Modelación in silico de actividad antimalárica de sustancias orgánicas
67
0
0
Texto completo
(2) “El propósito de la ciencia es buscar aquel conjunto de principios fundamentales a través de los cuales todos los hechos conocidos son comprendidos y por medio de los cuales se predicen nuevos resultados.” Tsung-Dao Lee.
(3) Dedicado a mi familia, en especial a mis padres, a ti mamá por ser mi guía en todo momento, por educarme y guiarme siempre por los caminos correctos. A todos por confiar en mí, este trabajo es también de todos. A mis amigos, a todos los que han transitado junto a mí durante estos años convulsos que son la Universidad, por el apoyo en las buenas y en las malas, por ser mi familia lejos de casa, por todos los recuerdos y momentos vividos. A Eduardo, por motivarme a alcanzar las metas más anheladas, apoyarme en mis locuras y comprenderme siempre..
(4) “La gratitud, como ciertas flores, no se da en la altura y mejor reverdece en la tierra buena de los humildes.” José Martí.. Agradezco a: Mi mamá Vivian Llerena León porque eres para mí como lo fue Martí para Fidel, una fuente inagotable de sabiduría y ejemplo y así mismo, la autora intelectual de todos mis logros; porque siempre me has enseñado que en la vida hay que ponerle ganas a las cosas y que todo requiere de esfuerzo y dedicación. Mi papá Antonio Roche Rodríguez por confiar en mí y apoyarme siempre que ha sido necesario, por brindarme buenos consejos y por ser un ejemplo para mí, porque también gracias a ti me he convertido en la persona que soy. A mis tutores, Oscar Martínez Santiago, por enamorarme de este mundo y de la ciencia en general, por aportar tus conocimientos desinteresadamente y enseñarme a ser una mujer de ciencia, por estar siempre ahí a pesar de la distancia y ver siempre la luz cuando yo solo veía sombras. A Evys Ancede Gallardo por acogerme casi en la recta final y ayudarme siempre, por tenerme paciencia, por tu apoyo incondicional, por todas las horas dedicadas a la realización de este trabajo, por transmitirme tus conocimientos y confianza. Mis abuelas, Coralia, por enseñarme que para lograr los objetivos hay que tener fe y luchar siempre, por ayudarme y protegerme, por hacerme fuerte y capaz de salir a flote. A Cari por sus siempre oportunos consejos, por confiar en mí y en la realización de este trabajo. A la memoria de mis abuelos Rolando y Antonio, por todos los mimos, porque, donde quiera que estén, este momento es la realización de sus sueños. A Eloy, porque has sido como mi otro padre, por la confianza que has depositado en mí, por apoyarme siempre, por los tantos consejos y experiencia. A mis tíos, Roly y Milaida por preocuparse y ocuparse siempre, tanto estando cerca, como desde la lejanía de su misión internacionalista. A mi tío Jose, en paz descanses, por enseñarme a amar tanto a la vida, a luchar por los sueños y a ser feliz a pesar de las cosas malas, porque sé que este momento sería para ti momento de júbilo y alegría. A mis hermanos Ernesto y Alejandro; y a su mamá Gladis, por estar siempre pendiente de mí y por ayudarme en todo lo que necesito..
(5) A mi novio Eduardo, por compartir conmigo todos estos años, impulsarme a cumplir sueños y cumplirlos conmigo, por confiar en mí, porque gran parte de la realización de este trabajo te lo debo a ti; por tu paciencia, por tu amor, por ser al mismo tiempo mi mejor amigo y por resaltar siempre lo mejor de mí, porque sencillamente no me imagino mi vida sin ti. A mis suegros, Nancy y Eduardo, a mi cuñada favorita Nancita, por demostrarme que el cliché de los suegros malvados es falso, por apoyarme, confiar y estar siempre pendientes de mí, por ser parte de mi familia. A la familia de Eduardo en general por considerarme parte de la familia, en especial a Guillermito, mi primo querido. A mis loquis Anabel, Elianet y Patricia, por estar siempre juntas durante estos 5 y hasta 6 años, por soportarnos las unas a las otras y por querernos; porque la amistad es de las cosas más bonitas de la vida y yo he tenido el placer infinito de compartirla con ustedes, porque nunca acabe. Gracias por ser mi soporte, por regalarme tantos buenos recuerdos y por estar siempre ahí. A mis amigos, a todos los que hecho en estos años y otro que venían de antes, a Yoslandy, Karla, Beisi, Arlettys, Peña, Carlos Raúl, Roberto, Julito, Leydi Laura, Denis, Nelson, Oslaidys, Dianelis, Liset, Lisset, Norma Anabel, Yoli (porque en la distancia no está el olvido) y muchísimos otros, la lista sería interminable, a todos muchísimas gracias por estar ahí. A Raúl, por ser mi ejemplo, mi guía y mi amigo, por ayudarme siempre desinteresadamente, por entenderme y acompañarme en las aventuras de la ciencia. Te deseo lo mejor en tu vida, pues tú te lo mereces. A mis compañeros de aula, por ser el mejor colectivo del mundo, por haber tenido la suerte de compartir con todos ustedes estos 5 años, por hacer de la universidad una experiencia única e inolvidable, por apoyarme siempre. En especial a mi grupito de estudio Dani y Norbell por hacer de esta obligación una tarea llevadera y acaparar buenos recuerdos, por ser mis amigos. No los olvidaré nunca. A los profesores del Departamento de Química, por por todos los conocimientos que me brindaron, por formarme como profesional, así como aquellos que no pertenecen al departamento, pero que de una forma u otra contribuyeron en mi formación profesional. De forma especial agradecer a Haydee Ulloa (Nenena), por tomarme de la mano en mi formación y brindarme tu cariño, por ser una excepcional pedagoga, digna de la admiración y el respeto de todos los que hemos tenido el orgullo de recibir tus clases. A Yolanda, por recibirme en tu casa para ayudarme en la realización de este trabajo y entregarme tu infinita sabiduría..
(6) A mis compañeros del Grupo de Diseño, en especial a Elizabeth Goya por todos sus consejos y su ayuda, por compartir también muchos buenos momentos. A los integrantes del grupo de diseño del CBQ a Omar, Rosario, etc., por la ayuda y comprensión que me brindaron, en espacial a Reinaldo por todos los oportunos consejos..
(7) Resumen La malaria es causante de millones de muertes al año y los fármacos utilizados convencionalmente presentan fenómenos de resistencia. Surge así la necesidad de desarrollar nuevos antimaláricos. En este trabajo se realizan regresiones sobre un conjunto químico (Malaria Box) para desarrollar relaciones lineales y no lineales entre las estructuras moleculares y sus actividades correspondientes. En este estudio se modeló la actividad antimalárica (expresada como el EC 50) de 317 entidades químicas, frente al parásito Plasmodium falciparum. Las estructuras químicas fueron codificadas mediante índices de derivada de grafos moleculares (GDIs). Estos fueron calculados con el módulo DIVATI del software TOMOCOMDCARDD. Posteriormente, para seleccionar descriptores con mayor variabilidad, basado en el cálculo de la entropía de Shannon, fue utilizado el software IMMAN. Los modelos de RLM fueron desarrollados con el software MobyDigs. Los modelos de regresión no lineal fueron desarrollados con el software WEKA, mediante Máquinas de Vectores de Soporte. Los resultados de esta modelación muestran superioridad estadística del modelo no lineal con respecto al lineal, con las mismas variables, lo cual sienta las bases para predecir la actividad antimalárica con el uso de GDIs y la técnica de regresión no lineal, para posteriores estudios de cribado virtual y propuesta de potenciales antimaláricos..
(8) Abstract Malaria causes millions of deaths every year and conventionally used drugs are generating resistance phenomena. Therefore it is necessary to develop new antimalarial drugs urgently. In this work, regression models are developed on a chemical set (Malaria Box) to find linear and nonlinear relations between molecular structures and their corresponding activities. In this study, the antimalarial activity (expressed as the EC50) of 317 chemical entities was modeled against the parasite Plasmodium falciparum. The chemical structures were encoded using discrete molecular graph derivative indices (GDIs). These were calculated using the DIVATI module of the TOMOCOMD-CARDD software. Later, to select descriptors with greater variability, based on the calculation of the Shannon‟s entropy, the IMMAN software was used. The RLM models were found with the MobyDigs software. Nonlinear regression models were found with the WEKA software, using Support Vector Machines. The results of this modeling suggest statistical superiority of the nonlinear model with respect to the linear model, using the same variables, which establish the bases to predict the antimalarial activity using the GDIs and the nonlinear regression technique, for further studies of virtual screening and proposal of antimalarial candidates..
(9) Tabla de contenido INTRODUCCIÓN ................................................................................................................. 1 1-. MARCO TEÓRICO ...................................................................................................... 6 1.1- Malaria ...................................................................................................................... 6 1.1.1- Historia de la enfermedad ............................................................................ 6 1.1.2- Etiología ............................................................................................................ 6 1.1.3- Epidemiología ................................................................................................. 7 1.1.4- Impacto económico de la Malaria .............................................................. 7 1.2- Química Grafo-Teórica ......................................................................................... 8 1.2.1- Introducción a la topología molecular ...................................................... 8 1.2.2- Descriptores moleculares ............................................................................ 8 1.2.3- Índices Topo-químicos. Índices de Derivada Discreta de Grafos Moleculares ................................................................................................................. 9 1.3- Elementos de Quimiometría ............................................................................. 11 1.3.1- Aplicaciones de DMs y Quimiometría..................................................... 11 1.3.2- Regresión lineal múltiple ........................................................................... 12 1.3.3- Regresión no-lineal ...................................................................................... 14 1.3.4- Validación Interna y Externa de modelos .............................................. 16 1.3.5- Compuestos ‘outliers’ y técnicas para la selección de los mismos ...................................................................................................................................... 18. 2-. MATERIALES Y MÉTODOS .................................................................................... 20 2.1- Bases de Datos Químicas ................................................................................. 20 2.1.1- Preparación de la Base de Datos ............................................................. 20 2.1.2- La diversidad química la base de datos................................................. 20 2.2- Herramientas Computacionales ...................................................................... 21 2.2.1- Software MobyDigs. Regresión Lineal Múltiple ................................... 21 2.2.2- Software WEKA. Regresión no-lineal múltiple ..................................... 22 2.2.3- Software DIVATI. Cálculo de los nuevos Índices de Derivada del Grafo ........................................................................................................................... 22 2.2.4- Software IMMAN. Análisis de Variabilidad ............................................ 23. 3-. RESULTADOS Y DISCUSIÓN ................................................................................ 26 3.1- Cálculos de los Índices de Derivada del Grafo ........................................... 26 3.2- Modelación de la Regresión Lineal Múltiple ................................................ 26 3.2.1- Selección de los mejores descriptores moleculares para la técnica de regresión lineal ................................................................................................... 26.
(10) 3.2.2- Modelos lineales ........................................................................................... 28 3.2.3- Discusión de los resultados de la regresión lineal ............................. 33 3.3- Aplicación de técnicas no lineales ................................................................. 35 3.3.1- Selección de atributos para la técnica no lineal .................................. 35 3.3.2- Modelos no lineales ..................................................................................... 36 3.3.3- Discusión de los resultados de la regresión no lineal ....................... 40 3.4- Comparación entre técnicas lineales y no lineales .................................... 41 CONCLUSIONES .............................................................................................................. 43 RECOMENDACIONES ..................................................................................................... 45 REFERENCIAS .................................................................................................................. 47 ANEXOS ............................................................................................................................. 54.
(11) Glosario AGs. Algoritmos Genéticos. AMs. Antimaláricos. CV. Cribado Virtual. DM. Descriptor Molecular. GDIs. Índices de Derivada del Grafo inspirados en las entropías de información. GSK. GlaxoSmithKline. ITs. Índices Topológicos. P. Plasmodium. QSAR Quantitative Structure Activity Relationships QSPR Quantitative Structure Property Relationships R 2. Coeficiente de correlación. R. Coeficiente de determinación o coeficiente de correlación al cuadrado. RLM. Regresión Lineal Múltiple. SVM. Support Vector Machines.
(12) Introducción.
(13) INTRODUCCIÓN “El ámbito iluminado por la ciencia está rodeado de un espacio en tinieblas tan extenso, que ha de parecer ridícula la pretensión de limitar la existencia al hábitat del conocimiento.” Juan Benet. Dentro de las enfermedades parasitarias, el paludismo o malaria es la más importante si se tienen en cuenta el número de individuos que enferman anualmente y su impacto socioeconómico (Komba et al., 2009). La malaria es una enfermedad infecciosa, producida por un protozoo intracelular del género Plasmodium (P). Existen 5 especies que pueden infectar al hombre, ellos son: P. falciparum, P. vivax, P. ovale, P. malariae. Recientemente, se ha descubierto que P. knowlesi (originario de los primates), puede infectar también a humanos, especialmente en la zona de Malasia y Borneo (González, 2005). Los parásitos se transmiten por la picadura de la hembra del mosquito Anopheles y la forma más frecuente y grave de la enfermedad se debe a P. falciparum. De forma general, las manifestaciones clínicas consisten en escalofríos y palidez cutánea, que se alterna con sensación de calor y rubefacción cutánea, sequedad e hipertermia. Frecuentemente, el paroxismo febril se acompaña de cefalea, mialgias y anemia. La infección por P. falciparum puede complicarse con un cuadro de malaria grave, caracterizada por una anemia importante, hipoglucemia, afectación renal, afectación cerebral con alteración de la conciencia y coma (Komba et al., 2009). La diversidad genética le confiere a Plasmodium la capacidad para evadir la respuesta. inmune. del. hospedador. y. producir. variantes. resistentes. a. medicamentos y vacunas, esto es en gran parte, responsable del éxito de la supervivencia de este parásito en la historia evolutiva, así como del fracaso de las medidas empleadas con el objetivo de erradicarlo (Jimenez-Diaz et al., 2009). El tratamiento ha sido posible durante muchos años gracias a la existencia de un número restringido de fármacos que presentan cada uno de ellos una serie de limitaciones de tipo farmacológico, aunque el mayor problema es la aparición de resistencias (Dockrell and Playfair, 1983). Los fármacos de mayor utilización como la cloroquina y la asociación de sulfadoxina + pirimetamina presentan beneficios limitados y en determinados casos son de una eficacia cuestionable debido a los 1.
(14) fenómenos de resistencia (González, 2005). Todo esto pone de manifiesto la imperiosa necesidad mundial de desarrollar nuevos antimaláricos (AMs) y el importante desafío que supone esta empresa. En la actualidad se han realizado grandes esfuerzos para lograr más eficiencia en la selección de compuestos antimaláricos. Tal es el caso de la utilización de cribados automatizados de alto rendimiento sobre la colección de productos de GlaxoSmithKline (GSK), a través de los cuales se han identificado más de 13 000 compuestos activos en cepas de P. falciparum (Gamo et al., 2010). Hay pocos ejemplos de la utilización previa de los métodos in silico con este propósito. Dentro de estas técnicas, el cribado virtual (CV) tiene la ventaja de ser más económica (ahorro en compra de reactivos y robotización), rápida, y permite tener en cuenta una. cantidad. de. compuestos. del. orden. de. billones,. cifra. impensable. experimentalmente. Por otra parte los estudios de Relaciones Cuantitativas de Estructura-Actividad (por sus siglas en inglés, QSAR) se han utilizado ampliamente en la modelación de disímiles propiedades moleculares de naturaleza física, química y biológica, son actualmente, el enfoque más utilizado en el diseño de nuevos fármacos (Lazarou et al., 2009). Este tipo de análisis es muy útil y generalmente se utiliza como principal herramienta en la selección de compuestos durante el protocolo de CV. Sin embargo, los estudios de Regresión Lineal Múltiple (RLM) y de regresión no lineal basados en técnicas de Máquinas de Soporte Vectorial (SVM) reportados en la literatura, han aportado resultados limitados, cuando estas técnicas son bien fundamentadas para describir la actividad biológica. Aunque se han desarrollado esfuerzos importantes para optimizar la búsqueda de nuevos antimaláricos mediante el uso de modernas herramientas de quimioinformática; actualmente no se cuenta con una estrategia que combine técnicas de regresión y clasificación en una metodología de trabajo adecuada para desarrollar un cribado virtual extenso sobre el universo de estructuras químicas conocidas. La creación y mejoría de las herramientas permite obtener productos de calidad superior. Por esta razón, se hace necesaria la búsqueda de nuevos métodos de modelación, ortogonales a los existentes, con herramientas más potentes para desarrollar nuevas relaciones matemáticas entre 2.
(15) las estructuras y sus respectivas actividades y que las mismas sirvan para posteriores estudios en el desarrollo de nuevos AMs. Además, se ha demostrado que los descriptores moleculares basados en la derivada discreta del grafo (GDIs) codifican adecuadamente la estructura química (Martínez Santiago et al., 2016). Este estudio se basa, fundamentalmente, en la identificación, optimización y evaluación de nuevas entidades químicas con mayor actividad antimalárica que los limitados fármacos que actualmente se utilizan en el tratamiento de esta enfermedad. Lo expuesto anteriormente permite identificar el siguiente problema científico: Problema científico: ¿Cómo determinar, mediante la realización de estudios de regresión lineal y no lineal, los modelos in silico que describan satisfactoriamente la actividad antimalárica de moléculas de naturaleza orgánica frente al parásito Plasmodium falciparum? Hipótesis: Es posible determinar relaciones matemáticas cuantitativas lineales y no lineales, entre medidas experimentales de la actividad frente al parásito Plasmodium falciparum (causante de la malaria) y descripciones de las correspondientes estructuras moleculares por medio del uso de los Índices de Derivada Discreta de Grafos Moleculares. Objetivo General: Determinar los modelos in silico de regresión que describan satisfactoriamente la actividad antimalárica de moléculas orgánicas contra el parásito Plasmodium falciparum y que puedan ser usados en posteriores estudios de cribado virtual y desarrollo de nuevas entidades químicas como candidatos prometedores para combatir la malaria. Objetivos específicos: 1. Calcular los Índices de Derivada de Grafos moleculares a 317 estructuras orgánicas diversas, para la identificación de los mejores atributos mediante el uso de algoritmos genéticos y entropía de Shannon, para su modelación matemática.. 3.
(16) 2. Determinar modelos de Regresión Lineal Múltiple que relacionen las estructuras del conjunto químico con sus respectivas actividades biológicas. 3. Determinar modelos de regresión no lineal basados en las técnicas de SVM, que relacionen las estructuras del conjunto químico con sus respectivas actividades biológicas. 4. Aplicar la técnica estadística de “dejar varios fuera” con el fin de rechazar del conjunto químico los valores outliers, para la realización de una segunda modelación, con el uso de las mismas herramientas estadísticas y de inteligencia artificial antes mencionadas. 5. Determinar los modelos in silico que describan satisfactoriamente la actividad antimalárica mediante la comparación de todos los modelos obtenidos. La novedad científica de este trabajo se fundamenta en la obtención de nuevos modelos matemáticos lineales y no lineales para la identificación de potenciales fármacos AMs mediante el uso de GDIs como estrategia de codificación de las estructuras químicas.. 4.
(17) Capítulo 1: Marco Teórico.
(18) 1- MARCO TEÓRICO “La ciencia es más que un simple conjunto de conocimientos: es una manera de pensar.” Carl Sagan. 1.1- Malaria 1.1.1- Historia de la enfermedad Las fiebres palúdicas fueron descritas por Hipócrates 400 años antes de J.C. No solamente se diagnosticaba la enfermedad, sino que se realizaban pronósticos acerca de su evolución a pesar del desconocimiento de su etiología. Los términos empleados más comúnmente, malaria y paludismo, también conservaban algunas imprecisiones. Al igual que la palabra malaria (mal aire) resultaba incorrecta al señalar como origen de la enfermedad la transmisión por el aire, el término paludismo (de “palus”, terreno pantanoso) podía hacer pensar que sólo se producía en aguas estancadas (Andriantsoanirina et al., 2009). La teoría bacteriana del paludismo apareció a raíz de las investigaciones de Klebs y Tommasi-Crudeli que describieron el “Bacillus malariae” en 1879 (Bell et al., 2009). Ellos observaron ciertos bacilos y filamentos muy largos en cultivos con agua y tierra procedente de las palúdicas Lagunas Pontinas italianas, así como en la sangre de conejos inoculados y en enfermos de malaria (Berghout et al., 2009). El protozoo fue descrito por primera vez en la sangre de un paciente por Charles Laveran en 1880, que observó al parásito en un frotis sin teñir de sangre fresca (Machado-Tugores et al., 2012). 1.1.2- Etiología La enfermedad en el hombre está causada principalmente por cuatro especies de Plasmodium: P. falciparum, P. vivax, P. ovale y P. malariae. De ellas, P. falciparum es el responsable del 95% de las muertes, pero la especie más prevalente es P. vivax. En cuanto a la distribución geográfica, P. falciparum y P. malariae se encuentran especialmente en Asia y África, P. ovale se encuentra de forma casi exclusiva en África y P. vivax predomina en Latinoamérica, India, Pakistán, Oceanía y, más raramente, en África (Komba et al., 2009).. 6.
(19) 1.1.3- Epidemiología Actualmente, la malaria representa la enfermedad parasitaria de mayor importancia mundial al generar entre 300-500 millones de casos/año en todo el mundo y cerca de 2 millones de muertes en el mundo (Schapira et al., 2000). P. falciparum es la especie de mayor abundancia global y el que provoca una mayor gravedad de la enfermedad (Machado-Tugores et al., 2012). La resistencia del parásito a los fármacos antipalúdicos es un peligro real y siempre presente para los avances conseguidos hasta ahora. Otra grave preocupación concierne al rápido aumento de la resistencia a los insecticidas. Si no se gestiona de un modo adecuado, esta resistencia constituye una amenaza potencial para futuros avances en el control del paludismo (Machado-Tugores et al., 2012). 1.1.4- Impacto económico de la Malaria El paludismo produce pérdidas económicas importantes, que, a largo plazo, han llevado a diferencias considerables entre los valores del Producto Interno Bruto (PIB) de los países con y sin paludismo (sobre todo en África). Los costos sanitarios de esta enfermedad incluyen gastos tanto personales como públicos en prevención y tratamiento. En algunos países con gran carga de paludismo, la enfermedad es responsable de: (Machado-Tugores et al., 2012) • Hasta un 40% del gasto sanitario público. • Un 30% a 50% de los ingresos en hospitales. • Hasta un 60% de las consultas ambulatorias. En la actualidad el gasto en el tratamiento de la enfermedad es muy superior al de las pruebas de diagnóstico rápido (PDR), pero se espera que disminuya por la estrategia de ampliar la prueba parasitológica a todos los casos sospechosos de malaria antes de usar el tratamiento. Con los precios actuales de las PDR y las terapias combinadas con artemisina (TCA) y el estricto cumplimiento del protocolo de tratamiento, el ahorro en materias primas podría llegar a 68 millones de dólares en el sector público de la región africana (M., 2012). De igual manera el acceso universal a las redes mosquiteras tratadas con insecticida en África en 2015,. 7.
(20) podría reducir entre 31 y 48 millones el número de casos de paludismo que acuden a centros de salud pública (M., 2012). 1.2- Química Grafo-Teórica 1.2.1- Introducción a la topología molecular Un grafo se expresa usualmente como un conjunto de vértices (V) interconectados por un conjunto de aristas (E), donde cada vértice vi del grafo representa un objeto i y la arista eij que conecta dos vértices (vi y vj), representa la relación entre estos dos objetos (Harary, 1971, Gorbátov, 1988, Stadler, 2005). En la química grafoteórica, los objetos del grafo pueden representar orbitales, átomos (o sus núcleos), enlaces, grupos de átomos, moléculas, o colecciones de moléculas. Las aristas de un grafo químico simbolizan las interacciones entre objetos químicos y se usan para definir enlaces químicos, reacciones, mecanismos de reacciones, modelos cinéticos, u otra relación o transformación de los objetos químicos. Estas representaciones de los grafos químicos tienen numerosas aplicaciones en la química, como son: 1) los índices topológicos (ITs) y otros índices estructurales para los estudios QSAR (Kier and Hall, 1990, Devillers and Balaban, 1999, Kier and Hall, 1976, Kier and Hall, 1986, Kier and Hall, 1999, Marrero-Ponce, 2004) 2) el enfoque de orbitales moleculares de Hückel (Graovac et al., 1977, Dias, 1993) 3) la enumeración de isómeros, percepción de simetría estructural y codificación de compuestos químicos (Pólya and Read, 1987, Fujita, 1991) 4) grafos cinéticos y de reacción (Temkin et al., 1996) y 5) el diseño de síntesis asistida por computadoras (Koča et al., 1989), entre otras muchas aplicaciones. De todas estas aplicaciones mencionadas, la de mayor interés para el presente trabajo es aquella relacionada con la utilización de los índices topológicos para los estudios QSAR. 1.2.2- Descriptores moleculares Los Descriptores Moleculares (DMs) juegan un rol fundamental actualmente en el desarrollo de las Ciencias Químicas, las Ciencias Farmacéuticas, las políticas de protección ambiental e investigaciones de la salud (Todeschini and Consonni, 2009). Los DMs son representaciones matemáticamente formales de las moléculas (u otras entidades químicas) y son obtenidos de aplicar un algoritmo 8.
(21) definido sobre una determinada representación molecular. La definición más usada y aceptada de un DM es: el resultado final de un procedimiento lógico y matemático en el cual se transforma la información química codificada en una representación simbólica de la molécula en un número (o conjunto de números) de utilidad (Todeschini and Consonni, 2009). La representación molecular es una figura, imagen, símbolo, mapa, idea, etc., a partir de la cual, la molécula como un ente fenomenológicamente real es sustituida o presentada, al seguir determinados procedimientos y reglas convencionales. La cantidad de información química derivada de dicha representación simbólica depende del tipo de representación utilizada (Testa and Kier, 1991, Jurs et al., 1995). 1.2.3- Índices Topo-químicos. Índices de Derivada Discreta de Grafos Moleculares Los Índices Topológicos (ITs) son descriptores moleculares derivados de una invariante grafo-teórica y codifican información estructural, contenida en la conectividad molecular. Una invariante grafo-teórica es aquella propiedad del grafo que no depende del orden de numeración de los elementos del mismo, las cuales pueden ser obtenidas por manipulación algebraica del grafo (Martínez-Santiago et al., 2014). Los ITs son representaciones numéricas de la estructura molecular. Estos valores numéricos son matemáticamente derivados de alguna forma directa y no ambigua de una representación gráfica de la estructura molecular, generalmente un grafo con hidrógenos suprimidos. Los mismos son sensibles a determinadas características estructurales, tal como, tamaño, simetría, ramificaciones y ciclos, y deben además codificar información acerca del tipo de átomos presentes y la multiplicidad de los enlaces en los que están implicados dichos átomos (Ivanciuc et al., 1993). Los. ITs. pueden. ser. agrupados. en. dos. grandes. categorías:. Índices. Topoestructurales e Índices Topoquímicos. Los ITs topoestructurales codifican información concerniente a distancia y adyacencia entre los átomos, mientras que los topoquímicos además de 9.
(22) cuantificar información topológica incluyen información que permite lograr una adecuada diferenciación atómica basada en propiedades características de los átomos y/o su estado de hibridación (Ivanciuc et al., 1993). Las aplicaciones de los ITs han estado dirigidas fundamentalmente hacia la predicción cuantitativa de propiedades químico-físicas y biológicas de compuestos orgánicos, en estudios que se han denominado QSPR y QSAR, respectivamente. Esta división, no es solo formal porque aunque el método en ambos tipos de estudio es similar, generalmente, la actividad biológica es una propiedad mucho más compleja que las propiedades químico-físicas debido a la gran cantidad de factores que influyen en la bioactividad de un compuesto químico. La aplicación de los ITs al diseño y selección de nuevas entidades químicas es probablemente una de las áreas más activas de investigación en la aplicación de tales descriptores moleculares a problemas biológicos (Kier and Hall, 1976, Kier and Hall, 2001, Martínez-Santiago et al., 2014, Todeschini and Consonni, 2009). Un tipo especial de índices topo-químicos (ITs) lo constituyen los derivados de calcular la derivada discreta del grafo molecular, además este concepto se ha extendido al relacionar la derivada de un grafo con las diferentes entropías propias de la teoría de información (Barigye et al., 2014). Las ecuaciones de definición de estos DMs se muestran en la tabla siguiente: Tabla 1. Ecuaciones de definición de los índices de derivada del grafo Descriptor Jenssent Joint. Ecuación de Definición (. ). (. ). Significado Derivada de un grafo. (. ). Derivada de un grafo inspirada en la entropía de unión. Mutual. (. Derivada de un grafo inspirada en la. ). entropía mutua Conditional. (. ). (. ). Derivada de un grafo inspirada en la entropía condicional. •. fi: intensidad de participación de un vértice en el suceso escogido.. •. fij: número de veces que aparece un par de vértices simultáneamente. 10.
(23) Un suceso, es una forma matemática de fragmentar las moléculas, el cual genera una matriz de incidencia, a partir de la cual luego de varios procedimientos algebraicos, se calculan los índices de derivada del grafo mediante la utilización de las ecuaciones antes expuestas (Barigye et al., 2014). 1.3- Elementos de Quimiometría 1.3.1- Aplicaciones de DMs y Quimiometría Recientes aplicaciones de índices basados en teoría de grafos, han demostrado un gran potencial de estos DMs en el diseño de nuevas entidades químicas, en búsquedas virtuales en bibliotecas químicas combinatorias y en evaluaciones a gran escala de similitud/diversidad química en extensas bases de datos de compuestos químicos. Los ITs son además, ampliamente usados en estudios de relación estructura-actividad/propiedad (Barysz et al., 1983, Ivanciuc, 2000). Desarrollar relaciones cuantitativas estructura-actividad es el paso final de un complejo proceso que comienza con una determinada descripción de la estructura molecular y finaliza con algunas inferencias, hipótesis y predicciones del comportamiento (biológico, químico-físico, medioambiental, etc.) de las moléculas en un sistema analizado. Un estudio QSAR se basa en el supuesto de que en la estructura molecular (su conectividad, sus características geométricas, estéricas y sus propiedades electrónicas) están contenidas las características responsables de las propiedades físicas, químicas y biológicas que muestran las sustancias y que esta información puede ser capturada en uno o más DMs (Todeschini and Consonni, 2009). La mayoría de las estrategias QSAR son enfocadas hacia la construcción de modelos basados fundamentalmente en métodos de clasificación o regresión, aunque de manera general muchos métodos quimiométricos son usados, en dependencia del problema bajo estudio. El término quimiometría surgió en la década del 70 y se define como la disciplina química que combina herramientas estadísticas como procedimientos para el análisis e interpretación de los datos químicos (Van de Waterbeemd, 1995, Brereton, 1990, Devillers, 1991, Frank, 1994). Las técnicas utilizadas para la recopilación, la elaboración, el análisis y la caracterización de conjuntos de datos, hoy en día, se interceptan no solo con varios campos de la Matemática y la 11.
(24) Estadística clásica sino también de la Inteligencia Artificial (IA) y otras ramas de la ciencia de la computación (Rumelhart et al., 1986, Vapnik, 1995). A continuación serán descritos algunos aspectos importantes de los métodos de regresión lineal múltiple (RLM) y Máquinas de Soporte Vectorial (SVM) que ayudarán al entendimiento de importantes resultados del presente trabajo. 1.3.2- Regresión lineal múltiple El análisis de regresión lineal múltiple es una técnica que se utiliza para desarrollar relaciones entre una única variable dependiente (criterio) y varias variables independientes (explicativas, predictoras) (Hair et al., 1999). Así mismo, la regresión múltiple remite a la correlación múltiple, que se representa por R. Sus fundamentos se hallan en la correlación de Pearson (Alzina, 1989). La ecuación (modelo) de regresión múltiple tiene la siguiente forma: (1.1) Si „a‟ es un valor constante, Y la variable dependiente, X1, X2,…Xn variables predictoras (independientes) y. coeficientes estimados para cada. variable independiente del modelo. Como puede observarse, la RLM puede utilizarse en la predicción de los valores de la variable dependiente, en base a una combinación de variables independientes y los coeficientes que ponderan las variables independientes en la ecuación 1.1 son mayormente determinados por el método de mínimos cuadrados (Frank, 1993, Draper and Smith, 1998). Un buen modelo no debe presentar ni demasiadas variables, ni debe olvidar las que sean verdaderamente relevantes. Es decir, debe cumplir el principio de la parsimonia y la selección del número óptimo de variables, según el cual un fenómeno debe ser descrito con el número mínimo de elementos posibles. El principio de parsimonia tiene aplicaciones de importancia en el análisis exploratorio de modelos de RLM, pues de un conjunto de variables explicativas que forman parte del modelo a estudiar, debe seleccionarse la combinación más reducida y simple posible, tener en cuenta la varianza residual, la capacidad de predicción y la multicolinealidad. Diversos procedimientos se han propuesto para seleccionar el número óptimo de variables a incluir en la ecuación. Los métodos más comunes de regresión se 12.
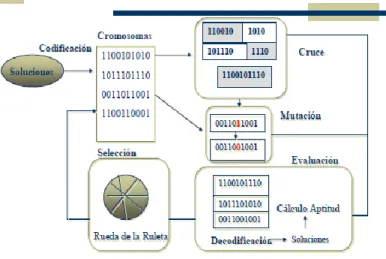
(25) basan en la adición o eliminación de una variable a la vez y siguen un determinado criterio (Draper and Smith, 1998), estos se conocen como: selección hacia adelante, eliminación hacia atrás; y selección paso a paso (Norusis, 1985). Este último método es uno de los más utilizados (es una combinación de los dos anteriores) y sigue un proceso de selección de variables paso a paso. Existen también otros métodos modernos que pueden ser aplicados con éxito a la búsqueda de combinaciones óptimas de las variables, como es el caso de los Algoritmos Genéticos. Según D. Goldberg los Algoritmos Genéticos (AGs) son: Algoritmos de búsqueda basados en los mecanismos de selección natural y genética natural. Combinan la supervivencia de los más compatibles entre las estructuras de cadenas, con una estructura de información ya aleatorizada, intercambiada para construir un algoritmo de búsqueda con algunas de las capacidades de innovación de la búsqueda humana (Goldberg, 1989a). Un AG no es más que un método de búsqueda que imita la teoría de la evolución biológica de Darwin (1859) para la resolución de problemas. Para ello, se parte de una población inicial de la cual se seleccionan los individuos más capacitados para luego reproducirlos y mutarlos, y de esa forma obtener finalmente la siguiente generación de individuos que estarán más adaptados que la anterior generación (Leardi et al., 1992). En la naturaleza todo el proceso de evolución biológica se hace de forma natural pero para aplicar el AG al campo de la resolución de problemas habrá que seguir una serie de pasos. Como premisa se debe conseguir que el tamaño de la población sea lo suficientemente grande para garantizar la diversidad de soluciones. Se aconseja que la población sea generada de forma aleatoria para obtener dicha diversidad. Los pasos básicos de un algoritmo genético son (Goldberg, 1989a, Leardi, 1994, Leardi, 2001): • Evaluar la puntuación de cada uno de los cromosomas generados. • Permitir la reproducción de los cromosomas donde los más aptos sean los que tengan más probabilidad de reproducirse. 13.
(26) • Con cierta probabilidad de mutación, mutar un gen del nuevo individuo generado. • Organizar la nueva población. Estos pasos se repiten hasta que se dé una condición de terminación. Se puede fijar un número máximo de iteraciones antes de finalizar el algoritmo genético o detenerlo cuando no se produzcan más cambios en la población (convergencia del algoritmo). Esta última opción suele ser la más habitual.. Figura 1. Esquema general de un AG simple 1.3.3- Regresión no-lineal La regresión lineal no siempre ofrece resultados satisfactorios cuando se aplica a determinados problemas, debido a que, en ocasiones, la relación entre Y y X no es lineal, sino que exhibe algún grado de curvatura. En la estimación directa de los parámetros de funciones no-lineales suelen utilizarse herramientas que conllevan mayor rigor de cálculo y que, normalmente, exhiben tiempos de cálculo mayor que en la RLM. Clasificador Máquinas de Soporte Vectorial Las Máquinas de Vectores de Soporte (SVM, por su nombre en inglés Support Vector Machine) son una moderna y efectiva técnica de inteligencia artificial, que ha tenido un formidable desarrollo en los últimos años. Estas herramientas son sistemas de aprendizaje que usan un espacio de hipótesis de funciones lineales en un espacio de rasgos de mayor dimensión, entrenadas por un algoritmo proveniente de la teoría de optimización. De forma general el algoritmo se enfoca 14.
(27) en el problema de aprender a discriminar entre miembros positivos y negativos de vectores n-dimensionales. Mediante una función matemática denominada kernel, los datos originales se redimensionan para buscar una separabilidad lineal de los mismos. De manera general, las SVM permiten hallar un híper plano óptimo que separe las clases (Chen et al., Hearst MA, 1998). Funciones Kernel Las funciones kernel son funciones matemáticas que se emplean en las SVM (Cortes C, 1995). Estas funciones son las que le permiten convertir lo que sería un problema de regresión no-lineal en el espacio dimensional original, a un problema más sencillo de regresión lineal en un espacio dimensional mayor. El tipo de kernel determina la transformación o mapeo que se le realizará a los datos. Entre los kernels más empleados por su implementación en diversos programas de modelación como Weka (Morate, 2001), se encuentran: El kernel Polinómico: (. ). (. ). (1.2). El kernel Gaussiano: (. ). (. ‖. ‖. (1.3). ). Y el kernel Universal de Pearson: (. (1.4). ) √. [. (. √‖. ‖ )]. Donde ω y σ controlan la altura y amplitud del pico de la función. WrapperSubsetEval como método de selección de variables: Esta técnica evalúa sets de atributos mediante el uso de una serie de entrenamiento. La validación cruzada se usa para estimar la exactitud de la serie de entrenamiento para un set de atributos determinado. Es el clasificador más utilizado para estimar la precisión de subconjuntos (Ron Kohavi, 1997).. 15.
(28) 1.3.4- Validación Interna y Externa de modelos Una condición necesaria para que sea válido un modelo de regresión es que el coeficiente de determinación (R2) esté cercano, tanto como sea posible, a uno y que el error estándar estimado (s) sea pequeño (capacidad de ajuste a los datos); sin embargo la consideración de estos únicos parámetros estadísticos no es suficiente, pues los valores de los mismos no necesariamente están relacionados con la capacidad del modelo de realizar buenas predicciones de una data futura (Todeschini and Consonni, 2009). Las técnicas de validación constituyen herramientas fundamentales a la hora de evaluar la capacidad predictiva de los modelos obtenidos por métodos multivariados de regresión y clasificación (Diaconis and Efron, 1983, Cramer et al., 1988, Golbraikh and Tropsha, 2002). A continuación se hace referencia con más detalle a las técnicas de validación más usadas y que son de especial interés para este trabajo. Validación interna La validación cruzada (VC) opera mediante la realización de un número de reducidas modificaciones al conjunto de compuestos de la data original y entonces calcula la precisión de las predicciones de cada uno de los resultados de los modelos (Wold, 1978, Stone, 1974). Es decir, se ajusta el modelo a los nuevos datos, se deja la parte omitida fuera, y estos se evalúan en el modelo para computar las predicciones de los casos que fueron excluidos. Este procedimiento se repite para cada conjunto de datos modificados. El poder predictivo del modelo puede expresarse como q2, denominado como la „varianza predictiva‟ o la „varianza de la validación cruzada‟, la cual es igual a (1-PRESS/SSY), o sea, que puede ser calculado acorde a la siguiente fórmula: ∑( ∑( donde,. ̂. ̂) ̅). (1.5). ̅ es la respuesta observada, estimada y media del i-ésimo caso,. respectivamente. Cuando se utiliza un solo compuesto en cada grupo de VC (lo cual da N grupos), el procedimiento se conoce como dejar “uno” fuera (LOO, acrónimo de Leave-One-Out). No obstante, Shao ha mostrado que desde el punto. 16.
(29) de vista teórico y práctico, el procedimiento de dejar „varios‟ fuera (LSO, acrónimo de Leave-Several-Out) es preferible al LOO (Shao, 1993). En la técnica de validación por Bootstrap, la talla original del conjunto de datos (n) es preservada en la serie de entrenamiento, a partir de la selección de m objetos (. ) que son asignados al conjunto de evaluación y estos son sustituidos por. m objetos repetidos de la serie de entrenamiento (Efron, 1982, Efron, 1987). El modelo es calculado en el conjunto de entrenamiento y las respuestas son predichas en el conjunto de evaluación. Las diferencias de los cuadrados entre las respuestas ciertas y las predichas son recogidas en el PRESS. Este procedimiento de elaboración del conjunto de entrenamiento es repetido miles de veces, los PRESS son sumados y el poder predictivo promedio es calculado (Efron, 1982, Wehrens et al., 2000). El método del revuelto [prueba de aleatoriedad (y-sc: y-scrambling)] es empleado para evaluar la correlación al azar (Tropsha et al., 2003, Wold and Erikson, 1995). En esta técnica, se calcula un modelo de regresión lineal para la verdadera variable respuesta (y) junto con un número de regresiones repetidas (200-300 veces) con las mismas variables, pero con la variable dependiente aleatoriamente revuelta (ỹ). Luego se calcula para cada modelo la varianza explicada q2LOO, y se evalúa la correlación entre la respuesta verdadera y la revuelta(Clark and Fox, 2004) de la siguiente manera: ( ̃) donde, la. (1.6). es la varianza explicada para el modelo obtenido con los mismos. predictores con el k-ésimo vector revuelto,. es la correlación entre los vectores. para la respuesta verdadera y la k-ésima revuelta. Un valor del intercepto cercano a cero implica que el modelo no es obtenido al azar mientras que un intercepto grande indica que los modelos aleatorios poseen el mismo desempeño que el modelo verdadero, por lo que se pudiera considerar aleatorio (Clark and Fox, 2004, Rücker et al., 2007). Validación externa La validación externa. permite evaluar si los modelos obtenidos son. generalizables a nuevos compuestos químicos y el “verdadero” poder predictivo de 17.
(30) los mismos (Tropsha et al., 2003). Para esto se divide la data en 2 conjuntos: la serie de entrenamiento (sirve para construir el modelo) y la serie de predicción (no utilizada en la selección de variables ni en el desarrollo del modelo, pero usada exclusivamente para evaluar el modelo tras su formación). 1.3.5- Compuestos ‘outliers’ y técnicas para la selección de los mismos Los „outliers‟ son puntos que se desvían significativamente del modelo desarrollado (no se ajustan al modelo) o son pobremente predichos por estos, que afectan los parámetros estadísticos del mismo (Gonzalez Diaz et al., 2002). Generalmente, la identificación de „outliers‟ busca un mejoramiento cualitativo del modelo. Un buen ejemplo ha sido mostrado por Golbraikh y colaboradores en la modelación de la toxicidad de compuestos carbonílicos alifáticos para T. Pyriformis (Golbraikh et al., 2001). En este estudio, para un total de 140 compuestos solo se obtuvo un moderado ajuste estadístico ( R 2 = 0.753). Sin embargo, al remover cinco outliers R 2 aumentó hasta 0.853 (Golbraikh et al., 2001). Existen varias técnicas para detectar la presencia de „outliers‟, tales como: los análisis de los residuales estandarizados, los residuales studentizados, el método de Leverage, la estadística DFITS, la distancia de Cook y el método de dejar “varios” fuera (Pyka and Planar., 1993).. 18.
(31) Capítulo 2: Materiales y Métodos.
(32) 2- MATERIALES Y MÉTODOS “Los conceptos y principios fundamentales de la ciencia son invenciones libres del espíritu humano.” Albert Einstein. 2.1- Bases de Datos Químicas 2.1.1- Preparación de la Base de Datos La base de datos que se utiliza en este trabajo ha sido nombrada Malaria Box, esta fue creada a partir de una recopilación de la información contenida en tres bases de datos diferentes (St Jude´s, Novartis y GSK), las cuales fueron sometidas a varios análisis que se exponen a continuación. La actividad antimalárica es reportada en EC50 (la concentración que aniquila el 50 % de la población del parásito) expresada en nM (10-9 mol/L). Previo a cualquier análisis, los compuestos en cada base de datos fueron procesados para 1- eliminar sales, 2- eliminar pequeños fragmentos, 3desprotonar bases/ protonar ácidos, 4- generar tautómeros canónicos, y 5eliminar duplicados. Esta preparación de la base de datos fue realizada mediante el uso de Pipeline Pilot 8.5. En este punto cualquier compuesto con peso molecular >1000 o con más de 20 enlaces rotativos no fue tomado en consideración. Finalmente, la base de datos St Jude´s incluyó 1523 estructuras únicas; la base de datos Novartis incluyó 5661 estructuras únicas; y la base de datos GSK incluyó 13257 estructuras únicas. Estos compuestos estructuralmente únicos constituyen la fuente compuesta para la selección de un subconjunto representativo de antimaláricos (Thomas Spangenberg et al., 2013). 2.1.2- La diversidad química la base de datos Dadas las limitaciones en el número de compuestos que pueden ser puestos a prueba en detalle, para maximizar el potencial impacto de Malaria Box, fue importante maximizar la diversidad estructural en los compuestos seleccionados. Un Análisis de Componentes Principales bidimensional (2D-PCA, por sus siglas en inglés) fue usado para evaluar la diversidad química. Para evitar el costo de resíntesis y para hacer los compuestos rápidamente disponibles para el seguimiento experimental,. el. siguiente. paso. consistió. en. seleccionar. compuestos. comercialmente disponibles. Finalmente, 200 compuestos como drogas y 200 20.
(33) como sondas, que cubren la diversidad química del conjunto comercial, fueron seleccionados para Malaria Box (Thomas Spangenberg et al., 2013). 2.2- Herramientas Computacionales 2.2.1- Software MobyDigs. Regresión Lineal Múltiple Los modelos de regresión QSAR se obtuvieron con el programa MOBYDIGS (versión 1.0 – 2004) (Todeschini et al., 2004). Los pesos de cada descriptor en la ecuación de regresión son determinados por el método de mínimos cuadrados. Este programa utiliza un Algoritmo Genético como método de selección de parámetros, lo que le permite evaluar un número elevado de variables. Para cada estudio QSAR, se determinó el tamaño (grado de libertad) deseado para los modelos a generar. El tamaño de la población para todos los estudios fue fijado a 100. El AG con un tamaño poblacional inicial de 100 rápidamente converge (200 generaciones) y alcanza un modelo QSAR en un número razonable de generaciones y por tanto un tiempo prudencialmente aceptable. Se optimizaron los modelos mediante el uso, como función objetivo (función de optimización), del algoritmo genético el estadístico q2LOO y se validaron mediante el empleo de las técnicas de re-muestreo [bootstrapping (q2boot)], revuelto [Y-scrambling: a (R2), a (q2)] y validación externa (q2ext). La selección del mejor modelo fue desarrollada en términos del mayor coeficiente de correlación al cuadrado [coeficiente de determinación, (R2)] o de la F-test (razón de Fisher a nivel-p [p(F)]) y la(s) ecuación(es) de la desviación estándar más baja. Se analizaron los parámetros estadísticos q2LOO (validación cruzada “dejar uno afuera”) y el q2boot para evaluar la calidad de los modelos.. Figura 2. Representación gráfica del programa MobyDigs 21.
(34) 2.2.2- Software WEKA. Regresión no-lineal múltiple WEKA por sus siglas en inglés (Waikato Environment for Knowledge Analysis) es una herramienta que permite la experimentación de análisis de datos mediante la aplicación, análisis y evaluación de las técnicas más relevantes, principalmente las provenientes del aprendizaje automático, sobre cualquier conjunto de datos del usuario. El mismo contiene herramientas para realizar transformaciones sobre los datos, tareas de clasificación, regresión, agrupamiento, asociación y visualización. Weka soporta varias tareas estándar de minería de datos, especialmente, preprocesamiento de datos, clustering, clasificación, regresión, visualización, y selección.. Figura 3. Interfaz gráfica del software WEKA El software Weka fue utilizado para obtener los modelos de regresión no lineal. En este caso se usó como clasificador las Máquinas de Vectores de Soporte (SVM) con variaciones en los tipos de kernels. Programas Informáticos desarrollados en el CAMD-BIR 2.2.3- Software DIVATI. Cálculo de los nuevos Índices de Derivada del Grafo Con el fin de facilitar, desde un punto de vista computacional, el cálculo de los DMs se utilizó una aplicación, escrita en el lenguaje de programación JAVA (Figura 4) denominado DIVATI (Acrónimo de DIscrete DeriVAtive Type Indices), un nuevo módulo del programa TOMOCOMD-CARDD (Marrero-Ponce et al., 2013) 1.0 (acrónimo de Topological Molecular COMputational Design Computed-Aided „Rational‟ Drug Design) (Marrero-Ponce et al., 2013). 22.
(35) Figura 4. Interfaz gráfica del programa TOMOCOMD-CARDD y su módulo DIVATI Este software facilita el cálculo de todas las familias de índices basados en la derivada discreta de un grafo, sobre pares (duplas), tríos (ternas) y cuartetos (cuaternas) de átomos en cualquier tipo de estructura química orgánica. Para los cálculos locales de grupos de átomos se aplican operadores matemáticos. Si al evaluar estos operadores se tienen en cuenta todos los átomos de la molécula el resultado será un cálculo global (total) de la molécula. 2.2.4- Software IMMAN. Análisis de Variabilidad Con la técnica de análisis de variabilidad (AV) basada en el cálculo de la Entropía de Shannon (SE) (Godden and Bajorath, 2000, Godden and Bajorath, 2002, Barigye et al., 2014), se estima la cantidad de información codificada por los diferentes parámetros moleculares, como entidades independientes, y luego se comparan los valores entrópicos de estos. Con motivo de realizar el análisis de variabilidad de los parámetros moleculares propuestos en el presente trabajo, se ha implementado una herramienta computacional fundamentada en los conceptos de la teoría de información la cual se denomina IMMAN (acrónimo de Information Theory based CheMoMetric ANalysis). Esta aplicación permite el cálculo de la Entropía de Shannon (SE) a los DMs. En la Figura 5 se muestra la interfaz gráfica del software (aplicación visual).. 23.
(36) Figura 5. Interfaz gráfica del programa IMMAN. 24.
(37) Capítulo 3: Resultados y Discusión.
(38) 3-. RESULTADOS Y DISCUSIÓN “En lo tocante a ciencia, la autoridad de un millar no es superior al humilde razonamiento de un hombre.” Galileo Galilei. 3.1- Cálculos de los Índices de Derivada del Grafo Las estructuras químicas se codificaron a través del cálculo de los respectivos Índices de Derivada del Grafo (GDI), inspirados en entropías de Teoría de Información (conocidas en idioma inglés como: Jenssent, Conditional, Joint y Mutual) sobre pares, ternas y cuaternas de átomos respecto a 6 eventos diferentes y ortogonales entre sí. Los átomos en cada molécula fueron diferenciados mediante el uso de diversas ponderaciones químicas, físicas y topológicas basadas en grados del vértice. Algunos ejemplos de rasgos atómicos usados en las diferentes formas de ponderación son: el estado intrínseco (I), el grado del vértice de Ivaniciuc (V), la electronegatividad de Pauling (E), la polarizabilidad (P), el volumen de Vander Wals (W), la carga (C), el grado de valencia (N), el área de superficie total polar (T), la conectividad excéntrica (Y) y grado del vértice Alikhanidi (L). Se cuantificaron además, índices globales y locales sobre heteroátomos (HT), donantes de hidrógeno (DH), halógenos (HL), carbonos metilos (MC) y enlaces insaturados (IS). Todos los cálculos fueron desarrollados en el programa informático DIVATI v1.0, nuevo módulo del programa TOMOCOMD-CARDD, donde están implementadas todas las familias de índices basadas en el concepto de Derivada Discreta de un Grafo (Marrero-Ponce et al., 2013). 3.2- Modelación de la Regresión Lineal Múltiple En este epígrafe se expondrán los resultados de la modelación, mediante el uso de la Regresión Lineal Múltiple, de la actividad antimalárica de los compuestos contenidos en la base de datos Malaria Box. 3.2.1- Selección de los mejores descriptores moleculares para la técnica de regresión lineal La degeneración se refiere a la capacidad de un DM para evitar la obtención de valores idénticos para moléculas diferentes. Basados en el criterio anterior es 26.
(39) posible que, los descriptores no posean degeneración (N) o presenten algún tipo de degeneración que puede ser baja (L), intermedia (I) o alta (A). El grado de degeneración de un descriptor puede ser evaluado por medio del cálculo de la Entropía de Shannon (H). La relación directa que existe entre el contenido de información de los resultados numéricos de los DMs y la entropía de Shannon de los mismos ha sido extensamente estudiada por Godden y colaboradores (Godden and Bajorath, 2000, Godden and Bajorath, 2002, Godden and Bajorath, 2003). Basándose en estas ideas, un análisis de variabilidad (AV) cuantifica el contenido de información y, por lo tanto, la variabilidad de los DMs, mediante el uso del cálculo de la entropía de Shannon como criterio cuantitativo. Para ello se introduce un procedimiento de discretización que emplea los histogramas de distribución de frecuencias. Sea p(a)i la probabilidad de que el caso a esté en el intervalo i, para un número de intervalos N se construye una función de distribución de probabilidades, P(A), a la cual se le aplica la ecuación: ∑. ( ). (3.1). De esta forma se obtiene la entropía de cada variable (DMs), la cual es elevada para variables de alta variabilidad y mínima para las de poca variabilidad en la data. Por tanto, esta técnica permite evaluar la calidad de los DMs como entidades independientes y se ha utilizado en la literatura para comparar el desempeño de conjuntos de DMs implementados de diferentes paquetes computacionales, así como en estudios de diversidad molecular (Godden and Bajorath, 2000, Barigye et al., 2013a, Barigye et al., 2013b, Barigye et al., 2014, Godden et al., 2000). La degeneración es un atributo no deseable para un DM usado en la caracterización de estructuras químicas diversas estructuralmente. Se debe esperar que para un conjunto de datos químicos tan diverso como es Malaria Box los DM que tienen mayor variabilidad aporten mejores resultados, debido a una descripción matemática a la realidad química de cada estructura de la base de datos. Con el objetivo de hallar los descriptores moleculares que mayor variabilidad posean, los anteriormente calculados fueron sometidos a un análisis 27.
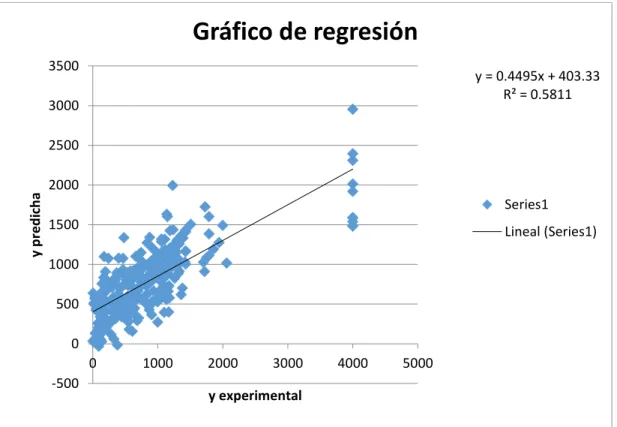
(40) con el software IMMAN, el cual reportó el valor de la entropía de Shannon (H), organizó los DMs en orden decreciente de H y permitió seleccionar los mejores atributos. 3.2.2- Modelos lineales Se desarrollaron modelos de RLM para la actividad antimalárica de las estructuras que componen el conjunto químico Malaria Box, con el programa MobyDigs v1.0. Este programa usa el algoritmo genético como método de selección de parámetros y el Q2Loo como función objetivo para escoger los mejores modelos. Además, el software determina varios parámetros estadísticos para evaluar la calidad de los modelos. De las 400 moléculas de la base de datos Malaria Box realmente se realizó la modelación sobre 317 ya que las 83 restantes no tenían definido el valor de la actividad antimalárica (Thomas Spangenberg et al., 2013). RLM con índices de derivada de Jenssent De los modelos de RLM desarrollados para los descriptores moleculares (basados en diferencias finitas de Jenssent) calculados se hizo una selección de las mejores variables. Los parámetros estadísticos del mejor modelo de regresión lineal obtenido, en este caso, para describir la actividad antimalárica expresada como EC50_nM de las moléculas de la base de datos Malaria Box y sus correspondientes gráficos de regresión y predicción se muestran a continuación: Parámetros estadísticos: Tabla 2. Parámetros estadísticos del modelo de RLM representado en la ecuación 3.2 del Anexo 1 (Jenssent) N. R2. R. Q2. Q2boot. F. 317. 0.3652. 0.6043. 0.2948. 0.2687. 11.55. Scv. y-sc. s. 572.713 603.6396 587.737. Donde, N es el número de compuestos, R² es el coeficiente de determinación, R es el coeficiente de correlación, s es la desviación estándar de la regresión, Q2 es el coeficiente de determinación obtenido a partir del método de validación cruzada (LOO), y-sc es la prueba de aleatoriedad (y-scrambling), Q2boot es el coeficiente de. 28.
(41) determinación de la validación por Bootstrap, Scv es la desviación estándar de la validación cruzada y F es el radio de Fisher. Gráficos de regresión y predicción:. Figura 6. Gráfico de predicción para el modelo de RLM representado en la ecuación 3.2 del Anexo 1. Figura 7. Gráfico de Regresión para el modelo de RLM representado en la ecuación 3.2 del Anexo 1 Como puede observarse en los gráficos anteriores y en los parámetros estadísticos, este modelo lineal, basado en la teoría de Jenssent, posee una pobre capacidad de ajuste a los datos experimentales. En busca de una mejora en la capacidad de ajuste y predicción del modelo de RLM pueden combinarse además otros descriptores moleculares basados en derivadas del grafo y teoría de la información (Conditional, Joint y Mutual) que son 29.
(42) ortogonales a los utilizados (Barigye et al., 2013c), que resulta en una recodificación de las estructuras, que tributa a una mayor cuantificación de la universalidad de información estructural. RLM basada en la combinación de las derivadas trabajadas (Conditional, Joint, Mutual y Jenssent) De la combinación de las mejores variables de las cuatro derivadas trabajadas (Conditional, Joint, Mutual y Jenssent) se obtuvo un modelo para describir la actividad antimalárica expresada como EC50_nM de las moléculas de la base de datos Malaria Box; los parámetros estadísticos y los correspondientes gráficos de regresión y predicción del mejor modelo se muestran a continuación: Parámetros estadísticos: Tabla 3. Parámetros estadísticos del modelo de RLM combinado, ecuación 3.3 (Anexo 2) N. R2. R. Q2. Q2boot. F. Scv. y-sc. s. 317. 0.3803. 0.63. 0.3072. 0.2743. 11.13. 497.294. 525.776. 511.711. Gráficos de regresión y predicción:. Figura 8. Gráfico de Regresión para el modelo de RLM representado en la ecuación 3.3 del Anexo 2. 30.
(43) Figura 9. Gráfico de predicción para el modelo de RLM representado en la ecuación 3.3 del Anexo 2 Como puede verse, los modelos de RLM obtenidos explican pobremente la variable dependiente; ya que todos los valores de ajuste (. ) se encuentran por. debajo de 0.5, al igual (como es de esperar) que los valores de. .. Al realizar un análisis de los resultados de los gráficos de regresión y predicción y los parámetros estadísticos del modelo puede observarse que posee una pobre capacidad de ajuste a los datos experimentales, debido a esto se realizó posteriormente la identificación de los compuestos outliers con el objetivo de mejorar el modelo estadísticamente. Identificación de outliers Un paso crucial en la construcción de modelos, resulta la detección de compuestos atípicos (“outliers”), que se definen como puntos que no se ajustan o son pobremente predichos; que afectan los parámetros estadísticos. Es decir, la identificación de los outliers busca un mejoramiento cualitativo y cuantitativo del modelo, y, aunque no es necesario justificar la extracción de estos puntos, se recomienda determinar la razón para su peculiaridad en aquellos casos en que sea posible (Verma, 2005). Existen varias técnicas para detectar la presencia de outliers, tales como: los análisis de los residuales estandarizados, los residuales. 31.
(44) studentizados, el método de Leverage, la estadística DFITS, la distancia de Cook y el método de “dejar varios fuera”. En este trabajo se eliminaron 60 outliers en busca de un mejoramiento cualitativo y cuantitativo del modelo, para esto se utilizó el método de “dejar varios fuera”. Se muestran a continuación, para el mejor modelo de regresión lineal múltiple obtenido luego de eliminar los outliers, con el objetivo de describir la actividad antimalárica expresada como EC50_nM de las moléculas de la base de datos Malaria Box, los parámetros estadísticos y sus correspondientes gráficos de regresión y predicción: Parámetros estadísticos: Tabla 4. Parámetros estadísticos del modelo de RLM sin los compuestos outliers, ecuación 3.4 (Anexo 3) N. R2. R. Q2. Q2boot. F. Scv. y-sc. s. 257. 0.5134. 0.76. 0.453. 0.4281. 18.16. 281.185. 298.130. 289.803. Gráficos de regresión y predicción:. Figura 10. Gráfico de Regresión para el modelo de RLM representado en la ecuación 3.4 del Anexo 3. 32.
(45) Figura 11. Gráfico de predicción para el modelo de RLM representado en la ecuación 3.4 del Anexo 3 Como puede observarse en los gráficos anteriores y en los parámetros estadísticos, el modelo lineal mejora un tanto la capacidad de ajuste a los datos experimentales con respecto al primer modelo expuesto, luego de eliminar los compuestos outliers. El valor de. es más cercano a uno,. aceptable y el valor de la diferencia:. −. presenta un valor. no excede a 0.3, lo que significa que. no existe un sobreajuste del modelo, ni presencia de variables irrelevantes, ni de outliers en la Data. 3.2.3- Discusión de los resultados de la regresión lineal Existen diferentes opiniones en relación con la interpretación de los parámetros estadísticos y con el establecimiento de los valores extremos mínimos que los mismos deben poseer para considerar “aceptados” o “validados” los modelos objetos de estudio. Con el objetivo de lograr agrupar el criterio de varios autores se tomó como referencia algunos artículos de revisión muy citados que plantean: : Es el parámetro utilizado por lo general para estimar el ajuste del modelo al comportamiento estudiado (en este caso RLM). Sus valores, se plantea, deben estar lo más cercanos posibles a 1.0, pero no debe considerarse a. como. parámetro único, debido a que existen muchas posibilidades de sobreajustes 33.
Figure


+6




Documento similar