Respiratory syncytial virus infection and immunity
15
0
0
Texto completo
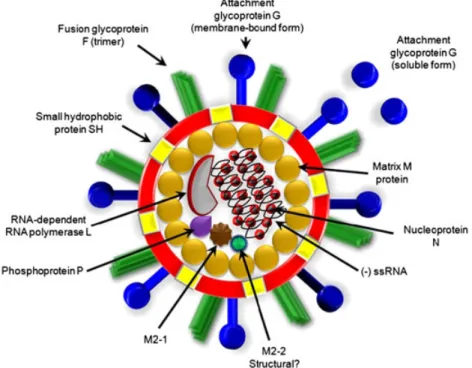
(2) Immunity triggered by RSV infection. 231. Figure 1. Structure of the respiratory syncytial virus (RSV) virion. RSV is an enveloped virus with eight known structural proteins. The fusion protein (F), the attachment glycoprotein (G) and the small hydrophobic protein (SH) are located on the surface of the virion. The ssRNA genome and the remaining viral structural proteins, which are the matrix (M), the nucleocapsid (N), the RNA-dependent RNA polymerase (L), the phosphoprotein (P) and the M2 gene product M2-1, reside inside the envelope. The exact location of the M2 gene product M2-2 is currently unknown. Despite extensive research efforts, no vaccines are currently available that are capable of inducing long-lasting protective immunity against this virus. Nevertheless, although a widely used prophylactic strategy based on a humanized neutralizing antibody has been shown to be effective at preventing RSV-induced damage, its high cost has limited the use of this treatment only for high-risk groups, such as preterm infants (<37 weeks of gestational age) and infants suffering from cardiovascular diseases and immunosuppression. Further, the only available antiviral drug for treating RSV replication, ribavirin, has shown questionable costeffectiveness [7]. Thus, it is essential to generate new treatments or ideally a vaccine for RSV that could be affordable by public health systems worldwide. Such an effort will require a better understanding of the pathology caused by RSV and its effects on the host immune system. In this review, we discuss the latest findings in the field of RSV infection, pathology and virulence that underscore the complexity of the immune response triggered by this pathogen. Furthermore, we describe new experimental strategies to prevent the pathology induced by the exposure to RSV early in life. Copyright © 2012 John Wiley & Sons, Ltd.. RESPIRATORY SYNCYTIAL VIRUS IMMUNITY. Protective immunity to respiratory syncytial virus and respiratory syncytial virus-induced immunopathogenesis In most healthy adults, symptoms elicited after RSV infection manifest as rhinitis, and the course of the disease resembles a common cold. However, RSV can cause severe inflammation of the respiratory tract in susceptible young children, the elderly and immunosuppressed individuals [5,8,9]. Further, in some susceptible patients, RSV may also produce damage in other tissues besides the lungs, such as the brain, heart and liver [10–12]. It has been estimated that between 0.5% and 3% of children younger than 2 years manifest severe respiratory symptoms after infection with RSV. Considering that RSV infects ~100% of infants by age 2 years, this apparently small percentage becomes very significant [2,3]. Similar to other respiratory viruses, RSV can repeatedly re-infect the host, with a significant percentage of children re-infected during their second year of life [2,3,13]. However, in most cases the severity of the pathology produced by RSV is gradually reduced after Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(3) 232 re-infection, diminishing significantly with the age of exposure [2,3]. Although numerous studies have shown an inverse correlation between the presence of neutralizing antibodies and re-infection [14,15], several other reports suggest that antibodies developed against RSV during natural infection contribute poorly to protection [16,17]. Lack of antibody-mediated protection could be due to the fact that RSV-blocking antibodies are usually elicited at very low frequencies after infection, requiring in some cases decades of repetitive exposure to the virus for their acquisition [16–18]. A possible explanation for this phenomenon is the poor immunogenicity of viral protein domains required for virulence. Additionally, other viral proteins are produced in large amounts, such as the secreted form of the G attachment glycoprotein, which could work as an antibody decoy promoting the production of ineffective antibodies to irrelevant viral epitopes (Figure 1) [19]. Thus, because of the absence of effective neutralizing antibodies, it is likely that RSV infection is mainly cleared by IFN-g secreting CD8+ and CD4+ T cells that promote virus clearance either directly by destroying infected cells or indirectly by limiting inflammation in the lungs [20–25]. Indeed, healthy individuals display T-cell activation levels that are sufficient to mediate viral clearance from the lungs, which promotes low levels of inflammation and hence reduced tissue pathology. The requirement of T-cell immunity for RSV clearance is underscored by the observation that HIV-1-infected patients display increased RSV titres for up to 199 days and mice depleted of CD4+ and CD8+ T cells display prolonged RSV replication [25,26]. A recent report suggests that the ratio of CD4+/CD8+ RSV-specific T cells may be key to defining the outcome of infection, with higher CD4+/CD8+ memory T cell ratios prevailing in the blood of healthy adults, as compared with those suffering primary RSV infection [27]. Such a finding may be useful in the future to predict disease outcome and assess the potential of experimental vaccines in animal models [28]. Unlike healthy adults, more susceptible hosts can display several harmful immune responses after RSV infection, such as increased inflammation in the lungs, locally impaired or damaging CD8+ T-cell responses and detrimental polarization of CD4+ T cells [24,29–32]. All these processes are likely to be driven both by viral determinants and host-cell mediators in response to RSV [22,33]. Copyright © 2012 John Wiley & Sons, Ltd.. P. A. González et al. Recent studies in mice suggest that RSV infection can desensitize alveolar macrophages to TLR ligands for several months after viral resolution [34]. This process could imprint the affected lungs with a phenotype that promotes airway pathology to subsequent viral or bacterial infections [34]. Furthermore, RSV-infected human epithelial cells have been shown to upregulate the expression of inhibitory surface molecules, such as PD-L1 that dampens T-cell activation [35]. Thus, RSV may increase pathology by remodelling the airways [36].. Respiratory syncytial virus interferes with T-cell function Although RSV antigens are presented and accessible to T cells during infection, as shown by the identification of immunogenic peptides for several RSV proteins in mice and humans, several studies suggest that T-cell function may be specifically impaired in the respiratory tract of those individuals suffering pathology during RSV infection [31,32,37,38]. This notion is supported by the observation that RSV-specific T cells remain functional in the periphery after viral infection, a phenomenon also seen for other respiratory viruses [31,37,38]. These findings have led to the concept that impaired antiviral T-cell immune responses in the lungs may be associated with both the tissue itself and the infecting pathogen. Despite this, CD8+ T cells in the lungs of RSV-infected individuals show reduced intracellular granzyme B content, limited secretion of IFN-g and failure to upregulate perforin expression upon antigen stimulation [31,38]. All these immune features are required for the destruction of virus-infected cells. It is noteworthy that production of IFN-g by CD8+ T cells confronted with RSV can be restored by adding exogenous IL-2, a phenomenon also observed for anergic T cells [31]. Consistent with this notion, in vivo IL-2 supplementation can significantly improve the effector function of lung CD8+ T cells recognizing RSV in infected animals [32]. These studies indicate that cytotoxic T cells recognizing RSV display an altered phenotype at the lungs and do not secrete key cytokines, such as IL-2 and IFN-g, which are required for the activation and full display of antiviral T-cell activity. Although it is tempting to relate reduced T-cell activation by RSV-infected dendritic cells (DCs) with subsequent host re-infections, this concept was recently challenged by respiratory Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(4) Immunity triggered by RSV infection viruses that block T-cell activation without necessarily re-infecting their hosts more frequently [13]. Other studies have shown that, despite controlling virus replication, RSV-induced T cells contribute to pathology because disease severity can be significantly reduced by depleting mice of these cells [25]. Consistent with this notion is the observation that RSV infection in neonate and adult mice can induce virus-specific CD4+ and CD8+ T cells, which are fully activated during infection yet display detrimental phenotypes that enhance disease severity during primary and secondary infection [29,39,40]. These observations suggest that RSV has evolved molecular mechanisms to dampen the antiviral T-cell immune response and also to promote in certain cases the induction of unwanted proinflammatory T cells.. Respiratory syncytial virus impairs dendritic cell function to modulate T-cell activation Pathogens can avoid T-cell immunity by dampening the function of DCs [41,42], which are professional antigen-presenting cells that sense pathogens through the engagement of pattern recognition receptors. Pathogen encounter generally leads to DC maturation and the secretion of immune-stimulating cytokines that contribute to the activation and polarization of T cells [43–45]. Upon exposure to RSV, most human and murine DCs display abortive levels of viral RNA synthesis, and only a fraction of cells become readily infected (Figure 2) [46–50]. Consistently, only negligible numbers of viral particles can be recovered from the supernatants of infected DCs. Thus, evidence suggests that these cells may be exploited by RSV to manipulate immunity rather than for viral replication. In fact, although DCs can recognize RSV molecular patterns via a variety of receptors, such as TLRs and RIG-1 [43,44], human and murine DCs undergo only weak maturation after virus encounter. Thus, only a modest upregulation of surface activation markers, such as CD40, CD80, CD86 and MHC (Figure 2) [46–49,51], is observed for DCs after RSV challenge. Nevertheless, DCs respond to the virus by secreting polarizing cytokines, such as IL-6 and IL-10, which can promote T-cell differentiation into phenotypes that are poorly effective at clearing the virus (Figure 2) [47,51]. Therefore, RSV seems to have developed Copyright © 2012 John Wiley & Sons, Ltd.. 233 molecular strategies to interfere with the function of DCs. Accumulating data suggest that in vitro infected human and murine DCs fail to efficiently prime T cells [47–49,52], probably because of the reduced capacity of RSV-infected DCs to secrete activating cytokines, such as IL-12, required to induce CD4+ T cells capable of driving the expansion of cytotoxic and memory CD8+ T cells (Figure 2) [22,38,46,47,49,53,54]. However, the identification of host and viral molecular determinants that account for the altered response shown by DCs to RSV infection remains elusive. Along these lines, a recent report showed that neonate and adult human DCs secrete different cytokine patterns in response to RSV, especially for the levels of TGF-b produced [55]. Cord blood-derived DCs secreted significantly more TGF-b1 in response to RSV infection than did DCs obtained from adult blood [55]. Furthermore, contrastingly different cytokine profiles were obtained in the co-cultures of these RSV-infected DCs with autologous T cells. Whereas co-cultures with adult DCs contained IL-2, IL-12, IFN-g and TNF-a, those with cord DCs contained IL-1b, IL-4, IL-6 and IL-17 [55]. This differential response of neonatal and adult DCs to RSV could contribute to increased infant susceptibility to developing inadequate virus-specific T-cell immunity and lung pathology. Consistent with this notion, neutralization of IL-17 in RSV-infected mice was recently shown to significantly reduce the production of mucus in the airways and decrease viral loads in the lungs [56]. Furthermore, IL-17 neutralization led to an increase in the number of RSV-specific CD8+ T cells, thereby reducing the production of Th2 cytokines in RSV-exacerbated allergic mice. In another study, IL-4 / mice displayed reduced peribronchial lymphocytic inflammation as well as increased levels of the Th1 cytokine IFN-g [57]. RSV infection leads to a sustained increase in the lung recruitment of both myeloid (mDCs) and plasmacytoid DCs (pDCs) in mice and humans. Further, as a result of infection, migration of these cells to the lymph nodes also takes place [58–62]. Plasmacytoid DCs accumulating in the lungs of RSV-infected mice secrete IFN-a and other type-I IFNs in response to the virus and are thought to contribute to controlling RSV-mediated lung pathology [59,63,64]. On the other hand, DCs subtypes different from plasmacytoid, such as monocyte-derived human DCs, fail to secrete these Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
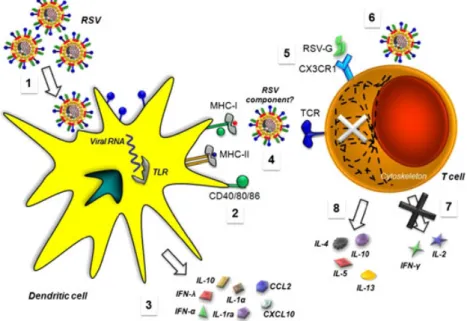
(5) 234. P. A. González et al.. Figure 2. Respiratory syncytial virus (RSV) interferes with DC and T-cell function. (1) RSV can infect DCs, as shown by the surface expression of viral encoded proteins, such as the fusion F protein and intracellular expression of viral RNA (nucleocapsid gene). (2) Upon infection with RSV, DCs mature as a result of the engagement of surface and internal pathogen recognition receptors, such as TLRs, lectins and RIG-I by viral determinants. (3) RSV-infected DCs secrete cytokines that can either promote CD4+ T-cell differentiation into Th2 phenotypes (e.g. IL-10 and IL-6) or inhibit their function (e.g. IFN-l and IFN-a) [49,53]. Furthermore, infection in neonates may promote the generation of detrimental CD8+ T cells. Additionally, DCs can secrete chemokines, such as CXCL10 and CCL2, that modulate immune cell migration. (4) RSV can impair DC–T cell interaction by interfering with immunological synapse assembly (detailed in Figure 3). (5) The soluble form of the RSV G glycoprotein can interfere with T-cell differentiation and migration by interacting with CX3CR1 receptors expressed on the surface of T cells. (6) Few reports have provided evidence for direct interaction between RSV and T cells at their surface, which has been described to interfere with T-cell cytoskeleton organization in response to activating stimuli. (7) RSV can impair T-cell activation and function in the lungs by reducing IFN-g secretion. (8) T cells activated in the context of an RSV infection can display Th2 phenotypes and secrete characteristic inflammatory cytokines. cytokines in high amounts in response to RSV, which would partly reduce DC maturation [65]. Previous experiments performed on human epithelial cell lines have reported that NS1/2 can interfere with STAT-2 function to modulate NF-kB and IRF-3 activity, leading to altered type-I IFN signalling [66,67]. A recent report showed that RSV can also interfere with STAT-1 and STAT-2 signalling in murine bone-marrow derived DCs [68]. In fact, NS1 could negatively modulate the capacity of human DCs to activate both CD4+ and CD8+ T cells by decreasing the proliferation of cytotoxic CD8+ T cells that migrate to the lungs and reducing the activation and proliferation of CD4+ Th17 cells with potential antiviral effects [39]. Furthermore, NS1 was seen to promote the capacity of DCs to activate IL-4-secreting CD4+ T cells while reducing the proliferation of total CD4+ T cells. Strikingly, nearly all these effects were shown to be independent of type-I IFN signalling. Copyright © 2012 John Wiley & Sons, Ltd.. Blockade of the chemokine CCL20 during RSV infection in mice reduced the frequency of mDCs in the lungs but did not alter pDC numbers or the recruitment of T cells [69]. Such a treatment ultimately led to reduced lung pathology and an enhanced Th1 effector response against RSV [69]. Similar data were obtained in CCR6 / animals, which support the notion that pDCs contribute positively to RSV clearance and that CCL20 may enhance this process [59,69].. Respiratory syncytial virus interferes with dendritic cell–T cell synapse Another mechanism by which RSV may alter T-cell activation and function is to impair the assembly of productive immunological synapses (IS) between RSV-infected DCs and antigen-specific CD4+ T cells (Figure 3(A)) [47,70,71]. Thus, by interfering with IS assembly, RSV could render DCs unable to productively prime T cells, even in the presence of cognate Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(6) Immunity triggered by RSV infection. 235. Figure 3. Respiratory syncytial virus (RSV) impairs the establishment of the immunological synapse between DCs and T cells. (A) DC–T cell activating immunological synapses (IS) are characterized by cell–cell interfaces containing a peripheral ring harbouring adhesion molecules and an inner ring enriched in TCR–pMHC complexes, co-stimulatory molecules and activating cytokine receptors. Upon engagement with their ligands, these molecules can induce intracellular activating signals that accumulate and lead to T-cell activation and differentiation. (B) However, RSV-infected DCs express viral proteins that may be targeted to the DC–T cell interface, either intracellularly or at the cell surface. Such proteins could impair the localization and/or function of host activating molecules that are required for T-cell activation during IS assembly, progress and termination. In addition, RSV proteins may promote the recruitment of host molecules at the IS that inhibit T-cell activation. Activating and inhibitory cytokines secreted by RSV-infected DCs are likely to further modulate the signalling events taking place within T cells that are required for an adequate cell activation and differentiation. Taken together, the outcome of T-cell stimulation will depend on the integration of both activating and inhibitory stimuli provided by virus-infected DCs. peptide-MHC (Figure 3(B)) [47]. We have recently observed that infected murine DCs show impaired IS formation and fail to induce activation of T cells (Figures 2 and 3) [47]. Under our experimental settings, inhibition of T-cell activation required cell– cell contact and did not seem to depend on the secretion of soluble mediators by infected DCs, as it has been suggested by other studies (Figure 2) [47,49,53]. Therefore, inhibition of immunological synapse assembly by RSV is likely to involve molecules expressed by DCs during the infective cycle which are recruited to the DC–T cell interface (Figure 3(B)) [47]. This possibility is consistent with a study showing that human epithelial cells expressing the RSV fusion (F) protein inhibit T-cell activation [72], although this phenomenon has not been described yet for DCs. Further research is required to analyse the mechanisms responsible for the RSV inhibition of DC–T cell synapse assembly, as well as the biological function of other RSV proteins and their contribution to modulating the capacity of DCs to prime T cells. Additional factors participating during antigen presentation, such as the quantity of surface cognate peptide-MHC, the length of DC–T cell Copyright © 2012 John Wiley & Sons, Ltd.. interactions and cytokines surrounding the DC–T cell environment can also influence the outcome of synapse assembly and efficiency and ultimately define the effector phenotype of the responding T cells [73–76]. Thus, the integration of activating and inhibitory signalling events in T cells, resulting from distinct stimuli and signalling pathways including TCR–peptide–MHC interaction, co-stimulatory molecules and cytokine receptors, will define the fate of antigen-specific T cells (Figure 3(B)). These observations emphasize that IS formation is a fine-tuned process that could be exploited at multiple levels either directly or indirectly by RSV virulence determinants to interfere with T-cell function. In fact, the diversity of T-cell phenotypes observed after exposure to RSV, such as pro-inflammatory phenotypes, Th2 commitment, partial T-cell activation or anergy, are in line with viral interference of DC–T cell IS (Figure 3(B)). Another aspect related to the inhibition of T-cell function, although poorly explored, is the direct effect of RSV on these cells. Although viral proteins interact at the surface of T cells, replication of viral RNA is not observed within these cells, suggesting abortive infection [77]. However, providing RSV to Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(7) 236 human CD4+ T-cell cultures modulates the cytoskeleton and interferes with the activation of these cells by an unknown mechanism (Figure 2) [52]. These findings suggest that RSV could inhibit the function of non-antigen-specific T cells after infection, which is consistent with the observation that RSV suppresses the activation of bystander murine T cells in vivo as well as in vitro [38,78].. Respiratory syncytial virus interferes with T-cell polarization Although controversial [13], the severity of RSV infection in humans and animal models has been in part associated with the expansion of CD4+ T cells that display phenotypes that are nonoptimal for virus clearance [39,40,79–81]. As part of this type of response, T cells recognizing RSV antigens often polarize towards a Th2 phenotype and secrete or promote the secretion of cytokines, such as IL-4, IL-5 and IL-13 [39,40,79,80,82–84]. These molecules can positively modulate the activation and recruitment of other immune cells, such as eosinophils, neutrophils and monocytes, into the lungs, which produce and secrete proinflammatory molecules contributing to pathology [85–87]. Concomitantly, cytokines such as IL-9 and IL-4 can modulate the immune response by dampening the activity of cytotoxic virus-specific CD8+ T cells (Figure 2) [21,31,37,38,88]. Contrarily, an increase in the number of Th1-polarized RSVspecific Tcells in the lungs can reduce RSV replication and decrease T-cell-associated pro-inflammatory cytokines [21,24]. Studies in animal models have shown that the natural establishment or induction of a balanced Th1 immunity with vaccination can considerably improve the host response to RSV infection [21,24,32]. These observations support the idea that pro-inflammatory responses and their associated cytokines are generally related to RSVinduced pathology. On the contrary, a properly balanced Th1/Th2 polarization is required for limiting virus replication in the infected tissue and reducing lung pathology. These findings have been confirmed by data obtained from vaccination and virus challenge studies in animal models for RSV [21,89–92]. Although CD8+ cytotoxic T cells are usually considered to be advantageous for viral clearance because they correlate positively with protective immunity in mice successfully vaccinated against Copyright © 2012 John Wiley & Sons, Ltd.. P. A. González et al. RSV, some reports suggest that naturally occurring CD8+ T cells specific to the virus may play detrimental roles during primary infection and secondary challenge with the virus [29,93]. This possibility is supported by the observation that depletion of CD8+ T cells during primary and secondary infections can reduce the severity of RSVinduced disease in infected animals [29]. It is thought that these detrimental virus-specific occurring CD8+ T cells may be secreting damaging pro-inflammatory cytokines that promote lung damage, similar to CD8+ T cells induced upon RSVenhanced allergic inflammation [94]. However, it is important to point out that pathological CD8+ T cells could be modulated by regulatory CD4+ T cells in such a way to reduce virus-induced illness without affecting viral clearance in the lungs [93,95]. Because RSV modulates CD4+ T-cell polarization, it was initially thought that the virus might also promote the expansion of regulatory T cells (Tregs) to downmodulate the activity of virusspecific cytotoxic T cells [49]. However, mice depleted of CD25+FoxP3+ regulatory T cells prior to infection display increased levels of proinflammatory chemokines and cytokines in the lungs after infection as compared with their nondepleted counterparts [96]. Furthermore, animals depleted of CD4+FOXP3+CD25+ natural Tregs show delayed recovery, enhanced weight loss and increased numbers of activated natural killer cells, which are thought to be beneficial for virus clearance [97]. This effect could be mediated by IL-10 secreted by these cells, which was recently shown to prevent excessive chemokine production and pro-inflammatory cytokines in the lungs after RSV infection [93]. Moreover, in one of these studies, Tregs were shown to differentially modulate the activity of RSV-specific CD8+ T cells to dominant and subdominant viral antigens [96,98]. These observations suggest that Tregs play a favourable role during RSV infection by downmodulating the production of pro-inflammatory cytokines that could contribute to decreasing lung inflammation and to shaping T-cell immunity against the virus. Whether Tregs participate during RSV infection in humans remains to be determined. However, the idea of expanding Tregs in infected individuals to decrease lung inflammation and promote effective antiviral CD8+ T-cell function seems to have some promising prospect. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(8) Immunity triggered by RSV infection. Respiratory syncytial virus interferes with T-cell migration In addition to modulating the polarization of T cells towards pro-inflammatory phenotypes, the RSV G glycoprotein can also alter T-cell migration into the lungs (Figure 2) [99,100]. The amino acid sequence of this glycoprotein contains an immune system-related CX3C chemokine domain that modulates T-cell migration and homing to the lungs. It is thought that this domain binds to CX3CR1 expressed on T cells [101,102]. Indeed, the CX3C motif of the RSV G protein has been suggested to be responsible for reducing the frequency of CX3CR1+CD4+ and CX3CR1+CD8+ T cells in the lungs of infected animals, as well as the frequency of IFN-g-secreting CX3CR1+ RSV-specific T cells in this tissue (Figure 2) [102]. This process is likely to be mediated by the soluble form of the RSV G protein diffusing from the site of infection (Figure 1) [103]. THERAPY AND PROPHYLAXIS FOR RESPIRATORY SYNCYTIAL VIRUS. Current available therapies for respiratory syncytial virus A strategy that has been approved and made commercially available to prevent RSV infection consists of the intravenous administration of a neutralizing humanized monoclonal antibody that inhibits the fusion capacity of the RSV F protein [104]. This approach is aimed at decreasing the probability of RSV infection, mainly in preterm and other susceptible infants. Along these lines are studies in mice showing that targeting the conserved domain within the RSV G protein, which harbours a CX3C-associated motif with monoclonal antibodies, can reduce both inflammation and virus titres in the lungs [18,105,106]. Consistent with this notion, these anti-RSV G protein antibodies are produced poorly after natural infection with RSV [16–18]. Combined with anti-RSV F neutralizing antibodies, monoclonal antibodies directed to the CX3C portion of the G protein could provide a more powerful method for preventing and treating RSV-associated pathology [18,104]. Although monoclonal antibodies have been proven to be effective at reducing RSV infection, it is still somewhat controversial whether antibodies generated against this virus during natural infection protect or not [14–17]. In any case, antibodies Copyright © 2012 John Wiley & Sons, Ltd.. 237 directed against RSV are likely to form immune complexes with the virus, which may be captured by Fcg receptors expressed on the surface of DCs and presented to T cells [16,17,107–110]. A recent report showed that RSV–antibody immune complexes captured by Fcg receptors on the surface of murine and human DCs can modulate the activation of CD4+ and CD8+ T cells against RSV [27]. Furthermore, it was observed that the ratio of murine CD4+/CD8+ T cells, which may be linked to adequate antiviral immunity, depends on whether these antibodies are neutralizing or not [27]. These studies will contribute to understanding the relationship between antiviral antibodies and T cells, a question that has been poorly assessed for viruses in general. On the other hand, replication of RSV in the lungs may be treated with ribavirin, an analogue of purine nucleotides. Ribavirin is thought to interfere with virus dissemination by promoting disruptive mutations [7]. However, clinical trials aimed at assessing the use of ribavirin against RSV have ultimately displayed insufficient power for determining whether RSV treatment with this drug is effective or cost-effective, so the drug is approved for use in preterm infants experiencing severe infection only in a few countries [7]. A newly developed strategy directed at inhibiting viral replication in the lungs is the administration of small interfering RNAs directed against RSV genes, such as the phosphoprotein gene P and nucleocapsid N for their degradation [111–113]. This later approach is currently under clinical evaluation [111].. Failure of an inactivated respiratory syncytial virus vaccine Only a few years after the virus was identified, a formalin-inactivated RSV preparation (FI-RSV) was used in a field trial during the mid-1960s as an initial attempt to vaccinate against the virus [114]. Unfortunately, after natural infection with RSV, children that had received this formulation showed an exacerbated pulmonary disease and suffered more severe symptoms than unvaccinated children [114,115]. Data explaining the failure of FI-RSV as a vaccine were obtained decades after intense research and suggested that the formalininactivated RSV administered prior to infection with the virus promoted immune polarization to an allergic-like response in the lungs, causing Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(9) 238 exacerbated disease [116]. In animal models of RSV infection, immunization with inactivated virus followed by challenge with infective RSV also induces massive eosinophil infiltration to the lungs [116–120]. In addition, immune complex deposition and complement activation has been observed in both mouse models and infected patients [117]. This is due to the establishment of unbalanced Th1–Th2 polarized responses, which are characterized by the secretion of pro-inflammatory cytokines that drive excessive infiltration of eosinophils and neutrophils into the lungs. Although modification of viral epitopes by formalin was initially thought to be responsible for the inadequate immune response displayed after immunization with FI-RSV [121], the development of low-affinity antibodies against this inactivated virus was recently suggested to contribute to the observed damaging immune response by impairing virus neutralization and thus efficient viral clearance [119].. Current vaccine development to prevent respiratory syncytial virus-induced damage Although a few passive prophylactic strategies are currently available for preventing infection of high-risk groups with RSV, cost-effective vaccines are highly demanded by public health systems to reduce hospitalization rates and the economic burden produced by this virus [5]. After the FI-RSV failed to produce immune protection, several new strategies aiming at inducing active prophylaxis against RSV infection were tried [114]. Thus, a formulation conferring protective immunity against RSV with minimal immunopathogenesis has been the goal of researchers around the world. However, to date no vaccine strategy capable of inducing active and long-lasting immunity to prevent RSV infection has reached the public. Given that the immune response against RSV is pivotal in the pathology to this virus, prophylactic treatments aiming to modulate this response in neonates are likely to be the best approach for developing an efficient vaccine. Such a vaccine should prevent pro-inflammatory T-cell polarization induced by RSV and overcome inhibition of naïve virusspecific T cells upon infection. This can be achieved by activating T cells with DCs induced to secrete balanced Th1/Th2-polarizing cytokines that promote an efficient immune response against the Copyright © 2012 John Wiley & Sons, Ltd.. P. A. González et al. virus. A traditional strategy that has worked for several pathogenic viruses and bacteria involves the development of attenuated strains displaying minimal pathogenicity that preserve sufficient immunogenicity. Such attenuated strains have the advantage of expressing most of the pathogen’s antigens, while maintaining virulence traits from the parental strain, strong enough to alert the immune system [122,123]. Using this strategy, attenuated RSV strains have been developed that are fully restricted for growth at body temperature and thus are safe [122]. Other RSV strains have been engineered to carry gene deletions or point mutations at key viral genes that encode immunemodulating regions, such as the attachment glycoprotein G [123]. Another approach involves the generation of recombinant RSV strains carrying host cytokines that promote Th1-type immune responses, such as IL-2 or GM-CSF, as a mechanism to deliver activating cytokines to the site of infection [90,91]. Similarly, RSV genes have been engineered to be expressed as chimaeras in other viruses, such as vaccinia virus, Sendai virus, parainfluenza and adenoviruses, which is a novel strategy for delivering viral antigens against RSV [92,124]. However, a possible disadvantage of these approaches is that they may display reduced safety or lack sufficient immunogenicity to establish protective immunity against RSV [124–126]. Alternatively, formulations consisting of recombinant protein subunits have been increasingly used for vaccine development. These formulations can be applied either alone or in combination with bacterium-derived or plant-derived adjuvants to increase viral antigen immunogenicity [89,127]. Although this approach benefits from increased safety, they may show reduced immunogenicity or unbalanced immune polarization [99,100,128], so they have not been developed further into clinical phases [125]. Another approach that requires further evaluation is the use of recombinant attenuated bacteria expressing RSV antigens [21,129–131]. Because bacteria express many pathogen-associated molecular patterns on their surface, they can induce strong Th1, Th2 or Th17 polarizing immune responses directed to the recombinant heterologous antigens that they express. Recently, our group has generated strains of Bacille Calmette Guérin (BCG, an attenuated strain of Mycobacterium bovis) expressing either the N or M2-1 RSV antigens (rBCG-RSV) that Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(10) Immunity triggered by RSV infection induce an immune response that protects against virus infection in a mouse model [21,28]. Unlike other strategies, this approach intends mainly to produce virus-specific T cells and not antibodies to the virus. We observed that rBCG-RSV produced RSV-specific protective Th1 immunity characterized by IFN-g-secreting T cells and simultaneously prevented the generation of T cells producing Th2-type cytokines [28]. Another advantage of this formulation is the capacity to prevent inflammation or cellular infiltration in the infected lungs [21]. Furthermore, rBCG-RSV-induced T-cell immunity led to undetectable viral loads in the airways of challenged animals [28]. These data suggest that T cells induced after rBCG-RSV immunization can confer immune protection against RSV and that these cells are refractory to the mechanisms used by the virus to downmodulate adaptive immunity [21]. Because BCG has been used worldwide for nearly a century as a routine vaccine against tuberculosis (more than four billion doses administered to humans since 1921, http://www.who.int/ vaccine_research/diseases/ari/en/index4.html), with a good safety record in newborns, it is possible that this approach might lead to an affordable and efficient vaccine against RSV [21,130]. Another study has used an attenuated Salmonella strain as a bacterial vector for the delivery of RSV antigens, which induces immunity in an animal model of infection [129]. Oral administration of this vaccine promoted a balanced Th1/Th2 immune response characterized by the expansion of RSVspecific antibodies and cytotoxic T cells, reducing lung viral loads in infected mice [129]. Although there are no data showing how such vaccines can affect DC function, the induction of protective immunity by recombinant bacteria expressing RSV antigens suggests that these formulations can modulate the immune response to produce virusspecific IFN-g secreting T cells and thus should be considered as potentially affordable and effective alternatives for immunizing against RSV. These animal immunization studies have provided compelling evidence for the feasibility of prophylactic priming with immunogenic formulations prior to RSV exposure that can elicit protective immunity capable of preventing infection and pathology by the virus [21,89,90,92,127,132]. Moreover, even if RSV virulence mechanisms are aimed at interfering with the establishment of an effective primary immune response, if virus-specific T cells Copyright © 2012 John Wiley & Sons, Ltd.. 239 are efficiently activated and differentiated by adequately primed DCs prior to natural infection, they may then be able to override inhibiting signals provided by virus-infected cells. This provides hope for the development of new effective vaccines against this widely disseminated pathogen.. CONCLUDING REMARKS Research throughout these past years has defined RSV as a virus capable of producing excessive inflammation in the lungs while simultaneously interfering with T-cell function by modulating the immunobiology of DCs. However, severe disease leading to hospitalization after RSV infection only occurs in a fraction of individuals who are particularly susceptible to developing severe lung pathology. Ultimately, this process may lead to the establishment of an immunopathogenic-like disease. The identification of host and viral determinants defining this higher susceptibility and severity of disease after infection remains elusive. However, host genetic factors are likely to play relevant roles during infection and, thus, identifying and associating these markers with increased susceptibility is fundamental for the design of useful prognosis kits intended to predict disease outcome. On the other hand, new immune components have recently been shown to participate during RSV-induced pathology, which could help to define what differentiates an efficient viral immune response from a pathologic one. In this regard, alteration of DC function by RSV is likely to be a key event at determining the nature and function of the T-cell response during infection. Accordingly, RSV has developed molecular strategies to interfere with the capacity of DCs to efficiently activate T cells, such as blocking DC–T cell IS assembly, although the mechanism by which this occurs is currently unknown. Furthermore, whether increased susceptibility to suffer severe pathology is mainly attributable to the DC–RSV interaction remains to be assessed. T-cell commitment after interaction with DCs will be defined in part by surrounding cytokines provided by the innate immune response. However, the question as to how these factors trigger detrimental T-cell immune responses, particularly in susceptible individuals, remains unanswered. Identifying the mechanisms by which these cytokines influence the outcome of T cells is crucial, as well as the signalling events taking place in T cells primed by RSV-infected cells. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(11) P. A. González et al.. 240 Addressing these questions should shed light on the mechanisms used by the virus to impair T-cell function, providing tools for predicting the severity of RSV-induced pathology and contributing to the design of effective and safe vaccines that induce balanced and robust protective immunity against RSV.. CONFLICT OF INTEREST The authors have no competing interest.. ACKNOWLEDGEMENTS The authors are supported by grants FONDECYT nº 1070352, FONDECYT nº 1085281, FONDECYT nº 1100926, FONDECYT nº 3070018, FONDECYT nº 3100090, FONDECYT nº 11075060, FONDECYT nº 1100926, FONDEF D06I1008, SavinMucoPath-INCO-CT-2006-032296 and Millennium Institute on Immunology and Immunotherapy. A. M. K. is a Chercheur Étranger D’Excellence, Chaire de la Région Pays de la Loire.. REFERENCES 1. Storey S. Respiratory syncytial virus mar-. 10. Eisenhut M. Extrapulmonary manifesta-. 19. Bukreyev A, Yang L, Fricke J, et al. The. ket. Nature Reviews. Drug Discovery 2010;. tions of severe RSV bronchiolitis. Lancet. secreted form of respiratory syncytial virus. 9: 15–16.. 2006; 368: 988.. G glycoprotein helps the virus evade anti-. 2. Glezen WP, Taber LH, Frank AL, et al. Risk. 11. Eisenhut M. Extrapulmonary manifesta-. body-mediated restriction of replication by. of primary infection and reinfection with. tions of severe respiratory syncytial virus. acting as an antigen decoy and through. respiratory syncytial virus. American Jour-. infection—a systematic review. Critical. effects on Fc receptor-bearing leukocytes.. nal of Diseases of Children 1986; 140: 543–546.. Care 2006; 10: R107.. Journal of Virology 2008; 82: 12191–12204.. 3. Henderson FW, Collier AM, Clyde WA Jr,. 12. Thorburn K, Hart CA. Think outside the. 20. Zhou J, Yang XQ, Fu Z, et al. Increased. et al. Respiratory-syncytial-virus infec-. box: extrapulmonary manifestations of. pathogenesis and inflammation of airways. tions, reinfections and immunity. A pro-. severe respiratory syncytial virus infec-. from respiratory syncytial virus infection. spective, longitudinal study in young. tion. Critical Care 2006; 10: 159.. in T cell deficient nude mice. Medical. children. The New England Journal of Medicine 1979; 300: 530–534.. 13. Le Nouen C, Hillyer P, Munir S, et al. Effects of human respiratory syncytial. Microbiology and Immunology 2008; 197: 345–351.. virus, metapneumovirus, parainfluenza. 21. Bueno SM, Gonzalez PA, Cautivo KM, et al.. clinical risk factors for severe respiratory. virus 3 and influenza virus on CD4+ T cell. Protective T cell immunity against respira-. syncytial virus (RSV) infection. Journal of. activation by dendritic cells. PLoS One. tory syncytial virus is efficiently induced. Pediatrics 2003; 143: S112–S117.. 2010; 5: e15017.. by recombinant BCG. Proceedings of the Na-. 4. Welliver RC. Review of epidemiology and. 5. Nair H, Nokes DJ, Gessner BD, et al.. 14. Kawasaki Y, Hosoya M, Katayose M, et al.. Global burden of acute lower respiratory. Role of serum neutralizing antibody in. infections due to respiratory syncytial. reinfection of respiratory syncytial virus.. virus in young children: a systematic review and meta-analysis. Lancet 2010; 375: 1545–1555. 6. van den Hoogen BG, de Jong JC, Groen J, et al. A newly discovered human pneumo-. Pediatrics International 2004; 46: 126–129. 15. Hall CB, Walsh EE, Long CE, et al. Immunity to and frequency of reinfection with respiratory syncytial virus. Journal of Infectious Diseases 1991; 163: 693–698.. tional Academy of Sciences of the United States of America 2008; 105: 20822–20827. 22. Bueno SM, Gonzalez PA, Pacheco R, et al. Host immunity during RSV pathogenesis. International Immunopharmacology 2008; 8: 1320–1329. 23. Gonzalez PA, Bueno SM, Riedel CA, et al. Impairment of T cell immunity by the. virus isolated from young children with. 16. Handforth J, Friedland JS, Sharland M.. respiratory syncytial virus: targeting viru-. respiratory tract disease. Nature Medicine. Basic epidemiology and immunopathol-. lence mechanisms for therapy and pro-. 2001; 7: 719–724.. ogy of RSV in children. Paediatric Respira-. phylaxis.. tory Reviews 2000; 1: 210–214.. 2009; 16: 4609–4625.. 7. Ventre K, Randolph AG. Ribavirin for res-. Current. Medicinal. Chemistry. piratory syncytial virus infection of the. 17. Dakhama A, Vitalis TZ, Hegele RG. Persis-. 24. Olson MR, Hartwig SM, Varga SM. The. lower respiratory tract in infants and. tence of respiratory syncytial virus (RSV). number of respiratory syncytial virus. young children. Cochrane Database of. infection and development of RSV-specific. (RSV)-specific memory CD8 T cells in the. Systematic Reviews 2007; 1: CD000181.. IgG1 response in a guinea-pig model of. lung is critical for their ability to inhibit. acute bronchiolitis. European Respiratory. RSV vaccine-enhanced pulmonary eosino-. Journal 1997; 10: 20–26.. philia. Journal of Immunology 2008; 181:. 8. Murata Y. Respiratory syncytial virus infection in adults. Current Opinion in Pulmonary Medicine 2008; 14: 235–240.. 18. Collarini EJ, Lee FE, Foord O, et al. Potent. 7958–7968.. 9. Martinez FD. Respiratory syncytial virus. high-affinity antibodies for treatment and. 25. Graham BS, Bunton LA, Wright PF, et al.. bronchiolitis and the pathogenesis of. prophylaxis of respiratory syncytial virus. Role of T lymphocyte subsets in the patho-. childhood asthma. Pediatric Infectious Dis-. derived from B cells of infected patients.. genesis of primary infection and rechal-. ease Journal 2003; 22: S76–S82.. Journal of Immunology 2009; 183: 6338–6345.. lenge with respiratory syncytial virus in. Copyright © 2012 John Wiley & Sons, Ltd.. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(12) Immunity triggered by RSV infection. 241. mice. The Journal of Clinical Investigation. PD-L1 expression inhibits CD8+ T cell. to human metapneumovirus and res-. 1991; 88: 1026–1033.. nonspecific antiviral activity. Journal of. piratory syncytial virus. American Journal. Infectious Diseases 2011; 203: 85–94.. of Respiratory Cell and Molecular Biology. 26. King JC Jr, Burke AR, Clemens JD, et al. Respiratory syncytial virus illnesses in. 36. Tourdot S, Mathie S, Hussell T, et al. Respi-. human immunodeficiency virus- and non-. ratory syncytial virus infection provokes. 47. Gonzalez PA, Prado CE, Leiva ED, et al.. infected children. The Pediatric Infectious. airway remodelling in allergen-exposed. Respiratory syncytial virus impairs T cell. Disease Journal 1993; 12: 733–739.. mice in absence of prior allergen sensitiza-. activation by preventing synapse assem-. tion. Clinical and Experimental Allergy 2008;. bly with dendritic cells. Proceedings of the. 38: 1016–1024.. National Academy of Sciences of the United. 27. Kruijsen D, Bakkers MJ, van Uden NO, et al. Serum antibodies critically affect. 2006; 34: 320–329.. virus-specific CD4(+)/CD8(+) T cell balance. 37. DiNapoli JM, Murphy BR, Collins PL,. during respiratory syncytial virus infec-. et al. Impairment of the CD8+ T cell. 48. Le Nouen C, Munir S, Losq S, et al. Infec-. tions. Journal of Immunology 2010; 185:. response in lungs following infection with. tion and maturation of monocyte-derived. 6489–6498.. human respiratory syncytial virus is spe-. human dendritic cells by human respira-. 28. Cautivo KM, Bueno SM, Cortes CM, et al.. cific to the anatomical site rather than the. tory syncytial virus, human metapneumo-. Efficient lung recruitment of respiratory. virus, antigen, or route of infection. Virol-. virus, and human parainfluenza virus. syncytial virus-specific Th1 cells induced. ogy Journal 2008; 5: 105.. States of America 2008; 105: 14999–15004.. type 3. Virology 2009; 385: 169–182.. by recombinant Bacillus Calmette-Guerin. 38. Vallbracht S, Unsold H, Ehl S. Functional. 49. de Graaff PM, de Jong EC, van Capel TM,. promotes virus clearance and protects. impairment of cytotoxic T cells in the lung. et al. Respiratory syncytial virus infection of. from infection. Journal of Immunology. airways following respiratory virus infec-. monocyte-derived dendritic cells decreases. 2010; 185: 7633–7645.. tions. European Journal of Immunology. their capacity to activate CD4 T cells. Journal. 29. Tregoning JS, Yamaguchi Y, Harker J, et al. The role of T cells in the enhancement of. 2006; 36: 1434–1442.. of Immunology 2005; 175: 5904–5911.. 39. Munir S, Hillyer P, Le Nouen C, et al. syncytial virus. 50. Johnson TR, Johnson CN, Corbett KS, et al.. respiratory syncytial virus infection sever-. Respiratory. interferon. Primary human mDC1, mDC2, and pDC. ity during adult reinfection of neonatally. antagonist NS1 protein suppresses and. dendritic cells are differentially infected. sensitized mice. Journal of Virology 2008;. skews the human T lymphocyte response.. and activated by respiratory syncytial. 82: 4115–4124.. PLoS Pathogens 2011; 7: e1001336.. virus. PLoS One 2011; 6: e16458.. 30. Beckham JD, Cadena A, Lin J, et al. Respi-. 40. Varga SM, Wang X, Welsh RM, et al.. 51. Boogaard I, van Oosten M, van Rijt LS,. ratory viral infections in patients with. Immunopathology in RSV infection is. et al. Respiratory syncytial virus differ-. chronic, obstructive pulmonary disease.. mediated by a discrete oligoclonal subset. entially activates murine myeloid and. Journal of Infection 2005; 50: 322–330.. of antigen-specific CD4(+) T cells. Immunity. plasmacytoid dendritic cells. Immunology. 31. Chang J, Braciale TJ. Respiratory syncytial. 2001; 15: 637–646.. 2007; 122: 65–72.. virus infection suppresses lung CD8+. 41. Bueno SM, Gonzalez PA, Carreno LJ, et al.. 52. Rothoeft T, Fischer K, Zawatzki S, et al.. T-cell effector activity and peripheral CD8+. The capacity of Salmonella to survive. Differential response of human naive and. T-cell memory in the respiratory tract.. inside dendritic cells and prevent antigen. memory/effector T cells to dendritic cells. Nature Medicine 2002; 8: 54–60.. presentation to T cells is host specific.. infected by respiratory syncytial virus.. Immunology 2008; 4: 522–533.. Clinical and Experimental Immunology 2007;. 32. Chang J, Choi SY, Jin HT, et al. Improved effector activity and memory CD8 T cell. 42. Tobar JA, Carreno LJ, Bueno SM, et al. Viru-. development by IL-2 expression during. lent Salmonella enterica serovar Typhimurium. 53. Chi B, Dickensheets HL, Spann KM, et al.. experimental respiratory syncytial virus. evades adaptive immunity by preventing. Alpha and lambda interferon together. infection. Journal of Immunology 2004; 172:. dendritic cells from activating T cells. Infec-. mediate suppression of CD4 T cells induced. 503–508.. tion and Immunity 2006; 74: 6438–6448.. by respiratory syncytial virus. Journal of. 33. Carreno LJ, Gonzalez PA, Bueno SM, et al.. 43. Kawai T, Akira S. Antiviral signaling. Modulation of the dendritic cell–T-cell. through pattern recognition receptors.. synapse to promote pathogen immunity and prevent autoimmunity. Immunotherapy 2011; 3: 6–11. 34. Didierlaurent A, Goulding J, Patel S, et al. Sustained. desensitization. to. bacterial. Journal of Biochemistry 2007; 141: 137–145.. 150: 263–273.. Virology 2006; 80: 5032–5040. 54. Bartz H, Turkel O, Hoffjan S, et al. Respiratory syncytial virus decreases the capacity. 44. Steinman RM, Hemmi H. Dendritic cells:. of myeloid dendritic cells to induce inter-. translating innate to adaptive immunity.. feron-gamma in naive T cells. Immunology. Current Topics in Microbiology and Immunology 2006; 311: 17–58.. 2003; 109: 49–57. 55. Thornburg NJ, Shepherd B, Crowe JE Jr.. Toll-like receptor ligands after resolution of. 45. Banchereau J, Steinman RM. Dendritic. Transforming growth factor beta is a major. respiratory influenza infection. The Journal. cells and the control of immunity. Nature. regulator of human neonatal immune. of Experimental Medicine 2008; 205: 323–329.. 1998; 392: 245–252.. responses following respiratory syncytial. 35. Telcian AG, Laza-Stanca V, Edwards MR,. 46. Guerrero-Plata A, Casola A, Suarez G,. et al. RSV-induced bronchial epithelial cell. et al. Differential response of dendritic cells. Copyright © 2012 John Wiley & Sons, Ltd.. virus infection. Journal of Virology 2010; 84: 12895–12902.. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(13) P. A. González et al.. 242 56. Mukherjee S, Lindell DM, Berlin AA, et al.. 66. Spann KM, Tran KC, Collins PL. Effects of. by membrane-bound and soluble ligands.. IL-17-induced pulmonary pathogenesis. nonstructural proteins NS1 and NS2 of. Cytokine & Growth Factor Reviews 2007; 18:. during respiratory viral infection and. human respiratory syncytial virus on. exacerbation of allergic disease. The American. interferon regulatory factor 3, NF-kappaB,. 77. Kauth M, Grage-Griebenow E, Rohde G,. Journal of Pathology 2011; 179: 248–258.. and proinflammatory cytokines. Journal of. et al. Synergistically upregulated interleu-. Virology 2005; 79: 5353–5362.. kin-10 production in cocultures of mono-. 57. Moore ML, Newcomb DC, Parekh VV,. 19–31.. et al. STAT1 negatively regulates lung. 67. Elliott J, Lynch OT, Suessmuth Y, et al.. cytes and T cells after stimulation with. basophil IL-4 expression induced by respi-. Respiratory syncytial virus NS1 protein. respiratory syncytial virus. International. ratory syncytial virus infection. Journal of. degrades STAT2 by using the Elongin–. Archives of Allergy and Immunology 2007;. Immunology 2009; 183: 2016–2026.. Cullin E3 ligase. Journal of Virology 2007;. 58. Lukens MV, Kruijsen D, Coenjaerts FE,. 81: 3428–3436.. 142: 116–126. 78. Ostler T, Pircher H, Ehl S. “Bystander”. et al. Respiratory syncytial virus-induced. 68. Jie Z, Dinwiddie DL, Senft AP, et al. Regu-. recruitment of systemic memory T cells. activation and migration of respiratory. lation of STAT signaling in mouse bone. delays the immune response to respiratory. dendritic cells and subsequent antigen pre-. marrow derived dendritic cells by respira-. virus infection. European Journal of Immu-. sentation in the lung-draining lymph node.. tory syncytial virus. Virus Research 2011;. Journal of Virology 2009; 83: 7235–7243.. 156: 127–133.. nology 2003; 33: 1839–1848. 79. Sutton TC, Tayyari F, Khan MA, et al. T. 59. Smit JJ, Lindell DM, Boon L, et al. The bal-. 69. Kallal LE, Schaller MA, Lindell DM, et al.. helper 1 background protects against. ance between plasmacytoid DC versus. CCL20/CCR6 blockade enhances immu-. airway hyperresponsiveness and inflam-. conventional DC determines pulmonary. nity to RSV by impairing recruitment of. mation in guinea pigs with persistent res-. immunity to virus infections. PLoS One. DC. European Journal of Immunology 2010;. piratory syncytial virus infection. Pediatric. 2008; 3: e1720.. 40: 1042–1052.. Research 2007; 61: 525–529.. 60. Gill MA, Palucka AK, Barton T, et al.. 70. Dustin ML. T-cell activation through. 80. Monick MM, Powers LS, Hassan I, et al.. Mobilization of plasmacytoid and myeloid. immunological synapses and kinapses.. Respiratory syncytial virus synergizes. dendritic cells to mucosal sites in children. Immunological Reviews 2008; 221: 77–89.. with Th2 cytokines to induce optimal. with respiratory syncytial virus and other. 71. Carreno LJ, Riquelme EM, Gonzalez PA,. levels of TARC/CCL17. Journal of Immu-. viral respiratory infections. Journal of Infec-. et al. T-cell antagonism by short half-life. tious Diseases 2005; 1991: 1105–1115.. pMHC ligands can be mediated by an. 81. Byeon JH, Lee JC, Choi IS, et al. Compari-. 61. Wang H, Peters N, Laza-Stanca V, et al.. efficient trapping of T-cell polarization. son of cytokine responses in nasopharyn-. Local CD11c+ MHC class II- precursors. toward the APC. Proceedings of the National. geal aspirates from children with viral. generate lung dendritic cells during respi-. Academy of Sciences of the United States of. lower respiratory tract infections. Acta. ratory viral infection, but are depleted in. America 2010; 107: 210–215.. the process. Journal of Immunology 2006;. nology 2007; 179: 1648–1658.. Paediatrica 2009; 98: 725–730.. 72. Schlender J, Walliser G, Fricke J, et al. Res-. 82. Bermejo-Martin JF, Garcia-Arevalo MC,. piratory syncytial virus fusion protein. De Lejarazu RO, et al. Predominance of. 62. Guerrero-Plata A, Kolli D, Hong C, et al.. mediates inhibition of mitogen-induced. Th2 cytokines, CXC chemokines and. Subversion of pulmonary dendritic cell. T-cell proliferation by contact. Journal of. innate immunity mediators at the mucosal. function by paramyxovirus infections.. Virology 2002; 76: 1163–1170.. level during severe respiratory syncytial. 177: 2536–2542.. Journal of Immunology 2009; 182: 3072–83.. 73. Maldonado RA, Irvine DJ, Schreiber R,. 63. Wang H, Peters N, Schwarze J. Plasmacy-. et al. A role for the immunological synapse. toid dendritic cells limit viral replication,. in lineage commitment of CD4 lympho-. pulmonary inflammation, and airway. cytes. Nature 2004; 431: 527–532.. virus infection in children. European Cytokine Network 2007; 18: 162–167. 83. Shimojo N, Katsuki T, Tateno N, et al. T helper lymphocyte response to respiratory. hyperresponsiveness in respiratory syncy-. 74. Rothoeft T, Gonschorek A, Bartz H, et al.. syncytial virus and its components in. tial virus infection. Journal of Immunology. Antigen dose, type of antigen-presenting. patients with respiratory allergy and non-. 2006; 177: 6263–6270.. cell and time of differentiation contribute. atopic controls. International Archives of. 64. Smit JJ, Rudd BD, Lukacs NW. Plasmacy-. to the T helper 1/T helper 2 polarization. Allergy and Immunology 2008; 147: 110–116.. toid dendritic cells inhibit pulmonary. of naive T cells. Immunology 2003; 110:. 84. Alwan WH, Kozlowska WJ, Openshaw PJ.. immunopathology and promote clearance. 430–439.. Distinct types of lung disease caused by func-. of respiratory syncytial virus. The Journal of. 75. Bromley SK, Peterson DA, Gunn MD, et al.. Experimental Medicine 2006; 203: 1153–1159.. Cutting edge: hierarchy of chemokine. 65. Munir S, Le Nouen C, Luongo C, et al.. receptor and TCR signals regulating T cell. 85. Castilow EM, Meyerholz DK, Varga SM.. Nonstructural proteins 1 and 2 of respira-. migration and proliferation. Journal of. IL-13 is required for eosinophil entry into. tory syncytial virus suppress maturation. Immunology 2000; 165: 15–19.. the lung during respiratory syncytial virus. of human dendritic cells. Journal of Virology 2008; 82: 8780–8796.. 76. Gonzalez PA, Carreno LJ, Figueroa CA, et al. Modulation of immunological synapse. Copyright © 2012 John Wiley & Sons, Ltd.. tional subsets of antiviral Tcells. The Journal of Experimental Medicine 1994; 179: 81–89.. vaccine-enhanced disease. Journal of Immunology 2008; 180: 2376–2384.. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(14) Immunity triggered by RSV infection 86. Fischer JE, Johnson JE, Kuli-Zade RK, et al. Overexpression of interleukin-4 delays. 243 site of infection. Journal of Virology 2010; 84: 10501–10509.. 105. Haynes LM, Caidi H, Radu GU, et al. Therapeutic monoclonal antibody treat-. virus clearance in mice infected with respi-. 96. Ruckwardt TJ, Bonaparte KL, Nason MC,. ment targeting respiratory syncytial virus. ratory syncytial virus. Journal of Virology. et al. Regulatory T cells promote early. (RSV) G protein mediates viral clearance. 1997; 71: 8672–8677.. influx of CD8+ T cells in the lungs of respi-. and reduces the pathogenesis of RSV infec-. 87. Johnson TR, Rothenberg ME, Graham BS.. ratory syncytial virus-infected mice and. tion in BALB/c mice. The Journal of Infec-. Pulmonary eosinophilia requires interleu-. diminish immunodominance disparities.. kin-5, eotaxin-1, and CD4+T cells in mice. Journal of Virology 2009; 83: 3019–3028.. immunized with respiratory syncytial. 97. Lee DC, Harker JA, Tregoning JS, et al.. ment with respiratory syncytial virus G. virus G glycoprotein. Journal of Leukocyte. CD25+ natural regulatory T cells are criti-. glycoprotein monoclonal antibody or F. Biology 2008; 84: 748–759.. cal in limiting innate and adaptive immu-. (ab’)2 components mediates reduced pul-. 88. Dodd JS, Lum E, Goulding J, et al. IL-9. nity and resolving disease following. monary inflammation in mice. The Journal. regulates pathology during primary and. respiratory syncytial virus infection. Journal. memory responses to respiratory syncytial. of Virology 2010; 84: 8790–8798.. virus infection. Journal of Immunology 2009;. tious Diseases 2009; 200: 439–447. 106. Miao C, Radu GU, Caidi H, et al. Treat-. of General Virology 2009; 90: 1119–1123. 107. Kalergis AM, Ravetch JV. Inducing tumor. 98. Fulton RB, Meyerholz DK, Varga SM.. immunity through the selective engage-. Foxp3+ CD4 regulatory T cells limit pul-. ment of activating Fcgamma receptors on. 89. Cyr SL, Jones T, Stoica-Popescu I, et al.. monary immunopathology by modulating. dendritic cells. The Journal of Experimental. Intranasal proteosome-based respiratory. the CD8 T cell response during respiratory. syncytial virus (RSV) vaccines protect. syncytial virus infection. Journal of Immu-. 183: 7006–7013.. BALB/c mice against challenge without. nology 2010; 185: 2382–2392.. Medicine 2002; 195: 1653–1659. 108. Herrada AA, Contreras FJ, Tobar JA, et al. Immune complex-induced enhancement. 99. Hancock GE, Speelman DJ, Heers K,. of bacterial antigen presentation requires. et al. Generation of atypical pulmonary. Fcgamma receptor III expression on den-. 90. Bukreyev A, Belyakov IM, Berzofsky JA,. inflammatory responses in BALB/c mice. dritic cells. Proceedings of the National Acad-. et al. Granulocyte-macrophage colony-. after immunization with the native attach-. emy of Sciences of the United States of. stimulating factor expressed by recombi-. ment (G) glycoprotein of respiratory syn-. nant respiratory syncytial virus attenuates. cytial virus. Journal of Virology 1996; 70:. eosinophilia. or. enhanced. pathology.. Vaccine 2007; 25: 5378–5389.. viral replication and increases the level of pulmonary antigen-presenting cells. Journal of Virology 2001; 75: 12128–12140.. 7783–7791.. America 2007; 104: 13402–13407. 109. Tobar JA, Gonzalez PA, Kalergis AM. Salmonella escape from antigen presenta-. 100. Tebbey PW, Hagen M, Hancock GE.. tion can be overcome by targeting bacteria. Atypical pulmonary eosinophilia is medi-. to Fc gamma receptors on dendritic cells.. 91. Bukreyev A, Whitehead SS, Prussin C,. ated by a specific amino acid sequence of. Journal of Immunology 2004; 173: 4058–4065.. et al. Effect of coexpression of interleukin-. the attachment (G) protein of respiratory. 110. Liu Y, Gao X, Masuda E, et al. Regulated. 2 by recombinant respiratory syncytial. syncytial virus. The Journal of Experimental. expression of FcgammaR in human den-. virus on virus replication, immunogenic-. Medicine 1998; 188: 1967–1972.. dritic cells controls cross-presentation of. ity, and production of other cytokines.. 101. Tripp RA, Jones LP, Haynes LM, et al.. Journal of Virology 2000; 74: 7151–7157.. CX3C chemokine mimicry by respiratory. 92. Voges B, Vallbracht S, Zimmer G, et al.. syncytial virus G glycoprotein. Nature. Recombinant Sendai virus induces T cell. Immunology 2001; 2: 732–738.. antigen–antibody complexes. Journal of Immunology 2006; 177: 8440–8447. 111. DeVincenzo. J,. Lambkin-Williams. R,. Wilkinson T, et al. A randomized, double-. immunity against respiratory syncytial virus. 102. Harcourt J, Alvarez R, Jones LP, et al.. blind, placebo-controlled study of an. that is protective in the absence of antibodies.. Respiratory syncytial virus G protein and. RNAi-based therapy directed against res-. Cellular Immunology 2007; 247: 85–94.. G protein CX3C motif adversely affect. piratory syncytial virus. Proceedings of the. CX3CR1+ T cell responses. Journal of Immu-. National Academy of Sciences of the United. 93. Weiss KA, Christiaansen AF, Fulton RB, et al. Multiple CD4+ T cell subsets produce. nology 2006; 176: 1600–1608.. States of America 2010; 107: 8800–8805.. immunomodulatory IL-10 during respira-. 103. Arnold R, Konig B, Werchau H, et al. Res-. 112. Zhang W, Tripp RA. RNA interference. tory syncytial virus infection. Journal of. piratory syncytial virus deficient in soluble. inhibits respiratory syncytial virus replication. Immunology 2011; 187: 3145–3154.. G protein induced an increased proinflam-. and disease pathogenesis without inhibiting. matory response in human lung epithelial. priming of the memory immune response.. 94. Jiang XB, Wang ZD, Zhu Y, et al. Inhibition of CD8+ T lymphocytes attenuates respiratory syncytial virus-enhanced allergic inflammation. Respiration 2009; 77: 76–84.. cells. Virology 2004; 330: 384–397.. Journal of Virology 2008; 82: 12221–12231.. 104. The_IMpact-RSV_Study_Group. Palivizu-. 113. Bitko V, Musiyenko A, Shulyayeva O, et al.. mab, a humanized respiratory syncytial. Inhibition of respiratory viruses by nasally. 95. Liu J, Ruckwardt TJ, Chen M, et al.. virus monoclonal antibody, reduces hospi-. administered. Epitope-specific regulatory CD4 T cells. talization from respiratory syncytial virus. 2005; 11: 50–55.. reduce virus-induced illness while pre-. infection in high-risk infants. Pediatrics. serving CD8 T-cell effector function at the. 1998; 102: 531–537.. Copyright © 2012 John Wiley & Sons, Ltd.. siRNA.. Nature. Medicine. 114. Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(15) P. A. González et al.. 244 infants despite prior administration of. 120. Kruijsen D, Schijf MA, Lukens MV, et al.. antigenic inactivated vaccine. American. Local. Journal of Epidemiology 1969; 89: 422–434.. responses. innate. and. regulate. adaptive. immune. inflammatory. 126. Crowe JE Jr, Collins PL, London WT, et al. A comparison in chimpanzees of the im-. cell. munogenicity and efficacy of live attenu-. 115. Kapikian AZ, Mitchell RH, Chanock RM,. influx into the lungs after vaccination with. ated respiratory syncytial virus (RSV). et al. An epidemiologic study of altered. formalin inactivated RSV. Vaccine 2011; 29:. temperature-sensitive. clinical reactivity to respiratory syncytial. 2730–2741.. and vaccinia virus recombinants that. (RS) virus infection in children previously. 121. Moghaddam A, Olszewska W, Wang B,. vaccinated with an inactivated RS virus. et al. A potential molecular mechanism. vaccine. American Journal of Epidemiology. for hypersensitivity caused by formalin-. 1969; 89: 405–421.. inactivated. 116. Connors M, Kulkarni AB, Firestone CY,. vaccines.. Nature. Medicine. 2006; 12: 905–907.. mutant. vaccines. express the surface glycoproteins of RSV. Vaccine 1993; 11: 1395–1404. 127. Etchart N, Baaten B, Andersen SR, et al. Intranasal immunisation with inactivated RSV and bacterial adjuvants induces. et al. Pulmonary histopathology induced by. 122. Collins PL, Murphy BR. New generation. mucosal protection and abrogates eosino-. respiratory syncytial virus (RSV) challenge. live vaccines against human respiratory. philia upon challenge. European Journal of. of. RSV-immunized. syncytial virus designed by reverse genet-. BALB/c mice is abrogated by depletion of. ics. Proceedings of the American Thoracic. formalin-inactivated. CD4+ T cells. Journal of Virology 1992; 66:. Society 2005; 2: 166–173.. Immunology 2006; 36: 1136–1144. 128. Huang Y, Cyr SL, Burt DS, et al. Murine host responses to respiratory syncytial. 123. Elliott MB, Pryharski KS, Yu Q, et al.. virus (RSV) following intranasal adminis-. 117. Polack FP, Teng MN, Collins PL, et al. A. Recombinant respiratory syncytial viruses. tration of a Protollin-adjuvanted, epitope-. role for immune complexes in enhanced. lacking the C-terminal third of the attach-. enhanced recombinant G protein vaccine.. respiratory syncytial virus disease. The. ment (G) protein are immunogenic and. Journal of Clinical Virology 2009; 44: 287–291.. Journal of Experimental Medicine 2002; 196:. attenuated in vivo and in vitro. Journal of. 129. Xie C, He JS, Zhang M, et al. Oral respira-. 7444–7451.. 859–865.. Virology 2004; 78: 5773–5783.. tory syncytial virus (RSV) DNA vaccine. 118. Murphy BR, Sotnikov AV, Lawrence LA,. 124. Collins PL, Purcell RH, London WT,. expressing RSV F protein delivered by. et al. Enhanced pulmonary histopathology. et al. Evaluation in chimpanzees of vac-. attenuated Salmonella Typhimurium. Human. is observed in cotton rats immunized with. cinia virus recombinants that express. formalin-inactivated respiratory syncytial. the surface glycoproteins of human res-. 130. Haas MJ, Besieging RSV. SciBX 2009; 2: 7–9.. virus (RSV) or purified F glycoprotein. piratory syncytial virus. Vaccine 1990;. 131. Bueno SM, Gonzalez PA, Kalergis AM.. and challenged with RSV 3–6 months after. 8: 164–168.. immunization. Vaccine 1990; 8: 497–502.. 125. Crowe JE Jr, Randolph V, Murphy BR. The. Gene Therapy 2007; 18: 746–752.. Use of genetically modified bacteria to modulate adaptive immunity. Current Gene Therapy 2009; 9: 171–184.. 119. Delgado MF, Coviello S, Monsalvo AC,. live attenuated subgroup B respiratory. et al. Lack of antibody affinity maturation. syncytial virus vaccine candidate RSV. 132. Zeng R, Zhang Z, Mei X, et al. Protective ef-. due to poor Toll-like receptor stimulation. 2B33F is attenuated and immunogenic in. fect of a RSV subunit vaccine candidate. leads to enhanced respiratory syncytial. chimpanzees, but exhibits partial loss of. G1F/M2 was enhanced by a HSP70-like. virus disease. Nature Medicine 2009; 15:. the ts phenotype following replication. protein in mice. Biochemical and Biophysical. 34–41.. in vivo. Virus Research 1999; 59: 13–22.. Research Communications 2008; 377: 495–499.. Copyright © 2012 John Wiley & Sons, Ltd.. Rev. Med. Virol. 2012; 22: 230–244. DOI: 10.1002/rmv.
(16)
Figure

Documento similar