary conditions or subjected to exercise for 6 months. In parallel, various treatments were orally administrated to the mice until the end of the experiments. Cryosections of gastrocnemius muscles were stained with hematoxylin and eosin (H&E) to visualize the architecture of the tissue. Gastrocnemius muscles from sedentary (a) and exercised (e) normal mice and gastrocnemius muscles from sedentary (b-d) or exercised (f-h) mdx mice treated with a vehicle (b, f), enalapril (c, g), or losartan (d, h) are shown. Images are representative of each experimental group (three independent experiments) and were chosen in a blinded fashion. Bar 50 μm.
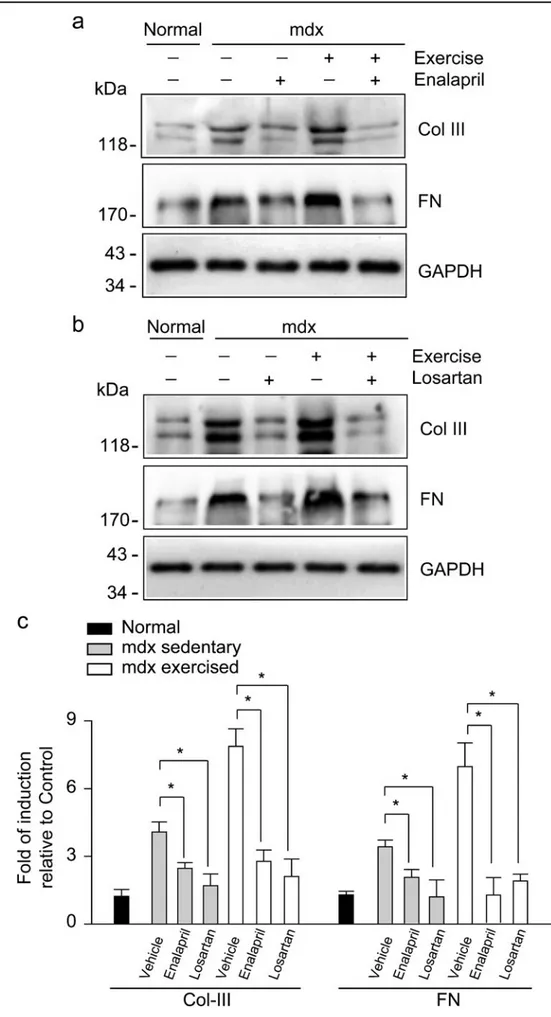
(6) 178 Fig. 3 Enalapril treatment decreases fibrosis in dystrophic skeletal muscle. Collagen III (Col III) and fibronectin (FN) protein levels were detected by Western blot analysis in extracts obtained from the gastrocnemius muscles of normal and of sedentary and exercised mdx mice treated with a vehicle, enalapril (a), or losartan (b) for 6 months. Dglyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein levels are shown as the loading control; molecular weight markers are shown in kiloDatons (kDa). c Densitometric analysis of Western blots shown in a, b. Values correspond to means ± SD of three independent experiments, with five mice being used for each experimental condition. * P<0.05. Cell Tissue Res (2013) 353:173–187.
(7) Cell Tissue Res (2013) 353:173–187 Fig. 4 ACE inhibition decreases ECM proteins in dystrophic skeletal muscle. Detection of collagen I (a-g), fibronectin (h-n) and collagen III (o-u) by indirect immunofluorescence analysis of cryosections of gastrocnemius muscles obtained from normal mice (a, h, o) and from sedentary (b-d, i-k, p-r) or exercised (e-g, l-n, s-u) mdx mice treated with a vehicle (b, i, p, e, l, s), enalapril (c, j, q, f, m, t) or losartan (d, k, r, g, n, u) for 6 months. Images are representative of each experimental group (three independent experiments) and were chosen in a blinded fashion. Bar 50 μm. 179.
(8) 180. types of collagen (Cabello-Verrugio et al. 2011, 2012b; Cohn et al. 2007; Graham et al. 2010; Marshall et al. 1989; Morales et al. 2011; Zhou and Lu 2010). Moreover, exercise has been described as exacerbating ECM accumulation (De Luca et al. 2003, 2008; Zeman et al. 2000). Therefore, we evaluated the effect of ACE inhibition by enalapril treatment on ECM protein levels in sedentary and exercised mdx mice. Figure 3a shows that ACE inhibition by enalapril reduced collagen III and fibronectin protein levels in the gastrocnemius muscles of sedentary and exercised mdx mice compared with mdx mice treated with a vehicle. The same effects were observed in sedentary and exercised mdx mice treated with losartan (Fig. 3b), as previously demonstrated (Cohn et al. 2007). The quantitative analysis of these experiments is depicted in Fig. 3c. In addition, the ECM amount was evaluated by immunofluorescence analysis (Fig. 4). The basal levels of collagen I and III and of fibronectin in the gastrocnemius muscle of normal mice (Fig. 4a, h) increased in sedentary mdx mice (Fig. 4b, i, p) and exercised mdx mice (Fig. 4e, l, s). ACE inhibition mediated by enalapril reduced collagen I and III and fibronectin levels in gastrocnemius muscle from sedentary (Fig. 4c, j, q) and exercised (Fig. 4f, m, t) mdx mice. The quantitative analysis of fibronectin detection by immunofluorescence is showed in Fig. S2b. The decrease of collagen I Fig. 5 ACE inhibition decreases collagen deposition in mdx skeletal muscle. Staining for collagen deposition (Sirius red) in gastrocnemius muscles from normal (a) or from sedentary (b-d) and exercised (e-g) mdx mice treated with a vehicle (b, e), enalapril (c, f), or losartan (d, g) for 6 months. Images are representative of each experimental group (four independent experiments) and were chosen in a blinded fashion. Bar 50 μm. Quantification of Sirius redpositive areas in sections shown in a-g for sedentary (h) or exercised (i) mdx mice. Values correspond to means ± SD of four independent experiments with four mice for each experimental condition (Veh vehicle, Enal enalapril, Los losartan). *P<0.05 relative to mdx treated with a vehicle. Cell Tissue Res (2013) 353:173–187. and III and fibronectin mediated by losartan in sedentary (Fig. 4d, k, r) and exercised (Fig. 4g, n, u) mdx mice is shown. Sirius red staining analysis showed that gastrocnemius muscle from sedentary mdx mice (Fig. 5b) had more collagen than that from normal mice (Fig. 5a), whereas exercised mdx mice presented the highest collagen levels (Fig. 5e). Enalapril treatment strongly reduced the amount of collagen observed in the gastrocnemius muscles of sedentary (Fig. 5c) and exercised (Fig. 5f) mdx mice compared with the muscles of mdx mice treated with a vehicle. The decrease of collagen mediated by losartan is also shown (Fig. 5d, g). The quantitative analysis of these experiments is shown in Fig. 5h, i. Together, these results suggest that the increase of fibrosis in dystrophic skeletal muscle under sedentary conditions or induced by exercise and evidenced by ECM protein accumulation is prevented by chronic treatment with an ACE inhibitor, thereby decreasing the development of fibrosis. Increment of CTGF expression in dystrophic skeletal muscles and pro-fibrotic activity of CTGF are reduced by enalapril The increase in ECM proteins observed in the skeletal muscles of mdx mice is concomitant with the induction of pro-fibrotic factors levels, such as CTGF (Sun et al. 2008)..
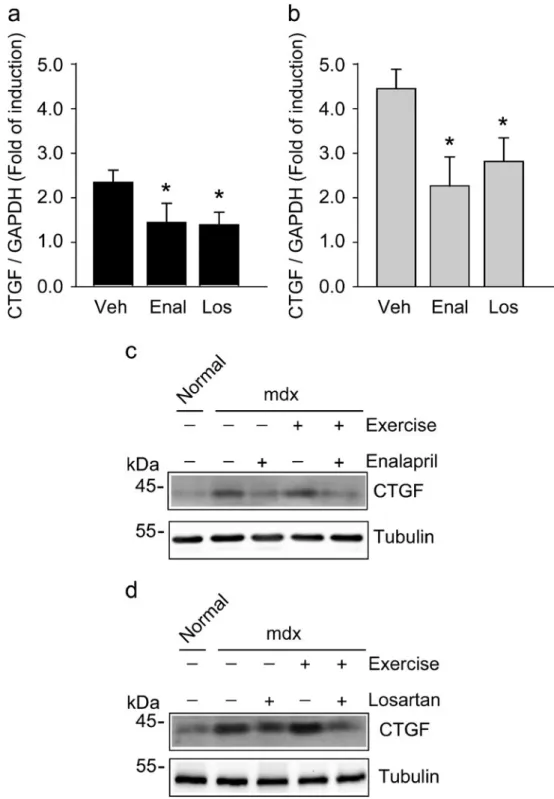
(9) Cell Tissue Res (2013) 353:173–187. Therefore, we decided to evaluate the effect of enalapril treatment on CTGF expression in sedentary and exercised mdx gastrocnemius muscles, as determined by reverse transcription plus qPCR (RT-qPCR). CTGF expression was reduced by enalapril in sedentary (Fig. 6a) and exercised (Fig. 6b) dystrophic mice. A similar diminution of CTGF expression was also seen after losartan treatment. These effects of enalapril and losartan were also observed at the protein levels of CTGF in mdx muscle (Fig. 6c, d). To Fig. 6 Increment of connective tissue growth factor (CTGF) expression in dystrophic skeletal muscles are reduced by enalapril. CTGF mRNA expression in gastrocnemius muscles of sedentary (a) or exercised (b) mdx mice treated with a vehicle (Veh), enalapril (Enal), or losartan (Los) for 6 months. mRNA levels of CTGF were determined by reverse transcription plus quantitative polymerase chain reaction (RT-qPCR) with GAPDH as the reference gene. Expression is presented as the fold of induction normalized to levels in normal mice and corresponds to the means ± SD of three independent experiments with four mice for each experimental condition. *P<0.05 relative to mdx mice treated with a vehicle. CTGF protein levels in gastrocnemius muscles from normal or from sedentary and exercised mdx mice treated with a vehicle (−), enalapril (c) and losartan (d) for 6 months were detected by Western blot analysis. Tubulin levels are shown as a loading control. Molecular weights are shown in kDa. 181. evaluate further the effect of ACE inhibition on CTGFdependent pro-fibrotic activity, we used a model of fibrosis induced by CTGF in the tibialis anterior muscle of normal mice by overexpressing CTGF. We have previously reported that CTGF overexpression by adenoviral infection produces the same muscle features as those observed in dystrophic muscle, such as damage and ECM increase (CabelloVerrugio et al. 2012b; Morales et al. 2011). The damage induced by CTGF overexpression in normal muscle (compare.
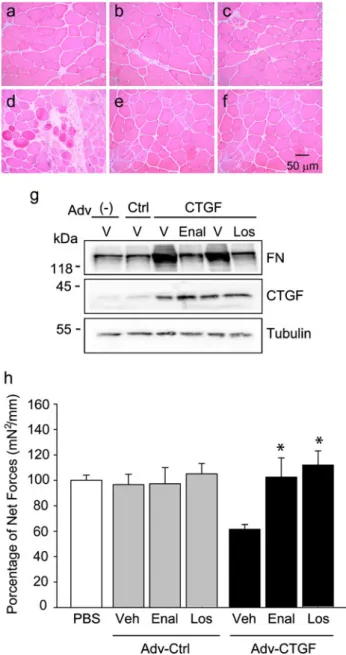
(10) 182. Cell Tissue Res (2013) 353:173–187. Fig. 7d with Fig. 7a) is decreased by enalapril (Fig. 7d) and losartan (Fig. 7f) treatments. CTGF-induced fibronectin expression was blunted by enalapril treatment and levels of CTGF were increased in muscles infected with the adenovirus carrying the sequence coding for murine CTGF (Fig. 7g). In addition, in order to validate our results, we were able to show (Fig. 7g) the diminution of CTGF-induced muscle damage and fibronectin levels by losartan, as we previously demonstrated (Cabello-Verrugio et al. 2012b). Then, we evaluated the effect of enalapril on muscle strength on muscles that overexpressed CTGF. Figure 7h shows that treatment with enalapril prevented the diminution of muscle strength observed in muscles that overexpressed CTGF. In addition, we decided to evaluate the activation of signaling pathways dependent on Ang-II in the model of skeletal muscle fibrosis induced by CTGF. Thus, we evaluated the p38 phosphorylation in tibialis anterior muscle overexpressing CTGF in normal mice. The result shown in Fig. S3 indicates that CTGF overexpression induces p38 phosphorylation, which is prevented by enalapril treatment. This result suggests that CTGF can activate Ang-II-dependent pathways that are sensitive to enalapril. We have previously demonstrated that CTGF induces ERK phosphorylation, which is also increased in mdx muscles (Cabello-Verrugio et al. 2012b). Therefore, we evaluated the effect of enalarpil treatment on ERK phosphorylation in sedentary and exercised mdx gastrocnemius muscles. The results shown in Fig. S4a indicate that enalapril administration decreases the phospho-ERK levels in both sedentary and exercised mdx mice. Results of the quantitatively analysis of these experiments are shown in the Fig. S4b. Thus, our results suggest that enalapril decreases the expression and fibrotic activity of CTGF in skeletal muscle thereby increasing the muscle strength. ACE inhibition does not change the expression and signaling of TGF-β1 in dystrophic and fibrotic skeletal muscle TGF-β1, a pro-fibrotic factor and inducer of CTGF expression and signaling, is augmented in dystrophic skeletal muscle (Andreetta et al. 2006; Zhou et al. 2006; Bernasconi et al. 1999). Therefore, we decided to evaluate the effect of ACE inhibition by enalapril treatment on TGF-β1 expression and activity in skeletal muscle from mdx mice. Figure 8a, b shows that the increase of TGF-β1 expression in gastrocnemius from sedentary and exercised mdx mice compared with that in normal mice was not affected by enalapril treatment. In addition, we showed that enalapril did not change the increase of TGF-β1 signaling as evaluated by nuclei positive for phospho-Smad3 in the gastrocnemius muscle from sedentary (Fig. 8d, e) or exercised (Fig. 8g, h) mdx mice (see quantitative analysis in Fig. 8j).. Fig. 7 ACE inhibition decreases the pro-fibrotic activity of CTGF in skeletal muscle. Tibialis anterior muscle (TA) of normal mice systemically treated with a vehicle (a, d), enalapril (b, e), or losartan (c, f) were infected with a control adenovirus (a-c) or with an adenovirus to overexpress CTGF (d-f). Cryosections of TA muscles infected for 5 days were stained with H&E. Three independent experiments were performed with two mice for each experimental condition. Images are representative of each experimental group and were chosen in a blinded fashion. Bar 50 μm. g Under the same conditions of infection, protein levels of fibronectin (FN) and CTGF were detected by Western blot analysis in extracts obtained from TA muscles. Tubulin levels are shown as the loading control. Treatment with a vehicle (V), enalapril (Enal), or losartan (Los) is shown. Molecular weights are shown in kDa. h Tetanic specific force of TA of normal mice systemically treated with a vehicle (Veh), enalapril (Enal), or losartan (Los) and injected with phosphate-buffered saline (PBS) or infected with a control adenovirus (Adv-Ctrl) or with an adenovirus to overexpress CTGF (AdvCTGF). Values are represented as percentages of specific isometric force (mN/mm2) shown by muscles injected with PBS. *,#P<0.05).
(11) Cell Tissue Res (2013) 353:173–187 Fig. 8 Enalapril treatment does not change TGF-β1 expression and activity in dystrophic skeletal muscle. TGF-β1 mRNA expression in gastrocnemius muscles from sedentary (a) or exercised (b) mdx mice treated with a vehicle (Veh), enalapril (Enal), or losartan (Los) for 6 months. mRNA levels of CTGF were determined by RT-qPCR with GAPDH as the reference gene. Expression is presented as fold induction normalized to levels in normal mice and correspond to the means ± SD of three independent experiments with four mice for each experimental condition. *P<0.05 relative to mdx mice treated with a vehicle. Detection of phospho-Smad3 through indirect immunofluorescence analysis in cryosections of gastrocnemius of normal (c) and of sedentary (d-f) or exercised (g-i) mdx mice treated with a vehicle (d, g), enalapril (e, h), or losartan (f, i). Images are representative of each experimental group and were chosen in a blinded fashion. Bar 50 μm. j Phospho-Smad3positive nuclei quantification of results from cryosections of gastrocnemius of normal and of sedentary and exercised mdx mice treated as in c-i. Values correspond to means ± SD from four independent experiments with four mice for each experimental condition (Veh vehicle, Enal enalapril, Los losartan). *,#P<0.05 relative to mdx mice treated with a vehicle. 183.
(12) 184. Our results were specific of ACE inhibition, because the treatment of mdx mice with losartan decreased both the TGF-β1 expression (Fig. 8a, b) and Smad3 phosphorylation (Fig. 8f, i) in skeletal muscle, as previously reported (Cabello-Verrugio et al. 2012b; Cohn et al. 2007). These results indicate that enalapril does not alter the increase of TGF-β1 expression and activity observed in both sedentary and exercised dystrophic skeletal muscle.. Discussion The purpose of the current study has been to assess the role of ACE inhibition on skeletal muscle fibrosis, its effects on pro-fibrotic factors expression and its role in muscle strength associated with dystrophic skeletal muscles under conditions of sedentarism or chronic exercise. The data obtained in this study are the first to show a decrease of fibrosis in dystrophic skeletal muscle under sedentary and exercised conditions by ACE inhibition with enalapril, as evaluated by a reduction in ECM protein collagen and fibronectin. In addition, the enalapril treatment has produced a significant increase in muscle strength. The data also indicate that enalapril reduces the expression of CTGF and its pro-fibrotic activity and the ERK signaling pathway, Fig. 9 Representation of the anti-fibrotic effect of inhibition of the renin angiotensin system on dystrophic skeletal muscle. In Duchenne muscular dystrophy (DMD), an increase occurs in transforming growth factor beta-1 (TGF-β1) and connective tissue growth factor (CTGF) expression and signaling. The administration of losartan decreases TGF-β1 expression and Smaddependent signaling, whereas the administration of enalapril or losartan both decrease CTGF expression and extracellular signal-related kinase (ERK)dependent activity. Thus, angiotensin II receptor type I blockade (by losartan) and ACE inhibition (by enalapril) produce a decrease of fibrosis associated with dystrophic skeletal muscle. Cell Tissue Res (2013) 353:173–187. whereas it did not have an effect on TGF-β1 levels and Smad-dependent signaling in dystrophic mice under sedentarism or chronic exercise (Fig. 9). A previous report of ACE inhibition as a systemic treatment for DMD has demonstrated that chronic treatment with enalapril has the ability to blunt Ang-II-dependent activation of pro-inflammatory and pro-oxidant pathways (Cozzoli et al. 2011). However, our results are the first direct evidence that enalapril decreases the fibrosis associated with a mild or severe phenotype of dystrophic skeletal muscle. Interestingly, our data obtained in vivo by using ACE inhibition by enalapril as an indirect participation of Ang-II are in agreement with our previously published results demonstrating that Ang-II induces pro-fibrotic effects in skeletal muscle cells (Cabello-Verrugio et al. 2011; Morales et al. 2012). Moreover, we have recently demonstrated that Ang-IIinduced pro-fibrotic effects in vitro are mediated by reduced nicotinamide-adenine-di-nucleotide phosphate (NAPDH)oxidase-induced reactive oxygen species (ROS) and an increase in NADPH-oxidase in the muscles of mdx mice (Cabello-Verrugio et al. 2011; Whitehead et al. 2010). Thus, even though our results and those from Cozzoli et al. (2011) are different, they are complementary in the mechanism, because Ang-II-induced ROS-dependent effects might produce and maintain an inflammatory response, activating.
(13) Cell Tissue Res (2013) 353:173–187. pathways such as nuclear factor kappa-B and/or the production of pro-inflammatory cytokines (Arthur et al. 2008; Wei et al. 2006, 2008). DMD is an inherited disorder that is characterized by the weakness of skeletal muscles and for which no effective treatment exists (Goldstein and McNally 2010). The involvement of the AT-1 receptor in the regulation of fibrosis in dystrophic skeletal muscle has been previously proposed, on the basis of the effects of losartan for 6–9 months, as a therapeutic tool for DMD in mdx mice (Cohn et al. 2007). Contrary to these results, Bish and colleagues (2011) have demonstrated that long-term losartan treatment (2 years) has no effect on muscle fibrosis in aged mdx mice. We have only used losartan as a control of an anti-fibrotic treatment for dystrophic muscle corroborating the results showed by Cohn et al. (2007). In addition, an evaluation of whether the RAS pathway inhibition effect is age- or mechanical-damagedependent is of interest. Evidence has been presented that the muscles of exercised mdx mice show an enhanced mechanical and inflammation-dependent degeneration phase (Burdi et al. 2009; Landisch et al. 2008; Okano et al. 2005). In this work, we have performed experiments with exercised mdx mice and demonstrated that RAS inhibition by ACE inhibition (enalapril) and AT-1 receptor blockade (losartan) partially recover muscle strength, concomitantly improving histological features observed in the exercised mdx mice. In addition, both enalapril and losartan reduce skeletal muscle fibrosis under exercise conditions. Because we have used the exercise model, the effect of losartan might depend on muscle activity and not on muscle age. Indeed,evidence is available that RAS has a role in impairing exercise performance (Wang et al. 2008). To study whether CTGF and TGF-β1, two of the main profibrotic factors, are involved in the mechanism through which enalapril reduces fibrosis, we have evaluated their expression and activity. Our results indicate that a correlation exists between CTGF expression, fibrosis and muscle strength: with increased CTGF, more fibrosis and less strength have been detected. Moreover, we have demonstrated that, in a model of CTGF-induced fibrosis, enalapril decreases the damage and ECM accumulation, acting as an inhibitor of CTGF activity. Since CTGF has been postulated to be an effector of TGF-β1dependent fibrosis, we need to develop new therapeutic tools that decrease CTGF without affecting other TGF-β1dependent effects on skeletal muscle, such as the proliferation of activated-cell satellites. In this context, only enalapril decreases the CTGF-profibrotic activity without affecting TGF-β1 expression and signaling, as shown in this report. Interestingly, only losartan and not enalapril, reduces TGF-β1 expression in skeletal muscle from mdx mice. In addition, we have recently described that losartan is a new molecule that reduces CTGF-dependent fibrosis in skeletal muscle (CabelloVerrugio et al. 2012b). Together, the results indicate that. 185. losartan is a broad-spectrum anti-fibrotic agent that affects CTGF and TGF-β1 expression and activities (CabelloVerrugio et al. 2012b; Cohn et al. 2007), whereas enalapril is a more specific anti-fibrotic drug that reduces CTGF expression. The mechanism involved in this differential effect on TGF-β1 and CTGF expression and activities must be studied further. Enalapril and losartan act by inhibiting RAS signaling at different levels. Whereas losartan inhibits RAS pathway activity, preventing Ang-II from binding to its AT-1 receptor, enalapril acts by inhibiting ACE activity, directly affecting the bioavailability of Ang-II and promoting the synthesis of Ang1–7, a new peptide with effects opposite to those of Ang-II (Iwai and Horiuchi 2009). Thus, enalapril action is upstream of losartan activity. Our results show that the effects of enalapril are more specific than those of losartan, which inhibits only CTGF expression and not TGF-β1 expression; however, it exhibits a similar improvement in muscle strength. In summary, we have shown that ARB and ACE inhibitors can reduce the damage and fibrosis associated with dystrophic skeletal muscle, partially recovering muscle strength. These beneficial effects have been observed with the differential expression of two main pro-fibrotic factors, namely CTGF and TGF-β1. Our results add new evidence for and reinforce the potential interest in RAS inhibition by ARB or ACE inhibitors in the therapy of DMD patients.. References Abraham D (2008) Connective tissue growth factor: growth factor, matricellular organizer, fibrotic biomarker or molecular target for anti-fibrotic therapy in SSc? Rheumatology (Oxford) 47 (Suppl 5):v8–v9 Andreetta F, Bernasconi P, Baggi F, Ferro P, Oliva L, Arnoldi E, Cornelio F, Mantegazza R, Confalonieri P (2006) Immunomodulation of TGF-beta 1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J Neuroimmunol 175:77–86 Arthur PG, Grounds MD, Shavlakadze T (2008) Oxidative stress as a therapeutic target during muscle wasting: considering the complex interactions. Curr Opin Clin Nutr Metab Care 11:408–416 Bauer R, Straub V, Blain A, Bushby K, MacGowan GA (2009) Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur J Heart Fail 11:463–471 Bernasconi P, Di Blasi C, Mora M, Morandi L, Galbiati S, Confalonieri P, Cornelio F, Mantegazza R (1999) Transforming growth factor-beta1 and fibrosis in congenital muscular dystrophies. Neuromuscul Disord 9:28–33 Bish LT, Yarchoan M, Sleeper MM, Gazzara JA, Morine KJ, Acosta P, Barton ER, Sweeney HL (2011) Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One 6:e20856 Bizario JC, Cerri DG, Rodrigues LC, Oliveira GL, Nomizo A, de Araujo DD, Fukuhara PS, Ribeiro JC, de Castro FA, Costa.
(14) 186 MC (2009) Imatinib mesylate ameliorates the dystrophic phenotype in exercised mdx mice. J Neuroimmunol 212:93–101 Brussee V, Tardif F, Tremblay JP (1997) Muscle fibers of mdx mice are more vulnerable to exercise than those of normal mice. Neuromuscul Disord 7:487–492 Burdi R, Rolland JF, Fraysse B, Litvinova K, Cozzoli A, Giannuzzi V, Liantonio A, Camerino GM, Sblendorio V, Capogrosso RF, Palmieri B, Andreetta F, Confalonieri P, De Benedictis L, Montagnani M, De Luca A (2009) Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J Appl Physiol 106:1311–1324 Cabello-Verrugio C, Acuna MJ, Morales MG, Becerra A, Simon F, Brandan E (2011) Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem Biophys Res Commun 410:665–670 Cabello-Verrugio C, Cordova G, Salas JD (2012a) Angiotensin II: role in skeletal muscle atrophy. Curr Protein Pept Sci 13:560–569 Cabello-Verrugio C, Morales MG, Cabrera D, Vio CP, Brandan E (2012b) Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J Cell Mol Med 16:752–764 Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC (2007) Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med 13:204–210 Cozzoli A, Nico B, Sblendorio VT, Capogrosso RF, Dinardo MM, Longo V, Gagliardi S, Montagnani M, De Luca A (2011) Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol Res 64:482–492 De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Fraysse B, Mirabella M, Servidei S, Ruegg UT, Conte Camerino D (2003) Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther 304:453–463 De Luca A, Nico B, Liantonio A, Didonna MP, Fraysse B, Pierno S, Burdi R, Mangieri D, Rolland JF, Camerino C, Zallone A, Confalonieri P, Andreetta F, Arnoldi E, Courdier-Fruh I, Magyar JP, Frigeri A, Pisoni M, Svelto M, Conte Camerino D (2005) A multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx mice. Am J Pathol 166:477–489 De Luca A, Nico B, Rolland JF, Cozzoli A, Burdi R, Mangieri D, Giannuzzi V, Liantonio A, Cippone V, De Bellis M, Nicchia GP, Camerino GM, Frigeri A, Svelto M, Camerino DC (2008) Gentamicin treatment in exercised mdx mice: identification of dystrophin-sensitive pathways and evaluation of efficacy in work-loaded dystrophic muscle. Neurobiol Dis 32:243–253 Goldstein JA, McNally EM (2010) Mechanisms of muscle weakness in muscular dystrophy. J Gen Physiol 136:29–34 Graham KM, Singh R, Millman G, Malnassy G, Gatti F, Bruemmer K, Stefanski C, Curtis H, Sesti J, Carlson CG (2010) Excessive collagen accumulation in dystrophic (mdx) respiratory musculature is independent of enhanced activation of the NF-kappaB pathway. J Neurol Sci 294:43–50 Gregorevic P, Plant DR, Leeding KS, Bach LA, Lynch GS (2002) Improved contractile function of the mdx dystrophic mouse diaphragm muscle after insulin-like growth factor-I administration. Am J Pathol 161:2263–2272. Cell Tissue Res (2013) 353:173–187 Iwai M, Horiuchi M (2009) Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2angiotensin-(1-7)-Mas receptor axis. Hypertens Res 32:533–536 Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA, Lowe DA (2008) Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 38:1290–1303 Leask A (2007) TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res 74:207–212 Marshall PA, Williams PE, Goldspink G (1989) Accumulation of collagen and altered fiber-type ratios as indicators of abnormal muscle gene expression in the mdx dystrophic mouse. Muscle Nerve 12:528–537 Morales MG, Cabello-Verrugio C, Santander C, Cabrera D, Goldschmeding R, Brandan E (2011) CTGF/CCN-2 overexpression can directly induce features of skeletal muscle dystrophy. J Pathol 225:490–501 Morales M, Vazquez Y, Acuña M, Rivera J, Simon F, Salas J, ÁlvarezRuf J, Brandan E, Cabello-Verrugio C (2012) Angiotensin IIinduced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int J Biochem Cell Biol 44:1993–2002 Moyer AL, Wagner KR (2010) Regeneration versus fibrosis in skeletal muscle. Curr Opin Rheumatol 23:568–573 Okano T, Yoshida K, Nakamura A, Sasazawa F, Oide T, Takeda S, Ikeda S (2005) Chronic exercise accelerates the degenerationregeneration cycle and downregulates insulin-like growth factor1 in muscle of mdx mice. Muscle Nerve 32:191–199 Pereira RM, dos Santos RA, da Costa Dias FL, Teixeira MM, Simoes e Silva AC (2009) Renin-angiotensin system in the pathogenesis of liver fibrosis. World J Gastroenterol 15:2579–2586 Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, SchultzeMosgau S, Stallmach A, Kinne RW, Wolf G (2009) TGF-beta and fibrosis in different organs—molecular pathway imprints. Biochim Biophys Acta 1792:746–756 Salas SP, Giacaman A, Vio CP (2004) Renal and hormonal effects of water deprivation in late-term pregnant rats. Hypertension 44:334–339 Sandri M, Podhorska-Okolow M, Geromel V, Rizzi C, Arslan P, Franceschi C, Carraro U (1997) Exercise induces myonuclear ubiquitination and apoptosis in dystrophin-deficient muscle of mice. J Neuropathol Exp Neurol 56:45–57 Sun G, Haginoya K, Dai H, Chiba Y, Uematsu M, Hino-Fukuyo N, Onuma A, Iinuma K, Tsuchiya S (2009) Intramuscular reninangiotensin system is activated in human muscular dystrophy. J Neurol Sci 280:40–48 Sun G, Haginoya K, Wu Y, Chiba Y, Nakanishi T, Onuma A, Sato Y, Takigawa M, Iinuma K, Tsuchiya S (2008) Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci 267:48–56 Vial C, Zuniga LM, Cabello-Verrugio C, Canon P, Fadic R, Brandan E (2008) Skeletal muscle cells express the profibrotic cytokine connective tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J Cell Physiol 215:410–421 Vilquin JT, Brussee V, Asselin I, Kinoshita I, Gingras M, Tremblay JP (1998) Evidence of mdx mouse skeletal muscle fragility in vivo by eccentric running exercise. Muscle Nerve 21:567–576 Wang P, Fedoruk MN, Rupert JL (2008) Keeping pace with ACE: are ACE inhibitors and angiotensin II type 1 receptor antagonists potential doping agents? Sports Med 38:1065–1079 Wei Y, Sowers JR, Nistala R, Gong H, Uptergrove GM, Clark SE, Morris EM, Szary N, Manrique C, Stump CS (2006) Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem 281:35137–35146.
(15) Cell Tissue Res (2013) 353:173–187 Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS (2008) Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am J Physiol Endocrinol Metab 294:E345–E351 Weller B, Karpati G, Carpenter S (1990) Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J Neurol Sci 100:9–13 Whitehead NP, Yeung EW, Froehner SC, Allen DG (2010) Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One 5:e15354. 187 Zeman RJ, Peng H, Danon MJ, Etlinger JD (2000) Clenbuterol reduces degeneration of exercised or aged dystrophic (mdx) muscle. Muscle Nerve 23:521–528 Zhou L, Lu H (2010) Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol 69:771–776 Zhou L, Porter JD, Cheng G, Gong B, Hatala DA, Merriam AP, Zhou X, Rafael JA, Kaminski HJ (2006) Temporal and spatial mRNA expression patterns of TGF-beta1, 2, 3 and TbetaRI, II, III in skeletal muscles of mdx mice. Neuromuscul Disord 16:32–38.
(16) Reproduced with permission of the copyright owner. Further reproduction prohibited without permission..
(17)
Figure



+5


Documento similar