EDGAR SEBASTIAN BOLIVAR BARBOSA
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN BIOLOGÍA BOGOTÁ D.C.
EDGAR SEBASTIAN BOLIVAR BARBOSA
Proyecto de Trabajo de Grado para optar por el Título de Licenciado en Biología Modalidad: Investigación-Innovación
Director
LUIS FRANCISCO BECERRA GALINDO
UNIVERSIDAD DISTRITAL FRANCISCO JOSE DE CALDAS Director externo
VINICIUS MARACAJA-COUTINHO
CENTRO DE GENÓMICA DE LA UNIVERSIDAD MAYOR DE CHILE
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN BIOLOGÍA BOGOTÁ D.C.
LA UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
En primer lugar quiero agradecer a mi familia por el apoyo incondicional durante toda la carrera, especialmente en la culminación de mi tesis.
Al Profesor Luis Francisco Becerra por darme la oportunidad de ingresar al grupo de investigación BIOMOL e introducirme a la Bioinformática.
Agradezco al Principal Investigador del Grupo LIB Laboratory of Integrative Bioinformatics profesor del Centro de Genómica y Bioinformática de la Facultad de Ciencias de la Universidad Mayor Vinicius Maracaja-Coutinho y al ingeniero Bioinformático del grupo LIB José Carlos Caris-Maldonado de la Universidad de Talca, por sus enseñanzas y colaboración en el transcurso y el desarrollo de la tesis.
CONTENIDO
LISTA DE FIGURAS ... 7
LISTA DE TABLAS ... 9
ANEXOS ... 10
RESUMEN ... 11
1. INTRODUCCIÓN ... 12
2. DEFINICIÓN DEL PROBLEMA ... 13
3. ANTECEDENTES ... 14
5. HIPÓTESIS ... 17
5.1. Hipótesis de co-expresión de genes codificantes y no codificantes en tejidos de Cannabis sativa ... 17
6. OBJETIVOS ... 18
6.1. OBJETIVO GENERAL ... 18
6.2. OBJETIVO ESPECÍFICOS ... 18
7. MARCO TEÓRICO ... 19
7.1.1. Clasificación Taxonómica de la Cannabis sativa. ... 20
7.1.2. Morfología de la Cannabis sativa ... 20
7.2.1. Políticas legales de cannabis en Colombia ... 21
7.2.2. Bioinformática en Colombia ... 22
7.3. CANNABINOIDES Y SU ROL EN ENFERMEDADES ... 22
7.3.1. Biosíntesis de los cannabinoides ... 25
7.4. TRANSCRIPTOMAS y RNA NO CODIFICANTE (ncRNAs) ... 26
7.4.1. Redes de co-expresión en genes y asignación funcional. ... 30
7.4.2. Experimentos RNA-seq... 31
7.5. DNA SECUENCIADO DEL GENOMA DE Cannabis sativa ... 31
7.5.1. Secuenciación genómica y su importancia ... 32
7.5.2. Genoma de Cannabis sativa y sus posibilidades. ... 33
7.6. DATOS PÚBLICOS Cannabis sativa ... 34
7.6.1. Navegador del genoma del cannabis (The Cannabis Genome Browser) ... 34
7.6.2. Extracción de datos de RNA-seq de Cannabis sativa ... 35
7.6.3. Control de calidad de secuencias y preparación de lecturas. ... 36
7.6.4.1. Predicción de genes ... 37
7.6.4.2. Ensamble de Transcriptomas. ... 38
7.6.5. Identificación de genes diferencialmente expresados (DEGs)... 38
7.6.6. Expresión de la biosíntesis de Cannabinoides en tejidos de Cannabis sativa. ... 39
7.6.7. Análisis diferencial ... 41
8.1. DELIMITACIÓN Y ÁREA DE ESTUDIO. ... 42
8.2. MINERIA DE GENOMAS DE REFERENCIA DE Cannabis sativa. ... 42
8.3. CONTROL DE CALIDAD FILTRADO DE RNA-seq DE Cannabis sativa ... 42
8.4. EVALUACIÓN Y SELECCIÓN DEL GENOMA DE REFERENCIA ... 44
8.5. RECONSTRUCCIÓN DEL TRANSCRIPTOMA DE REFERENCIA ... 46
8.5.1. Identificación de RNAs no codificantes largos LncRNAs ... 50
9. RESULTADOS ... 52
9.1. MINERIA DE DATOS PÚBLICOS ASOCIADO A LA CANNABIS SATIVA. 52 9.2. CONTROL DE CALIDAD Y TRIMMING DE SECUENCIAS. ... 54
9.3 ELECCIÓN DEL GENOMA DE REFERENCIA UTILIZADO ... 57
9.4 RECONSTRUCCIÓN DEL TRANSCRIPTOMA DE REFERENCIA ... 59
Los ensambles se corrieron por tejido individualmente contra los datos de RNA-seq, Los ensambles de los transcritos se pueden encontrar como ... 59
9.4.3. Identificación de los RNAs largos no codificantes (lncRNAs)... 61
9.4.3.1. Módulos de co-expresión. ... 62
10. CONCLUSIONES ... 69
11. RECOMENDACIONES ... 70
BIBLIOGRAFÍA ... 71
LISTA DE FIGURAS
Figura 1. Morfología Cannabis sativa ... 21 Figura 2. Ruta de los cannabinoides ... 25 Figura 3 Biosíntesis de cannabis (20) ... 26 Figura 4. Síntesis de proteínas. Imagen tomada de Instituto de Neurología de Buenos Aires (http://neurologiainba.com.ar) ... 27 Figura 5 Mecanismos de acción de LncRNAs en la célula (1) Los ncRNAs actúan sobre complejos de proteínas que están involucrados en la remodelación de la cromatina que conduce a la regulación local de la expresión génica; (2) Los ncRNAs se unen a la región promotora de genes e inhiben la transcripción de genes; (3) Los ncRNAs interactúan con factores de transcripción y actúan como co-represores o co-activadores de la transcripción (modulación de la actividad de la proteína). Mecanismos post-transcripcionales: (4) Los ncRNAs pueden actuar en el empalme o algunos interfieren con el proceso de empalme; (5) Los lncRNAs se procesan en miRNAs maduros (mediante las etapas de procesamiento que implican Drosha y Dicer) y (5a) inhiben la traducción del mRNA, o (5b) degradan el ARNm objetivo por RISC; (6) los ncRNAs pueden actuar como RNAs de interferencia pequeña endógena (siRNA) y puede ser escindido por RISC. Mecanismo post-traduccional: (7) Los lncRNAs pueden interactuar con proteínas diana que alteran la localización de proteínas y el papel organizativo en la célula. Biosíntesis de ncRNAs: (A) lncRNAs pueden ser generados por transcripción independiente, o (B) ncRNAs pueden ser generados a partir de intrones empalmados de genes codificadores de proteínas (intron derivado de miRNAs) (25). Figura y descripción tomada de Integrative bioinformatics of long non-coding RNAs: insights into function based on co-expression networks and in silico characterization ... 30 Figura 6. El uso de Cuffquant facilita la difusión de la carga de cálculo para lotes de muestras en varios equipos. Si no desea realizar análisis de expresión diferencial, puede ejecutar Cuffnorm en lugar de Cuffdiff. Cuffnorm produce tablas simples de valores de expresión que se pueden ver en R (por ejemplo) para agrupar muestras y realizar otro análisis de seguimiento. . Tomado de “Cufflinks Transcriptome assembly and differential expression analysis for
muestra el porcentaje no mapeado (Unmapped) y en Azul el porcentaje que ha sido
mapeado (mapped). ... 59
Figura 12. Parte de lectura de transcripto clout-SRR306861.sra.fastq-chemdog ... 60
Figura 13. Transcritos filtrados con AWK. VALORES ... 61
Figura 14 Expresión de lncRNAs y cRNAs en tejidos de Cannabis sativa. De azul celeste RNAs codificantes y de naranja se expresan los RNAs largos no codificantes. ... 62
Figura 15. Módulos de Co-expresión entre lncRNAs y proteínas codificantes ... 63
Figura 16 Correlación de expresión LOX1_finola Vs lncRNAs co-expresados ... 64
Figura 17 Correlación de expresión HLP1_finola Vs lncRNAs co-expresados ... 64
Figura 18 Correlación de expresión IPP_finola Vs lncRNAs co-expresados ... 65
Figura 19 Correlación de expresión OLS_finola Vs lncRNAs co-expresados ... 65
LISTA DE TABLAS
Tabla 1 Clasificación Taxonómica de la Cannabis sativa. ... 20
Tabla 2. Genes codificantes en diferentes organismos (Plan, 2014). Tomado de Integrative bioinformatics of long non-coding RNAs: insights into function based on co-expression networks and in silico characterization Eukaryotic. ... 29
Tabla 3 Genes involucrados en la síntesis de los Cannabinoides (“The Cannabis Genome Browser,” n.d.) ... 35
Tabla 4. Enlace de descarga de lecturas de cannabis sativa. ... 52
Tabla 5. Datos fastq ... 54
Tabla 6. Trimming de secuencia de referencia A. thaliana ... 56
Tabla 7. Predicción de genes con Augustos usando las proteínas y genes codificantes de Arabidopsis thaliana como referencia ... 57
Tabla 8. Genomas mapeados (Mapped) Vs no mapeados (Unmapped). ... 58
ANEXOS
Anexo 1. Listado de Cannabinoides extraída de “Chemistry and Analysis of
Phytocannabinoids and Other Cannabis Constituents”(Brenneisen, n.d.)... 75
Anexo 2. Sample description VS Mapping Purple Kush ... 79
Anexo 3. Sample description VS Mapping Cannatonic ... 80
Anexo 4. Sample description VS Mapping Chemdawg91 ... 82
Anexo 5. Sample description VS Mapping Finola1 ... 83
Anexo 6. Sample description VS Mapping LA Confidencial ... 84
Anexo 7. Sample description VS Mapping Pineapple Bannana Bubba Kush ... 86
Anexo 8 Correlación de expresión OLS_finola Vs lncRNAs co-expresados ... 87
Anexo 9 Correlación de expresión HDR_finola Vs lncRNAs co-expresados ... 88
Anexo 10 Correlación de expresión IPP_finola Vs lncRNAs co-expresados ... 89
Anexo 11 Correlación de expresión HLP1_finola Vs lncRNAs co-expresados ... 89
RESUMEN
La Cannabis sativa ha sido utilizada como fuente de fibra, aceite y proteínas y tiene una gran importancia médica para tratamiento de algunas enfermedades y síntomas como nauseas, ansiedad, como analgésico etc. Estos compuestos se encuentran en los cannabinoides como el THC y el CBD. El estudio de datos genómicos del cannabis ha sido entorpecido por leyes federales de los Estados Unidos con la Marihuana Tax ACt del año 1937 criminalizando la posesión o transferencia de cannabis. En 1953 Francis Crick y James Watson realizaron un gran avance para la biología molecular, El descubrimiento de la doble hélice del ADN. Esto genero estudios sobre la caracterización y función de los genes en tejidos y expresión de proteínas como también en estudios filogenéticos. La Cannabis sativa es una de las plantas domesticas con menores estudios y desarrollos genómicos a comparación del arroz, el trigo y la soya. Por el interés en sus propiedades psicoactivas. El cultivo selectivo de esta ha producido plantas de cannabis para usos específicos, incluyendo cepas de marihuana y cultivos de cáñamo de alta potencia para la producción de semillas y fibras.
Se realizó una minería para descargar los datasets del genoma de referencia de la Cannabis sativa, acudiendo a las bases de datos públicas de FTP de NCBI para el genoma de referencia de cannabis. Para los genomas de cannabis como Cannatonic, LA Confidential, Chemdawg 91, Purple Kush, Pineaple Banana Buba Kush se recurrió al SRA de NCBI y para la variedad de Finola se recurrió a la base de datos The Cannabis Genome Browser. El mapeo que se realizó de estos genomas con la secuenciación de referencia, mostro que la variedad Chemdawg91 presenta un porcentaje mayor de mapeo comparado con los otros genomas. Definiendo así nuestro genoma de referencia. Luego se realizó un ensamble de los transcritos de cada muestra contra el genoma de Chemdawg91 con cufflinks del cual saldrán transcriptomas finales en un archivo merged.gft los cuales fueron cuantificados para poder calcular la expresión de cada transcrito construido. Se identificaron los RNAs codificantes y lncRNAs, en el transcriptoma, el cual evidencio una menos dispersión de lncRNAs en los cuales se puede realizar un énfasis para realizar estudios sobre ellos. Se realizó una identificación de la co-expresión de RNAs codificantes y lncRNAs mostrando una relación con genes involucrados en la ruta de la Biosíntesis de Cannabinoides.
Este trabajo de investigación permite el estudio y análisis funcional del genoma de Cannabis sativa y poder realizar posteriores avances investigativos en el área terapéutica y medicinal, ya que la expresión relativa, debido a las distintas funciones metabólicas, las síntesis de las rutas metabólicas en los transcritos de Cannabis sativa es diferente. Debido a la diferenciación celular de tejidos
1. INTRODUCCIÓN
Desde los años de los 60’s se han realizado estudios genómicos y de transcriptomas para identificar la funcionalidad o historia evolutiva mediante el estudio de los RNAs codificantes como los RNAs no codificantes (ncRNAs) en células eucariotas (1). Algunos autores se refieren a los ncRNAs como “ruido” en la secuenciación de un tejido de una especie y que este puede ser desechado. Por otro lado hay autores que encontraron una expresión de estos ncRNAs están involucrados en la regulación transcripcional y post-transcripcional (2). Por estos motivos se han realizado estudios genómicos de plantas con gran importancia médica como alimenticia y de construcción como es la Cannabis sativa.
La Cannabis sativa es una planta con un largo historial de cultivo, como su variedad de usos. Es económicamente importante, ya que puede ser fuente de fibras, como también de semillas con gran variedad de nutrientes, componentes medicinales (nauseas, ansiedad, anti inflamatoria…) y considerada como una droga psicoactiva. El estudio genómico de la planta ha sido entorpecida, ya que para el año 1953 hubo un gran avance en el campo de la biología molecular que fue el descubrimiento de la doble hélice de James Watson and Francis Crick y la por políticas como la de Marihuana Tax Act de 1937, criminaliza la posesión o comercialización de cannabis bajo leyes federales de Estados Unidos. Por esta razón el conocimiento genético de la planta y su optimización es limitado a comparación del maíz, la soya, el arroz y el trigo (3).
En octubre de 2016 Phylos Bioscience publica un dataset de aproximadamente unas 850 variedades o cepas de cannabis por medio del Open Cannabis Project, con la colaboración de otros datasets genómicos disponibles en Medicinal Genomics, la Universidad Estatal de Michigan, NCBI, Sunrise Medicinal, la Universidad de Calgary, la Universidad de Toronto y la Academia de Ciencias Agrícolas de Yunnan, la cantidad de información genética excede las 1000 muestras de variedades únicas de cannabis. Estas son de libre acceso y están publicadas en BigQuery genomics cannabis dataset (3).
2. DEFINICIÓN DEL PROBLEMA
La Cannabis sativa es una planta con un largo historial de estudio, ya que hay una gran cantidad de literatura que resalta su importancia médica, por la fibra que es utilizada para la construcción y por sus compuestos segundarios como flavonoides, cannabinoides alcaloides, ligninanamidas y amidas fenólicas.
Debido a la discriminación de la marihuana en épocas donde hubo grandes avances científicos, como desarrollos en biología molecular, no se pudo realizar un estudio genético apropiado. Pero el año 2016, salen a la luz publicaciones en las cuales se muestran datasets de varias cepas con información genética secuenciada de la Cannabis sativa, El estudio del transcriptoma de la Cannabis sativa puede darnos evidencia de RNAs que pueden ser codificantes, los cuales ya tienen una función predeterminada en la expresión de un tejido o una proteína, y las no codificantes que pueden tener una función reguladora de la estructura de una cromosoma, expresión de una tejido o reguladora de genes asociadas a los cannabinoides.
Por consiguiente este trabajo de investigación abarca la siguiente pregunta:
3. ANTECEDENTES
Para la realización de este trabajo se tomaron como referencia los siguientes artículos internacionales y nacionales, relacionados con estudios genómicos de transcriptomas y análisis bioinformático.
En el año 2006 la revista Journal of Ethnopharmacology publica el artículo “Cannabinoids in medicine: A review of their therapeutic potential” por el autor .Ben Amar, el cual propone el uso de los cannabinoides en el área de la medicina para realizar tratamiento alternativo sobre el uso de cannabinoides para el tratamiento de enfermedades crónicas.
En el año 2013 la revista Methods in Molecular Biology publicó el artículo Genome-Wide Association Studies and Genomic Prediction por los autores Best, G., Unbiased, L., Clark, S. A., & Werf, J. Van Der. El cual sirvió como marco teórico para entender un poco el proceso de predicción de genes y la asociación que tienen estos genes entre sí.
En el año 2017 la revista PLoS ONE publicó el artículo “Terpene synthases from Cannabis sativa” de los autores Citation: Booth JK, Page JE, Bohlmann J, el cual habla los genes que están incluidos en la síntesis de los Cannabinoides en los tricomas de la cannabis sativa, el cual es abundante en la superficie de la inflorescencia de las hembras. Se realizó un análisis del transcriptoma de los tricomas de la variedad Finola y se revelaron las biosíntesis de los terpenos en todos los estados del cannabis.
Se hizo una revisión del manual del programa Bowtie2 del año 2016 con el cual se realizaron los mapeos de las secuencias y aplicar la ejecución de los comandos específicos.
La revista Marijuana and the Cannabinoids publica el artículo “Chemistry and Analysis of Phytocannabinoids and Other Cannabis Constituents”. El cual habla de los componentes químicos del Cannabis sativa, En el trabajo se crea un listado de aproximadamente 60 de los componentes químicos de las cannabinoides explicando la ruta para la síntesis de los cannabinoides y sus funciones médicas.
Se hizo una revisión del manual del programa TopHat2 publicado en la página de CCB (Center for computational Biology) este programa se utilizó para realizar los mapeos de los 6 genomas de la cannabis para determinar el genoma de referencia se acudió a él para la consulta para la aplicación y la ejecución de los comandos específicos.
transcritos de RNAs cortos y largos en la regulación de la expresión de un gen y expresión epigeneticas. Y cómo estos pueden tener diferentes roles como en la señalización, en la regulación de expresión de genes cis y trans y como modificadores de cromatina.
En el año 2012 la revista A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proceedings of the National Academy of Sciences
En Octubre del año 2016 Phylos Bioscience lanzó al público un dataset de aproximada 1000 genomas de Cannabis por medio de Open Cannabis Project, en combinación con otros datos genómicos disponibles en Medicinal Genomics, la Universidad Estatal de Michigan (Michigan State University), NCBI (Biotechnology Information’s Sequence Read Archive), Sunrise Medicinal, la Universidad de Calgary (University of Calgary), la Universidad de Toronto (University of Toronto) y la Academia de Ciencias Agrícolas de Yunnan (Yunnan Academy of Agricultural Sciences) tomadas de variedades únicas de Cannabis. Esto con el fin de incrementar el aporte científico de la Cannabis debido a que su historia entorpece el estudio de su genoma.
En el año 2013 la revista Genome Biology publico el artículo “TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions”. De los autores Daehwan Kim, Geo Perte, Cole Trapnell, Harold Pimentel, Ryan Kelley, and Steven L Salzberg. Este artículo habla de los usos que tiene TopHat2, el cual además de realizar alineaciones de secuenciación de novo, puede alinear lecturas a través de rupturas de fusión, que pueden ocurrir después de translocaciones genomicas. También produce alineaciones más precisas, en pseudogenomas y en genomas repetitivos.
En el año 2015 la revista Frontiers in Genetics publicó el artículo “Non-coding RNA: What is functional and what is junk?” de los autores Alexander F. Palazzo* and Eliza S. Lee. Este artículo habla sobre la presencia de RNA que no codifican y son considerados como ruido o adaptadores, resultado de la secuenciación de un tejido.
En el año 2012 la revista RNA Biology publica el artículo “Non-coding transcription characterization and annotation: A guide and web resource for non-coding RNA databases” de los autores Alexandre Rossi Paschoal, Vinicius Maracaja-Coutinho, João Carlos Setubal, Zilá Luz Paulino Simões, Sergio Verjovski-Almeida & Alan Mitchell Durham. Este artículo habla sobre la cantidad de transcritos de RNA no codificantes (ncRNA) y como gracias a las tecnologías de alta secuenciación acompañada de acercamientos bioinformáticos ha producido un incremento de estos datos y como ha salido a la luz la función del rol de los ncRNAs en procesos biológicos. Debido a esto ellos proponen una manera de cómo clasificar esta database y a realizar un sondeo rápido de la información que se necesita.
5. HIPÓTESIS
5.1. Hipótesis de co-expresión de genes codificantes y no codificantes en tejidos de Cannabis sativa
Hipótesis nula (Ho): Los RNAs largos no codificantes (lncRNAs) no tienen una co- expresión tejido especifica con las proteínas codificantes.
6. OBJETIVOS 6.1. OBJETIVO GENERAL
Generar un transcriptoma de referencia de la Cannabis sativa a partir de datasets públicos de secuenciación masiva de última generación.
6.2. OBJETIVO ESPECÍFICOS
Realizar un levantado de los datasets públicos de 6 genomas disponibles de la Cannabis sativa.
Evaluar los datasets de RNAseq públicos para la selección de un genoma de referencia para ser utilizado
Identificar los RNAs codificantes y los RNAs largos no codificantes (LncRNAs) de proteínas en el transcriptoma de Cannabis sativa.
7. MARCO TEÓRICO 7.1. GENERALIDADES DE LA Cannabis sativa
La Cannabis sativa es una planta que pertenece a la familia de las Cannabáceas y presenta una germinación anual. Esta planta tiene un largo historial de cultivo como también una gran variedad de su uso. La Cannabis sativa ha sido utilizada por años como fuente de fibra y aceite, con semillas ricas en proteína y por sus propiedades medicinales y psicoactivas (6). El Cannabis posee una gran cantidad de diversidad fenotípica, la cual se puede observar en la forma de la hoja, forma de la planta, el tamaño de la semilla, la calidad de la fibra, la ramificación, la altura, la fitoquímica, la fenología y la ecología.(7). Tiene un gran variabilidad en cuanto a la estrategia reproductiva con algunas poblaciones manteniendo el tipo ancestral, monóico, mientras que otras mantienen una estrategia dirigida, dioica y/o mixta. Algunas variaciones de sus rasgos están relacionadas con los múltiples usos domesticados. El número de especies aún está en debate, ya que algunos dicen que está compuesta por tres especies (Cannabis sativa, C. indica, C. ruderalis), otros autores dicen que está compuesta por una sola especie (Cannabis sativa L.) con un alta variación. Actualmente, según la mayoría de las convenciones científicas, El Cannabis está clasificado como una especie, Cannabis Sativa. Que fue originalmente descrita por Linnaeus (1753) (7).
El cáñamo es usado generalmente para la extracción de fibra o aceite. Estas plantas se describen como, altas, delgadas y carentes de producción de terpenoides o cannabinoides. La Cannabis sativa produce más de 421 compuestos químicos, incluyendo alrededor de 80 compuestos terpeno-fenólicos, llamados fitocanabinoides que no han sido detectados en ninguna otra planta (8). Las únicas propiedades medicinales de esta planta, está relacionada con la presencia de cannabinoides, debido a un grupo de más de 100 productos naturales que se pueden acumular en la flores femeninas de los brotes de Cannabis sativa siendo el ∆9 -Tetrahydrocannabinol (THC) el principal cannabinoide responsable del efecto psicoactivo (6).
7.1.1. Clasificación Taxonómica de la Cannabis sativa.
La Cannabis sativa está clasificada dentro de la familia de las Cannabaceae, (Tabla 1.).
Tabla 1 Clasificación Taxonómica de la Cannabis sativa.
Reino Plantae
Subreino Viridiplantae Infrareino Streptophyta Súperdivisión Embryophyta División Tracheophyta Subdivisión Spermatophyta Clase Magnoliopsida Súperorden Rosanae
Orden Rosales
Familia Cannabaceae Genero Cannabis L. Especies Cannabis sativa L. 7.1.2. Morfología de la Cannabis sativa
Figura 1. Morfología Cannabis sativa 7.2.1. Políticas legales de cannabis en Colombia
se prohíbe el consumo de sustancias psicoactivas y su porte, reconociendo por otra parte el porte de la dosis personal (13). En el año 2011 se elimina del Código Penal ley 1453, el porte de sustancias psicoactivas, reconociendo el porte de sustancias psicoactivas, pero prohibiendo su venta(12).
7.2.2. Bioinformática en Colombia
El poco progreso de la Bioinformática en Colombia se debe al bajo desarrollo de un programa integral de producción académica. Debido a esto no existe promoción de proyectos de investigación dirigidos que a la resolución de problemas propios y que involucren un componente de bioinformática directo y bien planificado y que generen iniciativas de proyectos de investigación en el área que sean tomados en cuenta por comités evaluadores que objetivamente valoren el impacto científico de dichas propuestas. La Producción científica-Bioinformática es deficiente, en número, comparada con nuestros referentes regionales inmediatos, Brasil, México y Argentina. Tales datos fueron obtenidos mediante una búsqueda de publicaciones relevantes en revistas científicas de campo, como los son: Bioinformatics, PLOS Computacional Biology, BMC Bioinformatics, BMC Genomics y Journal of Computacional Biology. Dichas revistas gozan de los más altos índices de impacto en este campo de acuerdo con el Journal Citation Reports 2007. (Benitez-Paez A, Cardenas-Brito S, 2010).
Haciendo una búsqueda se pudieron encontrar algunos grupos de investigación que realizan estudios de componente bioinformático que son pocos los grupos dedicados a ellos y aún menos los que han podido proyectar su trabajo a nivel internacional. Entre esos grupo se pueden mencionar al Grupo de Parasitología Molecular (GEPAMOL) de la Universidad del Quindío, el Centro de Bioinformática del Instituto de Biotecnología de la Universidad Nacional de Colombia, el Grupo de Investigación en Bioquímica Computacional de la Pontificia Universidad Javeriana y el Grupo de Análisis Bioinformático (GABi) del Centro de Investigación y Desarrollo en Biotecnología. Esto demuestra un gran campo de investigación computacional en Colombia. (Benitez-Paez A, Cardenas-Brito S, 2010). Además de la producción científica de estos grupos de investigación, GENICAFE y su proyecto del genoma del café tiene como objetivo principal un extenso análisis de genómica funcional y estructural del café colombiano
7.3. CANNABINOIDES Y SU ROL EN ENFERMEDADES
Hay cannabinoides que no son psicoactivos como el cannabidiol (CBD), cannabichromene (CBC) y el delta-9-tetrahydrocannabivarin (THCV) los cuales poseen una actividad farmacológica diversa que se encuentran con mayor presencia en algunas variedades o cepas de la Cannabis sativa (15). Entre los compuestos psicoactivos de la planta Cannabis sativa están presentes los cannabinoides, los cuales contienen aproximadamente 400 compuestos químicos diferentes, de los cuales 60 conforman el grupo de los cannabinoides. La estructura de los cannabinoides es carboxílica la cual presenta 21 carbonos y estos están formados por tres anillos (ciclohexano, tetrahidropirano y benceno) (13).
La Cannabis tiene productos con una enorme cantidad de químicos. El término “cannabinoides” representa un grupo de C21 compuestos terpenofenólicos que hasta son encontrados en Cannabis sativa (16). La cantidad de compuestos de cannabinoides y sus características farmacológicas se encuentran en el siguen listado en la Tabla 2.
El Cannabigerol (CBG) fue el primer cannabinoide identificado y es el precursor del ácido cannabigerolico (CBGA), el cual fue el primer cannabinoide biogenético formado en la planta (16). Este cannabinoide no es psicoactivo, tiene componentes antibacteriales, retardando o eliminando el crecimiento bacterial, reduce la inflamación, inhibe el crecimiento celular en células tumorales o cancerosas y promueve el crecimiento óseo (17) .
El cannabichromene (CBC) presenta C5 análogos su papel puede ser como efecto antiinflamatorio y antiviral y este contribuye a los efectos analgésicos del Cannabis estudios demuestran que el CBC ayuda a promover la neurogenesis (17).
El Cannabidiol (CBC) fue aislado en el año 1940 pero su estructura fue rectificada en 1963 por Mechoulam y Shvo (16), se han descrito 7 cannabinoides del tipo CBD.
El ácido Cannabidiolico (CBDA) y el CBD son los cannabinoides más abundantes en el cannabis de tipo industrial conocido como cáñamo (16). En el año 1955 el primer ácido cannabinoide descubierto fue el CBDA. El CBD tienen un potencial medico increíble, esto puede ser cierto si la proporción de CBD contra THC se aplica para tratar una condición particular (17).
concentración, la coordinación la percepción sensorial y temporal, el apetito y muchas funciones importantes (17). Los efectos secundarios leves de las dosis más grandes de THC pueden ser, ansiedad, exaltación, ardor en los ojos, boca seca, temblores, aumento de la frecuencia cardiaca, falta de la memoria de corto plazo, el consumo de THC en corto tiempo puede intensificar y alterar los efectos (17).
El Delta-8-tetrahydrocannabinol (∆8-THC) es el precursor del ácido ∆8-THCA considerados como mecanismos en la síntesis de THC y THCA. La posición de doble enlace de 8,9 es termodinámicamente más estable que la posición 9,10, este cannabinoide es menos activo que el THC casi un 20% (16).
El Cannabicyclol (CBL), es conocido por ser mecanismo generador de calor de CBC (16). El Cannabielsoin (CBE) son mecanismos formados por el CBD. El Cannabinol (CBN) y Cannabinodiol (CBND) son mecanismos oxidativos del THC y el CBD, su producción de cannabinoides depende del tiempo y las condiciones de almacenamiento (16). El Cannabitriol (CBT) es caracterizado por la adición de OH. Los cannabinoides Misceláneos se caracterizan por tener una estructura inusual (16).
Figura 2. Ruta de los cannabinoides 7.3.1. Biosíntesis de los cannabinoides.
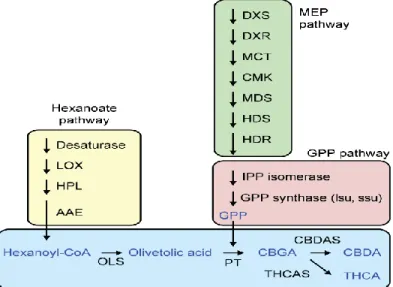
Las propiedades Medicinales del cannabis son altamente valoradas ya que sus cannabinoides son altamente activos. Estos metabolitos terpenofenólicos se encuentran únicamente en el cannabis y están principalmente en la resina producida por los tricomas glandulares de las inflorescencias de pistilo (19). La síntesis de los Cannabinoides (Figura3) Delta-9-tetrahydrocannabinol (THC) y ácido cannabidiolico (CBDA), también esta incluye los precursores de isoprenoides a través del hexanoato MEP (Vía plastidial de metileritritol) y GPP (Geranil difosfato) (6) (19).
sesquiterpenos sintasas (sesqui-TPS), respectivamente, que diversifican estos precursores en un gran número de diferentes mono- y sesquiterpenos (19).
El hexanoato surge por medio de la degradación de ácidos grasos, implicando desaturasa, lipoxigenasa (LOX) e hidroperóxido liasa (HPL). La activación de hexanoato mediante una enzima activadora de acilo (AAE) produce hexanoil-CoA, que es el sustrato para la enzima poliquétido sintasa que forma ácido olivetólico (OLS). La cadena lateral prenyl se origina en la vía MEP, que proporciona sustancias para la síntesis de GPP, y es añadido por una preniltransferasa aromática (PT). Los pasos finales son catalizados por las oxidociclasas THCAS y CBDAS (6).
Figura 3 Biosíntesis de cannabis (20) 7.4. TRANSCRIPTOMAS y RNA NO CODIFICANTE (ncRNAs)
Los transcriptomas que son una construcción de genes presentes en una célula, donde vamos a encontrar infinidad de tipos de ARN, mensajero el cual es pieza clave para la síntesis de proteínas. El proceso de transcripción es el siguiente:
El mRNA se transcribe a partir de genes.
Los transcritos de mRNA son entregados a ribosas ubicadas en el citoplasma de la célula.
Los ribosomas Traducen la secuencia en el mRNA y ensamblan aminoácidos.
transcriptoma se podrá determinar donde hay una activación de los genes y tejidos de un organismo (21). Al recopilar y comparar transcriptomas de diferentes tipos de celular, se puede tener una mayor comprensión de la constitución de un tipo de célula en específico. La función de la mayoría de los genes es desconocida. La búsqueda del transcriptoma de referencia puede permitir visualizar una lista de tejidos donde se expresa un gen, encontrando una posible función. Ejemplo: Si la bases de datos de transcriptoma muestra que los niveles de expresión de una gen del que no se conoce su función, son muy altos en células cancerosas que en células sanas es posible que la función del gen pueda estar en el crecimiento celular; Un gen desconocido se expresa en el tejido graso pero no en el tejido musculas o tejido ose, es posible que la función de este gen tenga que ver con almacenamiento de grasa o el metabolismo (21).
Figura 4. Síntesis de proteínas. Imagen tomada de Instituto de Neurología de Buenos Aires (http://neurologiainba.com.ar)
Las proteínas codificantes, no son las únicas unidades que están en nuestro genoma, también existe un buen porcentaje de otro tipo de moléculas que son llamadas RNAs no codificantes (ncRNAs) (22). La función de la mayoría de ncRNAs no es muy clara, pero se ha indicado que estos están involucrados en papeles como: la regulación de la expresión de un gen, el control de la estructura de la cromatina y de regulación post-transcripcional. Y esta puede estar durante la diferenciación celular del desarrollo de un tejido.
sugieren que los ncRNAs son tan importantes como las proteínas que están presentes en los procesos celulares. También se ha descubierto que los ncRNAs están involucrados en la regulación de la estructura tridimensional del cromosoma X, permitiendo acercar regiones del genoma, como lo hacen las proteínas que codifican conocidas como Xist (22). A partir de estos descubrimientos el número de ncRNAs ha aumentado al igual que el número de genes putativos (23).
Actualmente, para definir las familias específicas de ncRNAs se toma el plegamiento molecular estructural, suponiendo que la conformación estructural tiene una función similar en los procesos celulares. Al secuenciar, la similitud se utiliza para clasificar nuevas formas de ncRNAs, limitando la eficacia de la caracterización de las secuencias más divergentes. Para reflejar la cantidad de datos específicos y la complejidad para detectar datos de ncRNAs cortos y largos es necesario el uso de tecnologías de expresión de alto rendimiento. Se tomaron datos en los cuales secuencias menores que 200 nt se les tomara como RNAs cortos y RNAs largos como secuencias con un número mayor de 200 nt (24)
Al realizar estudios de alto rendimiento (high-throughput), se puede detectar en la complejidad de las células eucariotas, la presencia de estos ncRNAs. Una de las clases son los RNAs largos no codificantes (lncRNAs). Son moléculas de RNA de más de 200 bases de longitud, se creía que los lncRNAs era “ruido” de la transcripción. Aunque se podría decir que los lncRNAs están involucrados en funciones reguladoras en organismos complejos por el hecho de que los niveles de lncRNAs están involucrados en la regulación transcripcional y post-transcripcional (2). La transcripción de muchos genes en una célula eucariota son silenciados, pero, en muchos casos estos genes son transcritos en mRNA, el cual nunca es traducido. Varios mecanismos post-transcripcional están en posición de añadir un nivel de control sobre los sistemas que regulan la expresión génica de la célula eucariota, estos mecanismos son el resultado de secuencias de RNAs pequeños que no codifican (1).
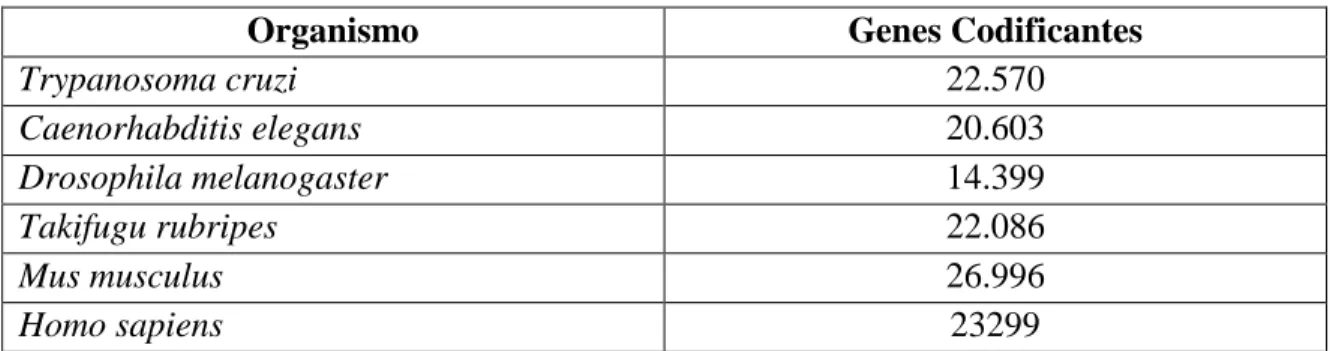
Tabla 2. Genes codificantes en diferentes organismos (25). Tomado de Integrative bioinformatics of long non-coding RNAs: insights into function based on co-expression
networks and in silico characterization Eukaryotic.
Organismo Genes Codificantes
Trypanosoma cruzi 22.570
Caenorhabditis elegans 20.603
Drosophila melanogaster 14.399
Takifugu rubripes 22.086
Mus musculus 26.996
Homo sapiens 23299
Este determina el grado de complejidad entre los organismos y que este no puede correlacionarse solo con el número de genes que codifican las proteínas. Evidenciando también una complejidad a lo largo de la evolución, ya que estaría directamente asociada con la expansión del contenido no codificante en organismo con un genoma complejo. El crecimiento en el número de regiones codificantes en comparación con las regiones no codificantes evidencia que hay una expansión del DNA no codificante y es mayor a la de las regiones codificantes en la historia evolutiva de las especies de diversas ramas evolutivas (25).
Figura 5 Mecanismos de acción de LncRNAs en la célula (1) Los ncRNAs actúan sobre complejos de proteínas que están involucrados en la remodelación de la cromatina que conduce a la regulación local de la expresión génica; (2) Los ncRNAs se unen a la región promotora de genes e inhiben la transcripción de genes; (3) Los ncRNAs interactúan con factores de transcripción y actúan como co-represores o co-activadores de la transcripción (modulación de la actividad de la proteína). Mecanismos post-transcripcionales: (4) Los ncRNAs pueden actuar en el empalme o algunos interfieren con el proceso de empalme; (5) Los lncRNAs se procesan en miRNAs maduros (mediante las etapas de procesamiento que implican Drosha y Dicer) y (5a) inhiben la traducción del mRNA, o (5b) degradan el ARNm objetivo por RISC; (6) los ncRNAs pueden actuar como RNAs de interferencia pequeña endógena (siRNA) y puede ser escindido por RISC. Mecanismo post-traduccional: (7) Los lncRNAs pueden interactuar con proteínas diana que alteran la localización de proteínas y el papel organizativo en la célula. Biosíntesis de ncRNAs: (A) lncRNAs pueden ser generados por transcripción independiente, o (B) ncRNAs pueden ser generados a partir de intrones empalmados de genes codificadores de proteínas (intron derivado de miRNAs) (25). Figura y descripción tomada de Integrative bioinformatics of long non-coding RNAs: insights into function based on co-expression networks and in silico characterization
Eukaryotic.
7.4.1. Redes de co-expresión en genes y asignación funcional.
utilizar estos recursos importantes para la anotación y la caracterización detallada de los transcriptomas (25). Los datos usados para este tipo de caracterización son los conjuntos de datos microarrays, los datos de RNA-seq (experimento de secuenciación de RNA), datos de ChIP-seq y CLIP/RIP-seq (ensayos de inmunoprecipitación de DNA y RNA) (25).
Las redes de co-expresión asignan una función a la exploración de datos génicos, esto permite un análisis consistente en la búsqueda de la expresión de diferentes transcripciones en conjunto, con el fin de identificar características particulares detrás de sus patrones de expresión (25). La identificación de los lncRNAs que tienen patrones de expresión están positivamente o negativamente asociados a los genes codificadores de proteínas pertenecientes a clases de ontologías de genes funcionales particulares (25).
7.4.2. Experimentos RNA-seq
Los experimentos de RNA-seq deben ser analizados con algoritmos robustos, eficientes, afortunadamente la comunidad bioinformática ha desarrollado un software para poder desarrollar dicho análisis. Las herramientas para realizar el análisis de RNA-seq tienen generalmente 3 categorías: 1) herramientas para lecturas de alineamiento; 2) Herramientas para ensamble de transcriptomas y anotación del genoma;3) herramientas para los transcriptomas y la cuantificación del gen (4). Para esto se ha desarrollado dos herramientas que cumplen con estas tres funciones y poder visualizar un análisis de los resultados. TopHat (4) el cual hace alineamientos de las lecturas del genoma y descubre los sitios de empalme del transcriptoma. Estos alineamientos se utilizan durante un análisis downstream de varias maneras. Cufflinks usa un mapa contra del genoma para ensamblar las lecturas en las trascripciones (4). Estas herramientas están ganando una amplia aceptación y se han utilizado en una serie de estudios recientes de transcriptomas.
7.5. DNA SECUENCIADO DEL GENOMA DE Cannabis sativa
Archive), Sunrise Medicinal, la Universidad de Calgary (University of Calgary), la Universidad de Toronto (University of Toronto) y la Academia de Ciencias Agrícolas de Yunnan (Yunnan Academy of Agricultural Sciences) disponen de más de 1000 muestras publicas siendo la mayoría tomadas a variedades de Cannabis únicas, estos datos se han reunido en un conjunto de datos publico llamado BigQuery genomics_cannabis dataset (3).
Aplicando estudios estandarizados de Bioinformática se analiza una nueva secuencia de Cannabis vía Google BigQuery, el cual podría ayudarnos a refrescar el entendimiento biológico y evolutivo de la Cannabis sativa. Open Cannabis Project divulga documentos públicos en el dominio en forma de secuencias de DNA, los contribuidores de este proyecto suben los datos en el formato que arrojo el secuenciador, esta secuenciación(3). Los resultados de estos análisis publicados fueron los siguientes:
- Alineaciones de secuencias de todas las muestras en Cannatonic que se pueden usar para crear ensamble de un genoma de calidad confiable de Cannabis
- Detección de variación genética.
Aunque hubo datos públicos sobre el Cannabis, la publicación de datos de parte de Phylos Bioscience sobre gran población de variedades permite comenzar un análisis bioinformático de los datos que están disponibles.
7.5.1. Secuenciación genómica y su importancia
La Secuenciación genómica se puede separar en dos categorías:
a) Secuenciación De novo; Este trata sobre la secuenciación del genoma de una especie que aún no ha sido secuenciada, generando un genoma de referencia (3). El desarrollo de las tecnologías de secuenciación de DNA han generado un avance en cuanto a la investigación genómica, estas tecnologías incluyen un analizador de genoma como Illumina Genome Analyzer, Applied Biosystems SOLiD System y Helicos BioSciences (27). La tecnología de Illumina ha demostrado que es posible la re-secuenciación del genoma humano completo y puede usarse para identificar con exactitud polimorfismos de nucleótidos únicos (SNPs) encontrados en el genoma, trazando una lectura sobre un genoma de referencia conocido.
Si se quiere realizar anotaciones minuciosas de una secuencia como inserciones, deleciones y variaciones estructurales, se realiza un ensamble De novo a cada genoma individual (27).
comparamos contra un genoma, generamos respuestas a posibles preguntas sobre mecanismos biológicos, como: ¿Porque algunos humanos son resistentes a las enfermedades transmisibles, mientras que los otros lo contraen?, ¿Por qué algunas enfermedades son genéticas?, ¿Por qué algunas plantas presentan mejores condiciones que otras, si estas presentan un ambiente de cultivo idéntico? (3). Para resolver esta clase de preguntas, necesitamos tener un conocimiento de la constitución genética de cada individuo y poder saber la relación causal entre organismos, las cuales puede ser, su estado genético, su estado fenotípico, o el contexto del entorno donde viven (3).
7.5.2. Genoma de Cannabis sativa y sus posibilidades.
Teniendo los datos genómicos del Cannabis disponibles, pueden ser posibles realizar avances en cuanto a conocimiento biológico de la planta, más detalle en la descripción y la relación entre variedades biológicas, para lograr esto es necesario realizar estudios entre los cuales se incluye:
a) Producción de un conjunto más detallado del genoma mismo. El ensamble de la variedad Cannatonic es el que produce los resultados genómicos, si se realiza un montaje más detallado es posible tener una mayor precisión de los datos genómicos(3).
b) Perfeccionar las anotaciones del genoma. El género cannabis tiene muchas especies relacionada para las cuales ya se han producido genomas de referencia, estos datos que estén relacionados pueden ser transferidos para ayudar a anotar un genoma de Cannabis, por ejemplo identificar la ubicación de los genes y sugerir cuales son las características funcionales de los genes (3).
c) Asociar observaciones genómicas/fenotípicas pareadas y mejorar los procesos de reproducción y domesticación. El origen en los datasets públicos utilizados son genómicos, el siguiente paso será agregar datos fenotípicos como: el tiempo de floración de cierta variedad, la biomasa, el rendimiento floral, permitiendo la variante promoción genómica a QTLs (Quantitative trait locus)(3). Locus cuya variación alélica está asociada con la diversificación de un carácter cuantitativo, con aquellos caracteres cuantificables que cambian de forma continua, con métodos como el GBLUP (Genomic best linear unbiased prediction) (3) que es una técnica que utiliza relaciones genómicas para evaluar la estimación genético de un individuo (28).
Es importante que para él manejo de las bases de datos, sea necesario el uso de herramientas informáticas con un enfoque al análisis de datos biológicos para llegar a identificar y tomar listado de los genes codificadores y los RNAS largos no codificantes (LncRNAs) de proteínas en el transcriptoma de la Cannabis sativa, ya que estos son de gran importancia pues con esta información se puede determinar la expresión de un gen por medio de los LncRNAS los cuales contribuyen a varios procesos biológicos. Por ejemplo la inactivación de un cromosoma X en los mamíferos, la cual se debe a una estructura tridimensional generada por LncRNAS. Algunos LncRNAS también pueden influir en la modificación de histonas y regular la expresión del sentido en transcripciones de Arabidopsis (29); estos LncRNAS también pueden regular el periodo de foto sensibilidad en la esterilidad del macho a través de la expresión diferencial mediante el aumento de la metilación en la región promotora del arroz (30). La identificación de los LncRNAs de un genoma que ha sido secuenciado por métodos computacionales es difícil debido a la baja expresión o mala conservación de la secuencia (31). Sin embargo la detección de los datos de secuenciación del transcriptoma es posible ya que con frecuencia los LncRNAS son poliadenilados. Los LncRNAS han sido identificados en genomas de mamíferos, incluyendo humanos, ratón y cerdo (Mortazavi A, William BA, McCue K, Schaeffer L, Wold B, 2008) como también se ha identificado en plantas como Arabidopsis, maíz y arroz (32).
7.6. DATOS PÚBLICOS Cannabis sativa
Los datos del genoma del cannabis se limitan a un transcriptoma de los datos. NCBI contiene 12,907 ESTs y 23 conjuntos de datos de RNA secuenciado (RNA-Seq) de lecturas de Illumina que no han sido montadas aun (6). Los archivos de cannabis fueron descargados de National Center For Biotechnology Information’s Sequence Read Archive (NCBI SRA) y The Cannabis Genomic Browser, el cual es una base de datos que contiene el transcriptoma de las cepas de cannabis canSat3 (Purple Kush) y finola1 (Finola) (3).
7.6.1. Navegador del genoma del cannabis (The Cannabis Genome Browser)
Tabla 3 Genes involucrados en la síntesis de los Cannabinoides (20)
DXR 1-desoxi-D-xilulosa 5-fosfato reductoisomerasa
MCT 4-diphosphocytidyl-methylerythritol 2-phosphate synthase CMK 4- diphosphocytidyl-2-C-methyl-D-erythritol kinase MDS 2-C-methyl-D-erythritol 2:4-cyclodiphosphate synthase
HDS 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase HDR 4-hydroxy-3-methylbut-2-enyl diphosphate reductase
IPP isopentenyl pyrophosphat
GPP synthase LSU geranyl diphosphate synthase large subunit GPP synthase SSU geranyl diphosphate synthase small subunit
OLS olivetol synthase PT prenyltransferase
THCA Δ9-tetrahydrocannabinolic acid CBDA cannabidiolic acid
7.6.2. Extracción de datos de RNA-seq de Cannabis sativa
pone a disposición de la comunidad investigativa datos de secuencias biológicas para mejorar la reproducibilidad y permitir nuevos avances mediante la comparación de datos.
7.6.3. Control de calidad de secuencias y preparación de lecturas.
Para poder trabajar con las secuencias disponibles los datos suelen estar en formato .fastq. Este formato contiene las secuencias y sus correspondientes valores de calidad de secuenciación. Para algunos archivos es necesario descargar la herramienta SRA toolkit, esta tiene una opción que permite convertir los archivos .sra a .fastq. El formato fastq almacena las secuencias y las cualidades Phred Quality Score (puntaje de calidad) en un solo archivo. Su objetivo es proporcionar de forma sencilla, una comprobación de control de calidad en datos de secuencias sin procesar los adaptadores de bases de datos de secuenciación de alto rendimiento. FASTQC realiza una revisión y determina si los datos presentan problemas los cuales se podrían tener en cuenta antes de realizar cualquier tipo de análisis. Este programa proporcionara unos gráficos de resumen y unas tablas para evaluar los datos obtenidos al correr el programa (Babraham Institute, 2016).
Para descartar datos de baja calidad es necesario utilizar herramientas informáticas como FASTX-toolkit las cuales filtran los datos y eliminan la baja calidad de bases, luego se hace una lectura de alineamiento haciendo un mapeo de estas lecturas para generar precisiones de presencia de contaminantes observando la cantidad de GC y la longitud de los genes, después se pueden aplicar métodos de normalización, también es posible evaluar la calidad global del conjunto de datos de RNA-seq mediante la comprobación de la reproducibilidad entre las distintas réplicas y los posibles efectos en baches encontrados. (FASTX-Toolkit, 2016). Durante el pre-procesamiento se deben eliminar regiones extremas con un contenido de bases anormal que no podrían ser mapeadas al genoma de referencia, este proceso se conoce como Trimming, el cuál escanea las lecturas de secuencias 5’ y elimina las bases 3’ hasta encontrar un parámetro establecido por el usuario. En el mercado se encuentran diferentes tipos de Trimming como por ejemplo: Solexaqa, Trimmommatic, ConDeTri (35).
7.6.4. Mapeos y ensamblaje de transcriptomas
Para realizar un análisis de forma apropiada, se debe hacer un mapeo de lecturas, que es una comparación contra un genoma para conservar todo aquello que alineó y las lecturas que no corresponden al rRNA (Rodríguez et al., 2017). Para realizar el mapeo de las lecturas a un genoma de referencia se utilizó Bowtie2 y TopHat2.
representa alineamientos de secuencias contra una secuencia de referencia, permitiendo la interoperación con un gran número de otras herramientas, lo que permite (38) a Bowtie2 interoperar con otras herramientas que soportan SAM, incluyendo SAMtools. (37).
TopHat2 mapea las lecturas de RNA-seq de Illumina contra un genoma, esto con el fin de identificar un empalme (Splice Junction) de exón-exón (39). El proceso de alineamiento consta de varios pasos. Si el archivo tiene una extensión GTF, TopHat extraerá las secuencias de la trascripción y utiliza Bowtie2 para realizar la alineación de las lecturas del transcriptoma, las únicas lecturas que se mapearan contra el genoma son las que no corresponden totalmente con el transcriptoma (39). Las lecturas que aún permanecen sin asignar se dividen en segmentos más cortos, que luego se alinean con el genoma. Los segmentos mapeados se utilizan para encontrar posibles sitios de empalme, las secuencias fijan un sitio de empalme donde ellas se enlazan a los segmentos que estaban sin asignar (39). En resumidas cuentas, TopHat realiza un alineamiento a lecturas de RNA-seq para referenciar genomas, usando Bowtie como alienador de lectura corta, luego este analiza los resultados de mapeo para identificar uniones de empalme entre exones(40).
Para ensamblar los archivos .SAM se utilizó Bedtools para recuperar las lecturas de cada contig (ensamble) que está asociado a cada una de las librerías.
7.6.4.1. Predicción de genes
Augustus en un programa predictor de genes para células eucariotas puede ser usado como un programa de ab initio lo que significa que basa su predicción puramente en la secuencia, este programa también puede incorporar indicios sobre la estructura genética procedente de fuentes extrínsecas tales como EST, MS/MS, alineaciones de proteínas y alineamientos genómicos sintéticos (41). La predicción de los genes se realizó mediante la transcritos de Arabidopsis thaliana ya que tienen una similitud con el genoma de Cannabis sativa (6).
Al correr Augustus se debe tener en cuenta que este cuenta con 2 argumentos principales: El archivo predeterminado o de consulta. El archivo que contiene la especie. El archivo de predeterminado o de consulta, contiene la secuencia de entrada de ADN y deben estar en formato (Multiple) fasta sin comprimir Ej.:
>name_of_sequence_1 agtgctgcatgctagctagct >name_of_sequence_2
gtgctngcatgctagctagctggtgtnntgaaaaatt
7.6.4.2. Ensamble de Transcriptomas.
Para el ensamble del transcriptoma, una cuantificación precisa del nivel de expresión de un gen de RNA-seq requiere de una identificación de la isoforma del gen dado producida en cada lectura. Esto depende del conocimiento de las variantes de corte y empalme de ese gen. Debido a que una muestra puede contener múltiples variantes de corte y empalme para un gen, Cufflinks, infiere en la estructura de empalme de cada gen (43).Cufflinks ensambla transcriptomas, estima la abundancia del transcriptoma y realiza un análisis para la expresión diferencial y la regulación en muestras de RNA-seq. Cufflinks acepta lecturas de RNA-seq alineado y ensambla los alineamientos en un set de transcriptos, después estima la abundancia relativa de los transcriptomas basado en cuantas lecturas puede soportar cada uno según los protocolos que se utilicen (44).
El número de lecturas de RNA-seq generado por el transcriptoma es directamente proporcional a la abundancia relativa de la muestra, sin embargo los fragmentos codificantes de DNA (cDNA) son generalmente seleccionados por su tamaño para la construcción de esta librería, ya que los transcritos más largos producen más fragmentos de secuenciación que los transcritos más cortos (43). Cufflinks cuenta las lecturas que corresponden a cada transcripción y luego normaliza el recuento de cada transcripción por la longitud de cada una, ya que se puede tomar dos transcripciones, A y B, las cuales presentan la misma abundancia. Si B es dos veces más largo que A, una librería de RNA-seq contendrá (en promedio) el doble de lecturas de B que de A, ya que dos secuencias de una misma librería pueden producir diferentes volúmenes de lecturas de secuenciación. (43).
Con el programa Cufflinks debemos ejecutar scripts que nos ayudaran a determinar ciertos parámetros como Cuffquant que estima la expresión de la isoforma (44). El programa ignora alineaciones que no son estructuralmente compatibles con el transcriptoma de referencia. Este programa hace una cuantificación de la expresión del transcriptoma; Cuffmerge es una herramienta que puede utilizar para combinar varios ensamblajes, este también ejecuta una herramienta, Cuffcompare quien filtra una serie de transfrags (fragmentos transcritos) que son probablemente “artifacts” (44).
7.6.5. Identificación de genes diferencialmente expresados (DEGs)
lecturas para el total rendimiento de la ejecución del programa. EL uso común de estos fragmento son kilobase de transcripto por millón de fragmentos mapeados (FPKM) con esto puede asegurar los niveles de expresión de diferente genes y transcripción que se puedan comparar a través de la ejecución del programa (43).
7.6.6. Expresión de la biosíntesis de Cannabinoides en tejidos de Cannabis sativa. Para lograr un análisis funcional del genoma de Cannabis sativa, es necesario examinar la expresión relativa de cada uno de los transcriptos representativos en los tejidos del cannabis, ya que diferentes organismos y tejidos tienen una diferente función fisiológica, por consiguiente la expresión de los genes es única, generando un patrón de expresión de genes altamente restringido (6). Con las plantas, los tejidos fotosintéticos se componen a menudo de unos conjuntos similares de tipos de células, como también las rutas metabólicas primarias y los procesos químicos, como la fotosíntesis (6). Debido a la anterior información, se pueden encontrar expresiones similares en los tejidos del cannabis (6). Las flores muestran un patrón de expresión génica consistente en la biosíntesis de cannabinoides y terpenoides en estos órganos (6).
Otra estrategia para realiza ensambles es utilizar Cuffmerge que es una programa que cumple las mismas funciones de Cufflinks pero además realiza anotaciones de un genoma de referencia, Cuffmerge puede integrar transcripciones de referencia basado en anotaciones para combinar la transcripción de referencia con transfrags (fragmento de transcripción) y produce un archivo donde se hace un análisis diferencial downstream (43).
Figura 7. Fusión de muestras ensambladas contra un transcriptoma de referencia. Los genes con baja expresión, utilizar Cuffmerge recupera el gen completo.
7.6.7. Análisis diferencial
8. METODOLOGÍA 8.1. DELIMITACIÓN Y ÁREA DE ESTUDIO.
Este proyecto se desarrolló por medio de un ordenador con sistema operativo Ubuntu y la información se guardó en un servidor de la Universidad Mayor de Chile y en ordenadores de las instalaciones del laboratorio de Biología Molecular ubicado en la sede B de la facultad de Ciencias y Educación de la Universidad Distrital Francisco José de caldas.
Toda la metodología del presente trabajo fue realizada desde la terminal de Ubuntu, ya que facilita el movimiento de los directorios y la ejecución de programas. Para la descarga de las secuencias es necesario contar con una máquina que tenga ciertas características, como las siguientes: Un buen procesador. Al correr un software de análisis o mapeo, utilizamos recursos, no solo de la red, sino también de la maquina con la que se esté ejecutando el programa, esto podría facilitar el análisis de las secuencias; gran capacidad de espacio en el disco duro del computador con que se esté trabajando. Las lecturas pueden llegar a pesar más de 40Gb.gusano.ddns.net, este tiene una capacidad de almacenamiento de aproximadamente 4,5 Tb.
8.2. MINERIA DE GENOMAS DE REFERENCIA DE Cannabis sativa.
Para el levantado de los datos se utilizó como referencia el genoma Cannabis descargado desde el servidor FTP de NCBI, estos serán convertidos a formato Fasta a partir de la herramienta SRAToolkit y poder realizar un control de calidad y su posterior análisis. Para el transcriptoma de referencia de Cannabis sativa se recurrió a los transcritos de las sub especies Cannatonic, LA Confidential, Chemdawg 91, Purple Kush, Pineaple Banana Buba Kush, los cuales se descargaron de los servidores de SRA NCBI y Genomes NCBI. Para la cepa de la subespecie Finola1, se recurrió a la base de datos The Cannabis Genome Browser (http://genome.ccbr.utoronto.ca/cgi-bin/hgGateway).
Se usaron datos de RNA-seq de diferentes tejidos de Cannabis sativa.
8.3. CONTROL DE CALIDAD FILTRADO DE RNA-seq DE Cannabis sativa
Los archivos descargados tienen una extensión .sra. Para poder trabajar con las secuencias y realizar un buen control de calidad, es necesario pasar de formato “.sra” a “.fastq”. Se usó SRA toolkit. Después utilizamos la siguiente línea de comandos:
Este se ejecutó para cada secuencia. La opción fastq-dump nos permite convertir el archivo .sra en .fastq, los archivos de salida se encuentran como archivos SRRRR306861.sra.fastq. Posteriormente se realizó el control de calidad de secuencias con el software, FastQC y conocer detalles de la calidad de cada corrida, con esto obtuvimos una impresión rápida de los datos y se pudo determinar si estos presentaban problemas, los cuales debemos tener en cuenta antes de realizar el análisis.
La línea de comandos que se ejecuto es la siguiente:
/Illumina$ fastqc SRR306861.sra.fastq (para cada secuencia .fastq.)
Una vez que se identificaron los contaminantes, estos fueron eliminados mediante un proceso conocido como trimming, utilizando el software Trimmomatic (versión 0.36), eliminar secuencias de baja calidad con un Phred de calidad Q=30. Esto también elimina los extremos de las secuencias que pueden presentar menor calidad debido a menos eficiencia en la polimerasa en el inicio y al final de la secuenciación.
Para este proceso se utilizó Trimmomatic versión 0.36. Se creó un script para la ejecución de cada uno de los archivos .fastq con la siguiente línea de comandos:
java -jar ~/tools/Trimmomatic-0.36/trimmomatic-0.36.jar SE –threads 11 –phred33 <path to SRRxxxxxx.fastq> output <SRRxxxxxx.sra_trim.fastq> ILLUMINACLIP:contaminants.fasta:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:7 MINLEN:15
Donde las funciones del comando son las siguientes:
java -jar ~/tools/Trimmomatic-0.36/trimmomatic-0.36.jar: software que se uso SE: Indica al programa Trimmomatic que se está analizando una librería single-end (45).
-threads 11: Indica que se utilizaron 11 procesadores
-phred33: especifica la codificación de las nucleobases generadas por la secuenciación.
Input <path to SRRxxxxxx.fastq>: dirección del archivo fastqc.
ILLUMINACLIP:contaminants.fasta:2:30:10: Corta el adaptador y otras secuencias específicas de las lecturas de Illumina, como contaminantes que se van a filtrar. Los valores “2:30:10” Inicialmente Trimmomatic buscará semillas (16 bases) permitiendo un máximo de 2 desajustes. Estas semillas se extenderán y recortarán si en el caso de las lecturas de pares terminales se alcanza una puntuación de 30 (aproximadamente 50 bases), o en el caso de una punta única se lee una puntuación de 10, (aproximadamente 17 bases).
LEADING: Corta las bases del inicio de una lectura, removiendo las bases por debajo de un umbral de calidad o N bases, (calidad bajo 3)(46) (LEADING:3).
TRAILING: Corta bases del final de una lectura, removiendo las bases por debajo de un umbral de calidad o N bases (baja calidad 3) (46) (TRAILING:3).
SLIDINGWINDOW: Realiza un corte de ventana deslizante, cortando una vez que la calidad media dentro de la ventana cae por debajo de un umbral, escaneando con una ventana de 4 bases, haciendo un corte cuando la calidad media por base cae por debajo de 7 (45) (SLIDINGWINDOW:4:7).
MINLEN: Arroja lecturas por debajo de una longitud especificada, en este caso por debajo de 14 bases de largo (46) (MILEN:15) para single read.
8.4. EVALUACIÓN Y SELECCIÓN DEL GENOMA DE REFERENCIA
Se genera un índice del genoma con Bowtie2-build, se utilizaron las herramientas Bowtie2 para generar los índices de los genomas de referencias. Las lecturas de referencia son mapeadas a los 6 genomas de cannabis seleccionados, contra el genoma disponible seleccionado para lograr obtener un genoma parecido al Cultivar de los datos de RNA-seq, se eligió como referencia el genoma que presentara mayor porcentaje de lecturas mapeadas. El mapeo fue realizado con Tophat2, para esto se utilizaron las coordenadas genómicas de genes predichos en el genoma, y tener el genoma de referencia ayudando en la definición de los genes presentes en el transcriptoma de cannabis. Los porcentajes de mapeo total fueron evaluados y se seleccionó la mejor referencia para continuar con los análisis.
Para el mapeo de los 6 genomas de Cannabis se ejecutó las siguientes líneas de comandos:
Cannatonic: tophat2 --b2-sensitive --library-type fr-unstranded -p 4 -G /home/carlos/Desktop/Projects-VM/Cannabis/Augustus-out/cannatonic-augustus.gff
-o ../02-mapping/${i}_cannatonic_thout
Chemdog: tophat2 --b2-sensitive --library-type fr-unstranded -p 4 -G /home/carlos/Desktop/ProjectsVM/Cannabis/Augustusout/chemdogaugustus.gff
-o ../02-mapping/${i}_chemdog_thout
/…/…/../Cannabis/genomes/index/chemdog/chemdog-index ${i}.trimmed.
Las funciones de la línea comandos son las siguientes:
tophat2: Programa de mapeo
--b2-sensitive: es un preset que es usado por defecto, aumenta la posibilidad de que informe la alineación correcta para una lectura que alinea muchas puntos, estableciendo un número máximo de repeticiones de puntos repetitivos, ajustando también el tamaño de lectura. También cubre una amplia área del espacio, sensibilidad y precisión. Si el preset termina en sensitive, demora su ejecución pero se compensa con precisión y mejores resultados (47).
--library-type fr-unstranded: La librería predeterminada no está entrelazada (fr-unstranded), este preset hace una lectura desde el extremo izquierdo del fragmento (en las coordenadas de la transcripción) mapea la hebra de la transcripción y los mismo, mapea con la hebra del extremo derecho a la hebra (48)
-p: Incremente la memoria de Bowtie2.
-G: Suministra a TopHat con un conjunto de anotaciones de modelos génicos y/o transcripciones conocidas. Con esta opción, TopHat primero extraerá las secuencias de transcripción y utiliza Bowtie para alinear y para hacer primero una lectura de la transcripción (49).
Para la predicción de genes se utilizó al programa AUGUSTUS, que es un predictor de genes para organismos eucariotas. Se usaron los genes codificantes de la proteínas de Arabidopsis thaliana como referencia, para identificar genes idénticos en la cepas de Cannabis sativa. El archivo arrojado por el programa es un archivo .gff.
La línea de comando de Augustus fue la siguiente:
augustus --strand=both --genemodel=complete --gff3=on --UTR=on --species=arabidopsis queryfilename
Las funciones de la línea comandos son las siguientes:
--strand=both: Reporta genes pronosticados en ambas cadenas. --genemodel=complete: Solo pronosticar genes completos(50).
--gff3=on: Formato que almacena características genómicas en un archivo de texto, este es utilizado con GMOD para el intercambio de datos y la representación de los datos genómico(51), el archivo de salida con formato gff3.
--UTR=on: Predice las regiones no traducidas, además de la secuencia codificante(51).
--species=arabidopsis queryfilename: especie modelo que se va a utilizar para la predicción).
El archivo GFF salió de augustus donde encontramos los genes que se predijeron.
8.5. RECONSTRUCCIÓN DEL TRANSCRIPTOMA DE REFERENCIA
Para la reconstrucción del transcriptoma de referencia utilizamos una versión modificada de la metodología utilizada anteriormente por nuestro grupo en el trabajo de Amaral y colaboradores (52)
Cufflinks ensambla individualmente cada trascrito de lecturas de RNA-seq que ha sido alienado al genoma de referencia del cannabis, ya que una muestra puede contener lecturas de múltiples variantes de empalme por cada gen. Primero se ensamblaron los transcritos de cada muestra en utilizando la herramienta Cufflinks con la siguiente línea de comandos:
cufflinks -p 8 -o clout-test --library-type fr-unstranded -F 0.05 -u –b ../../../../../../Cannabis/genomes/index/cannatonic/cannatonic-index.fa
SRR306861.sra.fastq_cannatonic_thout/accepted_hits.bam
Las funciones de la línea comandos son las siguientes:
Cufflinks: Programa utilizado.
-p: Indica la cantidad de partículas para alinear las lecturas, su valor es 8 -o: Hace un resumen estadístico en un archivo de salida <outprefix>