Brain metabolite clearance: impact on Alzheimer's disease
9
0
0
Texto completo
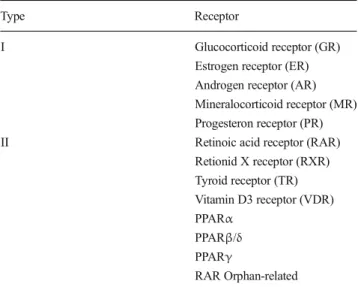
(2) 554. mitochondrial reactive oxygen species (ROS) (Caspersen et al. 2005; Manczak et al. 2006; Reddy and Beal 2008). As a consequence of these pathological changes, one of the main physiological characteristics of AD is cognitive impairment (Perl 2010), which is commonly associated with specific brain areas and reflects the loss of the neuronal network due to altered synaptic structure and functionality (Sheng et al. 20 12 ). Disru pt ion of t he n ec es sar y p res yn ap tic neurotransmitter-releasing machinery and/or altered expression of specific postsynaptic proteins, such as PSD95, are the basis of the synaptic impairment observed in AD (Sheng et al. 2012; Südhof 2012; 2013. Importantly, although neuronal network damage occurs across the whole brain, the affected brain areas usually include the temporal and parietal cortex, the hippocampus and the amygdala. Of these areas, the hippocampus, which is associated with memory and cognition, is one of the most critically involved regions (Shaerzadeh et al. 2013). Although AD was described more than a century ago and a large amount of effort and resources have been devoted to AD research, no effective treatments are currently available to halt or reverse this devastating disease (Langbaum et al. 2013). In 2012, Cramer et al. (2012) published an original study in which they provided substantial evidence of the benefits obtained after the administration of bexarotene to different AD mouse models (EML 2013; FDA 2013). Despite the initial expectation, some authors were skeptical and suggested that further analysis and experimentation should be performed before bexarotene can be confirmed as a novel AD drug (Crunkhorn 2012; LaFerla 2012; Strittmatter 2012). Indeed, since its publication, four groups have published “technical comments” regarding Cramer’s work, mainly indicating the impossibility of replicating some of the results and, more importantly, highlighting the lack of correlation between the molecular effects and the behavioral and cognitive rescue observed after bexarotene administration (Fitz et al. 2013; Price et al. 2013; Tesseur et al. 2013; Veeraraghavalu et al. 2013). However, apart from creating controversy, Cramer’s work has once again highlighted the critical role of metabolite clearance from the brain. Interestingly, although several authors have agreed that metabolic clearance plays a critical role in brain homeostasis, very few of them have indicated the importance of understanding the clearance-related mechanisms triggered after any therapeutic intervention. In fact, upon examining the results obtained by the different research groups (Fitz et al. 2013; Landreth et al. 2013; Price et al. 2013; Tesseur et al. 2013; Veeraraghavalu et al. 2013), it is clear that all of the groups have provided further evidence that bexarotene is able to trigger the retinoid X receptor (RXR):peroxisome proliferator-activated receptor (PPAR) signaling cascade, leading to increased expression of liver X receptor (LXR) target genes. However, it is quite obvious that all the authors have focused on the endpoint of RXR. Metab Brain Dis (2014) 29:553–561. stimulation, particularly on the LXR genes, thus underestimating the effect of PPAR stimulation on several cellular processes. In the present short review, we offer a comprehensive view of the events that are triggered after NR stimulation and how they might lead to increased metabolite clearance from the brain.. Nuclear Receptors (NRs) NRs are a complex transcription factor superfamily (Table 1) identified as being critical for normal cell physiology and capable of sensing the extracellular and intracellular media (Olefsky 2001). Their dysfunction has been associated with several pathological conditions such as cancer, insulin resistance and infertility, among others (Olefsky 2001; Gronemeyer et al. 2004). NRs exert their actions not only by direct binding to their ligands and by modulating gene expression but also through their interaction with several cell signaling pathways such as Wnt, phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) (Mulholland et al. 2005; Fuenzalida et al. 2007; Inestrosa and Toledo 2008). NRs can be divided into two main categories: Type I, the main characteristic of which is the formation of homodimers, such as the androgen, estrogen and progesterone receptors; and Type II, which form RXR heterodimers (Olefsky 2001; Mulholland et al. 2005; Zolezzi and Inestrosa 2013). The latter type includes the thyroid receptor, the RXR (homodimer), the vitamin D receptor, the retinoic Table 1 Nuclear receptors superfamily chart Type. Receptor. I. Glucocorticoid receptor (GR) Estrogen receptor (ER) Androgen receptor (AR) Mineralocorticoid receptor (MR) Progesteron receptor (PR) Retinoic acid receptor (RAR) Retionid X receptor (RXR) Tyroid receptor (TR) Vitamin D3 receptor (VDR) PPARα PPARβ/δ PPARγ RAR Orphan-related. II. The present table corresponds to a resumed list of NR family members. In this case we have focused on Type I and II category, which depends on the ability of each nuclear receptor to form homo- or heterodimers. Type II NR usually forms RXR heterodimers (based on Mulholland et al. 2005; and HGNC database gene names). PPAR, peroxisome proliferator-activated receptor.
(3) Metab Brain Dis (2014) 29:553–561. acid receptor, the LXR and the PPAR (Mulholland et al. 2005). Three different mammalian PPARs have been identified: PPARα, PPARβ/δ and PPARγ (Neher et al. 2012). Although all PPARs have been described in both the adult and developing brain (Heneka and Landreth 2007), PPARγ is the most studied isoform and has showed the most promising neuroprotective effects in different models of neurodegenerative disorders, such as AD (Mulholland et al. 2005; Zolezzi and Inestrosa 2013; Chen et al. 2012; Neher et al. 2012; Toledo and Inestrosa 2010).. RXR-LXR/PPAR According to the FDA, bexarotene is “a synthetic retinoid that exerts its biological action through selective binding and activation of the RXR receptors” (FDA 2013). Currently, three isoforms have been described for the RXR: α-γ (Mangelsdorf et al. 1992). Usually, these isoforms form heterodimers in the cell, constituting functional complexes, with both cytoplasmic and nuclear functions (Mulholland et al. 2005; Dawson and Xia 2012). Three mechanisms have been described for the RXR depending on its dimeric partner: i) non-permissive, ii) permissive and iii) conditional transactivation; thus, the function of the RXR in the RXR-NR complex depends on its partner. For example, if partnered with LXR and PPAR, which are both affected by permissive transactivation, the RXR will enhance LXR and PPAR activity, allowing for the recruitment of NR (LXR or PPAR) coactivators (and vice versa) (Dawson and Xia 2012). It is well known that LXR and PPARs function in a coordinated manner. Moreover, it has been demonstrated that the expression of ApoE, the key Aβ-clearance protein, and several ATP binding cassette (ABC) transporters depends on a transcriptional cascade triggered by the PPARs through LXR (Chawla et al. 2001; Kang and Rivest 2012). Based on this information, it is clear that the choice made by Cramer’s group to target the RXR was intended to enhance LXR activity via a PPARγ-mediated mechanism. Indeed, reduced soluble Aβ levels; increased ApoE, ABCA1 and G1 expression; and microglial activation were some of the main effects observed by Cramer’s group (2012). Interestingly, they also reported improved cognitive and behavioral scores; however, as declared by the authors, a “poor correlation with the removal of insoluble deposited forms of Aβ” was noted.. PPARs and Alzheimer’s disease Although the discussion regarding plaque burden is quite valid, the real impact of plaques remains a matter of discussion (Haass and Selkoe 2007; Meyer-Luehmann et al. 2008; Selkoe 2011; Lesné et al. 2013). Moreover, some authors have. 555. proposed that it is not only the number and size of the plaques that are important but also the intrinsic characteristics of the plaque (Carvajal and Inestrosa 2011; Selkoe 2011). Thus, along with the critical role of brain clearance, it appears that it is far more important to discuss the restoration of cognitive capacity versus the reduction in plaque burden, basically because this restoration implies the rescue of several compromised tissues and synapses as well as the re-establishment of the neuronal circuitry. These topics have been the scope of several years of research, in addition to crosstalk with several cell signaling pathways and improved cellular energy metabolism. There is evidence associating the pathophysiology of AD with free radicals (Smith et al. 1996; Miranda et al. 2000). In fact, through in vitro experimentation, it has been shown that one of the neurotoxic mechanisms of Aβ aggregates involves oxidative stress (Bhel et al. 1994; Abramov et al. 2004; Querfurth and LaFerla 2010). Since Santos et al. (2005) noted for the first time that PPAR activation was able to protect against Aβ insults by means of PPAR agonist administration, the relevance of this NR subfamily in neurodegenerative disorders, such as AD, has constantly increased. Importantly, PPAR-mediated protection against Aβ has been demonstrated both in vitro and in vivo using transgenic murine models of AD, such as APPswe/PSEN1DeltaE9 (Toledo and Inestrosa 2010). Indeed, several PPARγ agonists such as troglitazone and rosiglitazone, which are primarily used to treat type II diabetes, have been shown to delay Alzheimer’s development and promote cell survival through PPARγ activation (Watson and Craft 2003; Inestrosa et al. 2005; Zolezzi and Inestrosa 2013; Toledo and Inestrosa 2010). Interestingly, although PPARγ activity associated with oxidative stress response is well documented (Manji et al. 2012; Polvani et al. 2012), the interaction with several antioxidant and anti-inflammatory regulatory pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), nuclear factor erythroid 2-related factor (NRF2) and the Wnt/β-catenin pathway have also been noted (Martín et al. 2012). Additionally, it has been proposed that PPARγ also upregulates Bcl-2, which is an antiapoptotic protein and a Wnt target gene (Fuentealba et al. 2004), following survival pathways other than the traditional MAPK or Akt pathways. This process prevents neural degeneration and increases mitochondrial stability (Fuenzalida et al. 2007). Moreover, during the last few years, it has been further proposed that the administration of PPAR agonists induces additional effects regarding neuronal functionality, including neurite outgrowth, and has a direct effect on mitochondrial fusion-fission dynamics (Quintanilla et al. 2013; Zolezzi et al. 2013; Zolezzi and Inestrosa 2013). Similarly, along with the increased antioxidant capacity often described for the PPARs, which counteracts Aβ-induced oxidative damage to protect the synapses and mitochondria (Terry et al. 1991; Bhel et al..
(4) 556. 1994; Smith et al. 1996; Miranda et al. 2000; Abramov et al. 2004; Caspersen et al. 2005; Manczak et al. 2006; Reddy and Beal 2008; Lee et al. 2010; Querfurth and LaFerla 2010; Paula-Lima et al. 2011; Cavalluci et al. 2012), we have recently shown that PPAR agonists are also able to induce mitochondrial dynamic events through PPARγ-coactivator1α (PGC-1α). This process prevents mitochondrial dysfunction due to oxidative insults, suggesting that cell metabolism is protected and mitochondrial biogenesis is most likely increased (Zolezzi et al. 2013; Zolezzi and Inestrosa 2013). This latter finding is highly relevant considering that mitochondrial dynamics have recently been described as a critical mechanism associated with mitochondrial and cellular fate after critical insults (Manji et al. 2012). Such dynamics help to sustain cell metabolism, and successive fusion-fission cycles allow for the elimination of dysfunctional organelles and for the repair of mitochondrial DNA that could be damaged after a toxic challenge (Haemmerle et al. 2011; Hondares et al. 2011; Silva et al. 2013; Zolezzi et al. 2013) (Fig. 1). Moreover, as noted for antioxidant activity, the mitochondrial effects derived from PPAR activation could also be related to several cell signaling pathways, such as Wnt (Silva-Alvarez et al.. Fig. 1 Comprehensive RXR:PPAR-agonist mechanism of action. This scheme shows a more complete view of the mechanisms triggered after RXR:PPAR stimulation. We propose that the benefit observed after PPAR or RXR-agonist administration are obtained because a synergistic combination of these nuclear receptors. On the one hand, RXR stimulation will induce several PPAR-related effects, such as improved antioxidant activity achieved through increased peroxisome number and antioxidant enzyme activity; as well as improved mitochondrial function through a PGC-1α-mediated mechanism, preventing mitochondrial dysfunction due to increased oxidative stress. On the other hand, RXR-PPAR activation will lead to increased expression of ApoE and its transporters, ABCA1, ABCG1 and LRP1, favoring Aβ clearance from the brain. RXR, retinoid X receptor; PPAR, peroxisome proliferator-activated receptor; PGC-1α, PPARγ coactivator 1α; Mnf, mitofusina; Drp1, Dynamin related protein 1; mtTFA, mitochondrial transcription factor; CAT, catalase; ROS, reactive oxygen species, Aβ, amyloid β-peptide; ApoE, apolipoprotein E; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; LRP1/2, Lipoprotein related protein 1/2. Metab Brain Dis (2014) 29:553–561. 2013). Additionally, some widely known ethnobotanical drugs, which have shown interesting systemic effects against neurodegenerative disorders such as AD, have also been recently suggested to exert part of their actions through a PGC-1α-mediated mechanism, indicating a possible further connection with the PPARs (Silva-Alvarez et al. 2013; Zolezzi et al. 2013). On the other hand, PPARs have also demonstrated interesting effects regarding tau pathology, which is the other wellrecognized pathologic hallmark of AD. In summary, the cellular alterations derived from tau hyperphosphorylation are associated with altered cytoarchitecture and impaired cell and axonal traffic, leading ultimately to cell death and neurodegeneration (Barroso et al. 2013). Recent investigations have addressed the potential effects of PPARs against tau hyperphosphorylation and demonstrated that several PPAR agonists are able to protect neurons by reducing phospho-tau levels (Barroso et al. 2013; Cho et al. 2013; Dumont et al. 2012). Although the precise mechanisms associated with the reduced levels of phospho-tau observed after PPAR agonist treatment remain unclear, some authors have suggested that the agonists likely interact with the key components of several cellular signaling pathways, such as cyclin-dependent kinase (CDK) 5 and protein kinase B (Akt)/glycogen synthase kinase (GSK) 3β (Cho et al. 2013; Tokutake et al. 2012). In summary, all of these effects suggest that the cognitive rescue observed in in vivo assays of PPAR agonist administration, including Cramer’s work (which involved indirect activation of PPARγ), may have been derived from a combination of several concomitant effects. PPARs control oxidative damage by enhancing antioxidant defense through increased antioxidant enzyme activity; protect the neuronal network and promote neurite outgrowth, thus favoring new neuronal connections; and modulate the ROS production rate and neuronal energy balance. PPAR activation has a direct impact on mitochondrial health through the control of several mitochondrial fusion/fission-related proteins and mitochondrial DNA transcription factor expression via a PGC-1αmediated mechanism.. Blood-Brain Barrier (BBB) Although the importance of the neuronal component in the neurodegenerative process is clear, additional components of the brain—and more importantly, the health status of these components—are also quite critical. In this regard, several authors have indicated that the level of Aβ depends on the balance between the production rate of Aβ in the brain and the excretion rate of Aβ from the brain (Zlokovic 2008; Kaneyiko et al. 2013). Moreover, it is well known that excretion depends on the binding of Aβ to additional proteins, such as ApoE, which serves as a substrate for BBB transporters for the final.
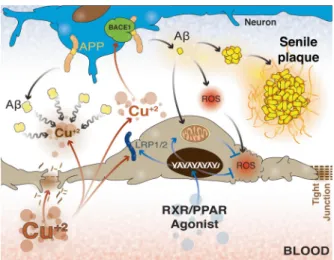
(5) Metab Brain Dis (2014) 29:553–561. elimination of Aβ from the brain (Zlokovic 2011). On the other hand, it has been recently revealed that the receptor for advanced glycation end products (RAGE) is associated with brain Aβ influx and that the RAGE-Aβ interaction leads to several pathologic changes not only in the brain but also at the BBB, affecting its permeability through several mechanisms including tight junction alterations (Deane et al. 2012; Kook et al. 2012) (Fig. 2). Transport across the BBB is highly specialized and depends on the expression of several transporters by endothelial cells. These transporters allow bidirectional traffic to maintain brain homeostasis (O’Donnell et al. 2006; Obermeier et al. 2013; Zolezzi and Inestrosa 2013). It is quite surprising that Cramer’s group has not addressed the impact of bexarotene on the BBB considering that traffic across the BBB plays a critical role in Aβ clearance in the brain (Zlokovic 2011). Indeed, increased Aβ accumulation around brain vessels has been proposed to induce further BBB alterations, causing micro-bleeding throughout the brain and favoring the development and spread of the disease (Iadecola 2013). Similarly, some authors have proposed that not only pathological but also environmental conditions might lead to alterations in the BBB, modifying its properties and affecting its permeability (O’Donnell et al. 2006; Zlokovic 2011; Obermeier et al. 2013). Interestingly, Singh et al. (2013) recently demonstrated that sub-cytotoxic concentrations of Cu2+ lead to increased. Fig. 2 Schematic representation of the main components of the bloodbrain barrier. As noticed, the main role of the BBB is to maintain and ensure the optimal microenvironmental conditions for normal neuronal activity. That task is carried out through a highly specialized transport system, constituted by several specific transport carriers, which maintain an appropriate balance between cellular metabolite accumulation in the intercellular space and the final excretion to the blood across the BBB. In AD, the Aβ production:excretion rate has been recognized as critical, and the variation of the expression levels of Aβ transporters, such as ABCt, LRP1/2 and RAGE, have a huge impact on Aβ levels in the brain. Cho, cholesterol; ABCt, ATP-binding cassette transporters; LRP1/2, low density lipoprotein receptor-related protein 1/2; Aβ, amyloid-β peptide; RAGE, receptor for advanced glycation end products; APP, amyloid precursor protein; BACE1, β-site APP cleaving enzyme 1; AA, amino acids. 557. Aβ production due to direct interaction with the APPsecretase processing machinery and to impaired Aβ clearance from the brain because of reduced LRP1 expression at the BBB. Both events offer a new perspective regarding Aβ accumulation as a result of combined mechanisms, which once again highlights the critical role of the BBB in neurodegenerative processes. In this regard, several authors have indicated that PPAR activation is able to induce changes in the BBB, protecting the brain as well as the BBB itself under different negative stimuli. In fact, PPARs have been associated with increased expression of ABCG2 (Hoque et al. 2012), protection against deprivation stimuli in BBB models (Mysiorek et al. 2009), protection during cerebral ischemia (Yin et al. 2010) and a decrease in the Aβ burden in murine models of AD (Kalinin et al. 2009). Regrettably, even though it has been suggested that PPAR activation might lead to increased ABCA1 and ABCG1 levels (Cramer et al. 2012), no studies have examined changes in the expression of BBB transporters after PPAR stimulation. We believe that the increased Aβ clearance is likely due to increased expression of specialized transporters at the brain-endothelial level. Indeed, considering the recent findings reported by Kaneyiko et al. (2013), regarding the importance of neuronal expression of LRP1 transporters for Aβ balance and degradation in the brain, it is possible that the role of the PPARs or NR agonists might involve a neuronal response also associated with efficient Aβ handling and excretion. Additionally, it is well recognized that traffic across the BBB, especially that required for sustaining brain homeostasis, is highly energy demanding (O’Donnell et al. 2006; Obermeier et al. 2013). Considering the main features of PPAR-derived effects, it is possible to suggest a direct link between PPARs and improved BBB functionality because of enhanced antioxidant protection and prevention of extended mitochondrial damage, which could compromise BBB energy metabolism (Zolezzi and Inestrosa 2013; Zolezzi et al. 2013). Moreover, Cramer reported that the main effects of bexarotene administration were observed several days after the beginning of the treatment, suggesting that the improvement of additional structures, such as the BBB, is quite possible and might lead to an increase in BBB trafficking.. PPAR side effects As with any type of drug, the use of NRs could also induce undesirable side effects. One of the most common events related to PPAR usage is abnormal growth and loss of differentiation in several tissues, which suggests pro-neoplastic activity (Sheaffer et al. 2005; Fuenzalida et al. 2007; Gonzales and Shah 2007). However, several contradictory studies have presented evidence indicating both pro-.
(6) 558. neoplastic and anti-tumoral activity, such as a pro-apoptotic effect (Harman et al. 2004; Hollingshead et al. 2007; Muzio et al. 2007). Interestingly, the final outcome seems to depend on the PPAR type and the expression level of each of the PPARs in a particular tumor (Sertizing et al. 2007), which highlights the importance of performing a full assessment of the genetic profile of each tumor prior to attempting any therapeutic approach. Similarly, although some authors have reported a decreased immune response because of PPAR overstimulation, others suggest that PPAR stimulation can be beneficial against some septic and infectious conditions (Drosatos et al. 2013; Philipson et al. 2013). As we have suggested previously, the controversy regarding the full range of effects observed for NR agonists, and particularly for PPARs, is due to the partial knowledge of the mechanisms of action of this family of drugs, such as the crosstalk between PPARs and several key points in critical cell signaling pathways. Further studies that properly assess the utility and feasibility of PPARs as therapeutic agents against several diseases are of the utmost importance.. Concluding remarks Bexarotene has emerged as a novel alternative for treating AD. However, despite the high expectations that arose after Cramer’s publication, some serious questions have emerged regarding the reproducibility of the originally described effects, particularly regarding the improvements in the behavioral parameters (LaClair et al. 2013). Moreover, the questions regarding the potential mechanisms involved in such a recovery are still awaiting answers. Aβ exerts effects on a wide range of brain structures, including the BBB, and although the critical role of Aβ clearance in AD is well accepted, studies regarding how different therapeutic alternatives affect the BBB are quite scarce (Iadecola 2013; Obermeier et al. 2013; Zolezzi and Inestrosa 2013). In the past, several authors have noted that a compromised BBB may not only be a consequence of the neurodegenerative process observed in AD but also serve as a starting point for the neuropathological changes that lead to AD (Iadecola 2013). Indeed, Singh et al. (2013) have recently demonstrated that chronic low levels of Cu2+ can interfere with brain Aβ levels, leading to reduced expression of LRP1 and limiting the clearance of Aβ through the BBB. The work of Singh et al. (2013) clearly suggests the new concept that BBB-related pathophysiological mechanisms should be further studied to properly approach any neurodegenerative disorder and that assays with old or novel therapeutic agents should include an assessment of BBBrelated effects (Fig. 3).. Metab Brain Dis (2014) 29:553–561. Fig. 3 New considerations regarding blood-brain barrier health. Although the association of AD-related Aβ accumulation and BBB functionality has been well known for a long time and some authors have clearly suggested a more critical role for the BBB in neurodegenerative processes, very few studies have focused on understanding the BBBrelated mechanisms as a cause of neuronal damage, and not only as collateral damage derived from primary neuronal or glial failure. Recently, experimental data showed that sub-cytotoxic concentration of Cu2+ can induce dramatic changes in the BBB structure, affecting the TJs and functionality, altering the expression of the Aβ transporters. Moreover, Cu2+ was also shown to induce APP β-processing leading to increased Aβ levels, and to act as a core for Aβ aggregation in the intercellular spaces, favoring senile plaque formation. These data provide strong evidence that primary damage of the BBB could be at the basis of a complex event, such as increased levels of Aβ and the neuronal alterations derived from Aβ brain accumulation. In the same way, the use of NR agonists, capable of restoring BBB transporter expression and protecting endothelial cells from Aβ-derived oxidative damage, might constitute interesting tools to counteract BBB failure. Aβ, amyloid-β peptide; APP, amyloid precursor protein; BACE1, β-site APP cleaving enzyme 1; LRP1/2, low density lipoprotein receptor-related protein 1/2; ROS, reactive oxygen species. We believe that although important progress has been made regarding AD physiopathology and AD therapy, partial explanations of the mechanisms of action of therapeutic agents, such as the RXR agonist bexarotene, are still quite common. Our objective was to provide a comprehensive view of the phenomena triggered after NR stimulation, including the potential effects on the BBB as a critical structure involved in brain homeostasis. Moreover, beyond the uncertainty regarding Cramer’s work, the results of our own research, along with previously and recently published data (Fantini et al. 2014), provide evidence that NR agonists have great potential not only for the treatment of AD but also for the treatment of other neurodegenerative disorders. Of course, further research is needed to assess every possibility derived from NR-agonist administration; however, as suggested by LaClair et al. (2013), it is extremely important that this future research considers a wider view of NRs to avoid partial explanations that could lead to the underestimation or misinterpretation of results obtained with this family of drugs..
(7) Metab Brain Dis (2014) 29:553–561 Acknowledgments We thank Dr. Waldo Cerpa for helpful discussions regarding this manuscript. This work was supported by FONDECYT N° 11130033 (to JMZ) and the Basal Center of Excellence in Science and Technology (PFB 12/2007) from CONICYT and SQM, the MIFAB Institute and Fundación Ciencia y Vida (to NCI). Graphic work was performed by Graphique-Science (http://graphiquescience.blogspot.com). Conflicts of interest The authors declare that they have no conflict of interest.. References Abramov AY, Canevari L, Duchen MR (2004) β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575 Alzheimer’s Association (2012) Alzheimer’s disease facts and figures. Alzheimers Dement 8:131–168 Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer’s disease. Lancet 377:1019–1031. doi:10.1016/S01406736(10)61349-9 Barroso E, del Valle J, Porquet D et al (2013) Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPARβ/δnull mice. Biochim Biophys Acta 1832:1241–1248 Bhel C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77:817–827 Carvajal FJ, Inestrosa NC (2011) Interaction of AChE with Aβ aggregates in Alzheimer’s brain: therapeutic relevance of IDN 5706. Front Mol Neurosci 4:19 Caspersen C, Wang N, Yao J et al (2005) Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19:2040–2041 Cavalluci V, D’Amelio M, Cecconi F (2012) Aβ toxicity in Alzheimer’s disease. Mol Neurobiol 45:366–378 Chawla A, Boisvert WA, Lee CH et al (2001) A PPAR gamma-LRXABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 7:161–171 Chen YC, Wu JS, Tsai HD, Huang CY, Chen JJ, Sun GY, Lin TN (2012) Peroxisome proliferator-activated receptor gamma (PPAR-γ) and neurodegenerative disorders. Mol Neurobiol. doi:10.1007/s12035012-8259-8 Cho DH, Lee EJ, Kwon KJ, Shin CY, Song KH, Park JH, Han SH (2013) Troglitazone, a thiazolidinedione, decreases tau phosphorylation through the inhibition of cyclin-dependent kinase 5 activity in SHSY5Y neuroblastoma cells and primary neurons. J Neurochem 126: 685–695 Cramer PE, Cirrito JR, Wesson DW et al (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335:1503–1506 Crunkhorn S (2012) Neurodegenerative disease: RXR agonist reverses Alzheimer’s disease. Nat Rev Drug Discov 11:271. doi:10.1038/ nrd3706 Dawson MI, Xia Z (2012) The retinoid X receptor and their ligands. Biochim Biophys Acta 1821:21–56 Deane R, Singh I, Sagare AP, Bell RD et al (2012) A multimodal RAGEspecific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122:1377–1392 Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, Homma S, Schulze PC, Goldberg IJ (2013) Peroxisome proliferatoractivated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail 6:550–562. 559 Dumont M, Stack C, Elipenahli C et al (2012) Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum Mol Genet 21:5091–5105 Eisai Manufacturing Ltd (EML), Targretin information; www.drugs.com/ uk/targretin-capsules-1302.html. Accessed 17 Jun 2013 Fantini J, Di Scala C, Yahi N, Troadec JD, Sadelli K, Chahinian H, Garmy N (2014) Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem Neurosci. doi:10.1021/cn400183w FDA, Drug approval package, Tagretin; www.accessdata.fda.gov/ drugsatfda_docs/nda/99/21055_Targretin.cfm. Accessed 17 Jun 2013 Fitz NF, Cronican AA, Lefterov I, Koldamova R (2013) Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924. doi:10.1126/ science.1235809 Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC (2004) Signal transduction during amyloid-β-peptide neurotoxicity: role in Alzheimer disease. Brain Res Brain Res Rev 47:275–289 Fuenzalida K, Quintanilla R, Ramos P et al (2007) Peroxisome proliferator-activated receptor γ up-regulates the Bcl-2 antiapoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem 282:37006–37015 Gonzales FJ, Shah YM (2007) PPARα: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 246:2–8 Gronemeyer H, Gustafsson JA, Laudet V (2004) Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3: 950–964 Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8:101–112 Haemmerle G, Moustafa T, Woelkart G et al (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1α. Nat Med 17:1076–1085 Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzales FJ, Peters JM (2004) Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat Med 10:481–483 Heneka MT, Landreth GE (2007) PPARs in the brain. Biochim Biophys Acta 1771:1031–1045 Hollingshead HE, Killins RL, Borland MG, Girroir EE, Billin AN, Willson TM, Sharma AK, Amin S, Gonzales FJ, Peters JM (2007) Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/ delta) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis 28:2641–2649 Hondares E, Rosell M, Díaz-Delfin J et al (2011) Peroxisome proliferatoractivated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 286: 43112–43122 Hoque MT, Robillard KR, Bendayan R (2012) Regulation of breast cancer resistant protein by peroxisome proliferator-activated receptor α in human brain microvessel endothelial cells. Mol Pharmacol 81:598–609 HUGO Gene Nomenclature Committee (HGNC) at http://www. genenames.org/genefamilies/NR, accessed 20 Jan 2014 Iadecola C (2013) The pathobiology of vascular dementia. Neurology. doi:10.1016/j.neuron.2013.10.008 Inestrosa NC, Toledo EM (2008) The role of Wnt signaling in neuronal dysfunction in Alzheimer’s disease. MolNeurodegener 3:9 Inestrosa NC, Godoy JA, Quintanilla RA, Koenig CS, Bronfman M (2005) Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents βamyloid neurodegeneration: role of Wnt signaling. Exp Cell Res 304:91–104.
(8) 560 Kalinin S, Richardson JC, Feinstein DL (2009) A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res 6:431– 437 Kaneyiko T, Cirrito JR, Liu CC, Shinohara M (2013) Neuronal clearance of Amyloid-β by endocytic receptor LRP1. J Neurosci 33:19276– 19283. doi:10.1523/JNEUROSCI.3487-13.2013 Kang J, Rivest S (2012) Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr Rev 33: 715–746 Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I (2012) Aβ1-42-RAGE interaction disrupts tight junctions of the bloodbrain barrier via Ca2+-calcineurin signaling. J Neurosci 32:8845– 8854 LaClair KD, Manaye KF, Lee DL et al (2013) Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener 8:18 LaFerla FM (2012) Preclinical success against Alzheimer’s disease with an old drug. N Engl J Med 367:570–574 Landreth GE, Cramer PE, Lakner MM et al (2013) Response to comments on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924. doi:10. 1126/science.1234114 Langbaum JB, Fleisher AS, Chen K et al (2013) Ushering in the study and treatment of preclinical Alzheimer disease. Nat Rev Neurol. doi: 10.1038/nrneurol.2013.107 Lee HP, Zhu X, Casadesus G et al (2010) Antioxidant approaches for the treatment of Alzheimer’s disease. Expert Rev Neurother 10:1201– 1208 Lesné SE, Sherman MA, Grant M et al (2013) Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 136:1383–1398 Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006) Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15:1437–1449 Mangelsdorf DJ, Borgmeyer U, Heyman RA et al (1992) Characterization of three RXR genes that mediate the action of 9cis retinoic acid. Genes Dev 6:329–344 Manji H, Kato T, Di Prospero NA et al (2012) Impaired mitochondrial function in psychiatric disorders. Nature Rev Neurosci 13:293–307 Martín A, Pérez-Girón JV, Hernanz R, Palacios R, Briones AM, Fortuño A, Zalba G, Salaices M, Alonso MJ (2012) Peroxisome proliferatoractivated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens 30:315–326 Meyer-Luehmann M, Spires-Jones TL, Prada C et al (2008) Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451:720–724 Miranda S, Opazo C, Larrondo LF et al (2000) The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Prog Neurobiol 62:633–648 Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC (2005) Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev 26:898–915 Muzio G, Maggiora M, Oraldi M, Trombetta A, Canuto RA (2007) PPARalpha and PP2A are involved in the proapoptotic effect of conjugated linoleic acid on human hepatoma cell line SK-HEP-1. Int J Cancer 121:2395–2401 Mysiorek C, Culot M, Dehouck L, Derudas B, Bordet R, Cecchelli R, Fenart L, Berezowski V (2009) Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr Neurovasc Res 6:181–193 Neher MD, Weckbach S, Huber-Lang MS, Stahel PF (2012) New insights into the role of peroxisome proliferator-activated receptors in. Metab Brain Dis (2014) 29:553–561 regulating the inflammatory response after tissue injury. PPAR Res. doi:10.1155/2012/728461 O’Donnell ME, Lam TI, Tran LQ (2006) Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 26:1234–1249 Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood-brain barrier. Nat Med 19:1584– 1596. doi:10.1038/nm.3407 Olefsky JM (2001) Nuclear receptor minireview series. J Biol Chem 276: 36863–36864 Paula-Lima AC, Adasme T, SanMartín C et al (2011) Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyRmediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal 14:1209–1223 Perl DP (2010) Neuropathology of Alzheimer's disease. Mt Sinai J Med 77:32–42 Philipson CW, Bassaganya-Riera J, Viladomiu M, Pedragosa M, Guerrant RL, Roche JK, Hontecillas R (2013) The role of peroxisome proliferator-activated receptor γ in immune responses to enteroaggregative Escherichia coli infection. PLoS One 8:e57812 Polvani S, Tarocchi M, Galli A (2012) PPARγ and oxidative stress: Con(β) catenating NRF2 and FOXO. PPAR Res. doi:10.1155/ 2012/641087 Price AR, Xu G, Siemienski ZB et al (2013) Comment on “ApoEdirected therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924. doi:10.1126/science. 1234089 Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362:329–44 Quintanilla RA, Godoy JA, Alfaro I, Cabezas D, von Bernhardi R, Bronfman M, Inestrosa NC (2013) Thiazolidinediones promote axonal growth through the activation of the JNK pathway. PLoS One 8(5):e65140. doi:10.1371/journal.pone.0065140 Reddy OH, Beal MF (2008) Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14:45–53 Salmon DP, Bondi MW (2009) Neuropsychological assessment of dementia. Annu Rev Psychol 60:257–282 Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC (2005) Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem 280:41057–41068 Selkoe DJ (2001) Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis 3:75–80 Selkoe DJ (2011) Resolving controversies on the path to Alzheimer’s therapeuthics. Nat Med 17:1060–1065 Sertizing P, Seifert M, Tilgen W, Reichrath J (2007) Present concepts and future outlook: function of peroxisome proliferator-activated receptors (PPARs) for pathogenesis, progression, and therapy of cancer. J Cell Physiol 212:1–12 Shaerzadeh F, Motamedi F, Minai-Tehrani D, Khodagholi F (2013) Monitoring of neuronal loss in the hippocampus of Aβ-injected rat: autophagy, mitophagy, and mitochondrial biogenesis stand against apoptosis. Neuromolecular Med. doi:10.1007/s12017-0138272-8 Sheaffer KL, Wada K, Takahashi H, Matsuhashi N, Ohnishi S, Wolfe MM, Turner JR, Nakajima A, Borkan SC, Saubermann LJ (2005) Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res 65:2251–2259 Sheng M, Sabatini BL, Südhof TC (2012) Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol 4:a005777 Silva DF, Selfridge JE, Lu J et al (2013) Bioenergetics flux, mitochondrial mass and mitochondrial morphology dynamics and AD and MCI cybrid cell lines. Hum Mol Genet. doi:10.1093/hmg/ddt247.
(9) Metab Brain Dis (2014) 29:553–561 Silva-Alvarez C, Arrázola MS, Godoy JA, Ordenes D, Inestrosa NC (2013) Canonical Wnt signaling protects hippocampal neurons from Aβ oligomers: role of non-canonical Wnt-5ª/Ca(2+) in mitocondrial dynamics. Front Cell Neurosci 7:97 Singh I, Sagare AP, Coma M, Perlmutter D, Gelein R, Bell RD, Deane RJ, Zhong E, Parisi M, Ciszewski J, Kasper RT, Deane R (2013) Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. PNAS. doi:10.1073/pnas. 1302212110 Smith MA, Perry G, Richey PL et al (1996) Oxidative damage in Alzheimer’s. Nature 382:120–21 Strittmatter WJ (2012) Old drug, new hope for Alzheimer’s disease. Science 335:1447. doi:10.1126/science.1220725 Südhof TC (2012) The presynaptic active zone. Neuron 75:11–25 Südhof TC (2013) Neurotransmitter release : the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690 Terry RD, Masliah E, Salmon DP et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–80 Tesseur I, Lo AC, Roberfroid A et al (2013) Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924. doi:10.1126/science.1233937 Tokutake T, Kasuga K, Yajima R, Sekine Y, Tezuka T, Nishizawa M, Ikeuchi T (2012) Hyperphosphorylation of tau induced by naturally secreted amyloid-β at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3β signaling pathway. J Biol Chem 287:35222–35233. 561 Toledo EM, Inestrosa NC (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry 15:272–285 Veeraraghavalu K, Zhang C, Miller S et al (2013) Comment on “ApoEdirected therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340:924. doi:10.1126/science.1235505 Watson GS, Craft S (2003) The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs 17:27–45 Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J, Jiang X, Wang Y, Chen YE (2010) Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci 30:6398–6408 Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201 Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimmer’s disease and other disorders. Nat Rev Neurosci 12: 723–38 Zolezzi JM, Inestrosa NC (2013) Peroxisome proliferator-activated receptors and Alzheimer’s disease: hitting the blood-brain barrier. Mol Neurobiol. doi:10.1007/s12035-013-8435-5 Zolezzi JM, Silva-Alvarez C, Ordenes D et al (2013) Peroxisome proliferator-activated receptor (PPAR) γ and PPARα agonists modulate mitochondrial fusion-fission dynamics: relevance to reactive oxygen species (ROS)-related neurodegenerative disorders? PLoS One 8:e64019. doi:10.1371/journal.pone.0064019.
(10)
Figure
Documento similar