Análisis de Modelos Termodinámicos para su Aplicación en la Recuperación de Proteínas Mediante Sistemas de dos Fases Acuosas Edición Única
112
0
0
Texto completo
(2) INSTITUTO TECNOLÓGICO Y DE ESTUDIOS SUPERIORES DE MONTERREY. CAMPUS MONTERREY DIVISIÓN DE INGENIERÍA Y ARQUITECTURA PROGRAMA DE GRADUADOS EN INGENIERÍA. Los miembros del comité de tesis recomendamos que el presente proyecto de tesis presentado por (la) el Ing. Gerardo Buentello Pérez sea aceptado como requisito parcial para obtener el grado académico de: Maestro(a) en Ciencias en Biotecnología Especialidad en Ingeniería de Bioprocesos. Comité de Tesis:. _________________________ Dr. Joaquín Acevedo Mascarúa Asesor. _______________________ Dr. Alejandro García Cuéllar Sinodal. __________________________ Dr. Marco Antonio Rito Palomares Sinodal. Aprobado:. _______________________ Dr. Francisco Ángel Bello Director del Programa de Graduados en Ingeniería Diciembre, 2007.
(3) Agradecimientos. Agradezco a mis asesores y sinodales, en especial al Dr. Joaquín Acevedo y al Dr. Alejandro García a quienes les quité más tiempo durante las asesorías.. A mis padres por apoyarme en las decisiones que he tomado hasta ahora.. A mis amigos salmones, a la bola, a Olga y Peña, a Majo, a Mo, a Karla Romoleroux, a Santiago Penedo, a Adrián y Karlita, a la gente con la que escalo, a mis compañeros y a todo aquel que con su compañía me haya enseñado algo..
(4) Resumen. Resumen En la actualidad la investigación sobre la producción de proteínas mediante procesos biotecnológicos se ha incrementado enormemente, por lo que el estudio de métodos de separación para la recuperación de dichos productos también. Dentro de estos métodos de recuperación, la técnica de extracción mediante dos fases acuosas (Aqueous two-phase systems, ATPS) ha sido popular debido a que estos procesos se operan a temperaturas y con sustancias que no desnaturalizan a las proteínas.. Sin embargo la puesta en marcha de extracciones mediante dos fases acuosas no es un procedimiento sencillo, ya que es necesaria una gran cantidad de experimentación para poder determinar el sistema adecuado que se usará y las condiciones óptimas con las que será operado el proceso. Debido a este problema, ha surgido la necesidad de estudiar mediante modelos termodinámicos a este tipo de sistemas, para que por medio de ellos se puedan hacer predicciones sobre lo que ocurrirá con el sistema y la partición de proteínas si se modifica algún componente del sistema o si se cambian las condiciones de operación.. En este trabajo se realizó un análisis sobre los modelos termodinámicos más utilizados en este tipo de sistemas. Estos modelos son el NRTL (No random two liquids), Wilson modificado y NRTL modificado. Los sistemas que se estudiaron fueron escogidos en base a la disponibilidad de datos experimentales en la literatura y a las características de los compuestos que los formaban. Se buscó que los compuestos tuvieran propiedades diferentes en cuanto a tamaño del polímero y el tipo de sal usada. Los sistemas seleccionados están compuestos por: PEG 4000-Agua-K2HPO4, PEG 4000-Agua-Li2SO4, PEG 6000-Agua-K2HPO4 y PEG 1000-Agua-K2HPO4.. Además se analizaron dos ecuaciones para la partición de las proteínas. La primera ecuación fue deducida por Haynes, mientras que la segunda por Grossmann. Las proteínas que se estudiaron fueron: lisozima, catalasa, IgG y BSA.. i.
(5) Resumen. Dentro de los objetivos se incluyó el análisis del ajuste que podían tener los modelos termodinámicos a los diferentes sistemas y se midió el grado de ajuste que obtuvo cada uno para poder compararlos entre sí. Además el ajuste de los modelos se hizo mediante dos métodos numéricos diferentes, uno utilizando el “Solver” de Excel y el otro el “Genetic algoritmh and direct serach” de Matlab, para poder comparar y analizar cuál de estos métodos es un mejor camino para el ajuste. También se analizó la robustez numérica y la eficiencia computacional que se alcanzaba con cada uno de los modelos y métodos usados. Finalmente se usaron los modelos ajustados para predecir la composición de las fases de los sistemas y la partición de las proteínas, analizando la desviación que se tuvo con cada uno de los modelos y métodos usados.. El modelo que obtuvo una menor desviación promedio en los diferentes sistemas fue el Wilson modificado. En general el método del “Solver” fue un mejor camino para ajustar los parámetros de interacción ya que con estos parámetros se obtuvieron menores desviaciones promedio. La robustez de los modelos termodinámicos es muy similar, aunque en cuanto a eficiencia computacional el modelo de NRTL supera a los demás, esto va de acuerdo con la simplicidad del modelo. Al comparar los modelos modificados (Wilson y NRTL) el de Wilson modificado resultó ser más eficiente.. Los resultados obtenidos demuestran que es posible obtener buenas predicciones de este tipo de sistemas y de la partición de algunas proteínas. Se pudo concluir, que el modelo de Wilson modificado satisfizo mejor los factores analizados, al igual que la ecuación de Grossmann para la partición de proteínas.. ii.
(6) Índice. Índice Resumen ..........................................................................................................................i Índice ............................................................................................................................ iii Nomenclatura ..................................................................................................................v Capítulo 1........................................................................................................................1 1. Introducción ............................................................................................................1 1.1. Sistemas de dos fases acuosas (ATPS: Aqueous two-phase systems)................1. 1.2. Objetivos .........................................................................................................4. 1.2.1. Objetivo General ......................................................................................4. 1.2.2. Objetivos específicos................................................................................4. 1.3. Descripción de la tesis......................................................................................5. Capítulo 2........................................................................................................................6 2. Equilibrio líquido-líquido en ATPS y partición de biomoléculas ..............................6 2.1. Equilibrio líquido-líquido en ATPS..................................................................6. 2.1.1. Modelos termodinámicos .........................................................................8. 2.1.1.1. NRTL (Non-random two liquid) ...........................................................8. 2.1.1.2. Wilson modificado .............................................................................12. 2.1.1.2.1 Contribución de largo alcance (LA) ..............................................15 2.1.1.2.2 Contribución combinatorial (Comb)..............................................18 2.1.1.2.3 Contribución de corto alcance (CA) ..............................................19 2.1.1.3. NRTL modificado ..............................................................................21. 2.1.1.3.1 Contribución de corto alcance (CA) ..............................................22 2.1.2. 2.2. Equilibrio líquido-líquido y ajuste de parámetros....................................24. 2.1.2.1. Equilibrio líquido-líquido ...................................................................24. 2.1.2.2. Ajuste de parámetros de interacción....................................................26. Partición de biomoléculas ..............................................................................27. Capítulo 3......................................................................................................................36 3. Evaluación de los modelos termodinámicos ...........................................................36 3.1. Ajuste de parámetros de interacción ...............................................................36. iii.
(7) Índice 3.2. Análisis numérico ..........................................................................................44. 3.3. Predicción de la composición de cada fase .....................................................49. 3.4. Predicción de la partición de proteínas ...........................................................55. Capítulo 4......................................................................................................................67 4. Conclusiones y trabajo futuro ................................................................................67 4.1. Conclusiones..................................................................................................67. 4.2. Trabajo futuro ................................................................................................69. Referencias....................................................................................................................70 Apéndices......................................................................................................................74 Apéndice A. Datos experimentales equilibrio de fases ...............................................74 Apéndice B. Propiedades físicas utilizadas para cálculos ...........................................75 Apéndice C. Valores iniciales de los parámetros de interacción .................................76 Apéndice D. Valores predichos para todos los sistemas .............................................77 Apéndice E. Datos experimentales coeficientes de partición ......................................99 Apéndice F. Características de las proteínas...............................................................99. iv.
(8) Nomenclatura. Nomenclatura a. actividad. A. constante de Debye-Hückel. Aij,Aji parámetro de interacción binario AE. energía Helmholtz molar de exceso. B. parámetro de tamaño finito de los iones. d. densidad de la fase. D. constante dieléctrica del agua. F. constante de Faraday. gij,gji. parámetro de interacción binario. gE. energía Gibbs molar de exceso. gideal. energía Gibbs molar de exceso ideal. m. g. energía Gibbs molar de mezclado. greal. energía Gibbs molar de exceso real. Gij,Gji parámetro de interacción binario hE. entalpía molar de exceso. I. fuerza iónica de la mezcla. k. constante de Boltzmann. kji. constante de partición. K. coeficiente de partición. L. moles o flujo molar. m. molalidad. M. peso molecular. n. número de moles. q. número efectivo de segmento. r. número de segmentos. R. constante universal de los gases. T. temperatura absoluta. To. temperatura de referencia, 298.15 °K. v. suma de los números estequiométricos del anión y catión. v.
(9) Nomenclatura va. número estequiométrico del anión. vc. número estequiométrico del catión. vi. volumen molar del líquido puro i. E. v. volumen molar de exceso. x. fracción molar. xji. fracción molar local. X. fracción hipotética efectiva. z. carga del ion. Símbolos griegos α,αij,αji parámetro no aleatorio binario Δφ. diferencia de potencial electrostático. γ. coeficiente de actividad. γ*. coeficiente de actividad normalizado a dilución infinita. λij. parámetro de interacción binario. τij,τji. parámetro de interacción binario. θ. fracción de segmento efectiva. Φ. fracción volumétrica. subíndices 1. PEG. 2. Agua. 3. Sal. a,a’,a” anión c,c’,c” catión m. especies neutras. ca. sal. i,j,k,l compuesto i, j, k o l LA. contribución de largo alcance. CA. contribución de corto alcance. Comb contribución de combinatorial p. proteína. vi.
(10) Capítulo 1. Capítulo 1. 1 Introducción. 1.1 Sistemas de dos fases acuosas (ATPS: Aqueous two-phase systems) Los sistemas de dos fases acuosas son usados tradicionalmente para la recuperación primaria de productos biotecnológicos; como lo son enzimas de uso industrial, proteínas recombinantes, drogas terapéuticas, etc. (Rito-Palomares, 2004). Se forman cuando se mezclan dos polímeros o un polímero y una sal con agua a ciertas condiciones.. Desde 1896 Beijerinck notó la incompatibilidad que había cuando se mezclaban agua y polímeros solubles, como agar con almidones o gelatina solubles. Después de mezclarse los componentes, estos se separaban en dos fases inmiscibles. Debido a esto, posteriormente se empezó a hacer investigación sobre otros sistemas similares que tuvieran la capacidad de formar fases inmiscibles, en donde los sistemas formados por polietilenglicol (PEG) y dextran han sido los más estudiados. Otros sistemas que son capaces de formar fases acuosas son los compuestos por PEG y sales como citratos, sulfatos, carbonatos, fosfatos, etc. Estos sistemas han sido populares debido a que tienen un costo económico menor que los sistemas que tienen dos polímeros.. 1.
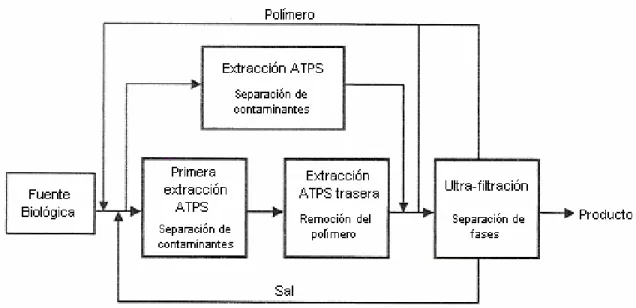
(11) Capítulo 1. La separación de compuestos químicos mediante la extracción o partición en fases inmiscibles, fue iniciada por Lyman C. Craig (Craig and Craig, 1956). Posteriormente a mediados de los años 50, Albertsson en Suecia demostró que la técnica de partición podría ser usada para la separación de materiales biológicos (Albertsson, 1986).. Un sistema de dos fases acuosas contiene principalmente agua, con una fase rica en el primer polímero y con la otra fase rica en el segundo polímero (o sal).. En la Figura 1 podemos ver un diagrama general de un proceso de recuperación utilizando dos fases acuosas. El proceso comienza con la fuente que contiene el material biológico para después pasar a una primera etapa de extracción mediante ATPS, posterior a la extracción se procede a separar la fase rica en polímero para poder ingresar a una etapa final en donde por medio de ultrafiltración se recupera el polímero y la sal para posteriormente ser recirculados al inicio del proceso. Después de la ultrafiltración se obtiene el producto listo para ingresar a las etapas de purificación por medio de cromatografía.. Figura 1 Diagrama general de un proceso de recuperación por sistema de dos fases acuosas (RitoPalomares, 2004).. 2.
(12) Capítulo 1 En la Figura 2a se pueden ver de manera esquemática las dos fases que se forman en un sistema ATPS, donde la fase superior es rica en el polímero 1 y la fase inferior es rica en el polímero 2 o la sal. Los sistemas de dos fases acuosas suelen representarse mediante curvas binodales (Figura 2b), en donde lo que se grafica son las composiciones o porcentajes peso del polímero 1 y del polímero 2 o la sal, las fases coexistentes se representan mediante la línea AB, donde A y B son las composiciones de las fases ‘(superior) y “(inferior), en equilibrio.. Figura 2 (a) Diagrama que representa las dos fases formadas en un sistema de dos fases acuosas (b) Diagrama representativo de la curva binodal de un sistema de dos fases acuosas polímeropolímero.. En la actualidad, la implementación de sistemas de dos fases acuosas es complicada debido al gran número de pruebas experimentales que se deben de llevar a cabo. Se siguen algunas reglas empíricas en su implementación, pero en algunas circunstancias es más bien un procedimiento de prueba y error. Este gran número de pruebas a su vez se traduce en tiempo y dinero invertido. Por este motivo ha surgido la necesidad de implementar modelos para el diseño de este tipo de procesos. Dentro de estos modelos de diseño es necesario utilizar modelos termodinámicos para predecir el equilibrio de fases y la partición de las proteínas, por lo que estos últimos vienen siendo la columna vertebral de todo el modelo. Debido a esto se vuelve de suma importancia la aplicación de modelos termodinámicos apropiados para este tipo de sistemas, por lo que ha sido necesario el estudio de los modelos termodinámicos ya existentes y de la obtención de algunos 3.
(13) Capítulo 1 nuevos. En la medida que se seleccione el modelo termodinámico más apropiado para los sistemas de dos fases acuosas, se hará más sencilla y confiable la implementación de un modelo de diseño en el que se prediga la partición de proteínas y el equilibrio de fases.. 1.2 Objetivos 1.2.1 Objetivo General. Analizar los modelos termodinámicos aplicados en los sistemas de dos fases acuosas, tomando en cuenta su capacidad de predecir los datos experimentales, la robustez numérica al ajustar sus parámetros y la capacidad de predecir la partición de proteínas.. 1.2.2 Objetivos específicos. . Ajustar los parámetros de interacción para todos los modelos termodinámicos en todos los sistemas por medio de dos métodos diferentes.. . Hacer el cálculo del equilibrio líquido-líquido con los parámetros de interacción ajustados, para todos los sistemas y con todos los modelos termodinámicos.. . Realizar pruebas para determinar la robustez de los diferentes modelos termodinámicos aplicados a los diferentes sistemas.. . Hacer un análisis sobre los modelos termodinámicos de acuerdo a los resultados obtenidos.. . Predecir la partición de las proteínas con los modelos termodinámicos que lo permiten (Wilson y NRTL modificados).. . Determinar cuál de los modelos termodinámicos es el más conveniente aplicar en la predicción de fases de sistemas de dos fases acuosas y en el cálculo de la partición de proteínas.. 4.
(14) Capítulo 1. 1.3 Descripción de la tesis La tesis se divide en cuatro capítulos. El primero de ellos es una introducción a lo que son los sistemas de dos fases acuosas. El segundo trata sobre la teoría involucrada en el estudio de los modelos termodinámicos aplicados a sistemas de dos fases acuosas; se discuten los modelos de NRTL, Wilson modificado y NRTL modificado y su implementación en el cálculo del equilibrio de fases y de la partición de proteínas. El tercer capítulo muestra los resultados que se obtuvieron en el estudio de los diferentes sistemas con los 3 modelos termodinámicos, además de la discusión de los mismos. En el último capítulo se concluye y se proponen algunas opciones de hacia donde se podrían dirigir los trabajos a futuro.. 5.
(15) Capítulo 2. Capítulo 2. 2 Equilibrio líquido-líquido en ATPS y partición de biomoléculas. 2.1 Equilibrio líquido-líquido en ATPS. Se han propuesto modelos termodinámicos (Prausnitz et al., 2000d) que intentan calcular las propiedades reales de las mezclas de líquidos con el fin de poder predecir el comportamiento de ciertas mezclas a ciertas condiciones de temperatura y concentración. Conforme las mezclas líquidas se vuelven menos ideales (moléculas más grandes, con carga, polares, etc.) los modelos termodinámicos tienen que incluir una mayor cantidad de factores que influyen al comportamiento de las mezclas. En estos modelos se trata de tomar en cuenta los factores más influyentes y se considera que los factores menos influyentes no existen. Debido a que la cantidad de sistemas que se pueden formar con la gran variedad de sustancias líquidas que existen es muy grande, se suelen aplicar modelos termodinámicos. para diferentes familias de sustancias con algunas propiedades. específicas como lo son polaridad, carga eléctrica, tamaño molecular, etc.. Los modelos termodinámicos son deducidos a partir de funciones de energía de Gibbs en exceso. Estas funciones son la diferencia entre el valor de la propiedad termodinámica. 6.
(16) Capítulo 2 (energía de Gibbs) para una disolución real y el valor de esa misma propiedad para una disolución ideal, en las mismas condiciones de temperatura, presión y composición. Cuando se tiene una disolución ideal la función de exceso tiene un valor de cero. La energía de Gibbs de exceso, gE, se define como: ideal g E g Treal , P , x g T , P, x. 2.1. en donde T, P y x representan respectivamente que la temperatura, presión y composición son constantes. Entonces, entre más distante de cero sea el valor de la función gE más alejado se estará de la disolución ideal. Debido a la capacidad de cuantificar el distanciamiento de la idealidad, estas funciones son buenas herramientas para predecir el comportamiento de disoluciones alejadas de la idealidad. En algunas ocasiones también se usa la propiedad residual, está es análoga a la propiedad en exceso sólo que considera constantes a la temperatura (T), el volumen (V) y la composición (x).. En el caso de los sistemas de dos fases acuosas (ATPS por sus siglas en inglés), las moléculas involucradas tiene algunas características que hacen que la mezcla sea fuertemente no ideal. El agua por una parte tiene la capacidad de formar puentes de hidrógeno, las cuales son fuerzas difíciles de modelar matemáticamente; los polímeros al ser moléculas extremadamente grandes están expuestos a una mayor cantidad de interacciones con moléculas vecinas además de su capacidad de formar puentes de hidrógeno; las sales electrolíticas son moléculas generalmente pequeñas, pero con la característica de que al mezclarse con agua se separan en sus respectivos cationes y aniones, quedando así como moléculas con carga eléctrica.. Debido a las características de las moléculas involucradas en los sistemas ATPS, algunos de los modelos termodinámicos que más se utilizan para predecir su comportamiento son el NRTL (Sé and Aznar, 2002), el Wilson modificado (Madeira et al., 2005) y el NRTL modificado (Wu et al., 1998a). Estos modelos fueron deducidos especialmente para. 7.
(17) Capítulo 2 sistemas fuertemente no ideales, pero cada uno de ellos toma en cuenta diferentes factores específicos.. 2.1.1 Modelos termodinámicos. En esta sección se estudiarán los modelos termodinámicos de NRTL, Wilson modificado y NRTL modificado. La selección de estos modelos termodinámicos se llevó a cabo de acuerdo a una revisión bibliográfica en donde se encontró que los modelos que se presentarán han tenido éxito en sistemas similares a los que se estudiarán aquí (fuertemente no ideales), además de ser muy populares entre este tipo de modelos termodinámicos.. 2.1.1.1 NRTL (Non-random two liquid) La forma más sencilla de la ecuación NRTL (Prausnitz et al., 2000c; Renon and Prausnitz, 1968b) se da cuando el sistema está formado únicamente por dos compuestos. Basado en algunas suposiciones hechas por Wilson, Renon llegó a la deducción del modelo NRTL en donde la suposición principal de este modelo radica en el concepto de composición local. Este concepto explica que para una mezcla de moléculas con interacciones fuertemente no ideales, las moléculas se agrupan de una manera no aleatoria en donde debido a las propiedades físicas de las moléculas se forman composiciones localizadas diferentes a la composición global. Este concepto se ayuda de la teoría de dos líquidos de Scott (Leland et al., 1969; Prausnitz et al., 2000e), la cual asume que hay dos tipos de células en una mezcla binaria: una para las moléculas tipo 1 y otra para las moléculas tipo 2, como se muestra en la Figura 3. Para las células que contienen a la molécula 1 en su centro, la energía de Gibbs residual, gij, es la sumatoria de todas las energías de Gibbs residuales de las interacciones experimentadas por la molécula 1 que se encuentra en el centro. De la misma manera ocurre para las células que contienen a la molécula 2 en el centro.. 8.
(18) Capítulo 2. Figura 3 Representación de los dos tipos de células de acuerdo a la teoría de dos líquidos para mezclas binarias de Scott y Leland (Leland et al., 1969).. La ecuación NRTL para la energía Gibbs en exceso es: G gE G x1 x2 21 21 12 12 RT x1 x2G21 x 2 x1G12. . 2.2. en donde. 12 . g12 g 22 RT. 2.3. 21 . g 21 g11 RT. 2.4. G12 exp 12 12 . 2.5. G21 exp 21 21 . 2.6. 12 21. 9. 2.7.
(19) Capítulo 2 El significado de gij representa. un parámetro energético de interacción. entre las. moléculas con subíndice i-j. El parámetro αij está relacionado con las distribución no aleatorizada de las moléculas en la mezcla, cuando este parámetro vale cero, esto quiere decir que la mezcla es completamente aleatoria, lo que reduciría la ecuación a la de Margules con dos subíndices. La ecuación de NRTL para el caso binario tiene 3 parámetros, pero el tratamiento de un gran número de sistemas binarios ha indicado que el parámetro α varía entre 0.2 y 0.47; cuando se tiene un número de datos escasos el valor de α se fija arbitrariamente en 0.3.. A partir de la ecuación (2.2) se pueden deducir las expresiones para los coeficientes de actividad:. 2 G 12G12 21 ln 1 x 21 2 x1 x2G21 x2 x1G12 2 2. 2.8. 2 G 21G21 12 ln 2 x 12 2 x2 x1G12 x1 x2G21 2 1. 2.9. Para sistemas en donde las no idealidades son moderadas, la ecuación de NRTL no presenta muchas ventajas respecto a otras, como la de Van Laar o la de Margules de 3 subíndices. Sin embrago, para sistemas que se apartan mucho de la idealidad, y especialmente para los sistemas parcialmente miscibles, esta ecuación proporciona una buena representación de los datos experimentales si se obtienen cuidadosamente los parámetros ajustables. Debido a que la selección de los parámetros de interacción es de suma importancia, se han realizado experimentos (Anderson et al., 1978) para corroborar la validez de algunos parámetros ajustables, y se ha demostrado que existen una gran cantidad. de. combinaciones. de. parámetros que. logran. ajustar. los. modelos. termodinámicos, estas combinaciones de parámetros se reducen en la medida que la calidad y/o cantidad de los datos experimentales aumenta y también cuando aumenta la adecuación del modelo termodinámico para la energía de Gibbs en exceso.. 10.
(20) Capítulo 2 La ecuación NRTL se puede expresar de una manera general para el caso en el que hay tres o más componentes involucrados, este caso se conoce como caso multicomponente.. Para una disolución de m componentes, la ecuación NRTL viene dada por: m m gE xi RT i 1. . ji. G ji x j. j 1 m. 2.10. G x. li l. l 1. donde. ji . g ji gii RT. 2.11. G ji exp ji ji . 2.12. ji ij. 2.13. Y el coeficiente de actividad de un componente i viene dado por: m. ln i . ji. G ji x j. j 1 m. G x. li l. l 1. m xr rj Grj m xG m j ij ij r 1m j 1 Glj xl Glj xl l 1 l 1 . 2.14. Las ecuaciones (2.11) y (2.12) contienen solamente parámetros obtenidos a partir de datos binarios. Renon (Renon and Prausnitz, 1968a, b) demostró que aun usando parámetros binarios para calcular el equilibrio de sistemas ternarios, es posible obtener buenas predicciones.. 11.
(21) Capítulo 2. 2.1.1.2 Wilson modificado Es importante antes de hablar del caso modificado de la ecuación de Wilson, tocar algunos puntos importantes de su ecuación original y empezar con el caso binario que es el más sencillo.. Wilson (Prausnitz et al., 2000c; Wilson, 1964) dedujo su expresión para la energía Gibbs en exceso para una mezcla binaria, ver ecuación (2.17); basándose en dos consideraciones moleculares. La primera consideración supone que la energía libre de mezclado tiene una relación similar a la de la ecuación de Flory-Huggins, ver ecuación (2.16); y la segunda que la distribución de las moléculas alrededor de una molécula central sigue la relación dada en la siguiente ecuación:. g ji x j exp x ji kT x ki g ki x k exp kT . 2.15. en donde xji es la fracción molar “local” de j alrededor de i y gji es proporcional a la energía de interacción entre j y i.. gm xi ln i RT i. gE x1 ln x1 12 x2 x2 ln x2 21 x1 RT. 2.16. 2.17. Los coeficientes de actividad deducidos a partir de la ecuación (2.17) son: 12 21 ln 1 ln x1 12 x2 x2 x1 12 x2 21 x1 x2 . 12 21 ln 2 ln x2 21 x1 x1 x x x x 1 12 2 21 1 2 . 12. 2.18. 2.19.
(22) Capítulo 2 En la ecuación (2.17) se define la energía de gibbs en exceso con referencia a una disolución ideal en el sentido de la ley de Raoult (Prausnitz et al., 2000a); esta ecuación obedece la condición de contorno que obliga a gE a anularse cuando se anulan x1 o x2, lo que quiere decir que en los límites donde xi=0 y xi=1 está ecuación se convierte a la de la ley de Henry y la ley de Raoult.. La ecuación de Wilson para el caso binario presenta dos parámetros ajustables, Λ12 y Λ21, que en la deducción de Wilson se relacionan con los volúmenes molares de los componentes puros y con unas diferencias de energías caracterizadas por:. 12 . v2 exp 12 11 v1 RT . 2.20. 21 . v1 exp 21 22 v2 RT . 2.21. donde vi es el volumen molar del líquido puro i, y las λ son energías de interacción entre las moléculas designadas por los subíndices. Las diferencias de las energías características son independientes de la temperatura, al menos para intervalos moderados.. La ecuación de Wilson proporciona una buena representación de energía Gibbs exceso para muchas mezclas miscibles, pero es particularmente útil para mezclas polares debido a las suposiciones moleculares que representan bien este tipo de interacciones.. El caso modificado de la ecuación de Wilson incorpora los modelos semiempíricos formulados para sistemas de electrolíticos concentrados; véase (Grigera, 1992; Maurer, 1983; Prausnitz et al., 2000h; Renon, 1986). Estos modelos corrigen la teoría de DebyeHückel a través de términos adicionales que tienen en cuenta las interacciones ion-ion y la disociación incompleta a altas concentraciones.. En estos modelos semiempíricos, se suele suponer que la energía Gibbs molar de exceso de las disoluciones de electrolito es la suma de dos contribuciones, una procedente de las. 13.
(23) Capítulo 2 fuerzas culombianas de largo alcance, LA, representadas por la teoría de Debye-Hückel (Prausnitz et al., 2000g) o su generalización, y la otra de las fuerzas de corto alcance, CA.. E E g E g LA g CA. 2.22. La mayoría de los modelos semiempíricos utilizan un término de Debye-Hückel para la contribución de largo alcance, pero para la contribución de corto alcance existen varias opciones, dentro de las cuales se encuentran las expresiones de composición local (UNIQUAC, NRTL, Wilson). La mayor parte de los modelos suponen la disociación completa de los electrolitos. Utilizando al menos dos parámetros binarios ajustables, estos modelos tienen un éxito razonable para disoluciones diluidas y moderadamente concentradas, hasta aproximadamente 6 molal.. Las fuerzas de largo alcance entre iones dominan a concentraciones diluidas de electrolito, mientras que las de corto alcance dominan entre todas las especies a concentraciones altas.. La ecuación de Wilson modificada también incluye otro término de contribución a la ecuación (2.22) el cual es llamado combinatorial. Este término lo que hace es tomar en cuenta los cambios de energía Gibbs de exceso debido al tamaño y la forma de las moléculas, resultando la siguiente ecuación: E E E g E g LA g CA g comb. 2.23. De la ecuación (2.23) se puede escribir la siguiente expresión para el coeficiente de actividad: ln i ln i ,LA ln i ,CA ln i ,Comb. 2.24. Los coeficientes de actividad son normalizados (Prausnitz et al., 2000b) al estado de referencia a dilución infinita:. 14.
(24) Capítulo 2. ln i* ln i ln iref j 1. 2.25. resultando de esta manera los coeficientes de actividad normalizados al estado de referencia a dilución infinita, en donde se toma como referencia xagua=1 y para toda xj≠agua=0.. 2.1.1.2.1 Contribución de largo alcance (LA) El cálculo de la contribución de largo alcance, gELA; considera a los electrolitos y al agua debido a que en los sistemas ATPS estos compuestos tienen la capacidad de interaccionar de esta forma. Antes de poder realizar el cálculo del coeficiente de actividad de largo alcance es necesario introducir algunos conceptos, éstos son presentados a continuación.. Primeramente, las medidas termodinámicas habituales no dan las propiedades de una especie iónica, sino la de los electrolitos neutros formados por cationes y aniones, entonces es importante introducir el criterio de electroneutralidad, el cual impone la condición de no poder variar independientemente la cantidad de sustancia de las especies iónicas. Si planteamos la disociación de un electrolito de la siguiente manera. M vc X va vc M zc va M za. 2.26. entonces MvcXva representa a un electrolito eléctricamente neutro que se disocia en vc iones positivos (cationes), cada uno con una carga zc, y va iones negativos (aniones), de carga za. Además las cargas se expresan en unidades normalizadas, en donde zc=1 para un protón.. Para que el criterio de electroneutralidad se cumpla, se tiene que cumplir que. 15.
(25) Capítulo 2. vc z c va z a 0 vc zc va z a. 2.27. al cumplirse este criterio nos aseguramos que el incremento o decremento en alguno de los iones este directamente relacionado con el cambio en su contraion .. La contribución de las fuerzas de largo alcance para el coeficiente de actividad del agua es calculado de acuerdo a (Haghtalab and Mokhtarani, 2001):. ln . agua LA. . 1 1 2 AM agua 1 2 2 1 BI 2 ln 1 BI 3 1 10 B 1 BI 2. 2.28. en donde A es la constante de Debye-Hückel, Magua es el peso molecular del agua, B es un parámetro que refleja el tamaño finito de los iones e I es la fuerza iónica de la mezcla en escala de molalidad.. El Valor de la constante B se puede calcular mediante la ecuación (2.29), según lo mostrado por Wu y Lin (Wu et al., 1998a): 6.359696d 0.5 B DT 0.5. 2.29. en donde d representa la densidad de la fase superior o inferior, D es la constante dieléctrica del agua con un valor de 78.41 y T la temperatura absoluta.. El valor de A puede ser obtenido en función de la temperatura absoluta (Chen et al., 1982) de acuerdo a: 2. T 273.15 T 273.15 T A 61.4453 exp 2.864468 exp 183.5379 ln 273.15 273.15 273.15 . 273.15 0.6820223T 273.15 0.0007875695 T 2 273.152 58.95788 T . . 16. . 2.30.
(26) Capítulo 2 y la fuerza iónica de la mezcla se puede calcular de acuerdo a la siguiente ecuación:. I (molkg 1 ) . 1 mi zi2 2 i. 2.31. en donde m representa la molalidad de la especie i en la fase y zi la carga de la especie i. Para el cálculo del coeficiente de actividad de un ion j se utiliza la siguiente ecuación (Haghtalab and Mokhtarani, 2001):. 1. ln . j LA. . Az 2j I 2 1 BI. 2.32. 1 2. Debido a que los electrolitos en las mezclas de sistemas ATPS son considerados fuertes (disociación completa), es necesario introducir la expresión para el coeficiente de actividad iónico medio. El coeficiente de actividad iónico medio, γca, para el electrolito i viene dado por:. ln ca i. . 1 vc ln c va ln a v. 2.33. en donde γc y γa son los coeficientes de actividad del catión y el anión respectivamente y v es igual a la suma de vc y va, v = vc + va.. Debido a que las contribuciones de largo alcance para la especies quedarán en la escala de molalidad por las unidades de la fuerza iónica, es necesario transformarlas a la escala de molaridad para poderlas sumar posteriormente con las demás contribuciones. Esto se puede hacer de acuerdo a la siguiente ecuación:. . ln ca ,m ln ca, x ln 1 0.001M agua vc va m. . 2.34. en donde las γca representan los coeficientes de actividad medios iónicos en escala molal (m) y mol (x) y m es la molalidad del electrolito en la fase.. 17.
(27) Capítulo 2. 2.1.1.2.2 Contribución combinatorial (Comb) El cálculo de la contribución combinatorial, gEcomb , parte de la entropía de mezclado atérmica de Guggenheim (Xu et al., 2003). La suposición de disolución atérmica y el modelo de red son tomados en cuenta, por lo que hE=0 y vE=0, entonces gEcomb= AE. Tomando en cuenta sistemas multicomponentes con soluciones acuosas en donde existen dos polímeros resulta la siguiente ecuación: E m g comb X 1 m X ni ln i ni qi ln i RT xi i1 i i 1. 2.35. donde α representa el factor no aleatorio, ni es el número de moles de las especies, Φi y xi son la fracción volumétrica y la fracción mol respectivamente, y Xi es la fracción hipotética efectiva de segmento del polímero, y está dada por: X i i Ci. 2.36. en donde si i = ion entonces Ci = zi de otra manera Ci = 1 además se tiene que. i . ni ri nr. 2.37. m. nr nk rk k 1. i . ni qi nq. 2.38. 2.39. m. nq nk qk. 2.40. 1 qi ri 1 1 ri . 2.41. k 1. 18.
(28) Capítulo 2 donde ri es el número de segmentos, θi es la fracción de segmento efectiva y qi significa el número efectivo de segmento.. Para entender lo que es un segmento del polímero (Prausnitz et al., 2000f), se puede poner como ejemplo a una mezcla de dos líquidos, en donde uno es el disolvente y el otro un polímero disuelto en él. Las moléculas de disolvente se consideran esferas simples, en cambio las moléculas de polímero se supone que se comportan como cadenas flexibles, es decir, como si estuvieran formadas por un gran número de segmentos móviles, cada uno con el mismo tamaño que una molécula de disolvente. De la ecuación (2.35) se puede obtener una expresión del coeficiente de actividad para la contribución combinatorial:. ln i ln. q Xi m X j 1 i qj xi j 1 . qi . m q r X i j i 1 j ln i j 1 qi r j . 2.42. 2.1.1.2.3 Contribución de corto alcance (CA) El cálculo de la contribución de corto alcance, gECA; se basa en las suposiciones de electroneutralidad local, repulsión de iones semejantes y en la agregación de estado hipotética de segmentos de polímero. Siguiendo el desarrollo de Wu y Chen (Wu et al., 1998b), la expresión de la contribución de corto alcance de la energía Gibbs de exceso puede ser extendida del caso de soluciones binarias de polímeros (Xu et al., 2003): E g CA 1 X a' X c' X m ln X j G jm X c ln X j G jc ,a 'c X a ln X j G ja ,c 'a RT m X X j c a ' j a c ' a " c " j a" c" 2.43. 19.
(29) Capítulo 2 en donde los subíndices m, c (o c’, c”) y a (o a’, a”) representan a las especies neutrales, cationes y aniones respectivamente. X representa la fracción hipotética efectiva de segmento, ver la ecuación (2.36); α es el factor no aleatorio y G y τ son parámetros de interacción representados por las siguientes relaciones:. aijT0. ij . ji . 2.44. T. a ji T0 T. 2.45. Gij exp ij ij . 2.46. G ji ,ki exp ji ,ki ji , ki . 2.47. De la ecuación (2.43) se pueden obtener los coeficientes de actividad, quedando de la siguiente manera:. . X c Gmc,a 'c ln m CA ln X j G jm X m' Gmm' X a ' qm X jG jm c a ' X a" X jG jc ,a 'c j m' j. a". a. c'. X aGma,c 'a X c' 1 X c" X jG ja ,c'a c". . j. 2.48. j. ln c CA X a ' ln X j G jc ,a 'c X m zc a ' X a" j m a". Gcm X j G jm j. a. c'. X c' X c" c". 20. X a Gca,c 'a. X j. j. G ja ,c 'a. . 1 zc. 2.49.
(30) Capítulo 2. . ln a CA X c ' ln X j G ja ,c 'a X m Gam za c ' X c" X j G jm j m c". j. c. a'. X a' X a". X c Gac ,a 'c. X. a". j. G jc , a 'c. . 1 za. 2.50. j. además se tiene que. am cm ca ,m. mc ,ac ma ,ca m ,ca. 2.51. 2.52. y. am cm ca,m. mc ,ac ma ,ca m ,ca. 2.53. 2.54. 2.1.1.3 NRTL modificado En el modelo NRTL modificado se utiliza la misma forma de modelo semiempírico utilizado en el caso de Wilson modificado, ver ecuación (2.23). Las contribuciones a la energía de Gibbs en exceso de largo alcance y combinatorial se expresan de la misma manera que en el modelo termodinámico anterior (ver, 2.1.1.2.1 y 2.1.1.2.2).. 21.
(31) Capítulo 2. 2.1.1.3.1 Contribución de corto alcance (CA). El término de la contribución de corto alcance sufre cambios en este modelo, el cual tiene como forma base la del modelo NRTL original propuesto por Renon y Prausnitz (Renon and Prausnitz, 1968b).. Posteriormente al modelo NRTL original se le hacen algunas modificaciones para el caso en el que se tienen sistemas acuosos con electrolitos. Esto fue hecho por Chen y Evans en 1986 (Chen and Evans, 1986). Después basándose en este modelo electrolítico, Wu, Lin y Zhu (Wu et al., 1998b); extienden el modelo para el caso de sistemas binarios de polímeros y sistemas de mezclas de polímeros con electrolitos. En este último modelo, las contribuciones de corto alcance toman en cuenta como suposición fundamental que la no idealidad en la entropía de mezclado es despreciable en comparación al calor de mezclado, resultando de esta manera la contribución de corto alcance para la energía de Gibbs en exceso:. E g CA Xm nq RT m. X G X G j. jm. jm. j. k. km. X a' Xc c a ' X a". k. X G X G. a". X c' Xa a c ' X c" c". ja ,c 'a. jc ,a 'c. k. kc ,a 'c. ja ,c 'a. j. k. jc ,a 'c. k. X G X G j. j. j. 2.55. ka ,c 'a. k. en donde los subíndices m, c (o c’, c”) y a (o a’, a”) representan a las especies neutrales, cationes y aniones respectivamente. X representa la fracción hipotética efectiva de segmento, ver la ecuación (2.36); y G y τ son parámetros de interacción representados por las siguientes relaciones: Gij exp ij ij . 2.56. G ji ,ki exp ji ,ki ji ,ki . 2.57. 22.
(32) Capítulo 2. ij . ji ,ki . Aij. 2.58. RT A ji ,ki RT. 2.59. De la ecuación (2.55) es posible deducir las expresiones para el coeficiente de actividad, quedando para cada una de las especies de la siguiente manera:. 1 ln mCA qm. X G X G j. jm. jm. j. k. km. k. k X k Gkm' km' X m 'Gmm ' mm ' X k Gkm ' m ' X k Gkm ' k k . X c Gmc ,a 'c X a' c a ' X a " X k Gkc ,a 'c. k X k Gkc,a 'c kc,a 'c mc,a 'c X k Gkc ,a 'c k . X a Gma,c 'a X c' a c ' X c " X k Gka ,c 'a. k X k Gka ,c'a ka,c'a ma ,c 'a X k Gka,c 'a k . a". k. c". k. X a' 1 ln cCA zc a ' X a" a". X G X G k. X c' a c ' X c" c". kc , a 'c kc , a 'c. k. k. kc , a 'c. k. k X k Gkm km X m Gcm cm X k Gkm m X k G km k k . k X k Gka,c'a ka ,c'a X aGca,c 'a k X k Gka,c'a ca,c'a k X k Gka,c 'a . 23. 2.60. 2.61.
(33) Capítulo 2. X c' 1 ln aCA za c ' X c" c". X G X G k. k. ka ,c 'a. k. X a' c a ' X a" a". ka ,c 'a ka ,c 'a. k. X cGac ,a 'c X k Gkc,a 'c k. k X k Gkm km X m Gcm am X k Gkm m X k G km k k k X k Gkc,a 'c kc,a 'c ac,a 'c X k Gkc,a 'c k . 2.62. en donde las ecuaciones (2.51) a (2.54) también se cumplen.. 2.1.2 Equilibrio líquido-líquido y ajuste de parámetros. 2.1.2.1 Equilibrio líquido-líquido. Para el cálculo del equilibrio líquido-líquido en sistemas multicomponentes es necesario resolver un sistema de ecuaciones simultáneas no lineales en donde hay que tomar en cuenta las ecuaciones de equilibrio (no lineales) y los balances de materia (lineales) correspondientes. Se puede elaborar una ecuación de equilibrio por cada componente que exista, por lo que para un sistema de m componentes se podrán obtener la misma cantidad de ecuaciones de equilibrio y también la misma cantidad de balances de materia.. Para el caso de sistemas en fase líquida la condición de equilibrio viene dada por la actividad, a, en donde para cada componente i se tiene que. ai' ai". 2.63. y. ai' i' xi'. 24. 2.64.
(34) Capítulo 2 2.65. ai" i" xi". en donde los superíndices ‘(prima) y “(doble prima) representan respectivamente a la fase superior e inferior, x a la fracción mol y γ al coeficiente de actividad.. Las ecuaciones del coeficiente de actividad, γ, están sólo en función de las composiciones de la mezcla líquida cuando la temperatura y la presión son constantes; por lo que la condición de equilibrio, si se sustituyen las ecuaciones (2.64) y (2.65) en la ecuación (2.63), se puede representar de acuerdo a. i' x1' ...xm' xi' i" x1" ...x m" xi". 2.66. Al resolver un problema de equilibrio líquido-líquido lo que se busca obtener son las coordenadas de las composiciones de xi’ y xi”. Si se observa la ecuación (2.66) se puede ver que se tendrían como incógnitas m composiciones de xi’ y m composiciones de xi”, además se sabe que la suma de las composiciones para cada fase debe de dar como resultado 1 por lo que se pueden generar dos ecuaciones más de acuerdo a ' i. " i. x x i. 1. 2.67. i. esto da como resultado un sistema con m ecuaciones de equilibrio, más dos ecuaciones de la sumatoria de composiciones con un número de 2m incógnitas por lo que es necesario incluir balances de materia para la solución, quedando de la siguiente manera el sistema final:. L' L" L xi' L' xi" L" xi. i 1...m 1. i' xi' i" xi". i 1...m. ' i. 1. " i. 1. x. 2.68 2.69 2.70. 2.71. i. x. 2.72. i. 25.
(35) Capítulo 2 en donde las dos nuevas incógnitas que se incluyen por los balances de materia, L’ y L”, representan respectivamente a la cantidad de moles de la fase superior y de la fase inferior, y L la moles totales del sistema.. Después de incluir los balances de materia el sistema queda con 2m incógnitas de composiciones xi más los valores de L’ y L”, dando un total de 2m + 2 incógnitas; y un número m de ecuaciones de equilibrio y m balances de materia, más las dos ecuaciones de la sumatoria de composiciones, resultando 2m + 2 ecuaciones; por lo que el sistema ahora puede ser resuelto.. En principio al tener un sistema de estas características el problema está resuelto, pero en muchas ocasiones el procedimiento numérico para hacerlo correctamente no es necesariamente sencillo. Para ver más sobre el equilibrio líquido-líquido ver (Prausnitz et al., 2000i).. 2.1.2.2 Ajuste de parámetros de interacción La selección de los parámetros termodinámicos de los diferentes modelos es crucial para el buen ajuste a los datos experimentales. Existen diferentes formas de ajustar esos parámetros y diferentes funciones objetivo utilizadas para la optimización (Vasquez and Whiting, 2000). Los parámetros que se suelen ajustar son 2 interacciones binarias por cada pareja de compuestos que se pueda formar, más 1 parámetro no aleatorio (αij) por cada compuesto. Por ejemplo en el caso de un sistema ternario se pueden formar 3 parejas diferentes, por lo que los parámetros ajustables serían 6 de las parejas más 3 por cada compuesto, teniendo en total 9. Sólo en el caso de los modelos modificados se agrega 1 parámetro aleatorio más que aparece en parte de la contribución combinatorial y de corto alcance, ecuaciones (2.41), (2.42) y (2.43).. 26.
(36) Capítulo 2 La función objetivo utilizada se basa en la condición de equilibrio termodinámico o de isoactividad, ecuación (2.66); pero se normaliza para obtener mejores resultados en la optimización. La función objetivo queda de la siguiente manera:. . x* ' x* i i j i i min FO * " i 1 j xi i j N. ". j. 2. 2.73. en donde γi* representa al coeficiente de actividad normalizado al estado de referencia a dilución infinita, j representa a las especies del sistema y N a los datos al equilibrio experimentales.. Para la minimización de la función objetivo se utilizaron dos métodos numéricos. Primeramente se usó la herramienta del “Solver” de Excel y posteriormente se utilizó un método basado en algoritmos genéticos, aplicado mediante el Toolbox “Genetic algoritmh and direct serach” de Matlab. Por lo que, cuando se mencione en los siguientes capítulos como método de ajuste al Solver o al algoritmo genético, se estará refiriendo a las herramientas de estos paquetes computacionales.. 2.2 Partición de biomoléculas El estudio de la partición de biomoléculas en ATPS tiene un grado de complejidad mayor que el de otras moléculas. Esto se debe principalmente a la compleja estructura molecular de este tipo de moléculas, sobre todo a las de mayor interés comercial (proteínas, enzimas, ácidos nucleicos, etc.). Entre más compleja sea la estructura de una molécula, son mayor la cantidad de interacciones que puede tener, más aún cuando cuentan con una gran variedad de grupos funcionales que tienen la capacidad de interactuar de muchas maneras.. Para el caso de sistemas ATPS se han estudiado diferentes efectos que tienen influencia en la partición de biomoléculas, siendo los principales: la composición del polímero en. 27.
(37) Capítulo 2 las fases, la adición de aditivos electrolíticos y no electrolíticos de bajo peso molecular, cambios en el pH, y la estructura y peso molecular de las biomoléculas.. La composición de polímero en las fases del sistema se puede manipular de acuerdo a la selección del polímero y de su peso molecular, pero esto sólo se podrá hacer sobre una fase ya que la composición de polímero en la otra fase queda fijada una vez que se tiene la de una de ellas. Se han hecho algunos estudios sobre el efecto que tiene la composición de polímero en las fases, pero la mayoría de estos han sido realizados exclusivamente para sistemas ATPS con PEG y Dextran, de donde se han podido obtener algunas reglas empíricas (Albertsson, 1986; D.Fisher and Sutherland, 1989).. Diamond y Hsu (A.D.Diamond and J.T.Hsu, 1989) pudieron derivar, a partir de la teoría de Flory-Huggins de termodinámica de polímeros; una relación cuantitativa entre el coeficiente de partición y la diferencia de composición de polímero en las fases que fue comprobada para algunos péptidos y proteínas de bajo pedo molecular (PM<25,000). La relación es la siguiente: ln K j k ji C pol (i ). 2.74. en donde Kj es el coeficiente de partición del péptido o proteína j; ΔCpol(i) es la diferencia entre las concentraciones del polímero i entre las dos fases (en los sistemas DEX-PEG, el PEG es usado generalmente como polímero i); y kji es una constante que depende del soluto que está siendo particionando y del sistema ATPS particular que se está usando. Para el caso en que se tienen biomoléculas de altos pesos moleculares, lo que se ha hecho es ajustar a relaciones empíricas, pero estas relaciones no ayudan a explicar la separación debido a su carencia de significado físico.. Otro efecto que se ha estudiado debido a su fuerte influencia en la partición de biomoléculas es el de la adición (como aditivos) o utilización de sales en los sistemas ATPS. La adición de sales afecta básicamente de dos maneras, hace que cambie la composición y por lo tanto las propiedades de las fases y hace que la biomolécula. 28.
(38) Capítulo 2 cambie sus propiedades, por lo que la interacción entre la biomolécula y el solvente también se verá afectada. Este último mecanismo en el que afecta la adición de sales se puede presentar, por ejemplo; cuando una proteína cambia su tamaño debido a la fuerza iónica del medio, cuando se dan cambios conformacionales por la presencia de ciertos aditivos o cuando se llegan a dar ligaduras entre el aditivo y la biomolécula. Se ha propuesto que la partición de la biomolécula depende mayormente de la interacción que tiene con el solvente, así como pasa en sistemas de compuestos orgánicos y agua, por lo que las sales tendrían un peso importante en esta dependencia. Las interacciones entre el solvente y las biomoléculas se pueden dividir básicamente en polares e hidrofóbicas. En la medida que el medio acuoso tenga la habilidad de interaccionar más con ambas regiones de las biomoléculas (polares e hidrofóbicas), la separación será mejor.. La manipulación del pH es otra manera de controlar la partición de biomoléculas. Esta manipulación generalmente se suele hacer mediante el uso de sales amortiguadoras, con las que en base a la relación de una sal y su sal conjugada se puede ajustar el pH. La mayoría de los estudios que se han hecho para sistemas de dos fases acuosas han sido fijando la composición del polímero y manipulando el pH del sistema, en algunas ocasiones adicionando sales como aditivos (Albertsson, 1986).. Eiteman y Gainer sugirieron que la diferencia de pH entre las fases para sistemas poliméricos de dos fases acuosas podría ser una medida de las propiedades fisicoquímicas que gobiernan la partición de solutos con carga en relación a aquellos con la misma estructura pero sin carga.. Hay dos factores fisicoquímicos que son los que afectan en mayor medida la partición de solutos en dependencia al pH en un sistema de fases acuosas. Uno es el cambio de las propiedades en el medio acuoso de las fases inducido por un cambio de pH determinado y/o el cambio correspondiente en la composición de las sales que conforman al amortiguador. El otro es el cambio (debido al pH) en el soluto, ocasionando cambios en las interacciones que tiene el soluto.. 29.
(39) Capítulo 2 La dependencia del coeficiente de partición de las proteínas respecto al pH generalmente es tratada en términos de una aparente diferencia de potencial electrostático entre las fases coexistentes en donde, debido a la influencia de esa diferencia, las proteínas cargadas se particionan. La dependencia del coeficiente de partición se desarrolló mediante una relación empírica (Johansson, 1985) entre el coeficiente de partición, Kp, y la carga neta de la proteína, zp, obteniéndose lo siguiente:. ln K p ln K o z p. 2.75. en donde Ko corresponde al valor del coeficiente de partición en el punto isoeléctrico, Kp y zp son el coeficiente de partición de la proteína y la carga neta de la proteína respectivamente y γ es definido como un factor que depende de la composición del polímero en el sistema, la sal usada como aditivo y la temperatura.. La carga neta de una proteína se considera como una carga global, esto debido a que las proteínas al ser moléculas complejas poseen una gran cantidad de grupos funcionales los cuales pueden estar cargados tanto positiva como negativamente, por lo que la carga neta sería la sumatoria de todos estos grupos individuales.. La ecuación (2.75) tiene un valor limitado. El carácter lineal de la relación ln Kp – zp generalmente no se ve en la práctica, como se puede ver en las curvas que se han obtenido experimentalmente (Figura 4). La existencia de una relación entre la carga neta del soluto, zp, y el coeficiente de partición, ln Kp, podría ser interpretada debido a las interacciones del soluto y el solvente.. La carga neta de una macromolécula depende de la cantidad de grupos iónicos que presente y esto está relacionado con la contribución de interacciones ion-dipolo que experimenta el soluto en las fases dentro de las contribuciones totales de energía de las interacciones del soluto con el solvente. El factor γ supuestamente está relacionado con la diferencia del potencial electrostático interfacial, pero podría ser visto como la diferencia de la capacidad del medio acuoso en las dos fases a participar en las interacciones de. 30.
(40) Capítulo 2 hidratación ion-dipolo con un soluto iónico siendo particionado. Debido a esto una relación entre ln Kp – zp, ya sea lineal o no, no puede ser considerada evidencia para decir que hay una dependencia directa entre la diferencia de potencial y la partición de un soluto cargado.. La mayor limitación de la ecuación (2.75) lo que la hace difícil de aceptar, no es precisamente el significado físico de los términos involucrados sino la implicación de que el coeficiente de partición del soluto con una carga neta de cero es independiente de la composición de sal del sistema.. Después que Johansson obtuviera esta relación empírica, ecuación (2.75), Albertsson (Albertsson, 1986), Haynes (Haynes et al., 1993) y Grossmann (Grossmann and Maurer, 1995), derivaron una similar basada en esa, las cuales son actualmente utilizadas para cálculos de predicción de proteínas.. La relación derivada por Albertsson (Albertsson, 1986) viene dada por:. ln K p ln K o . zpF RT. . 2.76. en donde F, R y T corresponden a la constante de Faraday, la constante universal de los gases y la temperatura absoluta, y Δφ es la diferencia del potencial eléctrico de las fases. Para el cálculo de la diferencia de potencial eléctrico se han usado varios métodos diferentes, algunos experimentales, otros basados en desarrollos teóricos.. 31.
(41) Capítulo 2. Figura 4 Gráfica del coeficiente ki contra pH, con diferentes aditivos de sales para hemoglobina humana (Zaslavsky, 1995).. Según lo desarrollado por Haynes (Haynes et al., 1993), la partición de proteínas se puede calcular de acuerdo a la siguiente expresión:. "p z p F ln K p ln ' p RT. 2.77. en donde los coeficientes de actividad, γp, que aparecen; pertenecen a la proteína. Correspondiendo los superíndices “ y ‘, a la fase inferior y superior respectivamente. Además proponen una relación basada en la teoría de potencial cuasi-electrostático para hacer el cálculo de la diferencia de potencial eléctrico. Aplicando dicha teoría obtienen la relación:. 32.
(42) Capítulo 2. " a ' RT " ' ln a zc z c z a F " za c' c . . . . 2.78. en donde los subíndices c y a corresponden a el catión y el anión respectivamente.. Esta últimas relación, ecuación (2.77), ha sido aplicada exitosamente con diferentes tipos de proteínas, ver Figura 5 , teniendo un ajuste aceptable al coeficiente de partición experimental.. Figura 5 Coeficientes de partición experimentales y calculados para una mezcla de proteínas diluidas en un sistema ATPS que contiene PEG 3350-Dextrano T-70 y KCl 50 mM a pH=7.5 y 25 °C. ● albúmina ▲ lisozima ■ quimiotripsina. Finalmente Grossmann (Grossmann and Maurer, 1995) hace un desarrollo similar. Ellos se dieron cuenta que la diferencia de potencial eléctrico puede ser calculada exactamente de la energía de Gibbs en exceso de la solución; suponiendo que no existe ningún campo eléctrico externo, que el sistema de dos fases es obtenido mediante la mezcla de componentes neutrales e introduciendo la condición de electroneutralidad para cada una. 33.
(43) Capítulo 2 de las fases coexistentes. Después aplicando la teoría del potencial cuasi-electrostático la relación que obtienen para el coeficiente de partición es la siguiente:. "p z p mk' ' ln K p ln ' ln " ln k" p z k mk k . 2.79. en donde el subíndice k representa al ion de referencia y m es la molalidad.. El tamaño de las biomoléculas es otro factor que afecta a la partición de las mismas, aunque no se comprende bien de que manera. Mediante experimentación se ha visto que para biomoléculas pequeñas como lo son los aminoácidos y glucósidos, es difícil determinar una tendencia en su partición debido a que se suelen separar uniformemente en las dos fases, en cambio para biomoléculas grandes como lo son las proteínas y los ácidos nucleicos, su partición tiende a ser más cargada hacia una de las fases. Esta tendencia se puede representar de manera gráfica según la Figura 6.. La correlación que se presenta en la Figura 6, sin embargo, no se manifiesta de manera generalizada para todas las biomoléculas. Esto se pudo observar de acuerdo a los experimentos realizados por Sasakawa y Walter (S.Sasakawa and H.Walter, 1972) a 16 diferentes tipos de hemoproteínas.. Esto hace notar que la relación que puede haber entre el peso molecular de algunas biomoléculas y el comportamiento de su partición, puede ser influenciado de una manera mayor por otros factores.. Existen dos implicaciones importantes para la aparente diferencia en la partición de biomoléculas pequeñas y grandes en sistemas de dos fases acuosas. Una es que la partición de biomoléculas grandes debe de ser tratada en términos de la teoría termodinámica para polímeros de Flory-Huggins. La otra implicación, aun más importante, es que el comportamiento de partición para macromoléculas biológicas no puede ser comprendido estudiando pequeños fragmentos de sus estructuras (Zaslavsky, 1995).. 34.
(44) Capítulo 2. Figura 6 Relación entre el peso molecular de la proteína (MW) y el coeficiente de partición Ko, en el punto isoeléctrico para el sistema DEX500-PEG6000 con 0.1 M NaCl o 0.05 M Na2SO4 y 0.01 M de fosfato o amortiguador de glicina a 20 °C (Zaslavsky, 1995).. La estructura de las biomoléculas también afecta la manera en la que interaccionan con su medio. La flexibilidad y conformación de las biomoléculas, sus ramificaciones, la cantidad de instauraciones que presente, los enlaces intramoleculares, las interacciones cercanas, etc.; son factores que afectan el comportamiento de la partición.. En la actualidad hay muchas cosas que se desconocen sobre la estructura de las biomoléculas, particularmente de las proteínas, sobre todo lo que corresponde a la estructura tridimensional de éstas (estructuras terciarias), por lo tanto es difícil hacer suposiciones y/o obtener conclusiones sobre el comportamiento de su partición basándonos en su estructura molecular.. 35.
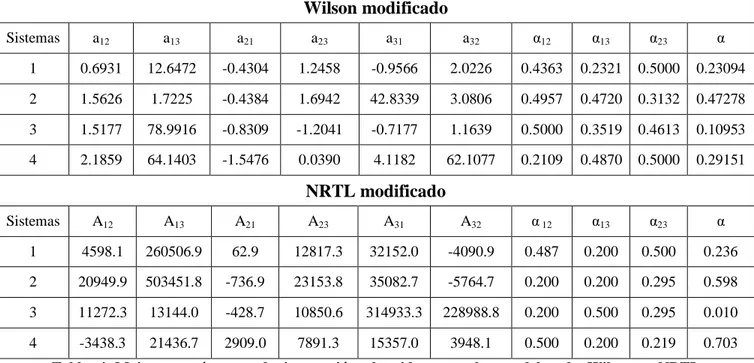
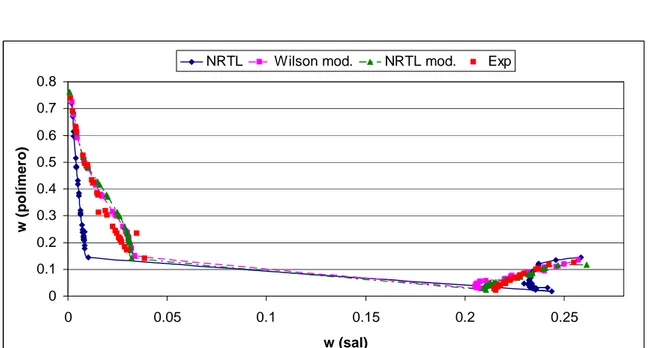
(45) Capítulo 3. Capítulo 3. 3 Evaluación de los modelos termodinámicos En este capítulo se presentará el análisis hecho a los modelos termodinámicos estudiados en el capítulo anterior aplicado a cuatro sistemas diferentes. El análisis se dividirá en cuatro secciones las cuales abarcan el ajuste de los parámetros termodinámicos, el análisis numérico, la predicción de la composición fases y la predicción de la partición de proteínas. Los sistemas estudiados del 1 al 4 se compusieron respectivamente de PEG 4000-Agua-K2HPO4, PEG 4000-Agua-Li2SO4, PEG 6000-Agua-K2HPO4 y PEG 1000Agua-K2HPO4, mientras que las proteínas estudiadas fueron la lisozima, catalasa, IgG y BSA.. 3.1 Ajuste de parámetros de interacción El ajuste de los parámetros de interacción se realizó de manera general de acuerdo a lo visto en la sección 2.1.2.2, pero tomando en cuenta algunos detalles particulares para cada uno de los modelos termodinámicos. Además debido a que el ajuste se realizó por dos métodos diferentes las consideraciones tomadas en cuenta cambian, principalmente en los valores de los parámetros de interacción αij.. 36.
(46) Capítulo 3 El ajuste de parámetros de interacción para el modelo NRTL es el más sencillo. Debido a que se tienen sistemas ternarios en todos los casos, los parámetros de interacción que se tienen son 6, por las parejas de interacción Aij y Aji que se pueden formar, más 3 debido a los parámetros no aleatorios αij, teniendo un total de 9 parámetros. Cuando se usa el Solver como método para el ajuste de los parámetros de interacción del modelo NRTL, lo que se hace es ajustar al mismo tiempo los 9 parámetros para la minimización de la función objetivo, ver ecuación (2.73). Los valores iniciales para las minimizaciones se presentan en los apéndices (ver Apéndice C. Valores iniciales de los parámetros de interacción). En el caso que se usa el algoritmo genético el procedimiento de ajuste de parámetros cambia un poco, debido a que los parámetros no aleatorios αij se quedan con un valor fijo de 0.3. Esto se debe a que al momento de incluir estos parámetros, los cuales tienen como restricción tener valores aproximados entre 0.2 y 0.5, el algoritmo genético no arroja buenos resultados, dejando el valor de la función objetivo en valores altos. El valor de 0.3 se elije de acuerdo a las recomendaciones hechas por Renon y Prausnitz (Renon and Prausnitz, 1968b). Estos valores predeterminados de αij también fueron fijados en valores de 0.2, 0.4 y 0.5, observando que se obtenían resultados muy similares que con el valor de 0.3, por lo que se prefirió dejar el valor recomendado por Renon y Prausnitz.. El procedimiento para ajustar los parámetros de interacción para los modelos Wilson y NRTL modificados es similar. En estos dos modelos también se hace el ajuste para sistemas ternarios, por lo que de la misma manera que en el modelo NRTL se tendrán 6 parámetros binarios de interacción ajustables, más otros 3 parámetros no aleatorios. Además de esos 9 parámetros se incluye un parámetro no aleatorio más, α, el cual tiene el mismo significado que los parámetros no aleatorios αij, pero que considera a todo el sistema en general, ya que afecta a todas las especies del sistema. Por lo tanto para estos dos modelos termodinámicos se tendrán 10 parámetros ajustables.. Para el ajuste de los parámetros de interacción de estos dos modelos termodinámicos se sigue un procedimiento un poco diferente. Cuando se usa el Solver como método para el. 37.
Figure



+7




Outline
Documento similar
Cedulario se inicia a mediados del siglo XVIL, por sus propias cédulas puede advertirse que no estaba totalmente conquistada la Nueva Gali- cia, ya que a fines del siglo xvn y en
No había pasado un día desde mi solemne entrada cuando, para que el recuerdo me sirviera de advertencia, alguien se encargó de decirme que sobre aquellas losas habían rodado
The 'On-boarding of users to Substance, Product, Organisation and Referentials (SPOR) data services' document must be considered the reference guidance, as this document includes the
In medicinal products containing more than one manufactured item (e.g., contraceptive having different strengths and fixed dose combination as part of the same medicinal
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
Package Item (Container) Type : Vial (100000073563) Quantity Operator: equal to (100000000049) Package Item (Container) Quantity : 1 Material : Glass type I (200000003204)
