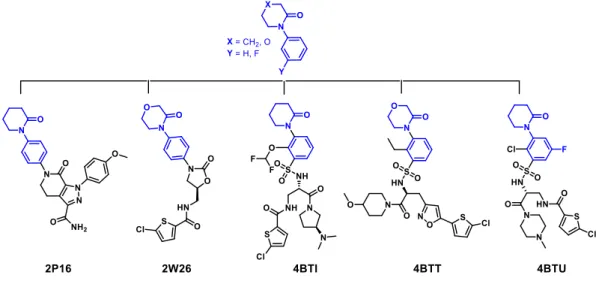
TítuloInnovative three step microwave promoted synthesis of N propargyltetrahydroquinoline and 1,2,3 triazole derivatives as a potential factor Xa (FXa) inhibitors: drug design, synthesis, and biological evaluation
39
0
0
Texto completo
(2) Molecules 2020, 25, 491. 2 of 39. pathways. However, compound 34 may target coagulation FXa mainly by the extrinsic and common pathway. Interestingly, the most active compounds in relation to the inhibition activity against FXa and coagulation parameters did not show toxicity at the performed coagulation assay concentrations. Finally, docking studies confirmed the preferential binding mode of N-propargyltetrahydroquinoline and 1,2,3-triazole derivatives inside the active site of FXa. Keywords: factor Xa inhibitors; microwave-assisted synthesis; N-propargyltetrahydroquinoline; 1,2,3-triazole; cell viability assay; coagulation parameters. 1. Introduction In the last few decades, statistics relating to a high number of cardiovascular syndromes that are a leading cause of heart problems and increasing death rates in Europe and the US have been published. A recent study carried out in Chile found the incidence risk rate for thromboembolic diseases among patients under general surgery is 55% and the principal cause of death in Chile is related to cardiovascular diseases [1,2]. With more than 24,000 deaths annually, cerebrovascular accidents (CVA) represent almost a third of all deaths [3,4] in Chile. Thrombosis is a common causal pathology for three widespread cardiovascular syndromes: acute coronary syndrome (ACS), venous thromboembolism (VTE), and strokes [5,6]. Furthermore, the latest statistical study from the Global Burden of Diseases, Injuries and Risk Factors (GBD) showed that 25% of people worldwide die from thrombosis related events [7]. The coagulation cascade is the process of the conversion of soluble fibrinogen to insoluble fibrin that terminates in production of a clot. This process is the property of plasma where factor X (FX), which is a serine endopeptidase enzyme, plays a vital role in the conversion of prothrombin to thrombin [8]. The clot composition is composed of activated platelets, red blood cells, and cross-linked fibrin protein. A clot, which blocks the bloodstream, can produce acute myocardial infarction (AMI), an ischemic stroke, and deep vein thrombosis (DVT) [9–11]. In the normal circulation system, blood remains in liquid form and does not coagulate. It is now well documented that endothelium cells have an important role in homeostasis maintenance and various pathological conditions. Under healthy conditions, it not only degrades thrombus and downregulates the overall process of thrombosis, but it also promotes thrombosis when damage to the endothelium of vessels occurs. The endothelial cell produces and releases certain factors such as PGI2 (Prostaglandin I2/Prostacycline), nitric oxide (NO), and adenosine diphosphate (ADP), which acts as the antiplatelet aggregating factors [12]. Thrombomodulin or CD141 is another important integral protein expressed in the membrane of the endothelial cells. This integral protein acts as a cofactor in the activation of protein C [13]. As the name suggests it modulates the activity of thrombin to work as the protein C activator. Once activated, protein C inhibits factor Va and VIIIa and thereby inhibits the platelet aggregation and clot formation [14]. Heparan sulphate a highly sulfated polysaccharide is also expressed on the surface of endothelial cells. Its analog heparin is the most commonly use anticoagulant drug in hospitals [15]. Binding of antithrombin with heparan sulfate leads to the conformational changes in the antithrombin. These changes in the antithrombin, generates the active form of antithrombin which not only inhibits the IXa and Xa, but also inhibits the thrombin, i.e., factor IIa. Tissue plasminogen activator is another protein expressed on the endothelial cells which line the internal lumen of blood vessels [16]. During an injury, at the site of action, endothelial cells lose the ability to act as an antiplatelet aggregating activity and became pro-coagulant. Injured endothelial cells release the Von Willebrand factor or factor VIII (FVIII), which is a large glycoprotein complex continuously produced by the endothelial cells [17]. VW in particular binds to the platelets to activate it and to factor VIII to begin the process of the intrinsic coagulation cascade [17–20]. Activated platelets activate the factor XII to.
(3) Molecules 2020, 25, 491. 3 of 42. During an injury, at the site of action, endothelial cells lose the ability to act as an antiplatelet aggregating activity and became pro-coagulant. Injured endothelial cells release the Von Willebrand factor or 2020, factor VIII (FVIII), which is a large glycoprotein complex continuously produced 3by the Molecules 25, 491 of 39 endothelial cells [17]. VW in particular binds to the platelets to activate it and to factor VIII to begin the process of the intrinsic coagulation cascade [17–20]. Activated platelets activate the factor XII to XIIa. XIIa.then XIIaactivates then activates the factor to XIa, which then activatesIXIXtoto IXa. IXa. IXa IXa along XIIa the factor XI toXIXIa, which then activates along with withCa Caions, ions, phospholipids, and factor VIII activates factor X to Xa (FXa). FXa again in the presence of Ca phospholipids, and factor VIII activates factor X to Xa (FXa). FXa again in the presence of Caions, ions, phospholipids,and andfactor factorVV catalyzes catalyzes the the conversion then phospholipids, conversion of ofprothrombin prothrombintotothrombin thrombin[21]. [21].Thrombin Thrombin then converts the fibrinogen to fibrin monomers [22]. Fibrin monomer is then deposited on the primary converts the fibrinogen to fibrin monomers [22]. Fibrin monomer is then deposited on the primary hemostaticplug, plug,which whichthen thencross-links cross-links and and stabilizes factor hemostatic stabilizes itself itself with withthe thehelp helpofoffactor factorXIIIa. XIIIa.Tissue Tissue factor producedby bythe thedamaged damaged endothelial endothelial cells cells also also activates extrinsic produced activates factor factor VII VIItotoVIIa VIIaleading leadingtotothethe extrinsic coagulationcascade. cascade.Factor FactorVIIa VIIaactivates activates factor factor IX IX and factors coagulation and IX IX to to IXa IXaand andXa Xawhich whichare arethe thecommon common factors involvedininthe theintrinsic intrinsiccoagulation coagulationpathway pathway[21,22] [21,22](Figure (Figure1). 1). involved. Figure 1. 1. Blood Blood coagulation coagulation cascade: cascade: extrinsic, intrinsic, and and common common pathway. pathway. Figure extrinsic, intrinsic,. Moreover,Factor FactorXa Xa isis noteworthy noteworthy as the sole extrinsic Moreover, sole and and common commonfactor factorofofintrinsic intrinsicand and extrinsic pathways that are involved in thrombin activation. Considering that one molecule of FXa catalyzes pathways that are involved in thrombin activation. Considering that one molecule of FXa catalyzes the the formation of 1000 thrombin molecules, enzyme plays a central roleininthe thecoagulation coagulationprocess process of formation of 1000 thrombin molecules, thisthis enzyme plays a central role of blood. Finally, serine protease enzyme generates main catalyzing reaction that results blood. Finally, this this serine protease enzyme generates thethe main catalyzing reaction that results in in clot clot production and wound closure [23,24]. Because of its vital role in the coagulation cascade, its production and wound closure [23,24]. Because of its vital role in the coagulation cascade, its inhibition is anapproach important for the development of anticoagulant/antithrombic agents. isinhibition an important forapproach the development of anticoagulant/antithrombic agents. Mortality associated to thrombin is very high, and the currently available prescription drugs to prevent thrombosis in cardiovascular patients include heparins and warfarin. Rivaroxaban and Apixaban are the recently approved orally active agents available for FXa inhibition, whereas Dabigatran inhibits the thrombin directly [25]. Warfarin is the vitamin K antagonist, which, if taken incorrectly, increases the chances of dangerous bleeding. Warfarin, which is available in orally active form, has several disadvantages. It is associated with drug interactions with over the counter drugs and has been associated with severe side effects such as blood in the urine, severe stomach and joint pain, vomiting of blood, and dizziness, among others [26]. Heparins on the other hand, which can only.
(4) prevent thrombosis in cardiovascular patients include heparins and warfarin. Rivaroxaban and Apixaban are the recently approved orally active agents available for FXa inhibition, whereas Dabigatran inhibits the thrombin directly [25]. Warfarin is the vitamin K antagonist, which, if taken incorrectly, increases the chances of dangerous bleeding. Warfarin, which is available in orally active form, has 2020, several disadvantages. It is associated with drug interactions with over the counter drugs Molecules 25, 491 4 of and 39 has been associated with severe side effects such as blood in the urine, severe stomach and joint pain, vomiting of blood, and dizziness, among others [26]. Heparins on the other hand, which can only be be administered parenterally, have high interpatientvariability variabilityinin metabolism, metabolism, and administered parenterally, have high interpatient and hence, hence,require require therapeutic drug monitoring for dose adjustment. Due to its short half-life and low bioavailability, therapeutic drug monitoring for dose adjustment. Due to short half-life and low bioavailability, frequent dosingofofheparin heparinisisalso alsoessential essential [27,28]. Both Both of these frequent dosing these agents agentsrequired requiredmonitoring monitoringofofclotting clotting time and dose adjustments prevent bleeding. time and dose adjustments toto prevent bleeding. discussed earlier,FXa FXaisisthe thepoint pointof ofjunction junction of the intrinsic AsAs discussed earlier, intrinsic and andextrinsic extrinsiccoagulation coagulationpathways. pathways. Inhibition FXacould couldtherefore thereforebe be useful useful to inhibit Inhibition ofofFXa inhibit intrinsic intrinsicand andextrinsic extrinsicpathways. pathways.Furthermore, Furthermore, unlikewarfarin, warfarin, FXa directly inhibit the critical factor involved in the cascade. Thecascade. coagulation unlike FXainhibitors inhibitors directly inhibit the critical factor involved in the The cascade is cascade the process whereby of the enzymes takes place at each which means coagulation is the processamplification whereby amplification of the enzymes takes placestep, at each step, which the concentration of theofFXa always lowerlower than the of theofthrombin. This allows for means the concentration theisFXa is always thanconcentration the concentration the thrombin. This allows the efficient and potent antithrombotic activity of such inhibitors at very low concentrations. On the for the efficient and potent antithrombotic activity of such inhibitors at very low concentrations. On the other hand, severalpreclinical preclinicaland andclinical clinicalinvestigations investigationshave havealready alreadydemonstrated demonstrateda alesser lesserrisk riskof other hand, several of bleeding the direct inhibition of FXa [22,29]. Nevertheless, latest clinical studies have bleeding withwith the direct inhibition of FXa [22,29]. Nevertheless, the the latest clinical studies have just just demonstrated that Rivaroxaban and Apixaban dose interruption could produce thromboembolic demonstrated that Rivaroxaban and Apixaban dose interruption could produce thromboembolic events. events. Moreover, the utilization warfarin combined with Rivaroxaban heightens chance of Moreover, the utilization of warfarinofcombined with Rivaroxaban heightens the chance the of hemorrhage hemorrhage in non-valvular atrial fibrillation patients [30–32]. in non-valvular atrial fibrillation patients [30–32]. recent years, several direct inhibitors have become available inmarketplace, the marketplace, and InIn recent years, several direct FXaFXa inhibitors have become available in the and others are indevelopment active development 2) [33,34]. Although new anticoagulants oral anticoagulants are areothers in active (Figure (Figure 2) [33,34]. Although these these new oral are more more efficacious than warfarin for the prevention of strokes and systemic embolism in patients efficacious than warfarin for the prevention of strokes and systemic embolism in patients with atrial with atrialthe fibrillation, the reversal agent alfa has only recently [35,36]. been approved [35,36]. fibrillation, reversal agent Andexanet alfaAndexanet has only recently been approved The development development of direct antidotes oral directare FXa inhibitors are still inbut the pipeline, but their expected ofThe antidotes for oral FXaforinhibitors still in the pipeline, their expected approval for approval for therapeutic purposes be further beneficial to anticoagulation therapeutic purposes will be further will beneficial to anticoagulation therapy [37,38].therapy [37,38].. Figure Currently clinically used FXa Inhibitorsand andthe theFood Foodand andDrug DrugAdministration Administration(FDA) (FDA)year yearof Figure 2. 2. Currently clinically used FXa Inhibitors of approval. approval.. NovelFXa FXainhibitors inhibitorsunder under development development are are mainly Novel mainly structural structural bioisosteres bioisosteres ofofexisting existingdrugs drugs (Figure3).3).They Theyshow showbetter betterefficacy efficacy and and have have aa higher (Figure higher safety safetyprofile. profile.Several Severalamidines, amidines,peptide, peptide, pyrrolidines, azetidines, and triazoles are amongthe thenewly newlyreported reportedcompounds compounds[39,40].xd [39,40].xd pyrrolidines, azetidines, and triazoles are among. Molecules 2020, 25, 491. 5 of 42. Within the approved FXa inhibitors, the phenyloxomorphonilo or phenyloxopiperidine scaffolds present in Apixaban and Rivaroxaban are important anchoring moieties because along with the S1 site, S4 is the most important binding region for factor Xa inhibitors [39]. These groups have also shown to be pivotal for potency in that these two drugs were the first approved FXa inhibitors [33,34].. Figure 3.3.(a) Rivaroxaban as as gold goldstandard standardreference referencecompounds. compounds.The The 4 binding Figure (a)Apixaban Apixabanand and (b) (b) Rivaroxaban S4 Sbinding scaffolds phenyloxopiperidine or phenyloxomorphonilo are highlighted in blue. scaffolds phenyloxopiperidine or phenyloxomorphonilo are highlighted in blue.. Consequently, FXa inhibitors are the critical milestones to be achieved in the clinical management Within the approved FXa inhibitors, the phenyloxomorphonilo or phenyloxopiperidine scaffolds ofpresent thrombosis. In recent years, various pharmacological activities N-heterocyclic in Apixaban and Rivaroxaban are important anchoring moieties of because along with compounds the S1 site, including tetrahydroquinoline (Figure 4d) have received immense importance because of their various pharmacological activities. Heterocyclic compounds containing nitrogen not only play a crucial role in system biology but are the scaffold essential for the development of new drugs. Tetrahydroquinolines (THQs) are found to be the basic skeleton of numerous natural and synthetic compounds with various biological properties. For example, alkyl- and arylalkylderivatives of THQ such as (−)-angustureine, (−)-cuspareine, (−)-galipeine, and (−)-galipinine, which were extracted from the angostura (Galipea.
(5) Molecules 2020, 25, 491. 5 of 39. S4 is the most important binding region for factor Xa inhibitors [39]. These groups have also shown to be pivotal for potency in that these two drugs were the first approved FXa inhibitors [33,34]. Consequently, FXa inhibitors are the critical milestones to be achieved in the clinical management of thrombosis. In recent years, various pharmacological activities of N-heterocyclic compounds including tetrahydroquinoline (Figure 4d) have received immense importance because of their various pharmacological activities. Heterocyclic compounds containing nitrogen not only play a crucial role in system biology but are the scaffold essential for the development of new drugs. Tetrahydroquinolines (THQs) are found to be the basic skeleton of numerous natural and synthetic compounds with various biological properties. For example, alkyl- and arylalkylderivatives of THQ such as (−)-angustureine, (−)-cuspareine, (−)-galipeine, and (−)-galipinine, which were extracted from the angostura (Galipea officinalis, Angostura trifoliata), tree have a variety of medicinal properties [41–43]. Different 1,2,3,4-tetrahydroquinolines with long alkyl chains at C-5 (Figure 4a) were extracted from a combined culture of Streptomyces nigrescens HEK616 and Tsukamurella pulmonis TP-B0596. These compounds were found to inhibit the growth of microorganisms [44]. Several 4-dioxygenated 3,4-dihydro 4-aryl-1,2,3,4-tetrahydroquinolin-2-(1H)-ones have been extracted and isolated from a variety of plants and fungi with a variety of medicinal properties [45]. For example, aflaquinolones A–G (Figure 4b), scopuquinolone B, aniduquinolones A–C, 6-deoxyaflaquinolone F, isoaflaquinolone E, 14-hydroxyaflaquinolone E, aspoquinolones A–D were also isolated from the various fungus belonging to the specie Aspergillus. These compounds have shown to have useful medicinal properties [46–48]. Moreover, some 1,4-disubstituted 1,2,3,4-tetrahydroquinoline derivatives (Figure 4c) were tested against HIV and few N-substituted tetrahydroquinoline derivatives were found to inhibit the reverse transcriptase enzyme of HIV-1 virus [49–52]. Previously, Quan et al. reported the synthesis of tetrahydroquinoline derivatives compounds having reversible factor XIa inhibitor activity in rats and rabbit thrombosis animal modes, whereas tetrahydroquinoline scaffold (Figure 4d) was assayed for its direct Factor Xa inhibition activity [52,53]. Similarly, 1,4-disubstituted-1,2,3-triazole derivatives (Figure 4e) have become an important pharmacophore due to its various biological properties and excellent stability. More recently, triazole has also been studied for its anticoagulant activity. For example, the hydrazone and sulfonamide derivatives of triazole were studied for their antiplatelet Molecules 2020, 25, 491 6 of 42 activity [54,55].. Figure 4. (a) 5-heptyl-1,2,3,4-tetrahydroquinoline [44]. (b) Aflaquinolone A [46]. (c) 1,4-disubstituted Figure 4. (a) 5-heptyl-1,2,3,4-tetrahydroquinoline [44]. (b) Aflaquinolone A [46]. (c) 1,4-disubstituted 1,2,3,4-tetrahydroquinoline [50] (d) Tetrahydroquinoline chemical structure. (e) 1,4-disubstituted-1,2,31,2,3,4-tetrahydroquinoline [50] (d) Tetrahydroquinoline chemical structure. (e) triazole chemical structure. 1,4-disubstituted-1,2,3-triazole chemical structure.. The synthesis of current antithrombotic agents is highly inefficient given that it has high costs and involves several synthetic steps, and specific catalysts. Considering the multiple-step synthesis development of commercially available FXa inhibitors [56,57], it makes the synthesis of these kind of inhibitors a challenge for the medicinal chemistry field. Since the first reported microwave utilization techniques in organic synthesis in 1986, microwave.
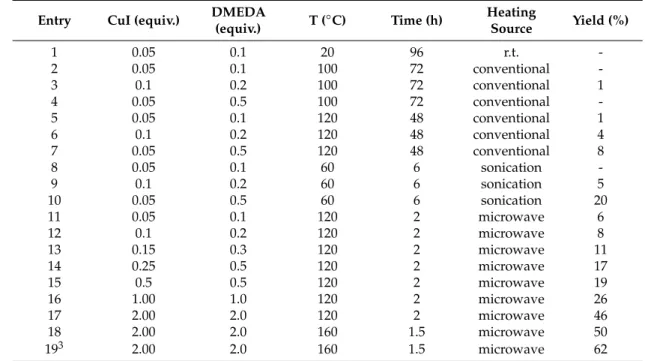
(6) Molecules 2020, 25, 491. 6 of 39. The synthesis of current antithrombotic agents is highly inefficient given that it has high costs and involves several synthetic steps, and specific catalysts. Considering the multiple-step synthesis development of commercially available FXa inhibitors [56,57], it makes the synthesis of these kind of inhibitors a challenge for the medicinal chemistry field. Since the first reported microwave utilization techniques in organic synthesis in 1986, microwave techniques have become a recognized laboratory system and they have an important synthetic approach in the field of chemistry [58]. Moreover, these methodologies are therefore being used as an important synthetic tool [59–63]. For these reasons, our group has been interested in using microwave approaches to achieve our synthetic goals [64–67] in the last few years. To this end, we have reported a modified Ullmann type procedure for related main core structures using microwave-assisted methodologies [67]. Currently, the utilization of microwave-assisted methodologies allows for the desired organic molecules in only three steps to be reached which involves Ullmann-Goldberg, N-propargylation, Mannich addition, Friedel-Crafts, and 1,3-dipolar cycloaddition type reactions. Hence, in our continuous search for decisive FXa inhibitors, several approaches in designing synthesis and characterization novel FXa inhibitors [68] have been undertaken. The present work includes the synthesis of novel tetrahydroquinoline and triazole derivatives by using microwave-assisted synthetic methodologies along with modern computer-aided drug designing and docking studies. Finally, in vitro and ex vivo assays have been performed in order to analyze the bioactivity of the molecules FXa. 2. Results and Discussion 2.1. Protein-Ligand Complexes Selection and Ligand Clustering First, the Protein Data Bank (PDB) (www.rcsb.org) [69] was searched for human FXa-ligand complexes available crystal structures, using the appropriate Uniprot accession number (P00742). A set Molecules 25, 491 7 of 42 of 130 2020, enzyme-ligand reported complexes were found for humans, and after substructure search using the phenyloxomorphonilo or phenyloxopiperidine scaffolds present in Apixaban and Rivaroxaban, the phenyloxomorphonilo or phenyloxopiperidine present retrieved in Apixaban and a set of 5 FXa-ligand complexes was compiled andscaffolds their structures from theRivaroxaban, PDB (Figure a5).set of 5 FXa-ligand complexes was compiled and their structures retrieved from the PDB (Figure 5).. Figure Virtual screening Figure 5. 5. Virtual screening of of interest interest fragment fragment in in FXa FXa PDB PDB crystallography crystallography data data base. base.. After structural the protein corresponding to the cluster (Root-meanAfter structural alignment alignmentagainst against the protein corresponding to center the cluster center square deviation connected component, RMSD CC), the root-mean square deviation for the α-carbon (Root-mean-square deviation connected component, RMSD CC), the root-mean square deviation for the for all residues and residues within 6within Å from center massofofmass all co-crystallized ligandsligands was α-carbon for all residues and residues 6 Åthe from theof center of all co-crystallized measured (Table 1). 1). was measured (Table Table 1. Summary of crystal structures of Factor Xa-ligand complexes used in this study.. PDB ID (Reference). Resolution (Å). RMSD Cluster Center (Å). RMSD Binding Site (6Å) 1. 2. Tanimoto Distance.
(7) Molecules 2020, 25, 491. 7 of 39. Table 1. Summary of crystal structures of Factor Xa-ligand complexes used in this study. PDB ID (Reference). Resolution (Å). RMSD Cluster Center (Å). 4BTI [70] 4BTU [70] 4BTT [70] 2W26 Rivaroxaban [34] 2P16 Apixaban [71]. 2.30 2.37 2.59 2.08 2.30. —– 0.440 0.530 0.276 0.290. 1. 1. RMSD Binding Site (6Å). All atoms within 6 Å from the center of mass of ligand alignment.. 2. —– 0.222 0.381 0.207 0.381 2. Tanimoto Distance 0.000 0.426 0.582 0.618 0.689. Tanimoto coefficient.. The alpha-carbon root-mean square deviation (CαRMSD) against the low-resolution FXa-ligand structure (4BTI) ranged from 0.276 to 0.530 Å, while the all-atom RMSD within 6 Å from the center of mass of all aligned ligands ranged from 0.207 to 0.381 Å, indicating that proteins have no significant conformational differences within their binding sites in comparison with the reference structure. The three-dimensional (3D) alignment of the ligands was used to generate a shape-based query, which was validated using a library of decoy [72,73]. The enrichment curve plots the number of active compounds recovered versus the proportion of the database screened. The AUC (area under the curve of the Receiver Operating Characteristic (ROC) plot) is defined as the probability that a randomly-chosen active compound has a higher score than a randomly-chosen inactive compound. The AUC of the probability obtained for the hypotheses is higher than 99% at ±95% confidence (Figure 6), suggesting the shape query hypothesis can be considered highly selective when using the actives that Molecules 2020, 25, 491correspond to each cluster. However, when using a more diverse collection of FXa 8 of 42 inhibitors from the extended database of useful decoys (DUD) [74], the AUC probability falls to near 77%. Although most actives rank higher than of most the decoy molecules, the query is considered Although most actives rank higher than most theof decoy molecules, the query is considered mildly mildly selective ofscoring the top solutions scoring solutions were retrieved. selective and onlyand 5% only of the5% top were retrieved.. Figure 6. Shape-based query for phenyloxomorphonilo or phenyloxopiperidine containing FXa Figure 6. Shape-based query for phenyloxomorphonilo or phenyloxopiperidine containing FXa inhibitors. (a) Three-dimensional (3D) structural alignment of ligands after protein complexes inhibitors. (a) Three-dimensional (3D) structural alignment of ligands after protein complexes superposition. (b) Shape-based query derived with color representing the combined shape with superposition. (b) Shape-based query derived with color representing the combined shape with chemical chemical features represented as spheres (hydrophobic = brown; rings = green; HB acceptor = red; HB features represented as spheres (hydrophobic = brown; rings = green; HB acceptor = red; HB donor = donor = blue). (c) ROC area under the curve. HB: hydrogen bonding blue). c) ROC area under the curve. HB: hydrogen bonding. After the generation of the shape-based query, the virtual screening protocol was applied to an in After the generation of the shape-based query, the virtual screening protocol was applied to an in house developed library. The shape similarity between the screened compounds was evaluated by the house developed library. The shape similarity between the screened compounds was evaluated by the Tanimoto Combo score method, which consists of the Tanimoto coefficient and the score retrieved from Tanimoto Combo score method, which consists of the Tanimoto coefficient and the score retrieved from the ROCS color force field, which represents the structural complementarity between the template and the ROCS color force field, which represents the structural complementarity between the template and the screened molecules. A final set of 28 molecules was finally prioritized and selected for synthesis the screened molecules. A final set of 28 molecules was finally prioritized and selected for synthesis (Scheme 1). (Scheme 1).. 2.2. Synthesis of Novel Derivatives The synthetic strategy applied for preparation of selected tetrahydroquinolines and 1H-1,2,3-triazole molecules was performed by using Ullmann-Goldberg, N-propargylation, Mannich addition, Friedel-Crafts, and 1,3-dipolar cycloaddition type reactions (Scheme 1)..
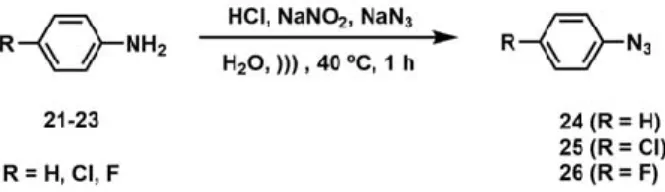
(8) Molecules 2020, 25, 491. 8 of 39. Molecules 2020, 25, 491. 9 of 42. 0 -dimethylethylenediamine Scheme1.1. General General synthetic compounds. (a) (a) N,NN,N’-dimethylethylenediamine Scheme synthetic pathway pathwayfor forselected selected compounds. ◦ (DMEDA),CuI, CuI,KK , PhMe, microwave irradiation (MW), 160 C. (b) propargylbromide, KCO CO3,3 ,KI, 3 PO (DMEDA), 3PO 4, 4PhMe, microwave irradiation (MW), 160 °C. (b) propargylbromide, K22 ◦ C. (c) formaldehyde, 1-vinyl-2-pyrrolidinone, InCl , CH CN, MW, 160 ◦ C. KI, CH CN, MW, 160 3 3 3 CH3CN, MW, 160 °C. (c) formaldehyde, 1-vinyl-2-pyrrolidinone, InCl3, CH3CN, MW, 160 °C. (d) HCl, (d) HCl, NaNO2 , NaNO3 , H2 O, 40 ◦ C. (e) (11–14), (24–26), copper nanoparticles supported on ZnO NaNO 2, NaNO3, H2O, ))), 40 °C. (e) (11–14), (24–26), copper nanoparticles supported on ZnO (CuNPs/ZnO), Et3 N, tetrahydrofuran (THF), 160 ◦ C. (CuNPs/ZnO), Et3N, tetrahydrofuran (THF), 160 °C.. 2.2. Synthesis of Novel Derivatives 2.2.1. Synthetic Methodology Improvements for Aniline Precursors The synthetic strategy applied for preparation of selected tetrahydroquinolines and 1H-1,2,3-triazole Through the performed application by of ausing methodology study, the performance of the Ullmann-type reaction molecules was Ullmann-Goldberg, N-propargylation, Mannich addition, has been improved. In previous research of our group, where the fluorine atom was not included, we Friedel-Crafts, and 1,3-dipolar cycloaddition type reactions (Scheme 1). observed a predictable behavior in the reaction and obtained a moderate yield, without the need to exceed the solvent boiling pointImprovements [75]. In this case, our knowledge of the Arrhenius equation, in 2.2.1. Synthetic Methodology for applying Aniline Precursors which the energy transmitted by microwaves affects the parameters of pressure, heating and especially Through the application of a methodology study, the performance of the Ullmann-type reaction time. The reaction time in all C-N couplings could be reduced for the synthesis of anilines when we has been improved. In previous research of our group, where the fluorine atom was not included, we increase the temperature to 160 °C [76–78]. observed a predictable behavior in the reaction and obtained a moderate yield, without the need to In a theoretical study of C-arylations with aryl halides [79], the authors have explained the reaction exceed the solvent boiling point [75]. In this case, applying our knowledge of the Arrhenius equation, in reactivity between the corresponding lactams and 4-iodoaniline compound [67]. According to this, the which the energy transmitted by microwaves affects the parameters of pressure, heating and especially reaction conditions were optimized by using lactam 1 (1.2 equivalents), aniline 5 (1 equivalent), CuI as time. The reaction time in all C-N couplings could be reduced for the synthesis of anilines when we catalyst and N,N’-dimethylethylenediamine (DMEDA) as ligand, in toluene as solvent using different increase the temperature to 160 ◦ C [76–78]. heating sources (Table 2). The corresponding isolated compound 6 was obtained in good yield [80] In a theoretical study of C-arylations with aryl halides [79], the authors have explained the reaction (Scheme 2). reactivity between the corresponding lactams and 4-iodoaniline compound [67]. According to this, the reaction conditions were optimized by using lactam 1 (1.2 equivalents), aniline 5 (1 equivalent), CuI as catalyst and N,N0 -dimethylethylenediamine (DMEDA) as ligand, in toluene as solvent using different heating sources (Table 2). The corresponding isolated compound 6 was obtained in good yield [80] (Scheme 2). Scheme 2. Synthesis of aniline derivative 6 [80]..
(9) Molecules 2020, 25, 491 Molecules 2020, 25, 491. 10 of 42 9 of 39. Table 2. Optimization of reaction conditions for the synthesis of compound 6. 1,2. Table 2. Optimization of reaction conditions for the synthesis of compound 6. 1,2. Heating DMEDA for selected Scheme 1. General synthetic pathway compounds. (a) N,N’-dimethylethylenediamine (%) (%) TT(◦(° C) Time(h) (h) CuI (equiv.) Entry Entry CuI (equiv.) DMEDA (equiv.) C) Time Heating Source YieldYield Source (equiv.) (DMEDA), CuI, K3PO4, PhMe, microwave irradiation (MW), 160 °C. (b) propargylbromide, K2CO3, KI, 1 CH3CN, 0.05160 °C. 0.10.1 1-vinyl-2-pyrrolidinone, 20 9696InCl3, CH3CN,r.t. r.t. 1 MW, 0.05(c) formaldehyde, 20 - HCl, MW, 160 °C. (d) 2 0.05 0.1 100 72 conventional 2 NaNO2, NaNO 0.05 3, H2O, ))), 40 0.1 72 nanoparticles conventional °C. (e) (11–14), 100 (24–26), copper supported on ZnO 3 0.1 0.2 100 72 conventional 1 (CuNPs/ZnO), Et 3N, tetrahydrofuran (THF), 160 °C. 3 0.1 0.20.5 100 7272 conventional 1 4 0.05 100 conventional 5 0.05 120 conventional 1 4 0.05 0.50.1 100 7248 conventional 2.2.1. Synthetic Methodology Improvements for Aniline Precursors 6 0.1 0.2 120 48 conventional 4 5 0.05 0.10.5 120 4848 conventional 1 7 0.05 120 conventional 8 Through the application of a methodology study, the performance of the Ullmann-type reaction 8 0.05 0.1 60 6 sonication 6 0.1 0.2 120 48 conventional 4 has been improved. In0.1 previous research where the atom was not included, we 9 0.2 of our group, 60 6 fluorinesonication 5 7 0.05 0.5 120 48 conventional 8 10 0.05 0.5 60 6 sonication 20 observed a predictable behavior in the reaction and obtained a moderate yield, without the need to 11 0.05 0.05 120 2 microwave 6 8 0.10.1 60 sonication exceed the solvent boiling point [75]. In this case, applying our 6knowledge of the Arrhenius equation, in 12 0.1 0.2 120 2 microwave 8 which by microwaves affects of pressure, heating and 9 the13energy 0.1 transmitted 0.20.3 60the parameters 62 sonication 5 0.15 120 microwave 11 especially time. The reaction time in all C-N couplings could be reduced for the synthesis of anilines when 14 0.25 0.5 120 2 microwave 17 10 0.05 0.5 60 6 sonication 20 we 15 temperature 0.5 to 160 °C [76–78]. 0.5 120 2 microwave 19 increase the 11 0.11.0 120 22 microwave 6 16 0.05 study 1.00 of C-arylations microwave In a theoretical with aryl120 halides [79], the authors have explained 26 the reaction 17 microwave 46 12 0.1 the2.00 0.22.0lactams and120 120 2 2compound microwave 8 the reactivity between corresponding 4-iodoaniline [67]. According50to this, 18 2.00 2.0 160 1.5 microwave 13 0.3by 1201 (1.2 equivalents), 21.5 microwave reaction conditions aniline 5 (1 equivalent), CuI as 193 0.15 were 2.00optimized 2.0 using lactam 160 microwave 62 11 catalyst and N,N’-dimethylethylenediamine as ligand, in mL). toluene asofsolvent using different 2 microwave 14 1 Conditions: 0.25lactam (1.2 equiv.), aniline 0.5 (1 equiv.),(DMEDA) 2 (3.5 17 K3 PO120 Yield isolated compound. 4 (2 equiv.), toluene 3 Reaction carried out using 1 equiv. of lactam and 1.2 equiv. of aniline. r.t.: room temperature. heating sources (Table 2). The corresponding isolated compound 6 was obtained in good yield [80] 15 0.5 0.5 120 2 microwave 19 (Scheme 2). 16 1.00 1.0 120 2 microwave 26. 17. 2.00. 2.0. 120. 2. microwave. 46. 18. 2.00. 2.0. 160. 1.5. microwave. 50. 2.00. 2.0. 160. 1.5. microwave. 62. 19. 3. Conditions: lactam (1.2 equiv.), aniline (1 equiv.), K3PO4 (2 equiv.), toluene (3.5 mL). Yield of isolated 2. aniline derivative 66 [80]. Scheme 2. Synthesis Synthesis oflactam aniline derivative [80]. compound. 3 Reaction carriedScheme out using 1 equiv. ofof and 1.2 equiv. of aniline. r.t.: room temperature 1. 2. Whenone oneofofthe theproposed proposedmechanisms mechanisms for Ullmann-Goldberg Ullmann-Goldberg type was When typereaction reactionwas wasanalyzed, analyzed,it it was noticedthat thatboth boththe thecatalyst catalystand and the the ligand ligand bind directly noticed directly to tothe thelactam lactamininthe theintermediate intermediateactivation activation step[81]. [81].This Thispremise premiseallowed allowed us to stoichiometry, changing the the limiting reagent to step to change changethe thereaction reaction stoichiometry, changing limiting reagent 1, obtaining satisfactorily 62% yield isolated under microwave conditions. tolactam lactam 1, obtaining satisfactorily 62% ofyield of compound isolated compound under heating microwave heating This modification resulted inresulted a 12% higher yield of theyield starting materials previously reported conditions. This modification in a 12% higher of the startingthan materials than previously yields (Table entry 1, 19). reported yields1,(Table entry 19). Althoughfor forpiperazinone, piperazinone,morpholinone morpholinone and thiomorpholinone Although thiomorpholinone derivatives, derivatives,the thesynthesis synthesiswas was carried out with a lower catalystloading loading(0.5 (0.5equiv.) equiv.)and andligand ligand(0.5 (0.5equiv.) equiv.)(Scheme (Scheme3).3). carried out with a lower catalyst. Scheme 3. Synthesis of Anilines Derivatives (7–9) [80].. The aniline derivatives 7, 8, and 9 were obtained in excellent yield (90%, 86%, and 94%, respectively) of isolated products under microwave reaction conditions in 1–2 h. However, the coupling reaction for compound 7 was carried out at 90 ◦ C, due to the deprotection side reaction. This methodological improvement allowed us to produce these interesting block buildings anilines in excellent yields, reducing reaction time and minimizing the energy consumption by using microwave technologies..
(10) The aniline derivatives 7, 8, and 9 were obtained in excellent yield (90%, 86%, and 94%, respectively) of isolated products under microwave reaction conditions in 1–2 h. However, the coupling reaction for compound 7 was carried out at 90 °C, due to the deprotection side reaction. This methodological improvement allowed us to produce these interesting block buildings anilines in Molecules 2020, 25, 491 10 of 39 excellent yields, reducing reaction time and minimizing the energy consumption by using microwave technologies. 2.2.2. Synthesis of N-propargylanilines 2.2.2. Synthesis of N-propargylanilines In the next step, N-propargyl anilines were synthesized according to the procedure developed In the next step, N-propargyl anilines were synthesized according to the procedure developed by by Rasool et al. with minor modifications such as temperature, time reaction and heating source Rasool et al. with minor modifications such as temperature, time reaction and heating source (Scheme 4) (Scheme 4) [82]. The reaction conditions were optimized using different heating sources (Table 3). [82]. The reaction conditions were optimized using different heating sources (Table 3). The best results The best results were achieved using the aniline derivatives (6–9), propargyl bromide, K2 CO3 as base, were achieved using the aniline derivatives (6–9), propargyl bromide, K2CO3 as base, KI as additive, in KI as additive, in acetonitrile as a solvent under microwave irradiation at 160 ◦ C. N-propargyl aniline acetonitrile as a solvent under microwave irradiation at 160 °C. N-propargyl aniline derivatives (11–14) derivatives (11–14) were obtained in good yields of isolated products (62–72%). However, the synthesis were obtained in good yields of isolated products (62%–72%). However, the synthesis of compound 13 of compound 13 had to be carried out at 90 ◦ C, due to the deprotection side reaction [83–86]. had to be carried out at 90 °C, due to the deprotection side reaction [83–86].. Scheme 4. Synthesis of N-allyl/propargyl aniline derivatives (11–14) (11–14) [80]. [80]. Table 3. Optimization of reaction conditions for the synthesis of compounds 11–14.1,2. Table 3. Optimization of reaction conditions for the synthesis of compounds 11–14.1,2. Yield (%) Entry Heating Source T (◦ C) Time (h) Yield 11 12 13 (%) 14. Entry 1 2 13 24 5 36 47. Heating Source. r.t. conventional r.t. conventional conventional conventional sonication conventional sonication microwave conventional. 20 80 80 90 40 80 160. T (°C) 20 80 80 90. 96 24 72 24 15 4 0.5. Time (h) 13 31 9636 2437 43 7255 2469. 19 11 40 13 47 49 31 57 36 67 72 37. 1210 13 29 1934 4037 45 4751 4970. 10 29 34 37. 914 15 309 3415 39 4330 6734. 1. Conditions: aniline derivatives (1.2 equiv.), propargyl 5 sonication 40bromide (1 equiv.), 15 K2 CO3 (1.543equiv.), 57KI (0.145equiv.), 39in acetonitrile (3.5 mL). 2 Yield of isolated compounds. r.t.: room temperature. 6. sonication. 80. 4. 55. 67. 51. 43. 69 of72 70 compounds 67 7 microwave 0.5 lower yields Noteworthy, the propargyl derivatives were 160 obtained with the isolated ◦ C, 4–15 h, 39–67%), and conventional 1 Conditions: using room temperature (96 h, 9–19% yield), sonication (40–80 aniline derivatives (1.2 equiv.), propargyl bromide (1 equiv.), K2CO3 (1.5 equiv.), KI (0.1 heating sources (80–90 ◦(3.5 C, 24–72 15–49%). In contrast, the microwave procedure showed the highest equiv.), in acetonitrile mL). 2 h, Yield of isolated compounds. r.t.: room temperature products yields (67–72%) within 30 min of reaction at 160 ◦ C. As a second reaction step, the microwave Noteworthy, the propargyl derivatives were obtained with the lower yields of isolated compounds methodology allowed us to produce the propargyl derivatives in an efficient and environmentally using room temperature (96 h, 9–19% yield), sonication (40–80 °C, 4–15 h, 39–67%), and conventional friendly approach. Molecules 2020, 25, 491 12 of 42 heating sources (80–90 °C, 24–72 h, 15–49%). In contrast, the microwave procedure showed the highest products yields (67–72%) within 30 min of reaction at 160 °C. As a second reaction step, the microwave 2.2.3. Synthesis of N-Propargyl Tetrahydroquinolines 2.2.3. Synthesisallowed of N-Propargyl Tetrahydroquinolines methodology us to produce the propargyl derivatives in an efficient and environmentally Different N-propargyl tetrahydroquinolines (17–20) were synthesized using acid-catalyzed friendly approach. Different N-propargyl tetrahydroquinolines (17–20) were synthesized using acid-catalyzed three-component cationic imino Diels–Alder reaction [87–89] (Scheme 5). three-component cationic imino Diels–Alder reaction [87–89] (Scheme 5).. . Scheme 5. Synthesis of N-propargyl tetrahydroquinoline derivatives (17–20). Scheme 5. Synthesis of N-propargyl tetrahydroquinoline derivatives (17–20).. The reaction conditions optimization involved the modification of the heating source [90,91]. Table 4. Optimization of reaction conditions for the synthesis of compounds 17–20.1,2..
(11) . Scheme 5. Synthesis of N-propargyl tetrahydroquinoline derivatives (17–20).. The reaction conditions optimization involved the modification of the heating source [90,91]. Molecules 2020, 25, 491. 11 of 39. Table 4. Optimization of reaction conditions for the synthesis of compounds 17–20.1,2.. The reaction conditions optimization involved the modification of the heating source Yield (%) [90,91]. Entry Heating T (°96 C) h to 15 Time (h) 4) through microwave-assisted Thus, the total reaction timeSource was reduced from min (Table 17 18 19 20 methodology and compound yields were 2.5-fold increase in comparison with conventional procedures. 32 34 36 were It must be1 considered that r.t. compound 18 was 20 not obtained,96 only decomposition products obtained [87]. Novel tetrahydroquinolines derivatives 17, 19, and 20 were obtained in excellent 2 conventional 80 24 47 45 52yields (83–89%) of 3 isolated products. conventional 80 72 53 54 59. 4 Table 4. Optimization conventional 90 for the synthesis 24 of compounds 59 56 1,2. of reaction conditions 17–20. 5 sonication 40 8 69 64 Entry 6. Heatingsonication Source. T. (◦ C). 80Time (h). 217. Yield (%) - 19 72 1872. 62 67 73 20. 83 microwave 160 96 0.5 - 34 89 36 17 r.t. 20 32 -85 1 Conditions: 2 80 24 formaldehyde 47 45 (1.1 equiv.), 52 conventional N-propargyl aniline derivatives (1 equiv.), 30%- in methanol 3 80 72 53 54 59 conventional 2 InCl3 (0.2 equiv.), KI (0.1 equiv.), in acetonitrile (3 mL). Yield of isolated compounds. 4 90 24 59 56 62 conventional 40 reduced 8 from 96 69h to 15 - min 64 Thus, 5 the total sonication reaction time was (Table 4)67 through 6 sonication 80 2 72 comparison 73 microwave-assisted methodology and compound yields were 72 2.5-fold increase in with 7 microwave 160 0.5 85 89 83. conventional procedures. It must be considered that compound 18 was not obtained, only 1 Conditions: N-propargyl aniline derivatives (1 equiv.), formaldehyde 30% in methanol (1.1 equiv.), InCl (0.2 3 decomposition products were obtained [87]. Novel tetrahydroquinolines derivatives 17, 19, and 20 were equiv.), KI (0.1 equiv.), in acetonitrile (3 mL). 2 Yield of isolated compounds. obtained in excellent yields (83–89%) of isolated products. 2.2.4. Synthesis of Aryl Azides 2.2.4. Synthesis of Aryl Azides The synthesis of the aryl azides were based on the reaction of diazonium salts with sodium azide The synthesis of the aryl azides were based on the reaction of diazonium salts with sodium azide in in water under sonication at 40 ◦ C (Scheme 6) [92]. The corresponding products were obtained in water under sonication at 40 °C (Scheme 6) [92]. The corresponding products were obtained in quantitative yields and they were used without further purification. quantitative yields and they were used without further purification.. Molecules 2020, 25, 491. 13 of 42. Scheme 6. Synthesis of aryl azides derivatives (24–26). Scheme 6. Synthesis of aryl azides derivatives (24–26).. 2.2.5. Synthesis of 1H-1,2,3-triazole derivatives 2.2.5. Synthesis of 1H-1,2,3-triazole derivatives The synthesis of novel triazoles were prepared by the 1,3-dipolar cycloaddition between the The synthesis of novel triazoles were prepared by the 1,3-dipolar cycloaddition between the N-propargyl amines (11–14) and the organic azide (24–26) catalyzed by copper nanoparticles supported N-propargyl amines (11–14) and the organic azide (24–26) catalyzed by copper nanoparticles supported on ZnO in tetrahydrofuran as solvent, triethylamine as base at 160 ◦ C under microwave irradiation on ZnO in tetrahydrofuran as solvent, triethylamine as base at 160 °C under microwave irradiation (Scheme 7) [93]. (Scheme 7) [93].. Scheme Synthesis 1H-1,2,3-triazolederivatives derivatives (27–38). (27–38). Compounds Compounds 27–29 Scheme 7. 7. Synthesis ofof1H-1,2,3-triazole 27–29 (X (X== CH CH22;; RR== H, H,Cl, Cl,F,F, respectively); 30–32 (X = N-Boc; R = H, Cl, F, respectively); 33–35 (X = S; R = H, Cl, F, respectively); respectively); 30–32 (X = N-Boc; R = H, Cl, F, respectively); 33–35 (X = S; R = H, Cl, F, respectively); 36–38 R= Cl, F, respectively). Novel 1H-1,2,3-triazole derivatives were obtained (X 36–38 = O; R(X= =H,O;Cl, F,H, respectively). Novel 1H-1,2,3-triazole derivatives (27–38) (27–38) were obtained in goodinto good to excellent yield (73–93%) using a lower loading of catalyst and in shorter reaction time (15 min) excellent yield (73–93%) using a lower loading of catalyst and in shorter reaction time (15 min) than than previously reported by assisted microwave methodologies. previously reported by assisted microwave methodologies.. 2.3. Theoretical Study In order to understand the relationship between the structure and the energetic behavior of all the series, including a thermochemical analysis associated to the yield of the reactions, an optimization and reactivity indexes calculation of electronic chemical potential (μ), chemical hardness (η), and electrophilicity (ω) was performed..
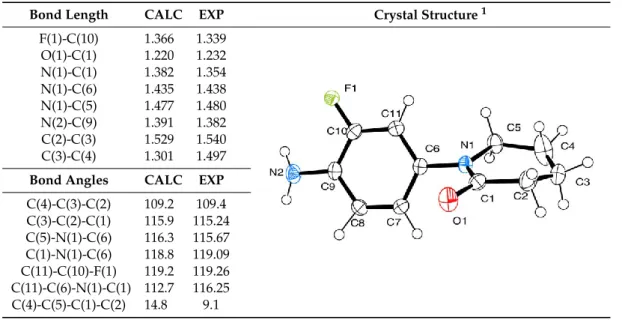
(12) Molecules 2020, 25, 491. 12 of 39. 2.3. Theoretical Study In order to understand the relationship between the structure and the energetic behavior of all the series, including a thermochemical analysis associated to the yield of the reactions, an optimization and reactivity indexes calculation of electronic chemical potential (µ), chemical hardness (η), and electrophilicity (ω) was performed. The electronic chemical potential (µ) has the same significance as the classic thermodynamics chemical potential but characterizes the tendency to escape of electrons from the equilibrium. This index its associated to the ionization potential and the electron affinity, and to the energies of Frontier Molecular Orbitals (FMOs) based on the Koopman’s theorem. The chemical hardness (η) is associated to the stability of the systems and the resistance of a molecule to exchange electron density. The same approximations as for electronic chemical potential were made. For electrophilicity (ω), a higher index value indicates a more electrophilic character for the molecule, as a lower index value, a more nucleophilic character has the molecule. Electrophilicity can also be associated with the energy stabilization of a molecule [79,94]. 2.3.1. 2020, X-Ray Molecules 25, and 491. Theoretical Optimization Correlation. 14 of 42. Structural parameters of molecules 6, 9, and 20 were optimized and compared with crystallographic data (Tables 5–7). The structures are well refined, with final R indices (I > 2σ(I)) of R1 = 0.0461, wR2 = 0.1292 for 6, R1 = 0.0343, wR2 = 0.0944 for 9 and R1 = 0.0518, wR2 = 0.1552 for 20. The crystallographic data and Table 5. Selected bond lengths and angles for molecule 6. structure refinement can be found in the Supplementary Material (Tables S27 to S29).. Bond Length F(1)-C(10) O(1)-C(1) Bond Length N(1)-C(1) F(1)-C(10) N(1)-C(6) O(1)-C(1) N(1)-C(1) N(1)-C(5) N(1)-C(6) N(2)-C(9) N(1)-C(5) C(2)-C(3) N(2)-C(9) C(3)-C(4) C(2)-C(3) C(3)-C(4) Bond Angles Bond Angles. C(4)-C(3)-C(2) C(4)-C(3)-C(2) C(3)-C(2)-C(1) C(3)-C(2)-C(1) C(5)-N(1)-C(6) C(5)-N(1)-C(6) C(1)-N(1)-C(6) C(1)-N(1)-C(6) C(11)-C(10)-F(1) C(11)-C(10)-F(1) C(11)-C(6)-N(1)-C(1) C(11)-C(6)-N(1)-C(1) C(4)-C(5)-C(1)-C(2) C(4)-C(5)-C(1)-C(2) 1. CALC. EXP. Crystal Structure 1. Table 5. Selected bond lengths and angles for molecule 6.. 1,366 1,220 CALC 1,382 1.366 1,435 1.220 1.382 1,477 1.435 1,391 1.477 1,529 1.391 1,301 1.529. 1,339 1,232 EXP 1,354 1.339 1,438 1.232 1.354 1,480 1.438 1,382 1.480 1,540 1.382 1,497 1.540. Crystal Structure 1. 1.301. CALC 1.497 EXP. CALC. 109.2 109.2 115.9 115.9 116.3 116.3 118.8 118.8 119.2 119.2 112.7 112.7 14.8 14.8. EXP. 109.4. 109.4 115.24 115.24 115.67 115.67 119.09 119.09 119.26 119.26 116.25 116.25 9.1. 9.1. ORTEP [95] drawing with thermal ellipsoids drawn at the 50% probability level. ORTEP: Oak Ridge Thermal. ORTEP Ellipsoid[95] Plot.drawing with thermal ellipsoids drawn at the 50% probability level. ORTEP: Oak Ridge Thermal Ellipsoid Plot. 1. Regarding bond angles for molecule (Tableand 5), the most significant difference occurs in the For the all studied molecules, bond6angles distances show a good correlation between dihedral angles, a difference between 3 torobustness 5 degrees,forshowing that the molecule a slightly calculated and with experimental, suggesting good the calculated structure andhas parameters, different around the centralbond C(6)-N(1) atoms, and a slightly more complex twist in the with noconformations significative difference between lengths. non-aromatic ring.bond angles for molecule 6 (Table 5), the most significant difference occurs in the Regarding dihedral angles, with a difference between 3 to 5 degrees, showing that the molecule has a slightly 6. Selected bond lengths and angles foramolecule different conformations Table around the central C(6)-N(1) atoms, and slightly 9. more complex twist in the non-aromatic ring. 1 Bond Length. CALC. EXP. F(1)-C(7) O(2)-C(2) N(1)-C(2) N(1)-C(5) N(1)-C(3) N(2)-C(8). 1.365 1.219 1.377 1.433 1.474 1.390. 1.364 1.231 1.352 1.436 1.479 1.383. Crystal Structure.
(13) Regarding bond angles for molecule 6 (Table 5), the most significant difference occurs in the dihedral angles, with a difference between 3 to 5 degrees, showing that the molecule has a slightly different conformations around the central C(6)-N(1) atoms, and a slightly more complex twist in the non-aromatic ring. Molecules 2020, 25, 491. 13 of 39. Table 6. Selected bond lengths and angles for molecule 9. Bond Length. Table 6. Selected for molecule 9. 1 CALC EXP bond lengths and angles Crystal Structure. F(1)-C(7) 1.365 1.364 Bond Length CALC EXP Crystal Structure 1 O(2)-C(2) 1.219 1.231 F(1)-C(7) 1.365 1.364 N(1)-C(2) 1.377 1.352 O(2)-C(2) 1.219 1.231 N(1)-C(5) 1.433 1.436 N(1)-C(2) 1.377 1.352 N(1)-C(3) 1.474 1.479 N(1)-C(5) 1.433 1.436 N(2)-C(8) 1.390 1.383 N(1)-C(3) 1.474 1.479 MoleculesC(1)-O(1) 2020, 25, 491 15 of 42 1.418 1.418 N(2)-C(8) 1.390 1.383 O(1)-C(4) 1.417 1.430 C(1)-O(1) 1.418 1.418 1 ORTEP [95] drawing with thermal ellipsoids drawn at the 50% probability level. O(1)-C(4) 1.417 1.430 Bond Angles CALC EXP For molecule 9, in addition to the good length correlation, between theoretical and experimental, Bond Angles CALC EXP thereC(4)-O(1)-C(1) was a favorable correspondence C(4)-O(1)-C(1) 110.6 109.4 110.6 109.4 between bond angles (Table 6). The main difference takes place in O(1)-C(1)-C(2) 116.7 the 115.6 the O(1) oxygen atom position, calculations showed that this atom is facing the same plane as the O(1)-C(1)-C(2) 116.7 115.6 C(3)-N(1)-C(5) 117.6 117.2 C(3)-N(1)-C(5) 117.6 117.2 fluorine F(1) atom. On the other hand, the crystal shows that the atoms are opposite from each other. 120.8 120.7 C(2)-N(1)-C(5) 120.8 120.7 The C(2)-N(1)-C(5) C(10)-C(5)-N(1)-C(2) dihedral angle was measured in order to visualize the difference between C(6)-C(7)-F(1) 119.0 118.3 C(6)-C(7)-F(1) 119.0 118.3 experimental and calculated structures (Figure S30, Supplementary Material). This behavior could be C(6)-C(5)-N(1)-C(2) 50.0 50.2 C(6)-C(5)-N(1)-C(2) 50.0 50.2 explained due to the hydrogen bonds C(4)-C(3)-C(2)-C(1) 18.7 16.0 interactions generated between the F(1) and O(2) atoms with the C(4)-C(3)-C(2)-C(1) 18.7 16.0 opposite hydrogen atoms (Figure S31, Supplementary Material). C(10)-C(5)-N(1)-C(2) 50.2in the molecule 131.7 C(10)-C(5)-N(1)-C(2) 50.2 131.7 1. ORTEP [95] drawing with thermal ellipsoids drawn at the 50% probability level.. Table 7. Selected bond lengths and angles for molecule 20. 1 Table 7. Selected bond EXP lengths and angles for molecule 20. Bond Length Crystal Structure CALC. F(1)-C(4) Bond Length CALC O(1)-C(10) F(1)-C(4)N(1)-C(2) 1.365 O(1)-C(10) N(1)-C(10) 1.219 N(1)-C(2) 1.432 N(1)-C(13) N(1)-C(10) 1.378 N(2)-C(5) N(1)-C(13) 1.474 N(2)-C(6) N(2)-C(5) 1.393 N(2)-C(6)N(3)-C(8) 1.465 N(3)-C(8)N(3)-C(18) 1.474 N(3)-C(15) 1.464 N(3)-C(18) O(3)-C(15) 1.372 N(3)-C(15) O(3)-C(15) O(2)-C(12) 1.219 O(2)-C(12) O(2)-C(11) 1.417 O(2)-C(11)C(8)-N(3) 1.418 C(8)-N(3) 1.474 Bond Angles Bond Angles CALC. 1.365 1.363 EXP 1.219 1.222 1.4321.3631.436 1.3781.2221.368 1.436 1.474 1.475 1.368 1.393 1.411 1.475 1.465 1.474 1.411 1.4741.4741.457 1.4641.4571.471 1.3721.4711.356 1.2191.3561.231 1.4171.2311.426 1.4181.4261.421 1.4741.4211.457 1.457 CALCEXP EXP. C(12)-O(2)-C(11) C(12)-O(2)-C(11)110.7 O(2)-C(11)-C(10) O(2)-C(11)-C(10)116.5 C(13)-N(1)-C(2) C(13)-N(1)-C(2)117.6 C(10)-N(1)-C(2) C(10)-N(1)-C(2)120.5 C(3)-C(4)-F(1) C(3)-C(4)-F(1) 116.5 C(5)-N(2)-C(19) 122.1 C(5)-N(2)-C(19) C(3)-C(2)-N(1)-C(10) 126.8 C(3)-C(2)-N(1)-C(10) C(12)-C(13)-C(10)-C(11) 18.2 C(12)-C(13)-C(10)-C(11) C(5)-N(2)-C(19)-C(20) 128.5 C(5)-N(2)-C(19)-C(20) C(9)-C(8)-N(3)-C(15) 153.8 C(9)-C(8)-N(3)-C(15) C(21)-N(2)-C(8)-N(3) 108.7 C(21)-N(2)-C(8)-N(3) 1. 110.7108.3108.3 116.5115.1115.1 117.6117.4117.4 120.5120.1120.1 116.5115.8115.8 117.8 122.1 117.8 137.7 126.8 137.7 13.1 18.2 55.3 13.1 128.5111.255.3 153.8158.0111.2 108.7 158.0. Crystal Structure 1. ORTEP [95] drawing with thermal ellipsoids drawn at a 50% probability level.. 1. ORTEP [95] drawing with thermal ellipsoids drawn at a 50% probability level.. ForBond molecule 9, for in addition good length between theoretical experimental, angles moleculeto20the also shows thatcorrelation, the most significant difference and between calculated and there was a favorable correspondence between (Table 6). Theofmain difference takes place experimental occurred in the dihedral anglesbond withangles a slight difference 5 degrees in the twist structure in the O(1) oxygen atom position, the calculations showed that this atom is facing the same plane as the on the non-aromatic ring but a large difference between the propargyl moiety position and the tetrahydroquinoline cycle. The last three angles shown (Table 7) were measured to demonstrate the different positioning of the tetrahydroquinoline cycle and the propargyl moiety. This could be due to the interactions generated between the propargyl and the carbonyl groups in the crystal (Figure S32, Supplementary Material)..
(14) Molecules 2020, 25, 491. 14 of 39. fluorine F(1) atom. On the other hand, the crystal shows that the atoms are opposite from each other. The C(10)-C(5)-N(1)-C(2) dihedral angle was measured in order to visualize the difference between experimental and calculated structures (Figure S30, Supplementary Material). This behavior could be explained due to the hydrogen bonds interactions generated between the F(1) and O(2) atoms with the opposite hydrogen atoms in the molecule (Figure S31, Supplementary Material). Bond angles for molecule 20 also shows that the most significant difference between calculated and experimental occurred in the dihedral angles with a slight difference of 5 degrees in the twist structure on the non-aromatic ring but a large difference between the propargyl moiety position and the tetrahydroquinoline cycle. The last three angles shown (Table 7) were measured to demonstrate the different positioning of the tetrahydroquinoline cycle and the propargyl moiety. This could be due to the interactions generated between the propargyl and the carbonyl groups in the crystal (Figure S32, Supplementary Material). 2.3.2. Energetic Behavior and Thermochemistry The electronic chemical potential for propargyl precursors 11–14 shows a mean of −3.0471 eV, with a maximum of −2.889 eV for molecule 11 and a minimum of −3.169 for molecule 13. For the arylazide compounds (24–26), the mean is −4.4049 eV, with a maximum of −4.277 eV for molecule 24 and a minimum of −4.490 eV for molecule 26. This implies that the reaction between precursors 11–14 and 24–26 present a medium polar character, and the electron density flux will take place from the propargylated molecule to the arylazide [94]. The Density Functional Theory (DFT) study for 1H-1,2,3-triazole derivatives showed that the highest chemical hardness is obtained in the molecules synthetized from precursor 24, thus being the more stable ones (average 2.013 eV). It is worth to notice that these derivatives (27, 30, 33, and 36, all from precursor 24) had the highest yield of all synthesized compounds (91.11 ± 1.55%). For the electrophilicity index, propargyl molecules 11–14 showed mean of 1.8301 eV, with a maximum of 1.908 eV for molecule 14 and a minimum of 1.667 for molecule 11. For arylazide compounds 24–26, the mean is 3.4799 eV, with a maximum of 3.657 eV for molecule 27 and a minimum of 3.234 eV for molecule 25. According to these values, propargyl precursors acts as nucleophiles and the arylazide precursors acts as electrophiles. This approach may not be entirely correct because the arylazide is a dipolar compound and the propargyl is a dipolarophile center. The reaction should go through a Huisgen cycloaddition, following a concerted mechanism with a nucleophilic attack to and from the propargylated molecule [96]. All energies for reactivity indexes and FMOs are summarized in Table 8. In order to understand the electrophilic behavior of arylpropargylated and arylazide precursors, the Fukui functions for molecules 13 and 24 were calculated. These molecules were chosen as an example due therefrom compound 33 was obtained. This reaction presented the best performance according to the products yield. Fukui functions helped us to understand the behavior of chemical reactions providing information on the local reactivity of atoms within the molecule, as shown in the Supplementary Material (Table S33 and S34). According to the f + function, N(9) nitrogen atom (molecule 24) was the most susceptible atom to experience a nucleophilic attack. However, the most susceptible atom for compound 13 was the C(18) carbon. For neutral or radical reagents or reactions, the f 0 function can be used. In this case, with a concerted mechanism, the function shows the same behavior previously discussed [97]. The calculations predicted that the mesomeric structure that reacts is the one with a triple bond between the nitrogen atoms (Figure S35, Supplementary Material). DFT calculations have been previously performed on similar systems, showing that the mechanism for the copper (I) catalyzed cycloaddition are in concordance with experimental and theoretical findings reported in this work. Moreover, previously published calculations also agrees with the mesomeric structure that actually reacts, showing that the reaction with and without catalyst should undergo the same mechanism, with the same sites being susceptible to nucleophilic attack [98]..
(15) Molecules 2020, 25, 491. 15 of 39. Table 8. Calculated energies (eV) for FMOs and reactivity indexes, for precursors and final compounds. Compound. HOMO. LUMO. GAP. HARD. (η). C. POT. (µ). ELEC. (ω). 11 12 13 14 27 30 33 36 28 31 34 37 29 32 35 38 24 25 26. −5.3917 −5.5550 −5.8366 −5.5593 −5.4418 −5.5892 −5.8339 −5.5982 −5.5354 −5.6674 −5.9278 −5.6973 −5.5147 −5.6524 −5.9082 −5.6750 −7.1049 −7.2350 –7.2467. −0.3859 −0.5519 −0.5010 −0.5954 −1.5467 −1.5913 −1.6205 −1.6003 −1.8583 −1.8931 −1.9249 −1.9111 −1.7138 −1.7557 −1.7843 −1.7682 −1.4487 −1.6602 –1.7339. 5.0058 5.0031 5.3357 4.9639 3.8951 3.9979 4.2134 3.9979 3.6771 3.7742 4.0028 3.7862 3.8009 3.8967 4.1239 3.9068 5.6562 5.5748 5.5128. 2.5029 2.5016 2.6678 2.4820 1.9475 1.9989 2.1067 1.9990 1.8386 1.8871 2.0014 1.8931 1.9005 1.9484 2.0620 1.9534 2.8281 2.7874 2.7564. −2.8888 −3.0534 −3.1688 −3.0774 −3.4942 −3.5903 −3.7272 −3.5993 −3.6968 −3.7802 −3.9264 −3.8042 −3.6142 −3.7040 −3.8462 −3.7216 −4.2768 −4.4476 –4.4903. 1.6671 1.8635 1.8819 1.9078 3.1347 3.2242 3.2970 3.2404 3.7167 3.7862 3.8514 3.8222 3.4367 3.5209 3.5872 3.5452 3.2339 3.5483 3.6575. The thermochemistry of the series was studied taking into consideration the sum of electronic and thermal enthalpies for both precursors and final compounds, in order to calculate the ∆H for each reaction (Table S36, Supplementary Material). All reactions have shown a highly exothermic behavior with a reaction enthalpy value of between −61.765 ± 0.295 kcal/mol. These energies were in agreement with previously calculated similar compounds [98]. It is important to note the most exothermic reaction is the one to obtain 33 (from 13 and 24) in accordance to its highest yield of reaction. 2.4. Biological Analysis 2.4.1. FXa Inhibition in Vitro Assay By using SensoLyte® 520 Factor Xa Assay Kit “Fluorimetric” FXa (bovine isolated enzyme) inhibition was measured. Upon FXa protease cleavage, this substrate generates the Rh110 (rhodamine 110) as free fluorophore, which can be detected at excitation/emission of 490 nm/520 nm, where the following values of inhibition FXa percentage and IC50 from the representative compounds of each series (compounds 6–38, Table 9), as well as for all final 1H-1,2,3-triazole derivatives [68,99] were obtained. The assay showed that only five of the novel compounds (27, 30, 31, and 34) exhibited inhibitory activity higher than 50% against FXa at 100 µM. According to these results, IC50 values were calculated using corresponding dilutions of each sample (Materials and Methods). It is important to note that none of the evaluated compounds showed fluorescence quench at the assayed wavelength avoiding the possibility to present a potential pan-assay interference compounds (PAINS) fragment [100–102]..
(16) Molecules 2020, 25, 491. 16 of 39. Table 9. FXa inhibition and IC50 values of novel compounds 6–38. Compounda. Inhibition FXa (%)b. IC50 ± SD [µM]c. 6 7 8 10 11 12 13 14 17 19 20 27 28 29 30 31 32 33 34 35 36 37 38. 19 18 20 15 17 14 25 24 25 27 15 52 33 46 63 73 21 41 60 28 34 34 39. 102.10 ± 0.14 41.71 ± 0.07 29.73 ± 0.09 67.92 ± 0.08 -. a. It was not possible to synthesize Compound 18. b Inhibition percentage at 100 µM. c Apixaban 97% inhibition, IC50 = 2.8 nM. Rivaroxaban 94% inhibition, IC50 = 0.70 nM.. 2.4.2. Anti FXa assay, Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT) ex Vivo Assay Ex vivo anticoagulation efficacy assays of compounds 27, 30, 31, 34, Apixaban, and Rivaroxaban (control samples) were evaluated by anti FXa (Table 10), activated partial thromboplastin time (aPTT) and prothrombin time (PT) assays by using a human plasma pool. Table 10. Anti FXa assay values of selected compound. Compound. Anti-FXa [IU/mL] a. 27 30 31 34. 0.29 ± 0.10 0.00 0.05 ± 0.09 0.42 ± 0.18. a. IU/mL: International Units per milliliter. Concentration 1 mM. Apixaban (500 nM, 1.50 ± 0.36 IU/mL) and Rivaroxaban (300 nM, 1.20 ± 0.23 IU/mL), respectively.. We know that chromogenic FX assay measures the amount of FX activity in a patient, however, chromogenic anti-FXa activity assay does not measure the patient’s FX. What it does instead is that it measures the ability of the patient’s plasma to repress exogenous FXa. The chromogenic anti-FXa assay is therefore used to determine the concentration of anticoagulants that inhibit FXa. What will need to be taken into consideration in the future is how anti-FXa monitoring will need to be increased. As more anticoagulants that are FX inhibitors are more freely available in the market, therapeutic compliance, together with diagnosing the causes of bleeding in a patient being treated with these anticoagulants, will need to be done much more often [103]. Widespread use of FXa inhibitors may require performing the ant-FXa assay as a complementary assay. If FXa inhibitors are more commonly used, this may mean that the anti-FXa assay as complementary assay will need to be done..
(17) is therefore used to determine the concentration of anticoagulants that inhibit FXa. What will need to be taken into consideration in the future is how anti-FXa monitoring will need to be increased. As more anticoagulants that are FX inhibitors are more freely available in the market, therapeutic compliance, together with diagnosing the causes of bleeding in a patient being treated with Molecules 2020, 25, 491 17 of 39 these anticoagulants, will need to be done much more often [103]. Widespread use of FXa inhibitors may require performing the ant-FXa assay as a complementary assay. If FXa inhibitors are more commonly used, this mean that theanti-FXa anti-FXalevels assaydemonstrated as complementary assay willrisk need to be done. Asmay an example, high the increasing of bleeding and low levels As an example, high anti-FXa levels demonstrated the increasing risk of bleeding and low levels of of anti-FXa established potential thromboembolic future events. anti-FXaAccording established thromboembolic future events.obtained values (0.3–0.7 IU/mL), selected topotential the therapeutic range of the anti-FXa According to the therapeutic range of the anti-FXa obtained values (Figure (0.3–0.77).IU/mL), selected compounds 27 and 34 were assay for PT and aPTT coagulation parameters compounds 27 and 34 were assay for PT and aPTT coagulation parameters (Figure 7).. Figure Figure7.7.(a) (a)PT PTand and(b) (b)aPTT aPTTcoagulation coagulationparameters parametersfor forcompounds compounds27 27and and34. 34.. parameter evaluated extrinsic common coagulation pathway. Oncontrary, the contrary, PTPT parameter evaluated the the extrinsic andand common coagulation pathway. On the aPTT aPTT parameter evaluated the intrinsic pathway andless its less sensitive thefinal final common common pathway. parameter evaluated the intrinsic pathway and its sensitive to tothe pathway. Compound simultaneouslyprolonged prolongedaPTT aPTTand andPT PTin in aa dose-dependent dose-dependent manner Compound 2727 simultaneously manner for forconcentrations concentrations up to 50 µM (Figure 6a,b). Meanwhile, compound 34 only prolonged PT in a dose-dependent manner up to 50 μM (Figure 6a and 6b). Meanwhile, compound 34 only prolonged PT in a dose-dependent (Figure(Figure 6a) for 6a) concentrations up to 50 up µM.to Apixaban Rivaroxaban prolonged both coagulation manner for concentrations 50 μM. and Apixaban and Rivaroxaban prolonged both parameters in a dose-dependent manner for concentrations up to 0.1 nM [104,105]. coagulation parameters in a dose-dependent manner for concentrations up to 0.1 nM [104,105]. These interesting results suggest compounds 27 inhibited targeting this coagulation These interesting results suggest thatthat compounds 27 inhibited FXa FXa targeting this coagulation factor factor both through both pathways order tothe increase thepotency possibleofpotency of their derivatives. Finally, through pathways in order toinincrease possible their derivatives. Finally, compound may target coagulation byand the common extrinsic pathway. and common pathway. 34 compound may target 34 coagulation FXa mainly byFXa the mainly extrinsic 2.4.3. Viability Assays 2.4.3. CellCell Viability Assays Cell viability viability assay was performed in HEK293 and cells using 3-(4,5-dimethylthiazol-2-yl) Cell assay was performed in HepG2 HEK293 and the HepG2 cells using the -5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) method (Figure 8). The compounds 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) were assayed under increasing concentrations, namely, 1 nM (a), 10 nM (b), 50 nM (c), 100 nM (d), 10 mM method (Figure 8). The compounds were assayed under increasing concentrations, namely, 1 nM (a), 10 (e), and 100 mM (f). In HEK293 cells, most of the assayed compounds only shows signs of toxicity (defined as the value obtained after subtracting two standard deviations from the average value of control wells treated only with vehicle) when concentrations are higher than 10 mM, such as compounds 30 and 31. Compound 31 showed a greater toxic effect in HepG2, reducing cell viability to 40% and 20% of control at 10mM and 100mM, respectively, while the rest of the compounds did not affect HepG2 cell viability..
Figure


![Figure 4. (a) 5-heptyl-1,2,3,4-tetrahydroquinoline [44]. (b) Aflaquinolone A [46]](https://thumb-us.123doks.com/thumbv2/123dok_es/7251417.435867/5.892.172.719.746.1064/figure-heptyl-tetrahydroquinoline-aflaquinolone.webp)
+7



Documento similar
Likewise, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion mole- cule-1 (ICAM-1), which are relevant in the subsequent leukocyte firm adhesion step, are
Generally, the electrophilic carboxylic center a is more reactive than the carbonate site b because it is less stabilized by resonance (Scheme 3-8). Two-step coupling via
Overall Rate of Evolution and Information Scores Phylogenetic information scores are calculated assuming an overall rate of evolution (that of the full data set comprising
As we commented in the previous chapter, a Kondo resonance appears on pristine TCNQ molecules adsorbed on that position. On the contrary, its appearance is not expected for
Through looking at the link between the chemical structure and the activity, our study proves that changes made to natural macamides at the level of the alkyl chain, the
The combination of SubPcs and SubNcs has been used as complementary absorbers in tandem (Figure 12), 55 wherein the SubNcs behave as an electron donor in PHJ
After filtration of the drying agent, the solvent was removed under reduced pressure, and the residue purified by column chromatography on silica gel (eluent: heptane/ethyl
A simple method to alleviate this problem consists of multiplying the pro- jection Fourier transform by the sign of the CTF (this approach is named phase flipping (Frank, 1996,
