Nuevas Metodologías para la síntesis de azaesteroides
145
0
0
Texto completo
(2)
(3) El presente trabajo se realizó en el Centro de Investigación de la Facultad de Ciencias Químicas de la Benemérita Universidad Autónoma de Puebla, bajo la dirección de la Dra. Sara Montiel Smith y con el apoyo otorgado por el CONACYT a través de la beca nacional número 359640 (CVU162058).
(4)
(5)
(6) Publicaciones Parte de los resultados obtenidos en este trabajo permitieron la publicación del artículo: . R. Martínez-Pascual, S. Meza-Reyes, J. L. Vega-Báez, P. Merino-Montiel, J. M. Padrón, A. Mendoza, S. Montiel-Smith, Novel Synthesis of Steroidal Oximes and Lactams and their biological evaluation as antiproliferatives agents, Steroids, 122 (2017) 24-33.. Asimismo, durante el periodo del doctorado se publicó un artículo de revisión bibliográfica y se presentaron diversos trabajos en Congresos Nacionales e Internacionales. . Roxana Martínez Pascual, Thiophosgene, Synlett, 26 (2015) 1776-1777. . “A Facile synthesis of 6-azasteroids”. Congreso Internacional, XV Congreso de la Sociedad Italo-Latinoamericana de Etnomedicina (11-15 Sep). Roxana Martínez Pascual, Sara Montiel Smith, José Luis Vega Báez, Módena, Italia, 2016.. . “Caracterización del acetato de 7-aza-6-oxo-B-homopregn-4-en-3β-ilo por RMN”. Congreso Nacional, Simposium de Resonancia Magnética del Posgrado en Ciencias Químicas. Roxana Martínez Pascual, Sara Montiel Smith, José Luis Vega Báez, México, Cd. de México, 2015.. . “Novedosa metodología para la formación de una B-homolactama a partir del Colesterol”. Congreso Internacional, 10ª Reunión Internacional de Investigación en Productos Naturales, Roxana Martínez Pascual, Omar Viñas Bravo, Sara Montiel Smith, José Luis Vega Báez, Jacinto Hernández Cruz, Mérida, México, 2014.. . “Obtención de oximas esteroidales mediante una novedosa metodología, aplicación a la pregnenolona y colesterol”. Congreso Nacional, 10a Reunión de la Academia de Química Orgánica, Roxana Martínez Pascual, Omar Viñas Bravo, Sara Montiel Smith, Socorro Meza, San Luis Potosí, México 2014..
(7) . “Síntesis de azaesteroides a partir de derivados colestánicos o espirostánicos”. Congreso Nacional, 40 Aniversario del ICUAP, 5° Encuentro Nacional Luis Rivera Terrazas, Roxana Martínez Pascual y Sara Montiel Smith, México, Puebla, 2014.. . “Síntesis de un nuevo azaesteroide tipo lactona bisnorcolánica a partir de diosgenina”. Congreso Internacional, 31 Congreso Latinoamericano de Química, CLAQ 2014. Roxana Martínez Pascual, Sara Montiel Smith, Omar Viñas Bravo Penélope Merino Montiel, José Luis Vega Báez, Lima, Perú, 2014.. . Uso de técnicas espectroscópicas para establecer la estructura del 3β-Acetato7-aza-B-homocolest-4-en-6-ona de manera inequívoca”. Congreso Nacional, Congreso Nacional de Química Analítica. Omar Viñas Bravo, Sara Montiel Smith, Penélope Merino, Puebla, México, 2014..
(8) Agradecimientos A la Dra. Sara Montiel Smith por aceptarme nuevamente en su laboratorio para continuar con mis estudios de doctorado. Sobre todo, por todo el apoyo que me ha brindado siempre, no sólo en el ámbito académico contribuyendo a mi formación profesional sino también en el aspecto personal. A la Dra. Socorro Meza, por ser siempre una persona amable, por su apoyo y confianza durante mi estancia en el laboratorio. Asimismo le agradezco por sus observaciones y comentarios acertados para el mejoramiento de este trabajo. A la comisión revisora, Dr. José Luis Vega Báez, Dra. Rosa Elba del Río Torres, Dra. Penélope Merino Montiel y Dr. Jorge R. Juárez Posadas por el seguimiento que realizaron al proyecto desde un inicio y hasta el final. Sus observaciones y comentarios fueron muy importantes para mejorar el presente trabajo. De manera especial quiero agradecer a Óscar, por haberme aceptado en su laboratorio durante una estancia corta en la Universidad de Sevilla, por compartir sus conocimientos respecto a las diversas técnicas de medición de actividad antioxidante, fue una experiencia muy enriquecedora para mi formación académica. Por otra parte y no menos importante, agradezco la hospitalidad y amistad brindada… eres una persona que deja huella en quienes te rodean por tu gran corazón, inteligencia y humildad. Al M. C. Vladimir Carranza Téllez por la obtención de los espectros de masas de alta y baja resolución. Al Dr. Ángel Mendoza por realizar los estudios de difracción de rayos X de los cristales obtenidos en este trabajo..
(9) Al Dr. José Manuel Padrón por realizar la evaluación de la actividad antiproliferativa a diversos compuestos sintetizados en este trabajo. A Martha, una de las grandes amistades que me llevó es la tuya, gracias por compartir no sólo la mesa de trabajo sino ratos muy agradables fuera del laboratorio. A Anabel y Laura, dicen que uno debe rodearse de gente positiva y con muchas competencias, ustedes son grandes mujeres con esas características, gracias por su amistad..
(10) Dedicatorias. Este trabajo está especialmente dedicado a mi hija Camila, el amor que siento por ti es incondicional, tu existencia me llena de felicidad y me motiva a seguir siempre adelante. Te amo. A mis padres, Ciro y Mari y a mis hermanos, Eric y Edgar, que siempre han estado conmigo en las buenas y en las malas, sin ustedes no lo habría logrado..
(11) i. RESUMEN En el presente trabajo se desarrollaron dos novedosas metodologías para introducir una función de tipo oxima en la posición 6 del esqueleto esteroidal. El primer protocolo consistió en una reacción de nitrosación modificada sobre el 4 (AcOH, Ac2O, NaNO2 y BF3OEt2) del colesterol y de la pregnenolona, lo que condujo a derivados 5-acetoxi-6-acetoxiimino en un sólo paso de reacción (Procedimiento A). La segunda metodología consistió en la funcionalización del anillo B del esterol de partida (colesterol, diosgenina o pregnenolona) formando derivados 3β,5-diacetoxi-6-nitroimino mediante una reacción clásica de nitrosación (AcOH, NaNO2 y BF3OEt2). En una etapa posterior, una hidrólisiscondensación in situ del grupo nitroimino con hidroxilamina en etanol a reflujo condujo a las 3β,5-diacetoxi-6-oximas correspondientes (Procedimiento B). En ambas rutas se prescindió del uso de sales contaminantes de cromo. Algunas estructuras novedosas fueron evaluadas in vitro como agentes antiproliferativos frente a seis líneas de tumores sólidos humanos, mostrando que los compuestos con dos grupos hidroxiimino son los más activos. El compuesto 6,23-dihidroxiimino derivado de diosgenina mostró ser el agente más activo con un valor de GI50 en el rango de 11-22 µM..
(12) ii. Los derivados 5-acetoxi-6-acetoxiimino obtenidos de colesterol y pregnenolona se hidrolizaron para obtener oximas de configuración Z que se utilizaron como precursoras para la síntesis de B-homolactamas mediante un rearreglo de primer orden de Beckmann. La síntesis desde la materia prima hasta el producto final fue de sólo tres pasos. Otra Bhomolactama análoga de la vespertilina se sintetizó a partir de la diosgenina en 5 pasos. Las lactamas derivadas de pregnenolona y diosgenina se evaluaron como agentes antiproliferativos mostrando actividad moderada.. Por otra parte, los derivados 5-acetoxi-6-hidroxiimino obtenidos de colesterol y diosgenina se utilizaron como materias primas de dos 6-azaesteroides. Como paso clave, se utilizó un rearreglo de segundo orden de Beckmann que permitió modificar las oximas correspondientes hacia derivados 3β-acetoxi-5,6-seconitrilos que sirvieron como precursores para introducir el nitrógeno en el anillo B esteroidal..
(13) iii. ABSTRACT Herein we report two novel methodologies to introduce an oxime functionality at C-6 of the steroidal framework. The first approach involved a modified nitrosation reaction at 4 of cholesterol or pregnenolone (AcOH, Ac2O, NaNO2 and BF3OEt2) affording the corresponding 5-acetoxy-6-acetoxyimino derivatives (Procedure A). The second method consisted in a two-step sequence starting from cholesterol, pregnenolone or diosgenin; in the first place, it was performed a traditional nitrosation reaction at 4 (AcOH, NaNO2 and BF3OEt2) in order to obtain the corresponding 3β,5-diacetoxy-6-nitroimine derivatives. Then, an in situ hydrolysis-condensation reaction of the nitroimino moiety with NH2OH in ethanol afforded the oxime functionality at C-6 (Procedure B). These methods avoided the use of hazardous oxidant agents in the process. The new steroidal oximes were assayed in vitro as potential antiproliferative agents against a panel of 6 human tumor cell lines. The 6,23-dihydroxyimino derivative exhibited the highest activity with GI50 values ranging from 11 to 22 µM..
(14) iv. Moreover, the 5-acetoxy-6-acetoxyimino derivatives obtained from procedure A were hydrolyzed in a basic medium in order to obtain Z-oximes, which subsequently were converted into B-homolactams through a Beckmann rearrangement. Furthermore, a vespertilin type B-homolactam was synthesized from diosgenin in a five-step sequence. Lactams derived from diosgenin and pregnenolone were evaluated as antiproliferative agents showing moderate activity.. Finally, 6-azasteroids were obtained from 5-acetoxy-6-hydroyimino derivatives. In this synthetic pathway the key reaction was an abnormal Beckmann rearrangement which allowed to convert the oximes into 3β-acetoxy-5,6-seconitriles. The nitrile functionality was used to introduce the nitrogen into the B ring of the steroidal framework..
(15) v. Tabla de contenido RESUMEN ............................................................................................................................................ i ABSTRACT ..........................................................................................................................................iii LISTA DE ABREVIATURAS................................................................................................................... 1 1.. INTRODUCCIÓN ......................................................................................................................... 3. 2.. ANTECEDENTES .......................................................................................................................... 6 2.1.. Oximas esteroidales con actividad citotóxica. ................................................................... 6. 2.2.. Oximas esteroidales como inhibidores de la enzima aromatasa. .................................... 13. 2.3.. Los azaesteoides ............................................................................................................... 15. 2.4.. Síntesis y actividad biológica de los aza-homoesteroides ................................................ 24. 3.. OBJETIVOS ...................................................................................................................................... 31. 4.. DISCUSIÓN DE RESULTADOS ........................................................................................................ 32. 5.. 6.. 4.1.. Síntesis de oximas esteroidales en la posición 6. ............................................................. 32. 4.2.. Reacción normal de Beckmann de oximas esteroidales................................................... 59. 4.3.. Reacción Anormal de Beckmann de oximas esteroidales ................................................ 63. 4.4.. Síntesis de 6-azaesteroides............................................................................................... 69. 4.5.. Evaluación biológica de oximas y lactamas esteroidales. ................................................ 77. METODOLOGÍA .............................................................................................................................. 79 5.1.. Cromatografía .................................................................................................................. 79. 5.2.. Puntos de fusión ............................................................................................................... 79. 5.3.. Polarimetría ...................................................................................................................... 79. 5.4.. Resonancia Magnética Nuclear........................................................................................ 79. 5.5.. Espectrometría de masas ................................................................................................. 80. 5.6.. Espectroscopia infrarrojo ................................................................................................. 80. 5.7.. Disolventes y materias primas ......................................................................................... 80. 5.8.. Rayos X ............................................................................................................................. 80. PARTE EXPERIMENTAL .................................................................................................................. 81. 6.1.. Procedimiento general para obtener: el diacetato de (6E)-acetoxiiminocolesta-3β,5-. diilo (146a) y el diacetato de (6E)-6-acetoxiimino-20-oxo--pregna-3β,5-diilo (146b) .............. 81 6.2.. Procedimiento para obtener el (6E)-6-hidroxiiminocolest-4-en-3β-ol (147). ................... 83. 6.3.. Procedimiento para obtener el (6E)-6-hidroxiimino-5-metoxi-5-colestan-3β-ol (149). 84.
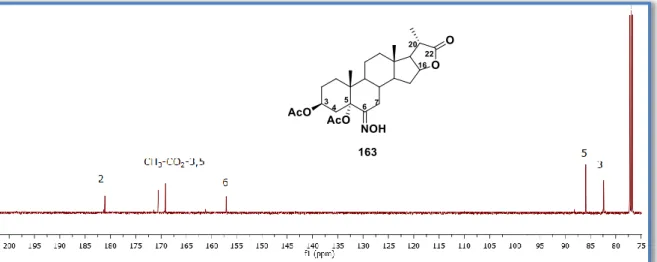
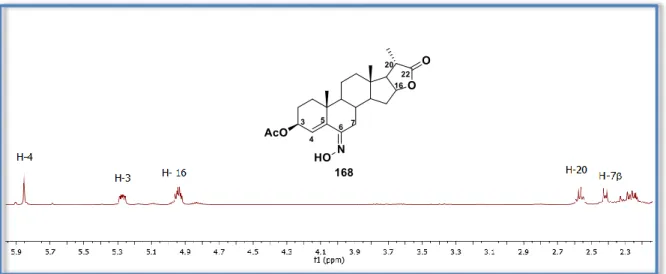
(16) vi. 6.4.. Procedimiento para obtener el (6E)-6-hidroxiimino-5-metoxi-5-colestan-3-ona (152). 85. 6.5.. Procedimiento para obtener el (6E)-6-hidroxiimino-colest-4-en-3-ona (4a).................... 86. 6.6. Procedimiento general para obtener: el acetato de (6Z)-hidroxiiminocolest-4-en-3-ilo (153a), el acetato de (6Z)-6-hidroxiimino-20-oxo-pregn-4-en-3β-ilo (153b) y el (6Z)- 3-acetoxi6-hidroximiinopregn-4-en-20,16-carbolactona (168). ............................................................... 87 6.7.. Procedimiento para obtener el (6Z)-6-hidroxiiminocolest-4-en-3β-ol (154). ................... 89. 6.8. Procedimiento general para obtener: acetato de 6a-aza-6-oxo-B-homocolest-4-en-3β-ilo (172a), el acetato de 6a-aza-6,20-dioxo-B-homopregn-4-en-3β-ilo (172b) y el (20S)-3β-acetoxi6a-aza-6-oxo-B-homopregn-4-en-20,16β-carbolactona (172c). .................................................. 90 6.9.. Procedimiento general para obtener la mezcla 155a/161a o 155b/161b. ...................... 92. 6.10. Procedimiento general para obtener: el diacetato de (6E,23E)-(25R)-6,23dihidroxiiminoespirostan-3,5-diilo (160a) y el acetato de (6E,23E)-(25R)-3-hidroxi-6,23dihidroxiiminoespirostan-5-ilo (160b). ...................................................................................... 93 6.11.. Procedimiento para preparar la 3-O-bencildiosgenina (164)....................................... 95. 6.12. Procedimiento para obtener la anti-(25R)-3β-bencil-5-acetoxi-6-nitroimin-5espirostan-23-ona (165). .............................................................................................................. 96 6.13. Procedimiento para obtener el (6E,23E)-(25R)-3β-bencil-6,23dihidroxiiminoespirostan-5-ilo (167). ........................................................................................ 97 6.14. Procedimiento para obtener la (6E)-(20S)-3,5-diacetoxi-6-hidroxiiminopregna20,16β-carbolactona (163)........................................................................................................... 99 6.15.. Procedimiento para obtener el 3-O-bencilcolesterol (170). ....................................... 100. 6.16. Procedimiento general para obtener: el acetato de (6E)-3β-hidroxi-6nitroiminocolestan-5-ilo (145a), diacetato de (6E)-6-nitroiminocolestan-3β,5-diilo (145b), diacetato de (6E)-6-nitroimino-20-oxopregna-3β,5-diilo (145c) y el acetato de (6E)-3β-bencil-6nitroiminocolestan-5-ilo (145d). .............................................................................................. 101 6.17. Procedimiento general para obtener: el acetato de (6E)-3β-hidroxi-6hidroxiiminocolestan-5-ilo (171a), diacetato de (6E)-6-hidroxiiminocolesta-3β,5-diilo (171b), diacetato de (6E,20)-6,20-dihidroxiiminopregna-3β,5-diilo (171c) y el acetato de (6E)-3βbencil-6-hidroxiimincolesta-3β,5-diilo (171d). ......................................................................... 104 6.18. Procedimiento general para obtener: el 3β-acetoxi-5-oxo-5,6-secocolestan-7-nitrilo (173), el 3β-bencil-5-oxo-5,6-secocolestan-7-nitrilo (174) y el (20S)-3-acetoxi-7-ciano-5-oxo5,6-secopregna-20-16β-carbolactona (176). ............................................................................. 107 6.19.. Procedimiento para obtener el (6E)-6-hidroxiimino-5-metoxi-5-colestan-3-ona (152). 109. 6.20.. Procedimiento para obtener el 3-oxo-5-metoxi-5,6-secocolest-4-en-7-nitrilo (178). 110. 6.21. (179).. Procedimiento para obtener la 5-metoxi-3-oxo-5,6-secocolest-4-en-7-carboxamida 111.
(17) vii. 6.22. Procedimiento general para obtener: el acetato de 7-carboxamida-5-oxo-5,6secocolestan-3β-ilo (179), la 3β-bencil-5-oxo-5,6-secocolestan-7-carboxamida (180) y la (20S)3-acetoxi-7-carboxamida-5-oxo-5,6-secopregna-20,16β-carbolactona (181) ........................ 112 6.23.. Procedimiento para obtener el 6-aza-colesta-3,5-dieno (183). ................................. 115. 6.24. (184).. Procedimiento para obtener la (20S)-6-azapregna-3,5-dien-20,16β-carbolactona 116. 7.. CONCLUSIONES ............................................................................................................................ 117. 8.. REFERENCIAS ................................................................................................................................ 120.
(18) 1. LISTA DE ABREVIATURAS ATFA. Ácido trifluoroacético. CC. Cromatografía en Columna. CCF. Cromatografía en Capa Fina. COSY. Correlation Spectroscopy. DEPT. Distortionless Enhancement by Polarization Transfer. DEG. Dietilenglicol. DHT. Dehidrotestosterona. DMP. 3,5-Dimetilpirazol. DMF. N,N-Dimetilformamida. DMSO. Dimetilsulfóxido. DPPE. 1,2-Bis(difenilfosfino)etano. EMC. Etilmetilcetona. HMBC. Heteronuclear Multiple Bond Coherence. HMPA. Hexametilfosforoamida. HBP. Hiperplasia Benigna Prostática. HRFAB-MS. Espectrometría de masa de alta resolución (por FAB). HSQC. Heteronuclear Single Quantum Correlation. Hz. Hertz. IC50. Concentración Inhibitoria media. IR. Infrarrojo. J. Constante de acoplamiento. mCPBA. Ácido m-cloroperbenzoico. NBA. N-Bromoacetamida. NMO. N-óxido de N-metilmorfolina. MsCl. Cloruro de metansulfonilo. Pf. Punto de fusión. ppm. Partes por millón. p-TsCl. Cloruro de p-toluensulfonilo. PCC. Clorocromato de piridinio. PDC. Dicromato de piridinio. QSAR. Quantitative Structure-Activity Relationship. TBAF. Fluoruro de tetra-n-butilamonio. TBDPSCl. Cloruro de terc-butildimetilsililo. THF. Tetrahidrofurano.
(19) 2 TMS. Tetrametilsilano. . Desplazamiento químico. . Doble enlace.
(20) Introducción. 1. INTRODUCCIÓN Los esteroides son moléculas lipofílicas utilizadas por los organismos como mensajeros químicos, actúan en un amplio rango de tejidos e influyen en muchos aspectos de los seres vivos, como la diferenciación sexual, la fisiología reproductiva, la osmorregulación y como intermediarios metabólicos. Los sitios más importantes en que son secretados son los ovarios, los testículos, las glándulas suprarrenales y la placenta. Cuando se secretan al medio ambiente pueden influir sobre otros organismos, como lo hacen las feromonas.1 Desde el punto de vista estructural, los esteroides son compuestos que poseen un esqueleto en común, tres anillos de seis miembros y uno de cinco miembros fusionados (núcleo del ciclopentanoperhidro-fenantreno). La Unión Internacional de Química Pura y Aplicada determinó nombrar los anillos como A, B, C y D, mientras que la numeración de los carbonos se debe realizar como se observa en la Figura 1. Es frecuente encontrar en los carbonos 10 y 13 grupos metilo y en la posición 17 una diversidad de cadenas laterales.2 Existen cuatro grandes familias de hormonas esteroidales: progestinas, andrógenos, estrógenos y corticoides (Figura 2). El colesterol (1a) es el precursor común de estas familias y las enzimas citocromo P450 y deshidrogenasas están implicadas en su biosíntesis a través de reacciones de hidroxilación y dehidroxilación-oxidación.3. Figura 2. Familias de compuestos esteroidales. Los esteroides, además de estar implicados en diversas funciones fisiológicas normales, también favorecen algunos efectos patológicos como la proliferación de células malignas. 3.
(21) 4. Introducción. provocando el cáncer hormonalmente dependiente.1 Una de las terapias utilizadas para este tipo de enfermedad consiste en regular la concentración de los estrógenos o andrógenos a través de la inhibición de una o más enzimas implicadas en la biosíntesis de estas hormonas; como ejemplos exitosos en los cuales se ha utilizado esta estrategia terapéutica se pueden mencionar los siguientes casos: para el tratamiento del cáncer de mama, se administran inhibidores de la aromatasa (CYP19), dicha enzima es la encargada de catalizar la transformación de la testosterona al estradiol; por otra parte se emplean inhibidores de las enzimas 5-reductasas en el tratamiento de la hiperplasia benigna prostática (HBP) y alopecia, modulando en este caso la concentración de la dihidrotestosterona (DHT). Más recientemente, se ha utilizado un inhibidor de la enzima 17-hidrolasa/C-17,20 liasa (CYP 17) para el tratamiento de cáncer de próstata.3 Gracias a la investigación de diversos grupos científicos que han enfocado su trabajo en la síntesis de moléculas que actúen como inhibidores enzimáticos de los procesos de esteroidogénesis, ha sido posible encontrar moléculas prometedoras para el tratamiento de estos padecimientos. En este contexto, los azaesteroides han mostrado poseer un amplio espectro de actividades biológicas interesantes para el desarrollo de nuevos fármacos como terapias para el cáncer u otras enfermedades.4 En la literatura se encuentran reportadas una serie de actividades biológicas en los azaesteroides, por ejemplo, inhibición de las enzimas 5α-reductasas 5 inhibidores PI-PLC (Fosfatidilinositol-Fosfoinositida Fosfolipasa C),. 6. antiinflamatoria,. 7. antiparasitaria,. 8. antifúngica,9 neurobloqueadores musculares10 y como agentes citotóxicos.11 Los azaesteroides son compuestos naturales o sintéticos en los cuales un átomo de carbono del núcleo esteroidal se ha reemplazado por un átomo de nitrógeno. Este cambio hace que la molécula sufra alteraciones en cuanto sus propiedades físicas y químicas y la actividad biológica que presentan. En la Figura 3 se muestran dos ejemplos de azaesteroides, uno natural aislado de plantas de tomate, la tomatidina 2 y otro sintético, la finasterida 2, un potente inhibidor de la enzima 5-reductasa. Dentro de los heteroesteroides, los azaesteroides son los más comunes, una posible razón, es que el grupo –NH- tiene aproximadamente el mismo tamaño que un grupo metileno lo que provoca que no haya una gran distorsión en la forma tridimensional del esteroide.12.
(22) Introducción. Dentro. de. la. azaesteroides. familia. de. destacan. azahomoesteroides.. El. los los. prefijo. “homo”, según las reglas de IUPAC,2 se utiliza cuando alguno de los anillos del. núcleo. expandido.. esteroidal En. la. ha. literatura. sido se. encuentran descritas actividades como antiproliferativa, antileucémica y antiandrogénica1318. para esta clase de compuestos. Con el propósito de desarrollar terapias más selectivas. contra el cáncer y sabiendo que las moléculas esteroidales están implicadas en el crecimiento celular, algunos grupos de investigación sintetizaron compuestos híbridos formados por una fracción esteroidal y un grupo citotóxico. El objetivo de esta estrategia es que el esteroide sirva como portador de la fracción citotóxica y que esta se libere en las células cancerosas, disminuyendo los efectos adversos de las quimioterapias actuales. 19,20 Algunos de estos híbridos están constituidos por azahomoesteroides y agentes alquilantes de nitrógeno que suelen emplearse en quimioterapias para tratar la leucemia, el cáncer de mama, testículo y pulmón.14 Esta combinación de compuestos ha mostrado ser exitosa, logrando reducir los efectos tóxicos de los agentes alquilantes, al aumentar su lipofilicidad y sus propiedades fisicoquímicas. De hecho, los híbridos pueden penetrar más fácilmente la bicapa de la membrana celular y llegar a su sitio de acción, el núcleo.21 Las oximas esteroidales son otro tipo de metabolitos bioactivos naturales o modificados que se han reportado como antiproliferativos, 22 inhibidores de las enzimas 5-reductasas5 y aromatasa.23 Además de la actividad biológica que presentan son importantes precursores sintéticos de azaesteroides con actividad biológica diversa. De lo anteriormente expuesto se destaca la importancia de los esteroides modificados conteniendo nitrógeno en su núcleo básico o fuera de este. Como tal, el núcleo ciclopentanoperhidrofenantreno es considerado como una molécula privilegiada24 al servir como líder para el diseño de moléculas bioactivas capaces de combinarse con distintos sitios activos y de esta manera contribuir al desarrollo de nuevas terapias contra diversas enfermedades, entre ellas el cáncer. En el presente trabajo se describen nuevas metodologías para la síntesis de oximas, lactamas esteroidales y azaesteroides. Las oximas y lactamas con estructuras novedosas fueron evaluadas como agentes antiproliferativos presentando de moderada a buena actividad biológica.. 5.
(23) Antecedentes. 2. ANTECEDENTES 2.1. Oximas esteroidales con actividad citotóxica. Los productos naturales encontrados en organismos marinos han sido una fuente importante de metabolitos secundarios biológicamente activos. De esponjas marinas de las morfo especies Cinachyrella alloclada y Cynachirella apion se aislaron las oximas esteroidales 4a y 5b25 (Figura 4). Este tipo de metabolitos llamaron la atención porque 4a se reportó como citotóxico contra líneas celulares de leucemia (P-388), cáncer de pulmón (A-549), colorrectal (H-29) y mieloma (MEL-28).26 Más adelante, se reportó el aislamiento de 6a de Cinachyrella australiensis, el cual fue reportado como antiviral contra la hepatitis.22,27 A partir de estos resultados, otros grupos de investigación se enfocaron en sintetizar familias de oximas esteroidales con el propósito de determinar la relación estructura-actividad de este tipo de compuestos.22,28. El grupo de investigación que reportó el aislamiento de la oxima 5b describió además su obtención a partir de β-sitosterol (1b), para corroborar la estructura propuesta del metabolito aislado y evaluar su actividad biológica.25 El primer paso consistió en la acetilación del alcohol de C-3 mediante el tratamiento de Ac2O en piridina (Esquema 1). Posteriormente, el 5 se epoxidó utilizando mCPBA, obteniéndose el compuesto 7b. La oxidación del epóxido 7b con CrO3 generó el derivado 5-hidroxi-6-cetona (8b). La hidrólisis básica de 8b condujo al derivado 3β-hidroxietilcolest-4-en-6-ona (9b). La condensación del carbonilo de 9b con NH2OHHCl y la subsecuente oxidación del alcohol del C-3 condujo al derivado 6-hidroxiimino-4-en-3-ona (5b), el cual no mostró actividad citotóxica.. 6.
(24) Antecedentes. Esquema 1. Síntesis de 5b a partir de β-sitosterol. En el 2001, Rodríguez y colaboradores26 evaluaron la actividad citotóxica de una serie de oximas esteroidales sobre cuatro líneas celulares cancerosas (P-388, A-549, HT-29, MEL28); los derivados hidroxiimino sintetizados variaron en la cadena lateral (tipo colesterol, sitosterol, gorgosterol, androstano o ausencia de cadena lateral) y en el grado de oxidación del anillo A.. Esquema 2. Oximas esteroidales sintéticas reportadas por Rodríguez y col.. Las observaciones realizadas en función de la cadena lateral fueron las siguientes: El derivado con cadena lateral colestánica 4a posee buena actividad citotóxica presentando valores de IC50 en un rango entre 1.5 a 2.5 µg/mL en las líneas celulares evaluadas. Las oximas esteroidales con cadena lateral de tipo sitosterol (5b), gorgosterol (11c) o carentes de cadena lateral (12, 13 y 14) tienen baja actividad con respecto a 4a (Esquema 2). Por otra parte se determinó que entre más alto sea el estado de oxidación en el anillo A mayor citotoxicidad presenta la molécula, siempre y cuando se mantenga la cadena lateral del tipo. 7.
(25) Antecedentes. colestánica. Los compuestos 15, 16 y 17 mostraron poseer de 5 a 10 veces más actividad citotóxica que 4a en las líneas celulares evaluadas. Para realizar la síntesis de los compuestos 4a, 5b y 11c se partió de colesterol (1a), βsitosterol (1b) y gorgosterol (1c) siguiendo la estrategia sintética usada para la síntesis de 5b según se describió en el Esquema 1, pero con una ligera modificación en el último paso de la oxidación (reactivo de Jones vs MnO2) (Esquema 3).. Esquema 3. Síntesis de las oximas esteroidales 4a, 5b y 11c.. La obtención de 12 se realizó a partir de la dehidroepiandrosterona (18), cuyo grupo cetónico se protegió usando HO(CH2)2OH en medio ácido y con una posterior acetilación se generó 19 (Esquema 4). Al acetal 19 se le aplicó la metodología utilizada para síntesis de 4a, 5b y 11c.. Esquema 4. Obtención de la oxima esteroidal tipo androstánica 12.. 8.
(26) Antecedentes. El derivado 6-hidroxiimino 14 carente de cadena lateral, se obtuvo a partir de 18. En este caso, se realizó una reducción de Wolf-Kishner generando el derivado 20, posteriormente se llevó a cabo la misma estrategia sintética para la obtención de 4a (Esquema 5).. Esquema 5. Síntesis de la oxima esteroidal 14, carente de cadena lateral.. Para la obtención de los derivados con insaturaciones en el anillo A (15, 16 y 17) se siguió la ruta que se muestra en el Esquema 6. Partiendo de colesterol (1a), se protegió el hidroxilo del C-3 con TsCl, luego la epoxidacion del 5 generó 21 que se trató con CrO3 y subsecuentemente se realizó una eliminación del grupo tosilato con LiBr en DMF, formando la cetona 22. Una reacción de osmilación sobre el 2 condujo a la mezcla de dioles diastereoisoméricos 23a-b.. Esquema 6. Síntesis de oximas esteroidales con insaturaciones en el anillo A. Por separado, 23a-b se trataron con Ac2O y piridina para proteger selectivamente los alcoholes en C-2 y C-3 y poder realizar una reacción de eliminación del alcohol del C-5;. 9.
(27) Antecedentes. luego, la desprotección de los ésteres en medio básico permitió obtener los derivados 24ab. El grupo cetónico contenido en 24a-b se transformó hacia las oximas correspondientes (25a-b) mediante el tratamiento con NH2OHHCl. Por último, la oxidación con el reactivo de Jones generó los productos 15 y 16. El compuesto 17 mostrado en el Esquema 2, se obtuvo cuando se hizo reaccionar la mezcla de los dioles 25a-b con MnO2 en cloroformo. En el 2008, Cui y colaboradores22 reportaron la síntesis y evaluación citotóxica de una serie de oximas esteroidales a partir de colesterol (1a), β-sitosterol (1b) y estigmasterol (1d) con grupos hidroxiimino en el C-7, C-3 y/o C-6, los productos fueron probados frente a diversas líneas cancerosas celulares como de hígado (Sk-Hep-1), de pulmón (H-292), de próstata (PC-3) y de ovario (Hey-1B). Encontraron que la actividad biológica de esta familia de compuestos está relacionada significativamente con la ubicación del grupo hidroxiimino y del tipo de cadena lateral concluyendo que la oxima en el anillo B (C-6 o C-7) es esencial para que los compuestos presenten bioactividad, que el grupo hidroxi en el C-3 da mejores resultados en comparación a un grupo cetónico y que los compuestos más activos son aquellos con cadena lateral tipo colestánica. La síntesis de los derivados 6-hidroxiiminos reportados por Cui22 se realizó mediante la siguiente secuencia: primero, la oxidación del alcohol del C-3 y del 5 con PCC generó los análogos con un sistema dicetónico, 26a-b y 26d (Esquema 7); posteriormente, la reducción selectiva de la cetona del C-3 con NaBH4 en presencia de CoCl26H2O condujo a los alcoholes 27a, 9b y 27d; luego, la condensación del grupo carbonilo del C-6 contenido en 27a y sus análogos con NH2OHHCl introdujo el grupo hidroxiimino generando 28a, 10b y 28d. Cabe destacar que en el último paso se obtuvo la oxima Z en una pequeña proporción. Las dicetonas 26a-b y 26d también sirvieron como intermediarios para la obtener los análogos 3,7-dihidroxiiminos, 29a-b y 29d, o 3-hidroxiimino, 6b, 30b y 30d, variando los equivalentes de NH2OHHCl/NaOAc (Esquema 7).. 10.
(28) Antecedentes. Esquema 7. Síntesis de derivados 3- y 6-hidroxiimino. Para la obtención de derivados 7-hidroxiimino, Cui y colaboradores22 partieron de colesterol (1a) y estigmasterol (1d). Como primer paso, se protegió el alcohol del C-3 formando el éster acético, luego se realizó una oxidación alílica del 5 con CrO3 en piridina por 25 h a temperatura ambiente, generando los derivados 31a y 31d, ambas cetonas se sometieron a una hidrólisis básica para desproteger el alcohol del C-3, formando 32a y 32d (Esquema 8). El tratamiento con NH2OHHCl en NaOH de 32a y 32d generó los derivados 3β-hidroxi-7hidroxiiminos (33a y 33d). La síntesis de los análogos 3β-acetoxi-7-hidroxiiminos, 34a y 34d, se realizó a partir de 31a y 31d, con NH2OHHCl y NaOAc.. Esquema 8. Síntesis de derivados 7-hidroxiiminos. En el 2014, Basal y colaboradores29 sintetizaron una serie de compuestos esteroidales 6hidroxiimino-4-enos y sus derivados O-alquilados. Las oximas y sus respectivos éteres de oxima mostraron tener de moderada a buena actividad antiproliferativa contra diversas líneas celulares como melanoma, leucemia, colón y cáncer renal. La síntesis de los productos se. 11.
(29) Antecedentes. realizó a partir del precursor 35, el cual condujo al derivado androstánico 6-hidroxiimino-4en-3-ona 12 mediante la misma estrategia sintética descrita por Rodríguez.26 Para la formación de los productos O-alquilados 36 y 37 se trató a 12 con la amina correspondiente en su forma de clorhidrato en medio básico (KOH y K2CO3). Posteriormente, los productos se trataron con ácido oxálico en Et2O anhidro, formando 38 y 39 (Esquema 9).. Esquema 9. Síntesis de oximas O-alquiladas. La introducción del grupo oxima en el C-23 de diversas sapogeninas también se ha descrito en. la. literatura,. Iglesias. y. colaboradores. reportaron. la. síntesis. de. 23-. hidroxiiminosapogeninas a partir de los acetatos de diosgenina (40a), epiesmilagenina (40b) y episarsasapogenina (40c)30,31 Para ello, primero realizó una reacción de nitrosación usando NaNO2, BF3OEt2 y AcOH, con lo cual se generaron los derivados 23-oxo sapogeninas 41ac, como productos mayoritarios. Posteriormente, el grupo carbonilo se condensó con el NH2OHHCl en medio básico lo que condujo a las 23-hidroxiimino sapogeninas, 42a-c (Esquema 10).. Esquema 10. Síntesis de 23-oximas derivadas de sapogeninas.. 12.
(30) Antecedentes. Se ha reportado que la diosgenina (40a) muestra citotoxicidad;32 sin embargo, la actividad antiproliferativa de 23-oximas no ha sido reportada. Muy recientemente, se describió la síntesis de la oxima derivada de la 23-acetildiosgenina (45) y se evaluó su actividad antiproliferativa resultando poseer moderada actividad (10.6 µM) para dos líneas celulares (HeLa y CaSki).33 Para obtener 45, se realizó una reacción de acetólisis de la diosgenina (40a), lo que condujo el derivado epoxicolestánico 43. La hidrólisis básica con una solución de KOH en etanol al 10% generó el derivado 23-acetil 44. Por último, la acetilación del alcohol del C-3 y el subsecuente tratamiento con NH2OHHCl en piridina a reflujo formó 45 (Esquema 11).. Esquema 11. Síntesis de oximas a partir de diosgenina.. 2.2. Oximas esteroidales como inhibidores de la enzima aromatasa. El cáncer de mama es una de las causas de muerte más comunes en mujeres, en el año 2012, se estimaron 1.67 millones de nuevos casos (25% de todos los casos de cáncer). 34 En México, el cáncer de mama es el más frecuente en la población femenina desde el año 2006. En el año 2013, el Instituto Nacional de Estadística y Geografía (INEGI), reportó que es la principal causa de muerte por cáncer en mujeres mayores de 20 años (29.5%). En las mujeres, alcanza su punto máximo en las del grupo de 60 a 64 años. 35 Los estrógenos, además de estar involucrados en numerosos procesos fisiológicos, son hormonas que se relacionan con el crecimiento y proliferación de células epiteliales cancerosas. En la mayoría de los casos, el crecimiento de los tumores malignos está relacionado con la alta concentración de estrona (E1) y estradiol (E2) en los tejidos mamarios. La formación de E1 y E2 se da a partir de la testosterona y de la androst-4-en-3,17-diona junto con una enzima denominada aromatasa. Es por ello que compuestos que inhiban la enzima aromatasa se pueden utilizar en el tratamiento para el cáncer de seno hormonalmente dependiente después. 13.
(31) Antecedentes. de una cirugía o radiación. Algunos fármacos aprobados clínicamente para realizar está función son los compuestos esteroidales, exemestano (Aromasin®), formestano (Lentaron®) y los no esteroidales, anastrozol (Arimidex®) y letrozol (Femara®).36 En este contexto, en 1992, Holland y colaboradores 37 reportaron que los derivados 6Ehidroxiimino 12 y 47 poseen actividad como inhibidores de la aromatasa. La síntesis de 12 se efectuó partiendo de 35 mediante el protocolo antes mencionado en el Esquema 4. Mientras que el compuesto 47, se obtuvo a partir de la oxima 46 usando MnO2 en cloroformo, con lo que se oxidó selectivamente el carbonilo del C-3. Los valores de las constantes de inhibición de la enzima aromatasa placentaria humana fueron de Ki = 4.41 µM dm-3 para 12 y una Ki = 0.08 µM dm-3 para 47 (Esquema 12).. Esquema 12. Síntesis de oximas con actividad inhibitoria de la enzima aromatasa. En el 2011, Pokhrel y colaboradores36 reportaron la síntesis de una serie de derivados del androstano con insaturaciones en los anillos A y B, entre ellos, se describe la síntesis del derivado 17-oxiimino 52, el cual mostró una excelente actividad de inhibición de la enzima aromatasa del 93.8%, valor que supera al reportado por el formestano (74.2%). Para la obtención de 52, primero se transformó la dehidroepiandrosterona (18a) hacia 48 mediante la condensación del carbonilo de C-17 y el NH2OHHCl en medio básico (Esquema 13). Posteriormente, se realizó una oxidación de Oppenauer sobre el alcohol del C-3 generando la cetona ,β-insaturada 49. La oxidación del 4 de 49 formó el epóxido 50, el cual se trató con HCl concentrado a baja temperatura obteniéndose el derivado clorado 51. Por último, se redujo el carbonilo del C-3 de 51 con NaBH4 para obtener el alcohol correspondiente (52).. 14.
(32) Antecedentes. Esquema 13. Síntesis de oximas en C-20 con actividad inhibitoria de la enzima aromatasa.. 2.3. Los azaesteoides Como se mencionó previamente, los azaesteroides son un grupo de compuestos naturales o sintéticos que se caracterizan por poseer un átomo de nitrógeno en lugar de un átomo de carbono dentro o fuera del ciclopentanoperhidrofenantreno. Cuando el reemplazo se da en el núcleo esteroidal se les denomina azaesteroide endonuclear, mientras que, cuando el átomo de nitrógeno se encuentra en un anillo extra o en la cadena lateral se le conoce como azaesteroide exonuclear. Como ejemplos de azaesteroides naturales endonucleares podemos mencionar dos: la samandrina (53), aislada del veneno de la salamandra muculosa Laurenti38 y el compuesto A25822B (54), que pertenece a un amplio rango de productos naturales conocidos como A255822 aislados del hongo Geotrichum flavo-brunneum (Figura 5).39 Este último se ha reportado que posee una fuerte actividad antifúngica.40. Como ejemplo de azaesteroide exonuclear se puede mencionar al bromuro de pancuronio (55), el cual es un compuesto que se utiliza clínicamente como relajante muscular y se aplica junto con otros fármacos en la anestesia general.10 Como ejemplo de azaesteroide sintético. 15.
(33) Antecedentes. endonuclear se encuentra la finasterida (2), un fármaco utilizado en el tratamiento de la hiperplasia benigna prostática (Figura 6).5 Una extensa investigación se ha conducido sobre todo a los azaesteroides sintéticos ya que son moléculas que presentan un amplio espectro de actividades biológicas.. La hiperplasia benigna de próstata (HBP) es un agrandamiento no canceroso de la glándula prostática cuya prevalencia aumenta progresivamente con la edad, puede ser causado por altos niveles de dehidrotestoterona (DHT). Los síntomas asociados a este padecimiento son la micción vacilante o intermitente, disminución de la fuerza o adelgazamiento del calibre del chorro urinario, disuria (dolor, molestia o sensación urgente que se presenta al orinar), frecuencia urinaria, nicturia (aumento en la frecuencia urinaria por las noches) o urgencia por ir al baño. La incidencia de HBP es aproximadamente del 70% a los 70 años edad y se vuelve universal con el avance de la edad.41 Si el problema es avanzado la cirugía es el tratamiento más frecuente; sin embargo, debido a los efectos secundarios han surgido otras terapias medicinales que pueden retardar o eliminar la cirugía. La enzima 5-reductasa ha emergido como la diana para tratar la DHT ya que la elevada actividad de esta enzima implica altos niveles de DHT, por lo tanto la inhibición de dicha enzima es un tratamiento lógico para este padecimiento. El mecanismo que se propone para explicar la transformación de la testosterona en DHT por la catálisis de la 5-reductasa involucra la formación de un complejo binario entre la enzima y el NADPH, seguido por la formación de un complejo ternario (I) con el sustrato, la testosterona (Esquema 14). Un residuo electrofílico (E+) presente en el sitio activo interacciona con el sistema enona favoreciendo la formación de un enolato, luego un hidruro del NADPH se transfiere a la DHT por la cara Re del carbocatión deslocalizado llevando a una reducción selectiva del C-5 (II). El enolato se tautomeriza a su forma cetónica tomando un protón del medio acuoso que entra en el C-4, lo que conduce a un complejo E-NADP+-. 16.
(34) Antecedentes. DHT. El complejo binario NADP+-enzima se forma después de la liberación del DHT (III) y finalmente la salida del NADP+ deja a la enzima libre para que continúe su proceso catalítico (Esquema 14).42. Esquema 14. Mecanismo de reducción de la testosterona a DHT.. Los métodos modernos de biología molecular han servido de apoyo para identificar dos tipos de enzimas 5-reductasas: la tipo I y la tipo II. 43 , 44 La enzima tipo I se expresa principalmente en los folículos y en la piel mientras que la tipo II se presenta en los tejidos genitales. Más recientemente se detectó una isoenzima tipo III, que se caracteriza por estar presente en muchos tejidos a la vez en los mamíferos, como el del páncreas, cerebro, células cancerosas de próstata y tejidos de piel.45 Un gran número de moléculas se han sintetizado como agentes inhibidores de las enzimas 5reductasas, en un principio se diseñaron compuestos conteniendo el sistema 4-en-3-ona y con una cadena lateral 17β con funciones oxigenadas, sin embargo no dieron buenos resultados in vivo debido a su rápida conversión a su forma reducida 4,5-dihidro. Por lo anterior, se pensó en compuestos que mimetizaran a la testosterona pero que no fueran fácilmente reducibles por la enzima. Un grupo de científicos de Merck sintetizó una gran familia de derivados 3-ceto-4-azaesteroides que dieron excelentes resultados. A partir de este exhaustivo estudio surgió el primer inhibidor aprobado para su uso clínico, la finasterida (2), la cual es una molécula de tipo 4-aza-3-ona, cuya inhibición se da muy fuertemente sobre la isoenzima tipo II; la dosis clínica de 5 mg por día reduce los niveles de DHT en un 65-80%46 La forma más probable en la que la finasterida inhibe la conversión de testosterona a DHT se explica mediante la trasferencia de un hidruro del NADPH para reducir el doble enlace del C-2 de la finasterida a cambio del C-5 de la testosterona. 47 Más adelante, se desarrolló otro potente inhibidor de tipo 4-azaesteroide, la dutasterida (57), que resultó. 17.
(35) Antecedentes. activo frente a ambas isoenzimas y fue aprobado por la “Food and Drug Administration” en 2002 para el tratamiento de la HBP (Figura 7).5 En la búsqueda de nuevos inhibidores de la enzima 5-reductasa, el grupo Glaxo sintetizó una nueva serie de moléculas de tipo 3-ceto-6-aza esteroides, encontrando muy buenos resultados. Por ejemplo, el compuesto 58 (Figura 8) exhibió un valor de IC50 = 9 nM para la isoenzima tipo I, 17 veces más que la finasterida (2, Figura 6).5 Otra actividad biológica que ha sido reportada para moléculas de tipo 6-aza es como inhibidores. PI-PLC.6. La. fosfatidilinositol. fosfolipasa C (PI-PLC) es una enzima que se localiza en el lado interno de la membrana celular y se. encarga. de. catalizar. la. hidrólisis. del. fosfatidilinositol (4,5)-bifosfatasa. Los productos generados de la hidrólisis son segundos mensajeros: el inositol (1,4,5)-trifosfato y diacilglicerol. El primero libera Ca2+ del almacenamiento intracelular para aumentar la concentración de Ca2+ libre intracelular; mientras que el segundo, activa el Ca2+ y la proteína quinasa C. La activación del Ca2+ y de la quinasa C da como resultado una serie de cambios celulares, como síntesis de DNA, proliferación celular y actividad neuronal. Las funciones anormales de la fosfatidilinositol fosfolipasa se han relacionado con la enfermedad del Alzheimer, o con la presencia de tumores malignos. Por lo anterior, la inhibición de la PIPLC puede servir como un tipo de terapia para el tratamiento del cáncer. En este contexto, se han reportado una serie de compuestos de tipo 6-azaesteroidal con actividad para inhibir la PI-PLC, por ejemplo, el 3β-hidroxi-6-azacolesterol (59, Figura 9) se reportó como inhibidor de la enzima PI-PLC (IC50 = 1.8 µM) y mostró actividad antiproliferativa contra células cancerosas de colón, siendo la presencia del grupo amino en la posición 6 muy importante para la actividad.6 La tuberculosis es una enfermedad provocada por una bacteria llamada Mycobacterium tuberculosis, el metabolismo del colesterol es una fuente potencial de energía y uno de los. 18.
(36) Antecedentes. medios en que la bacteria produce una serie de metabolitos secundarios muy importantes para ella. El bloquear la biosíntesis de estos metabolitos a través de la presencia de inhibidores representa una alternativa para el desarrollo de nuevas terapias anti-tuberculosas. Se sabe que la enzima 3β-hidroxiesteroide deshidrogenasa (3β-HSD) es la encargada de catalizar la oxidación y la isomerización del colesterol en colest-4-en-3-ona y también cataliza la transformación de dehidroepiandrosterona y pregnenolona a sus respectivas cetonas ,βinsaturadas. En este contexto, Sampson y colaboradores48 evaluaron la actividad inhibitoria de la enzima M. tb 3β-HSD de una serie de 6- y 4-azaesteroides. Los compuestos 59 y 60 mostraron ser los más eficaces de todos, teniendo un IC50 = 0.5 µM y 9 µM, respectivamente. La síntesis de 6-azaesteroides, generalmente implica reacciones de ruptura oxidativa del anillo B, introducción del nitrógeno y ciclocondensación para regenerar el anillo B. La primera síntesis reportada de 6-azaesteroides fue descrita por Jacob y colaboradores en 1960.49 Esta consistió en la ruptura oxidativa de 31a con O3 en AcOH, posteriormente se trató con H2O2 y medio básico, lo que condujo al 5,7-seco esteroide (61). Después se efectuó una reducción catalítica para hidrogenar el 3 conduciendo a 62. El grupo ácido de 62 se condensó con NH3 en etanol a 0 oC y una ciclación in situ generó la lactama 63. Finalmente, el azaesteroide con un grupo imino en el C-5 (64) se obtuvo luego de una reducción con hidruro de litio y aluminio en éter etílico a partir de 63 (Esquema 15).. Esquema 15. Primera síntesis reportada de un 6-azaesteroide. En ese mismo año, Lettré y colaboradores50 reportaron una estrategia similar que consistió en la oxidación del 5 de la cetona ,β-insaturada 31a con O3 y el posterior tratamiento reductivo con Zn y AcOH del ozónido intermediario para generar 65. El grupo aldehído de. 19.
(37) Antecedentes. 65 se oxidó al ácido carboxílico y este se transformó a su forma activada 67, usando SOCl2. El cloruro de ácido 67 tras tratamiento con NaN3 condujo a la acil azida 68. El rearreglo de Curtius de 68 generó un isocianato intermediario que sufrió una ciclocondensación en el mismo medio regenerando el anillo B. La subsecuente hidrogenación catalítica en medio ácido formó el azaesteroide 70. Cabe mencionar que las condiciones de condensación simultáneamente eliminaron el grupo acetato del C-3 (Esquema 16).. Esquema 16. Síntesis del 6-azaesteroide 70 realizada por Lettré. En 1993, Frye y colaboradores51 reportaron la síntesis de una serie de 6-azaandrost-4-en-3onas con distintos sustituyentes en el C-17 con el propósito de evaluar su actividad como inhibidores de las isoenzimas 5-reductasas I y II, usando una estrategia similar a la de Lettré y colaboradores.50 Como primer paso, se protegió el alcohol del C-3 de 71a, posteriormente se realizó la ozonólisis del 5 obteniendo un aldehído intermediario cuya oxidación generó el derivado ceto-ácido 72. El grupo ácido contenido en 72 se transformó al isocianato 73. Las condiciones de condensación y ciclación se optimizaron usando gel de sílice y calor, con esto se evitó la eliminación del grupo oxigenado del C-3, lo que condujo a 74 (Esquema 17). Por último, reacciones de protección-desprotección y una oxidación del alcohol del C3 generaron los derivados azaesteroidales con distintos sustituyentes R en la cadena lateral (76). Los análogos 76a-c mostraron una potente inhibición a las isoenzimas I y II con una muy buena eficacia in vivo en rata.. 20.
(38) Antecedentes. Esquema 17. Síntesis de 6 azaesteoides con actividad en la inhibición de 5-reductasas.. En 1995, Haffner52 investigó la actividad de inhibición de la enzima 5-reductasa de dos 6azaesteroides con un grupo metilo ( y β) en el C-7 (81a-b), derivados del colesterol (1a). La introducción del grupo metilo en el C-7 se realizó utilizando la olefinación de Peterson sobre el compuesto 77, generando el alqueno 78 cuya reducción catalítica formó la mezcla diastereoisomérica de 79. La mezcla se sometió a condiciones muy similares a las descritas por Lettré para acceder a la acil azida 80. El paso de ciclación se dificultó usando gel de sílice y calor, por lo que se recurrió a un medio ácido acuoso, THF y calor (Esquema 18). Los productos 81a-b mostraron excelentes valores de Ki en un rango de 0.8-7.9 nM.. Esquema 18. Síntesis de 6-azaesteroides con un grupo metilo en el C-7. En 1996, Rahier y Taton53 reportaron la síntesis de las 6-aza-B-homolactamas 85 y 86. Para ello se trató a 82 con mCPBA conduciendo a un epóxido intermediario y su subsecuente. 21.
(39) Antecedentes. apertura en medio básico condujo al triol 83. La oxidación con Pb(OAc)4 en benceno de 83 formó el ceto-aldehído 84 que se sometió a una aminociclocondensación reductiva utilizando NH4OAc y un exceso de NaBH3CN en MeOH por 16 h lo que condujo a los productos finales (Esquema 19). Los productos sintetizados se evaluaron como inhibidores de la enzima 5, 7-esterol 7-reductasa, los resultados obtenidos mostraron buenos valores de actividad. Esta enzima es la encargada de catalizar la producción de colesterol a partir de 7dehidrocolesterol en presencia de NADPH y el estudio se realizó con el propósito de entender el mecanismo de acción entre la enzima y sustrato a través del diseño de inhibidores (6-azaesteroides) que simulen intermediarios catiónicos involucrados en el mecanismo de la reducción.. Esquema 19. Síntesis de las B-homolactamas 85 y 86 por Rahier y Taton.53 En 1996, Fang y Sharp54 reportaron un proceso para producir una serie de estructuras de tipo 6-aza-androstenonas análogas a 91, variando la cadena de C-17. La ruta sintética empleada fue diferente con respecto a las reportadas hasta ese momento. Primero, se realizó la ruptura del anillo B de 71b mediante una técnica novedosa utilizando un agente nitrosante (HO3SONO) formando el derivado seconitrilo 87. Posteriormente, se realizó una epoxidación del doble enlace en el C-3 para generar 88. El rearreglo del epóxido en presencia de paladio (0) permitió obtener el compuesto dicetónico 89. Seguido de una hidrólisis parcial se formó el derivado 90. Finalmente, mediante un rearreglo de Hoffman y subsecuente ciclación se generó el azaesteroide 91 (Esquema 20). Los productos mostraron ser potentes inhibidores de la enzima 5-reductasa.. 22.
(40) Antecedentes. Esquema 20. Síntesis de 6-aza-4-en 3-onas propuesta por Fang y Sharp.54 En el 2007, del Río y colaboradores55 reportaron la síntesis de una nueva sal de oxaziridinio 97 partiendo del colesterol (1a) y su uso como agente quiral en la oxidación enantioselectiva de sulfuros a sulfóxidos, con un exceso enantiomérico superior al 99%. Para su síntesis se protegió el alcohol en el C-3 con t-butildifenil éter formando el compuesto 92. Posteriormente, se realizó una epoxidación del 5, obteniendo una mezcla de epóxidos diastereoméricos /β. Subsecuentemente, la mezcla se trató con el reactivo de Jones a 50 °C generando el 5,6-secocetoácido 93. El isocianato 94 se obtuvo a partir de 93, utilizando un protocolo muy similar al de Lettré.50 La ciclocondensación del isocianato con la cetona del C-5 contenidos en 94, se realizó con alúmina básica y éter de petróleo, conduciendo al azaesteroide 95. Para finalizar, se efectuó una epoxidación del 5, obteniendo la oxaziridina 96, cuyo tratamiento en medio ácido formó la sal 97 (Esquema 21).. Esquema 21. Síntesis de la sal de oxaziridinio 97 a partir de colesterol.. 23.
(41) Antecedentes. En 2005, Kasal56 reportó una novedosa ruta para obtener 6-azapregnanolonas a partir del 7noresteroide 98, a los productos se les realizó un estudio para evaluar su actividad neuronal, los resultados no fueron satisfactorios para este tipo de derivados. La síntesis se describe a partir de 98, cuya oxidación con HClO4 en NBA generó una halohidrina intermediaria 99. El tratamiento básico de 99 condujo al epóxido 100, el cual se sometió con BF3OEt2 generando la cetona 101. La condensación con NH2OHHCl, en medio básico condujo a la mezcla de oximas E y Z 102 y 103. El tratamiento con MsCl en piridina del isómero E, promovió la reacción de Beckmann generando el 6-azaesteroide 104. La hidrólisis básica y posterior oxidación formó el derivado dicetónico 105. La reducción con H2IrCl5 condujo a los productos deseados 106 y 107 (Esquema 22).. Esquema 22. Síntesis de 6 azaesteoides propuesta por Kasal57 y colaboradores.. 2.4. Síntesis y actividad biológica de los aza-homoesteroides Los azahomoesteroides son una clase de heteroesteroides en los que alguno de los anillos del núcleo esteroidal ha sido expandido. Como se mencionó anteriormente, estos compuestos han sido reportados como antiproliferativos, antileucémicos y antiandrogénicos.13-18. 24.
(42) Antecedentes. En este contexto, en la literatura se encuentran descritas numerosas síntesis de una serie de compuestos híbridos formados por una A, B o D homolactama esteroidal unida a un agente alquilante. La evaluación biológica de estos nuevos híbridos arrojó que son potentes antileucémicos con más baja toxicidad y mayor actividad antineoplásica en comparación al agente alquilante.11,14,18 Adicionalmente, se observó que la fracción NHCO es crucial para que sean más selectivos a las células malignas.58 Algunos estudios sugieren que la porción de la lactama por sí sola proporciona actividad antileucémica a los híbridos.59 Para formar estos derivados se une la porción 3β de esteroide con el agente alquilante, en el Esquema 23 se observa un ejemplo en el cual se unió el 3β-acetoxi-17a-aza-D-homo-androst-5-eno7,17-diona (108) con el clorambucilo (109), el híbrido fue reportado como un potente antileucémico que mostró baja toxicidad,60 por tanto se utilizó como un fármaco prototipo para sintetizar otros análogos con apoyo de estudios QSAR.. Esquema 23. Ejemplo de híbrido formado por un azaesteroide y un agente alquilante.. Más aún, los azahomoesteroides han sido reportados como antiproliferativos para diversos tipos de carcinoma como cervical, de pulmón, gástrico, nasofaringeal, entre otros.13,15-17 Las rutas sintéticas para obtener aza-homolactamas generalmente implican la formación de una oxima intermediaria que por rearreglo de Beckmann se transforma a la lactama deseada. En el 2003, el grupo de Nikolaropoulos61 describió la síntesis de la B-D-dihomolactama 116 partiendo del acetato de dehidroepiandrosterona 18a. Inicialmente se protegió el carbonilo del C-17 formando el acetal (19). Posteriormente se realizó una oxidación alilíca en el C-7, generando la cetona 111, que a su vez se convirtió al derivado 7-hidroxiimino 112 por condensación con NH2OHHCl en piridina. El tratamiento de 112 con SOCl2 promovió el rearreglo de Beckmann conduciendo a la B-homolactama 113. A continuación, se desprotegió el carbonilo de C-17 y se formó la oxima correspondiente 115. El rearreglo de. 25.
(43) Antecedentes. Beckmann de la oxima generó la lactama en el anillo D (116) (Esquema 24). Posteriormente se acopló por el C-3 con un agente alquilante nitrogenado conduciendo a 117 con el propósito de estudiar su actividad citotóxica.62. Esquema 24. Síntesis de una B,D-dihomolactama esteroidal. En el 2004, Pavlovic13 y colaboradores reportaron la síntesis de dos B-homolactamas, una tipo enamina 121a y otra 7-aza-6-ona 121b partiendo de colesterol (1a). Como primer paso de la ruta se efectuó una oxidación del 5 del colesterol, formándose estereoselectivamente el epóxido 118. El calentamiento a reflujo en DMSO condujo a la enona 119 cuyo tratamiento con NH2OHHCl en EtOH a reflujo formó la mezcla de oximas E y Z (120a y 120b), en 80% y 5% de rendimiento, respectivamente. Para aumentar el rendimiento de la oxima Z, se favoreció una isomerización EZ con h generada por una lámpara de mercurio en presencia de EtOH, piridina y NH2OHHCl. El rearreglo de Beckmann de las oxima E y Z condujeron a las lactamas deseadas 121a y 121b (Esquema 25). Se evaluó la actividad antiproliferativa de las oximas y de las B-homolactamas obtenidas contra dos líneas celulares: HeLa y K562. Los compuestos que presentaron mayor actividad fueron los derivados 121a y 121b contra K562 (IC50 = 5.01 y 6.01 µM, respectivamente).. 26.
(44) Antecedentes. Esquema 25. Síntesis de B-homolactamas propuesta por Pavlovic y col.13 En 2011, Huang y colaboradores63 reportaron la síntesis de las A-homolactamas 125a y 126a (Esquema 26). Una vez más, el precursor fue el derivado con un grupo hidroxiimino 124, que se obtuvo mediante una ruta de cuatro pasos a partir de colesterol (1a). En el último paso de la ruta, se obtuvo una mezcla de productos, la 4-azahomolactama 125a fue el mayoritario (45%) mientras que la 3-azahomolactama 126a fue el minoritario (21.5%). Al compuesto 125a le introdujeron un grupo oxima, semicarbazona y tiosemicarbazona en el C-6. A los productos se les evaluó la actividad antiproliferativa in vitro, el compuesto que presentó mejores resultados contra HeLa (IC50 = 6.5 µM) fue un derivado de 125a con un grupo tiosemicarbazona en el C-6.. Esquema 26. Síntesis de A-homolactamas con actividad citotóxica.. 27.
(45) Antecedentes. En 2011, Komarnytsky64 y colaboradores reportaron un procedimiento para la síntesis de 6aza-brasinoesteroides partiendo de estigmasterol (1d). La síntesis se inició con la mesilación del alcohol en C-3 y posterior tratamiento con NaHCO3, luego una oxidación generó el 6cetoesteroide 127. El tratamiento con NaBr en ácido p-TsOH formó una doble ligadura entre los carbonos 2 y 3 (128). Para la introducción de los dioles en la cadena lateral y en el anillo B se realizó una dihidroxilación asimétrica con RuCl3 en agua. Después de la protección de los alcoholes formados se obtuvo a 129, luego se procedió a la formación de la lactama en el anillo B, para ello, se trató con NaN3 en una mezcla de los ácidos MeSO3H y AcOH. Por último, la desprotección de los alcoholes condujo al compuesto deseado 131 (Esquema 27). Al análogo de brasinoesteroide obtenido se le evaluó la actividad como hormona de crecimiento, sin embargo se observó que la actividad disminuye dramáticamente al cambiar oxígeno por nitrógeno en el anillo B.. Esquema 27. Síntesis de azaesteroides análogos de hormonas de crecimiento vegetal. En el 2014, el grupo de Huang65 diseñó la síntesis de una familia de B-homoazaesteroides con distintos sustituyentes en el C-3 y variando la posición del nitrógeno con el propósito de encontrar nuevos agentes antiproliferativos con menor toxicidad. Los azahomoesteroides que presentaron una actividad similar a la del cis platino fueron 137, 139, 140 en líneas celulares como HeLa y GNE-2. De manera general, el compuesto que presentó mejores valores de actividad fue el 140 (Esquemas 28 y 29). La estrategia sintética utilizada para obtener la serie de lactamas 6-aza-7-ona (137-140) se realizó usando al intermediario dicetónico 26a, que se obtuvo a partir de colesterol (1a) y. 28.
(46) Antecedentes. PCC en CH2Cl2 según se describió en el Esquema 7. La reducción selectiva de 26a con NaBH4 y NiCl26H2O condujo al alcohol 132, el cual se protegió en forma del éster acético y se trató con NH2OHHCl en medio básico para obtener la oxima 133. La reacción de Beckmann de 133 con SOCl2 a 0 °C y la subsecuente hidrólisis básica generó la Bhomolactama 135. La oxidación sobre el alcohol intermediario 135 formó el derivado 3ceto-B-homolactama 136. Por último, el tratamiento de 136 con distintos nucleófilos dio una variedad de derivados con diferentes sustituyentes en el C-3 (Esquema 28).. Esquema 28. Síntesis de B-homoazaesteroides sustituidos en el C-3.. Para obtener los derivados 7-aza se partió de la oxima intermediaria 31a, obtenida a partir de colesterol (1a) mediante una secuencia de 2 pasos descrita en el Esquema 8. Posteriormente, se realizó el rearreglo de Beckmann de la oxima y una hidrólisis básica condujo a la B-homolactama 141, la oxidación con reactivo de Jones generó el compuesto 142. Por último, el tratamiento con dos distintos nucleófilos permitió obtener los derivados 143 y 144. Los productos finales presentaron baja actividad antiproliferativa.. 29.
(47) Antecedentes. Esquema 29. Síntesis de B-homolactamas con actividad citotóxica.. 30.
(48) Objetivos. 3. OBJETIVOS Objetivo General Sintetizar nuevos compuestos esteroidales con grupos oxima, lactama y 6-azaesteroides con potencial actividad biológica antiproliferativa. Objetivos Particulares. 1. Desarrollar u optimizar metodologías para la síntesis de azaesteroides y oximas esteroidales. 2. Proponer mecanismos de reacción que justifiquen la formación de los intermediarios y productos. 3. Evaluar la actividad antiproliferativa de los principales intermediarios y productos nuevos. 4. Caracterizar a los intermediarios y productos finales mediante los distintos métodos espectroscópicos y físicos. 31.
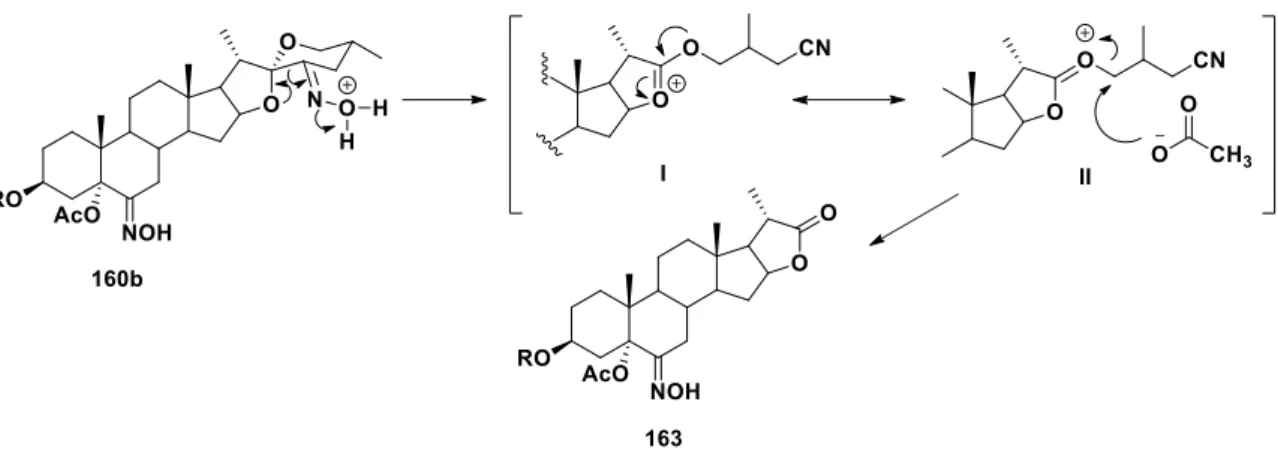
(49) Discusión de Resultados. 4.. DISCUSIÓN DE RESULTADOS. 4.1. Síntesis de oximas esteroidales en la posición 6. Como se describió en el capítulo de antecedentes, esencialmente se encuentran reportadas dos rutas para acceder a oximas en el C-6. La primera consiste en la generación de un epóxido entre los carbonos 5 y 6, y a partir de este la formación de una cetona en el carbono 6 que se condensa con el NH2OHHCl. Esta secuencia puede implicar hasta 5 pasos, ya que es necesario proteger el grupo alcohol del C-3 de los materiales de partida.37 La ruta más corta, reportada por Cui y colaboradores,22 consiste en una secuencia de 3 pasos: la oxidación del 5 para obtener un derivado de tipo 3,6-diona, luego, una reducción selectiva de la cetona en el C-3 con una sal de cobalto y NaBH4 y por último una condensación entre la cetona y el NH2OHHCl. Usando este protocolo, la oxima E se obtiene en un mayor rendimiento (75%) en comparación con la ruta de 5 pasos (menores al 50%). En el contexto de la síntesis de oximas, Iglesias y colaboradores30 propusieron que las 23hidroxiimino sapogeninas son intermediarias en la formación de los respectivos derivados 23-nitroimino cuando la materia prima correspondiente se hace reaccionar con NaNO2, BF3OEt2 en AcOH (Esquema 30). El mecanismo que se sugiere involucra la formación del enol I que reacciona con el ion nitrosonio formado a partir del NaNO2 y el BF3OEt2, generando el intermediario II. La tautomerización nitroso-oxima de II conduce al derivado 23-hidroxiimino III, que reacciona con otra molécula de NO+ para formar IV, el cual se rearregla hacia VI.. Esquema 30. Propuesta de mecanismo de reacción en la transformación de sapogeninas a sus respectivos 23-nitroimino derivados. 32.
(50) Discusión de Resultados. En este mismo reporte, los autores describen también la reacción de nitrosación del acetato de colesterilo (1e), formando en este caso, el diacetato de 6-nitroiminocolestano-3β,5α-diilo (145a) en un 77% de rendimiento (Esquema 31). La introducción de los grupos OAc y NNO2 en las posiciones 5 y 6 del producto se explican mediante la adición electrofílica del ion nitrosonio al 5 y la posterior introducción del grupo acetoxi al C-5 por la cara menos impedida del esteroide (I). Luego de la tautomerización nitroso-oxima se forma la respectiva oxima intermediaria (II), la cual reacciona nuevamente con otro ion nitrosonio y subsecuentemente se rearregla para dar el derivado 6-nitroimino (145a).. Esquema 31. Posible mecanismo de reacción para explicar la transformación de acetato de colesterilo en el derivado 3β,5-diacetoxi-6-nitroimino (145a).. Con estos antecedentes, se propuso sintetizar un derivado 6-hidroxiimino con un menor número de pasos con respecto a las metodologías ya reportadas, buscando aislar la oxima intermediaria II, mostrada en el Esquema 31, mediante la adición Ac2O al medio de reacción descrito por Iglesias. Para realizar este estudio no era posible utilizar como materia prima a una sapogenina, debido a que es bien conocido que cuando estas se hacen reaccionar con la mezcla Ac2O y BF3OEt2, se promueve la apertura del anillo F.66 Utilizando colesterol como material de partida, se realizaron dos ensayos variando la cantidad de Ac2O y AcOH (Esquema 32). En la primera prueba, se adicionaron 20 mL de AcOH, 2.5 mL de Ac2O, 2.5 mL de BF3OEt2 y 1.4 g de NaNO2 a 1 g de colesterol (1a), con lo que se obtuvo el derivado 6-nitroimino 145a y el compuesto esperado, el diacetato de (6E)-6-acetoxiiminocolesta-. 33.
(51) Discusión de Resultados. 3β,5-diilo (146a), en un 40% y 28% de rendimiento, respectivamente (Prueba A, Tabla 1). Cuando se ensayó con una mayor cantidad de Ac2O (15 mL) y menor proporción de AcOH (5 mL) se aumentó el rendimiento de 146a (65% de rendimiento), las cantidades de los otros reactivos no se modificaron y la temperatura se mantuvo entre 15-20 °C (Prueba B, Tabla 1). Es importante mencionar que las condiciones de reacción permitieron esterificar in situ el alcohol del C-3, debido a la presencia del Ac2O y el ácido de Lewis (BF3OEt2).67. Esquema 32. Reacción de colesterol con Ac2O, BF3OEt2 y NaNO2.. Tabla 1. Condiciones de nitrosación del colesterol Colesterol (g). BF3OEt2 (mL). AcOH (mL). Ac2O (mL). NaNO2 (g). A. 1. 2.5. 20. 2.5. 1.4. Rendimiento (%) 145a/146a 28/40. B. 1. 2.5. 5. 15. 1.4. 13/65. Un posible mecanismo para explicar la formación del derivado 146a involucra la formación de la oxima intermediaria I (Esquema 33), esta tiene la posibilidad de reaccionar con dos electrófilos, un ion nitrosonio para generar 145a o atacar nucleofílicamente al grupo acetilo del Ac2O que se encuentra en exceso, lo que explica la obtención de 146a (Esquema 33).. 34.
Figure


+7

Documento similar
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
Package Item (Container) Type : Vial (100000073563) Quantity Operator: equal to (100000000049) Package Item (Container) Quantity : 1 Material : Glass type I (200000003204)
Entre nosotros anda un escritor de cosas de filología, paisano de Costa, que no deja de tener ingenio y garbo; pero cuyas obras tienen de todo menos de ciencia, y aun
Tejidos de origen humano o sus derivados que sean inviables o hayan sido transformados en inviables con una función accesoria.. Células de origen humano o sus derivados que
De hecho, este sometimiento periódico al voto, esta decisión periódica de los electores sobre la gestión ha sido uno de los componentes teóricos más interesantes de la
Ciaurriz quien, durante su primer arlo de estancia en Loyola 40 , catalogó sus fondos siguiendo la división previa a la que nos hemos referido; y si esta labor fue de
