The herpes simplex virus type 2 glycoprotein D and host IRE 1α/XBP 1 pathway modulate the functions of dendritic cells after infection with this virus
168
0
0
Texto completo
(2) 1. A mi Tunga la Pilo y el Flaco Pancha y Ju Nelly y Migueles Osvaldo, Yiyi y Gloria Killo y Tomás A mi tutor Sol y Maite. “Feet on the ground and eyes on the horizon” @Opiado.
(3) 2. AGRADECIMIENTOS Las siguientes líneas están llenas de un agradecimiento que nace desde el corazón para quienes fueron participes de este largo y continuo proceso de aprendizaje (y entrenamiento). Este grado académico es la culminación de la construcción de mi proyecto de vida como científico y de mi formación formal. Primero quiero agradecer a mi guía durante esta formación doctoral, gracias al Dr. Pablo González por entregarme herramientas que me han permitido madurar como científico, como profesional y como persona. Gracias por el espacio y la libertad para pensar, ejecutar y crear. Por las oportunidades de realizar actividades de difusión con la sociedad y de vinculación con organismos gubernamentales, por estos años de ciencia en el mesón y en espacios públicos que visibilizan nuestro trabajo. Doy gracias a esta Casa de estudios, en particular por su profundo sentido universitario, democrático y dialogante. Es un orgullo también haber defendido la camiseta de la UC en la selección de volleyball. A la dirección de postgrado de esta Facultad al Dr. Lima y Dr. Bustamante y en especial a María Teresa y Elizabeth por esa preocupación en cada etapa, siempre sincera que supera lo administrativo. A la familia que hemos construido con mi compañera de vida María José, mi Tunga es el silencio en medio de estruendos, el calor en los huesos fríos, enzima de mi vida haces que todo sea energéticamente favorable y súper espontáneo. Gracias por tu apoyo, consejo y sabia compañía. A mis padres la Pilo y el Flaco por entregarme el sustrato inicial, por apoyarme en cada proceso de mi vida en las victorias, los empates y las derrotas. Gracias por educarme en las letras, la música, el arte y la vida. Por renunciar a privilegios e invertirlos en mi instrucción entendiendo la lógica mercantilista de nuestro sistema educativo, para tener esa cosa llamada educación, esas fueron las herramientas que me llevaron a caminar por las rutas que soñé y por las que aún no imagino. El fruto de este trabajo es también para mi hermana Francisca para quien he tratado de ser la mejor versión de mí, y hoy también para nuestra Julieta. Por su cariño y preocupación a mi tía Gloria y mis primas. A quienes entraron a mi vida dando cariño y consejo Nelly, Miguel Ángel y Yiyi son parte importante de esta etapa. A mis amigas y amigos el cariño, la energía, el transitar en esta vida son muchos los que van conmigo y es imposible individualizarlos en tan poco espacio. Cambiar de ciudad no fue fácil, pero la once tenía sabor a infancia y a Conce con mi amigo Osvaldo. He de agradecer en particular al Dr. Juan Carlos Vera y en su memoria dedicar este grado logrado también con sus enseñanzas. A mis amigos del café Andrés Vega y Felipe Aliaga, a los 3841 coffee brothers y por supuesto a Segundo, Salma, Gerardo y Fabían de Café Altura parte de otra ciencia que me mantiene despierto. A mis provincianos queridos, su apoyo y compañía al instalarme en Santiago, tío Roberto y Marco gracias totales. Para el desarrollo de este trabajo fue muy importante el entrenamiento que recibí de Natalia Muñoz, Pablo Céspedes y Roberto Gómez del equipo del Dr. Kalergis. Agradecer al Dr. López-Lastra también por su apoyo y a miembros de su laboratorio Karla Pino, Jenniffer Ángulo, Fernando Lowy, Jorge Vera y Estefania Castillo, donde estuve durante mi primer año de doctorado. A quienes son parte de la primera camada de nuestro laboratorio, con quienes construimos historias a diario y hemos ido creciendo en conjunto a Paula, Togno, Ignacio, MAFs, Francisco, Diana, Luisa, María Paz, Tomás, José y Nico. Y también a quienes acompañé en sus unidades de investigación María Ignacia y Vania. Finalmente, agradecer a las fuentes de financiamiento que aportaron a mi manutención al Astillero Marco Talcahuano, a la Beca Doctorado Nacional de Conicyt folio 21130749 y al Instituto Milenio en Inmunología e Inmunoterapia P09/016. Además, este trabajo de tesis doctoral fue financiado con aportes del proyecto Fondecyt 1140011..
(4) 3. INDEX. FIGURE INDEX ..................................................................................................................................... 5 TABLE INDEX ....................................................................................................................................... 6 THESIS ABSTRACT ............................................................................................................................. 7 RESUMEN DE TESIS ............................................................................................................................ 9 1.- THEORETICAL BACKGROUND ............................................................................................... 11 1.1.- STRUCTURE OF HERPES SIMPLEX VIRUSES (HSVS) .................................................................... 12 1.2.- REPLICATION CYCLE OF HSVS ................................................................................................... 18 1.3.- HSVS EVADE EARLY ANTIVIRAL RESPONSES ............................................................................. 23 1.4.- DENDRITIC CELLS AND HSV INFECTION .................................................................................... 24 1.5.- THE ADAPTIVE IMMUNE RESPONSE ELICITED TO HSV-2 ........................................................... 26 1.6.- VACCINE STRATEGIES AGAINST HSV-2 ..................................................................................... 30 1.6.1.- HSV-2 deleted for Us6 (glycoprotein D) a vaccine candidate ........................................... 33 1.7.- THE UNFOLDED PROTEIN RESPONSE ........................................................................................... 34 1.8.- HERPES SIMPLEX VIRUS INFECTION AND THE UNFOLDED PROTEIN RESPONSE .......................... 38 1.9.- THE UPR IRE-1/XBP-1 PATHWAY AND DENDRITIC CELL FUNCTION ........................................ 42 2.- HYPOTHESIS STATEMENT ....................................................................................................... 46 2.1.- BROAD HYPOTHESIS PUBLISHED IN THE HYPOTHESIS AND THEORY SECTION OF THE JOURNAL FRONTIERS IN IMMUNOLOGY ............................................................................................................. 46 2.2.- HYPOTHESES ASSESSED IN THIS THESIS ..................................................................................... 52 3.- CHAPTER 1 ..................................................................................................................................... 53 US6 GENE DELETION IN HERPES SIMPLEX VIRUS TYPE 2 ENHANCES DENDRITIC CELL FUNCTION AND T CELL ACTIVATION ........................................................................... 54 3.1.- ABSTRACT .................................................................................................................................. 55 3.2.- INTRODUCTION ........................................................................................................................... 56 3.3.- MATERIALS AND METHODS ....................................................................................................... 59 3.3.1.- Mice .................................................................................................................................... 59 3.3.2.- Virus Preparation ............................................................................................................... 59 3.3.3.- DC infection with HSV and viability................................................................................... 61 3.3.4.- DC maturation .................................................................................................................... 63 3.3.5.- DC-DC co-cultures ............................................................................................................. 63 3.3.6.- Electron microscopy ........................................................................................................... 63 3.3.7.- The Unfolded protein response (UPR) ............................................................................... 64 3.3.8.- DC-T cell antigen presentation assays ............................................................................... 65 3.3.9.- DC migration and T cell activation in vivo ........................................................................ 66 3.3.10.- Injection of DCs inoculated in vitro with viruses and genital challenge with virulent HSV-2 ............................................................................................................................................. 67 3.4.- RESULTS ..................................................................................................................................... 69.
(5) 4. 3.4.1.- HSV-2 ΔgD-2 does not induce DC death ............................................................................ 69 3.4.2.- Maturation of virus-inoculated DCs ................................................................................... 72 3.4.3.- DCs inoculated with WT and ΔgD-2 HSV-2 produce defective viral particles .................. 74 3.4.4.- WT but not ΔgD-2 HSV-2 induces increased XBP-1 splicing in DCs ................................ 77 3.4.5.- DCs inoculated with the ΔgD-2 activate CD8+ T cells and CD4+ T cells in vitro ............. 78 3.4.6.- ΔgD-2 promotes increased migration of DCs into LNs and T cell activation in vivo ........ 80 3.4.7.- DCs infected with ΔgD-2 prime an immune response and confer protection against HSV-2 challenge ........................................................................................................................................ 83 3.5.- DISCUSSION ................................................................................................................................ 88 3.6.- SUPPLEMENTARY FIGURES ......................................................................................................... 93 3.7.- SUPPLEMENTARY TABLE ............................................................................................................ 96 3.8.- CONFLICT OF INTEREST .............................................................................................................. 97 3.9.- AUTHOR CONTRIBUTIONS .......................................................................................................... 97 3.10.- FUNDING ................................................................................................................................... 97 4.- CHAPTER 2 ..................................................................................................................................... 98 IRE-1Α RNASE ACTIVITY SUPPORTS HERPES SIMPLEX VIRUS TYPE 2 REPLICATION IN DENDRITIC CELL CULTURES ................................................................................................. 99 4.1 ABSTRACT ................................................................................................................................... 100 4.2.- INTRODUCTION ......................................................................................................................... 101 4.3.- MATERIALS AND METHODS ..................................................................................................... 106 4.3.1.- Mice .................................................................................................................................. 106 4.3.2.- Virus propagation ............................................................................................................. 106 4.3.3.- DC infection with HSV, viability and caspase-3 activity assay ........................................ 107 4.3.4.- Conventional RT-PCR for XBP-1 splicing ....................................................................... 108 4.3.5.- Lipid droplets assay in infected DCs ................................................................................ 108 4.3.6.- DC-T cell antigen presentation assays and ex vivo lymphocyte proliferation assay ....... 109 4.3.7.- Statistical analysis ............................................................................................................ 110 4.4.- RESULTS ................................................................................................................................... 110 4.4.1.- Pharmacological inhibition of IRE1a promotes cell viability in HSV-2-infected DCs and inhibits viral replication in these cells ......................................................................................... 110 4.4.2.- Inhibition of IRE-1α enables HSV-2-infected DCs to activate CD8+ T cells in vitro ...... 113 4.4.3.- HSV-2 infection elicits accumulation of lipid droplets within DCs .................................. 115 4.5.- DISCUSSION .............................................................................................................................. 117 5.- THESIS DISCUSSION ................................................................................................................. 121 6.- CONCLUDING REMARKS ........................................................................................................ 131 7.- HIGHLIGHTS ............................................................................................................................... 133 8.- APPENDIX..................................................................................................................................... 134 8.1.- PUBLICATIONS GENERATED IN THIS THESIS AND PHD TRAINING ............................................ 134 8.2.- SCIENTIFIC MEETINGS ATTENDED DURING THIS THESIS .......................................................... 141 REFERENCES .................................................................................................................................... 142.
(6) 5. FIGURE INDEX. Figure 1: Structure of herpes simplex viruses and overall organization of the viral genome...14 Figure 2: Replicative cycle of herpes simplex viruses………………………………………..22 Figure 3: Herpes simplex viruses negatively modulate dendritic cell function……………....29 Figure 4: Three different branches coordinate the UPR in mammals....……………………...41 Figure 5: ΔgD-2 HSV-2 is attenuated in DCs…………………………………………………71 Figure 6: HSV-2 ΔgD-2 induces DC maturation and cytokine secretion……………………..73 Figure 7: DCs inoculated with ΔgD-2 or WT virus mainly release defective HSV particles...76 Figure 8: Differential UPR in WT- and ΔgD-2 virus-inoculated DCs………………………..79 Figure 9: DCs pulsed with ΔgD-2 activate CD4+ and CD8+ T cells in vitro…………………81 Continuation legend Figure 9………………………………………………………………..….…….82 Figure 10: ΔgD-2 promotes DC migration and CD4+ and CD8+ T cell activation in vivo..…..84 Figure 11: ΔgD-2 increases the relocation of CD103+ migrating DCs to draining LNs………85 Figure 12: Transfer of DCs infected with ΔgD-2 confer protection against intravaginal lethal challenge with HSV-2…………………………………………………………………………87 Supporting Figure 1. ΔgD-2 HSV-2 is attenuated in DCs, unlike WT HSV-2….……………93 Supporting Figure 2. Phenotype of bystander DCs overlaid on virus-inoculated DCs…….....94 Supporting Figure 3. IL-6 secretion by DCs inoculated with the indicated viruses determined by ELISA…………………………………………………………………………95 Figure 13: IRE-1α inhibition with 4µ8c limits virus yield from HSV-2-infected DC cultures……………………………………………………………………………………….112 Figure 14: IRE-1α inhibition with 4µ8c in HSV-2-infected DC cultures promote virus-specific CD8+ T cell activation………………………………………………………...114 Figure 15. HSV-2 induces lipid droplets in DCs…………………………………………….116 Figure 16. Visual abstract of conclusions and highlights……………………………………133.
(7) 6. TABLE INDEX. Table 1: Nonstructural functions of HSV tegument proteins…………………………………17 Table 2: HSV-2 mutants tested as attenuated virus vaccines in animal models………………51 Table 3: Primers used to generate the ΔgI and ΔgJ deletant viruses………………………….96.
(8) 7. THESIS ABSTRACT. Herpes simplex viruses and humans have co-existed for tens of thousands of years and this long relationship has translated into the evolution and selection of viral determinants for evading the host immune response and reciprocally, the evolution and selection of host immune components to limit infection and pathology. Herpes simplex virus type 2 (HSV-2) is highly prevalent in the human population and is the main etiological agent of genital ulcers and neonatal encephalitis. Moreover, HSV-2 is neurotropic and resides latently in neurons in the sacral ganglia, occasionally reactivating either clinical or subclinical recurrent infection. Currently there are no vaccines against this viruses and available therapies do not cure infection. Because dendritic cells are key immune cells that initiate and regulate antiviral responses, and HSV-2 interferes with their function, we studied the interaction between these cells and a mutant HSV-2 virus that confers protective immunity against challenge with wild-type virus in murine infection models. Furthermore, based on preliminary results generated at the beginning of this thesis and available literature, we generated a hypothesis for a novel theory that proposes that the outcome of the interaction between DCs and mutant HSV-2 vaccine candidates may correlate with protection in vivo. Finally, we focused on the unfolded protein response in infected DCs and how this cellular process modulates HSV-2 replication. In the First Chapter of this thesis, we describe that an HSV-2 mutant that lacks the glycoprotein D gene (∆gD-2) was attenuated in DCs, unlike wild-type HSV-2 (WT) or other HSV-2 glycoprotein mutants. DCs inoculated with ΔgD-2 activated virus-specific CD8+-T cells (gBT-I) and antigen-specific CD4+-T-cells (OT-II), in vitro. Also, mice inoculated with ΔgD-2 displayed increased CD103+-DC migration to lymph nodes and displayed both, activated CD8+ and CD4+ T cells. Mice primed with DCs inoculated with ΔgD-2 in vitro displayed significantly reduced infection and pathology after genital challenge with virulent HSV-2 compared to ∆gH2 primed mice, indicating that these cells can elicit a protective immune response with this virus. Taken together, these results suggest that ΔgD-2 activates DCs to promote antigen presentation and that this characteristic likely contributes to the immunogenicity and protective capacity of.
(9) 8. this candidate vaccine when administered alone. When studying the interaction of ΔgD-2 with DCs, we found that it elicited a different unfolded protein response (UPR) as compared to the WT virus, which we further studied in the Second Chapter of this thesis. Importantly, we also found that the pharmacological inhibition of the RNase activity of the UPR signaling molecule IRE-1α in DCs, hampered HSV-2 replication in DC cultures. Overall, this effect increased DC viability and supported the activation of virus-specific CD8+ T cells in vitro. Furthermore, we found that DCs infected with HSV-2 produced significant amounts of triglycerides (lipid droplets), which has been associated with decreased antigen presentation in these cells. Remarkably, MHC-I expression in DC cultures infected with HSV2 was restored to basal levels upon treatment with an IRE-1α inhibitor. These results indicate that HSV-2 induces an IRE-1α UPR in DCs that is detrimental for their function. Taken together, we identified both, viral land host factors that modulate outcome of the interaction between DCs and HSV-2, and thus could be harnessed to design novel vaccine strategies or therapies to limit infection and dissemination produced by this virus..
(10) 9. RESUMEN DE TESIS. Los virus herpes simple y los humanos han coexistido durante decenas de miles de años y esta larga relación se ha traducido en la evolución y selección de determinantes virales que evaden la respuesta inmune del hospedero y recíprocamente, la evolución y selección de componentes inmunes del hospedero para limitar tanto la infección como la patología asociada a la infección. El virus del herpes simple de tipo 2 (VHS-2) es altamente prevalente en la población humana y es el principal agente etiológico de úlceras genitales y de encefalitis neonatal. Además, el VHS-2 es un virus neurotrópico que reside de manera latente en las neuronas del ganglio sacro, ocasionalmente las reactivaciones desarrollan manifestaciones tanto clínicas como subclínicas. Actualmente no hay vacunas contra este virus y las terapias disponibles no curan la infección. Debido a que las células dendríticas (CDs) son células inmunitarias que inician y regulan las respuestas antivirales, y además como VHS-2 interfiere con su función, estudiamos la interacción entre estas células y un virus mutante de VHS-2 que confiere inmunidad protectora contra el desafío con aislados clínicos en modelos de infección murino. Así, sobre la base de los resultados preliminares generados al comienzo de esta tesis y la literatura disponible, generamos una hipótesis para una teoría novedosa la cual propone que el resultado de la interacción entre las CDs y cepas mutantes candidatos a vacuna, pueden correlacionarse con la protección conferida por estos mutantes in vivo. Finalmente, nos enfocamos en la respuesta a proteínas mal plegadas en las CDs infectadas y cómo este proceso celular modula la replicación de VHS-2. En el primer capítulo de esta tesis, describimos un mutante de VHS-2 el cual carece del gen que codifica para la glicoproteína D (ΔgD-2), este virus mutante resulto ser atenuado en CDs, a diferencia del VHS-2 de tipo silvestre (WT) y otros VHS-2 mutantes para otras glicoproteínas. Las CDs infectadas con ΔgD-2 son capaces de activar in vitro a células T CD8+ virus-específicas (gBT-I) y células T CD4+ antígeno-específicas (OT-II). Además, ratones inoculados con ΔgD-2 mostraron una mayor migración de CDs CD103+ a los ganglios linfáticos y activación de poblaciones de células T tanto CD8+ como CD4+. Por otro lado, la transferencia.
(11) 10. adoptiva de CDs infectadas in vitro con ΔgD-2 a ratones hembra demostró disminuir tanto la infección como la patología después del desafío genital con VHS-2 en comparación con ratones que recibieron CDs infectadas con un mutante para la glicoproteína H (∆gH-2), lo que indica que CDs cargadas con el virus ∆gD-2 in vitro pueden estimular una respuesta inmune protectora contra este virus. Tomados en conjunto, estos resultados sugieren que ΔgD-2 activa a las CDs promoviendo la presentación de antígenos y que esta característica probablemente contribuye tanto a la inmunogenicidad y como a la capacidad protectora de este candidato a vacuna cuando se administra sola. Como resultado del estudio de la interacción de ΔgD-2 con CDs, encontramos que este mutante desencadenó una respuesta a proteínas mal plegadas (UPR) mediada por la vía IRE-1α/XBP-1 diferente a la generada por el virus WT, abordamos este estudio en el segundo capítulo de esta tesis. De manera importante, encontramos que la inhibición farmacológica de la actividad de ARNasa de la molécula de señalización de UPR IRE-1α en las CDs, afectó la replicación de VHS-2. En general, observamos que al inhibir IRE-1α luego de la infección con VHS-2 aumenta la viabilidad de CDs, recuperando también la activación de células T CD8+ virus-específicas in vitro. Además, encontramos que las CDs infectadas con VHS-2 aumentan significativamente la síntesis de triglicéridos (gotas de lípidos), este proceso se ha asociado con una disminución en la presentación de antígenos en estas células. Sorprendentemente, la expresión de MHC-I en cultivos de CDs infectadas con VHS-2 se restauró a niveles basales tras el tratamiento con un inhibidor de IRE-1α. Estos resultados indican que el VHS-2 activa al sensor de UPR IRE-1α, lo cual resulta perjudicial para la función de las CDs. Tomados en conjunto los resultados aquí expuestos, identificamos tanto factores virales como del hospedero que modulan el resultado de la interacción entre las CDs y el VHS-2, esta evidencia experimental puede contribuir al diseño de nuevas estrategias de vacunas o terapias que limiten la infección y diseminación de este virus..
(12) 11. 1.- THEORETICAL BACKGROUND. The Herpesviridae family consists of over 100 double-stranded DNA viruses divided in alphaherpesvirinae, betaherpesvirinae and gammaherpesvirinae subfamilies (Alba, Das, Orengo, & Kellam, 2001). Only eight species of herpesviruses are known to commonly infect humans: i) herpes simplex virus type 1 (HSV-1 or human herpes virus 1: HHV-1), ii) herpes simplex virus type 2 (HSV-2, HHV-2), iii) varicella zoster virus (VZV, HHV-3), iv) Epstein Barr virus (EBV, HHV-4), v) cytomegalovirus (CMV, HHV-5), vi) human herpes virus 6, vii) human herpes virus 7 and viii) Kaposi’s sarcoma-associated herpes virus (KSV, HHV-8). The remainder are animal herpesviruses infecting a wide variety of mammalian species (Louten, 2016). Importantly, all members of the Herpesviridae family cause lifelong latent infections in their hosts with sporadic reactivations (Fields, Knipe, & Howley, 2013). Herpes simplex virus type 2 (HSV-2) infects nearly 500 million people worldwide and is the main cause of genital ulcers in symptomatic individuals (Paz-Bailey, Ramaswamy, Hawkes, & Geretti, 2007; Smith & Robinson, 2002). Importantly, infection may be transferred to neonates during birth, which may lead to life-threatening encephalitis (Looker et al., 2015). Although antivirals limit HSV-2 replication in the newborn, serious long-term neurologic sequelae may follow, despite treatment (Z. A. Brown et al., 1997; Steiner & Benninger, 2013; K. N. Ward et al., 2012). HSV-2 is persistent in humans, establishing latency in neurons with symptomatic or asymptomatic reactivations that shed infectious viral particles and significantly contribute to the spread of HSV-2 in the population (Celum et al., 2010; Corey, 2007). Importantly, the biology of herpetic infections supports the epidemiologic observations.
(13) 12. regarding an association between HSV-2 and human immunodeficiency virus type 1 (HIV-1) acquisition. More than 30 epidemiologic studies have demonstrated the risk of acquiring the human immunodeficiency virus (HIV) is 2- to 4-fold higher among individuals that are HSV-2seropositive (Freeman et al., 2006; Suazo, Tognarelli, Kalergis, & Gonzalez, 2015; Wald & Link, 2002). Thus, in vivo and in vitro studies suggest that mucosal HIV-1 shedding is more frequent and in greater amounts during mucocutaneous HSV-2 replication, including subclinical mucosal reactivations. (Corey, Wald, Celum, & Quinn, 2004; Cowan et al., 2006; Suazo, Tognarelli, et al., 2015; Wald & Link, 2002). Although oral antivirals limit the extent of HSV reactivations, reduce virus shedding and shorten the duration of herpetic lesions, these drugs do not resolve persistent infection (Johnston et al., 2012; Whitley & Gnann, 1992). Furthermore, some clinical manifestations of HSV-2 infection, such as herpetic lesions, resolve only moderately with the available antivirals in the form of topical creams and thus, better drugs for limiting HSV-2 are needed. In order to prevent infection with this virus or its clinical manifestations, a vaccine that blocks or hampers viral reactivation and virus shedding would be beneficial. Although important efforts have been undertaken for developing a vaccine against HSV-2 and HSV-1, regretfully these attempts have not been successful so far.. 1.1.- Structure of herpes simplex viruses (HSVs) The herpes simplex virion consists of four components: a) an electron-dense core containing the viral DNA, b) an icosahedral capsid surrounding the core, c) an amorphous, at times eccentric layer of proteins, called the tegument that surrounds the capsid, and d) an outer lipid bilayer envelope exhibiting glycoproteins on its surface (F. Liu & Zhou, 2007) (Figure 1). The most detailed analysis of the whole virion consists on cryo-electron tomography that defines.
(14) 13. its structure at a 7 nm resolution. This study defined that the virion is a spherical particle with an average diameter of 186 nm, which extended to 225 nm with the spikes included. The nucleocapsid was located in an eccentric position with one edge of the nucleocapsid close to the envelope and the other, the distal pole, 30 to 35 nm from the envelope (Grunewald et al., 2003). The tegument showed a particulate structure with some 7-nm filaments apposed to the membrane. The tegument cap on the distal pole was connected to the envelope by 4 nm wide linkers (Laine et al., 2015). The virion core contains the double-strand DNA (dsDNA) genome in a lineal form, wrapped as a toroid in a liquid crystalline state (Phage-like packing) (Booy et al., 1991). A small fraction of the virion DNA may be circular. The core does not contain highly basic proteins, such as histones, that would neutralize the negative charges of the viral DNA to allow proper folding within the capsid, but contains the polyamines spermidine and spermine. These polyamines appear to be tightly bound to the DNA and cannot be exchanged with exogenouslyadded labeled polyamines. Disruption of the envelope with nonionic detergents and urea did remove the spermidine, but not the spermine. It has been reported that spermine contained in the virion is sufficient to neutralize approximately 40% of the DNA phosphate charges (Gibson & Roizman, 1971). The viral DNA contains at least 152 kbp and variability in size is due to variations in the number of reiterations of specific terminal and internal sequences (see Figure 1). The genomes of HSV-1 and HSV-2 consist of two covalently linked segments, designated as L (long) and S (short). Each segment is composed of unique sequences (UL or US, respectively) flanked by relatively large inverted repeat elements (TRL-IRL and IRS-TRS) (Cortini & Wilkie, 1978; Roizman, 1979)..
(15) 14. Figure 1. Structure of herpes simplex viruses and overall organization of the viral genome. A. Left: Transmission electron microscopy photography from HSV-2 infected dendritic cells. Red arrow indicates the four elements in the HSV-2 virion. An electron opaque core containing the viral DNA, an icosahedral capsid surrounding the capsid (black), the tegument that surrounds the capsid (orange) and an outer lipid bilayer envelope (blue). Adapted from (A. Retamal-Diaz et al., 2017). Right: Schematic illustration of HSV-2. The HSV-2 is an enveloped, double stranded DNA virus (~154 kpb) that encodes >74 genes; the viral genome is enclosed in an icosahedral capsid of approximately 125 nm surrounded by a complex mesh of viral proteins termed the tegument. HSV-2 is enveloped by a lipid membrane that harbors numerous glycoproteins protruding to the outer side of the virion (gB, gC, gD, gE, gG, gH, gI, and gL within others). Adapted from (Retamal-Díaz, Suazo, Garrido, Kalergis, & González, 2015). B. The linear double-stranded DNA is represented, with a scale at the bottom. The unique portions of the genome (UL and US) are shown as heavy solid black lines, and the major repeat elements (TRL, IRL, IRS, and TRS) are shown as boxes. For each pair of repeats the two copies are in opposing orientations. As indicated, TRL, UL, and IRL are regarded as comprising the L region, and IRS, US, and TRS are regarded as comprising the S region..
(16) 15. There is a directly repeated sequence of some 254 bp at the genome termini (a sequence), with one or more copies in the opposing orientation (a’ sequence) at the internal joint between IRL and IRS (Dolan, Jamieson, Cunningham, Barnett, & McGeoch, 1998). The HSV virion proteins have been mainly named based on serial numbering of the virion proteins on electrophoretic gels (e.g. VP16, viral protein 16), on the order of the open reading frame (ORF) encoding them (e.g. UL8), or as infected cell proteins (ICPs; e.g. ICP5). Mass spectrometry analyses of purified extracellular virions has demonstrated the presence of several host proteins within the viral particles (Loret, Guay, & Lippe, 2008), although it is unclear if any these host proteins are essential for the virion structure. (Newcomb & Brown, 2012; Stegen et al., 2013). Cryo-electron microscopy studies have shown that the HSV nucleocapsid is composed of an outer layer arranged in a T = 16 symmetry and an intermediate layer organized in a T = 4 lattice (Schrag, Prasad, Rixon, & Chiu, 1989); the capsid is comprised of 162 capsomers, including 140 hexons, 11 pentons and a portal. The same study concluded that the outer and intermediate layers of the tegument are organized so that channels along their icosahedral twofold axes coincide, forming a direct pathway and potential channel between the DNA in the capsid and the exterior of the virion (Schrag et al., 1989). The outer shell of the capsid is composed of four viral proteins, VP5 (UL19), VP26 (UL35), VP23 (UL18), and VP19C (UL38). VP5, the major capsid protein is present in five copies in each penton capsomere and six copies in each hexon capsomere in this icosahedral shell. VP26 is present in six copies as a ring on top of the VP5 subunits on each hexon. Triplexes made up of one VP19C molecule and two VP23 molecules link adjacent capsomeres as a result of the triplex, forming a pseudotrimer with each subunit interacting in a roughly similar manner.
(17) 16. with two capsomeres (Okoye, Sexton, Huang, McCaffery, & Desai, 2006). The capsid also contains the UL6 protein, which forms a dodecamer thought to form a portal through which viral DNA is packaged and VP24 (UL26) and the protease that processes the scaffolding during DNA encapsidation (Sheaffer et al., 2001). Additionally, the UL16 protein may be a capsid-associated protein because it plays a role in DNA encapsidation (Gao, Hay, & Banfield, 2017). The space between the undersurface of the envelope and the surface of the capsid is designated as the tegument and it is largely unstructured (except for the fibrils that connect the tegument with envelope). However, some apparent icosahedral structure around the pentons exists and is comprised of at least 18 viral proteins (namely, UL48: VP16, UL41: vhs protein, UL49: VP22, UL36: VP1-2, UL14: UL14, UL17:UL17, UL46: VP11/12, UL47: UL47, Us11: US11, Us9: US9, Us10: US10). Additionally, tegument proteins participate in some nonstructural functions (e.g. viral cycle, cell cycle-arrest, immune evasion, as other), we describe these functions in Table 1. Recent studies are starting to define the protein-protein interactions both, within the tegument and between the tegument, as well as with capsid and envelope proteins. Interestingly, highly purified HSV virions contain cellular and selected viral gene transcripts in the tegument. These are likely packaged in this region of the virion because three RNA binding proteins (US11, UL47, and UL49) were identified in the tegument (Sciortino, Taddeo, Poon, Mastino, & Roizman, 2002). The envelope consists of a lipid bilayer with potentially as many as 13 distinct viral glycoproteins implanted in it. The virion envelope glycoproteins are gB (VP7 and VP8.5, encoded by the UL27 gene), gC (VP8, UL44), gD (VP17 and VP18, US6), gE (VP12.3 and VP12.6, US8), gG (US4), gH (UL22), gI (US7), gK (UL53), gL (UL1), and gM (UL11). The presence of gJ (US5) and gN (UL49.5) in virions has not been demonstrated. Virion envelopes.
(18) 17. Table 1. Nonstructural functions of HSV tegument proteins. Gene. UL48. UL41. Protein name. Function. References. VP16. - Transcriptional activator of immediate-early gene products (alpha genes). - Initiating the lytic program through the assembly of the transcriptional regulatory VP16-induced complex composed of VP16 and two cellular factors, HCF and Oct-1.. (K. M. Johnson, Mahajan, & Wilson, 1999; La Boissiere, Hughes, & O'Hare, 1999).. Virion host shutoff protein (vhs). - Contains a domain with endoribonuclease activity at early times of lytic infection degrade many constitutively expressed host mRNA and facilitating ordered expression of different classes of viral genes. - Binds translation initiation factors eIF4H, eIF4AI, and eIF4AII, thereby may interact directly with the translation initiation complex and thus digest specifically mRNAs. - During animal infections, vhs plays a key role in inhibiting the interferonmediated antiviral response as well as other components of innate and adaptive immune responses. - Participates in both accumulation and translation of viral mRNA at late time of lytic infection. - Modulates the RNase activity of vhs protein to ensure necessary level of key host mRNAs and proteins. - Plays a role in microtubule reorganization that occurs after viral infection by stabilizing microtubule network. - Participates in the capsid transport toward the host nucleus, in the routing of the capsid at the nuclear pore complex and subsequent uncoating. - Within the host nucleus, acts as a deneddylase and promotes the degradation of nuclear CRLs (cullin-RING ubiquitin ligases) and thereby stabilizes nuclear CRL substrates, while cytoplasmic CRLs remain unaffected. These modifications prevent host cell cycle S-phase progression. - Participates later in the secondary envelopment of capsids. Indeed, plays a linker role for the association of the outer viral tegument to the capsids together with the inner tegument protein. - Contributes to the nuclear transport of the viral transcriptional activator VP16 during the early phase of infection. - Seems to be important for efficient nuclear targeting of capsids. - The UL51-UL14 complex regulates final viral envelopment for efficient viral morphogenesis. - UL17 is the second constituent of the capsid vertex-specific component required for DNA packaging and retention.. (Fenwick & McMenamin, 1984; F. E. Jones, Smibert, & Smiley, 1995) (Doepker, Hsu, Saffran, & Smiley, 2004). (Mbong et al., 2012) (Elliott & O'Hare, 1998; Yedowitz, Kotsakis, Schlegel, & Blaho, 2005). UL49. VP22. UL36. VP1-2 (Large tegument protein). UL14. UL14. UL17. UL17. UL46. VP11/12. UL47. UL47. - Modulates the activity of vhs-RNase activity. - Participates in the primary envelopment of virions in the perinuclear space by interacting with two nuclear egress proteins UL31 and UL34.. (Z. Liu et al., 2014; Shu, Taddeo, Zhang, & Roizman, 2013). US11. - Inhibits autophagy by hijack host proteins PKR/EIF2AK2, this interaction also prevents the interferon-induced shut down of protein synthesis following viral infection. - Downmodulates the host RLR signaling pathway via direct interaction with host DDX58 and IFIH1. - May also participate in nuclear egress of viral particles through interactions with host NCL and regulation of the viral UL34 mRNA.. (Lussignol et al., 2013; Xing, Wang, Lin, Mossman, & Zheng, 2012) (Greco et al., 2012). US11. - Mediated cell survival by the activation of the host PI3K/AKT pathway. - Inhibits of TMEM173/STING-mediated type I interferon production.. (Sandbaumhuter et al., 2013) (Gastaldello et al., 2012) (Desai, 2000). (Oda, Arii, Koyanagi, Kato, & Kawaguchi, 2016; Yamauchi et al., 2008) (Toropova, Huffman, Homa, & Conway, 2011). (Wagner & Smiley, 2011) (Deschamps & Kalamvoki, 2017)..
(19) 18. also contain at least two (UL20 and US9), and possibly more (UL24, UL43, and UL34) nonglycosylated intrinsic membrane proteins (Rajcani & Vojvodova, 1998; P. L. Ward, Campadelli-Fiume, Avitabile, & Roizman, 1994). Herpes simplex virus infections stimulate phospholipid synthesis, and the de novo synthesized phospholipids are inserted into nuclear and cytoplasmic membranes to maintain membrane integrity in the course of nuclear and cellular expansion, to supply membrane constituents for the envelopment of capsids by budding at nuclear membranes, and Golgi membranes to provide membranes for the synthesis of vacuoles for virus exit from the cell (Sutter et al., 2012; Wild et al., 2012).. 1.2.- Replication cycle of HSVs Herpes simplex viruses have numerous proteins and glycoproteins presented on their exterior (Akhtar & Shukla, 2009; Rajcani & Vojvodova, 1998). Of the many glycoproteins expressed on the surface, at least five viral glycoproteins have been implicated in viral entry, namely glycoproteins B (gB), gC, gD, gH and gL (Avitabile, Forghieri, & Campadelli-Fiume, 2009; Turner, Bruun, Minson, & Browne, 1998). gB acts both as a viral attachment protein and fusion protein by binding to heparan sulfates (HS) on the surface of virus-susceptible host cells (Atanasiu et al., 2013), and also binds to paired immunoglobulin-like type 2 receptor (PILR) alpha (Arii et al., 2010; Satoh et al., 2008). A similar role in virus attachment has been described for gC, although only for HSV-1 (Herold, WuDunn, Soltys, & Spear, 1991). After gB-mediated attachment to the cell surface, gD binds to either of its receptors: nectin-1 (PVRL1; poliovirus receptor-related 1) expressed on the surface of most host cells or, alternatively to HVEM (Herpesvirus Entry Mediator, TNFRSF14), which is mainly expressed in immune cells.
(20) 19. (Matsushima et al., 2003; Steinberg et al., 2013). Moreover, 3-O sulfate HS has also been suggested as a potential receptor for gD, although its physiological relevance requires more study (J. Liu et al., 2002). Binding of gD to its receptors is thought to induce conformational changes in this molecule that lead to the activation of the protein complex formed by gH/gL (Lazear et al., 2014). The activated gH/gL complex would then promote changes in gB that would activate the fusogenic properties and mediate the fusion of viral and host lipidic membranes (Fusco, Forghieri, & Campadelli-Fiume, 2005; Krummenacher et al., 2005). Alternatively, herpes simplex viruses can enter cells through via endocytic pathway in which also requires fusion of the enveloped particles with the vesicular membrane (late endosome) for the release of the viral nucleocapsid proximal to the nucleus. (Milne, Nicola, Whitbeck, Eisenberg, & Cohen, 2005; Anthony V Nicola, McEvoy, & Straus, 2003). In both cases, fusion of membranes derives in the entry of the viral capsid and accompanying viral proteins (tegument) into the cytoplasm (Dohner et al., 2002). Once released into the cytoplasm, the capsid is argued to associate to microtubules, at least in neurons, through the two tegument proteins VP1-2 (encoded by the UL36 gene) and UL37. Through these proteins, the capsid travels to the outer nuclear membrane to bind to the host nuclear pore complex (NPC) to release the viral DNA into the nucleus through nuclear pores (Dohner et al., 2002; Granzow, Klupp, & Mettenleiter, 2005). A role for nucleoporin Nup358 has been associated to the docking of VP1-2 and the facilitation of DNA release through this macromolecular complex (Copeland, Newcomb, & Brown, 2009). Once released into the nucleus, the viral DNA is transcribed thanks to the host RNA-polymerase II (Holland, Anderson, Shipman Jr, & Wagner, 1980; Kops & Knipe, 1988). However, not all genes are expressed synchronously but rather in four consecutive waves. First, immediate early genes.
(21) 20. (alpha) are transcribed, many of which encode for immune evasion determinants and factors needed for controlling cell translation (Silva et al., 2012). Then, follows the transcription of early genes (beta) that play roles in DNA replication (W. Wu et al., 2013). Finally, early late and late genes (gamma-1 and gamma-2) are transcribed, which mainly encode structural virion components, such as capsid, tegument and surface proteins (S. Chen et al., 1992; Rajcani, Andrea, & Ingeborg, 2004). Nevertheless, these proteins encode important immune evasion determinants (see below). For newly synthesized virions, capsid proteins migrate to the nucleus to assemble with viral DNA and acquire at this place a thin layer of tegument proteins. Unlike other viruses, HSV does not alter nuclear pores on exit, but rather undergoes envelopment in the inner nuclear membrane to form an enveloped capsid (Hofemeister & O'Hare, 2008). The capsid then travels into the perinuclear space and immediately fuses with the outer nuclear membrane thanks to glycoproteins gB and gH, exposing a tegument-recovered capsid into the cytoplasm (Farnsworth et al., 2007). Once in the cytoplasm, the capsid is further coated with additional tegument proteins and is once again enveloped in the trans Golgi network (Turcotte, Letellier, & Lippe, 2005). From here, virions are exported in vesicles to the cell surface and released for infection of other cells (Figure 2). However, HSV can also propagate directly to adjacent cells through cell-cell interactions. This type of infection is mediated through a process called virological synapse and provides the virus a safe haven from neutralization with antibodies or complement (see below) (Hook, Lubinski, Jiang, Pangburn, & Friedman, 2006; Lubinski, Lazear, Awasthi, Wang, & Friedman, 2011). HSV a non-lymphotropic virus has been shown to infect T cells through infected fibroblasts in vitro, importantly this process requires the surface expression of.
(22) 21. glycoprotein D of HSV (Aubert, Yoon, Sloan, Spear, & Jerome, 2009; D. C. Johnson, Webb, Wisner, & Brunetti, 2001)..
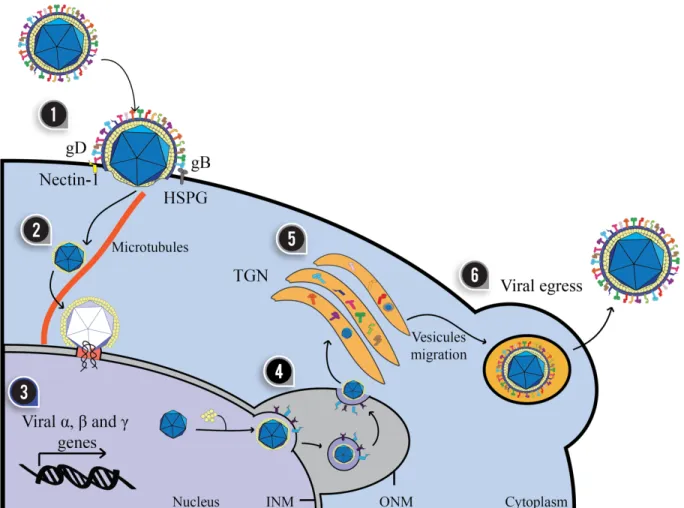
(23) 22. Figure 2. Replicative cycle of herpes simplex viruses. The infection of a target cell is a process that involves at least four viral glycoproteins on the surface of the virion. (1) Glycoprotein B (gB) binds heparan sulfate proteoglycans (HSPG) on the cell surface. For HSV-1, the gC glycoprotein also participates in this process. Then, gD binds to Nectin-1 or HVEM on the cell surface, triggering the activation of the gH/gL complex and the fusion of the virus and host membranes with gB. (2) Within the cell, the viral capsid is transported to the nucleus by the interaction of tegument proteins with microtubules (demonstrated in neurons). (3) Once in the nucleus, the viral genes are expressed sequentially with the transcription of immediately early genes (α), early genes (β) and late genes (γ). (4) Virus production requires the replication of the viral genome which is encapsidated in the nucleus and partially coated by tegument proteins that mediate the exit of the capsid into the perinuclear space through the inner nuclear membrane (INM) and the outer nuclear membrane (ONM). (5) The capsid is then released into the cytoplasm where it is re-invaginated into the Golgi apparatus where it acquires its surface glycoproteins. (6) Finally, the virion leaves the cell by exocytosis. Modified from (RetamalDíaz et al., 2015) ..
(24) 23. 1.3.- HSVs evade early antiviral responses The innate immune response provides an immediate nonspecific line of defense against pathogens. Upon infection, HSV-2 stimulates the innate system to produce interferon alpha (IFN-α), through its interaction with epithelial cells and plasmacytoid dendritic cells (pDCs) in the genital tissue (Donaghy et al., 2009). Recognition of the virus occurs mainly via Toll-like receptors (TLRs), including TLR9 (Lund, Linehan, Iijima, & Iwasaki, 2006), TLR2 (Kurt-Jones et al., 2004), and TLR3 (S. Y. Zhang et al., 2007). Cytoplasmic DNA sensors, such as stimulator of IFN genes (STING), also mediates the production of IFN in response to HSV infection, as demonstrated in cell cultures and in knockout mouse models (Ishikawa, Ma, & Barber, 2009). However, HSV also encodes viral determinants that downmodulate IFN activity, such as through the degradation of IRF3, which is mediated by the viral immediate early infected cell protein 0 (ICP0) that prevents the IFN-β mRNA accumulation within cells (Melroe, DeLuca, & Knipe, 2004). In addition, expression of ICP0 alone can prevent TLR2-dependent NF-κB activation in cell cultures, which suggests that the virus can downregulate the immune response at several points in the inflammatory pathway (van Lint et al., 2010). Relevant components in the innate response against HSV are natural killer (NK) cells and NK T cells; both cells are early sources of IFN-γ after HSV infection. In vivo studies have demonstrated the necessity of these innate immune modulators in mediating the control of herpesvirus infection and furthermore, provide a link between innate and adaptive arms of the antiviral immune system (Ashkar & Rosenthal, 2003). Local delivery of TLR ligands in a vaginal model of HSV-2 infection was shown to induce potent innate immunity against genital HSV-2, with high levels of type I IFNs and protection accompanied by a significant increase in NK cell number in the genital tract (Ashkar et al., 2004). Nevertheless, soluble and immune.
(25) 24. mediators responsible for this innate immune response, as well as the viral determinants that induce these components upon natural infection have been poorly studied. Less studied is the role of these immune and viral determinants in the context of genital infection with HSV-2. Increased knowledge in this area should help developing new therapies and vaccines that ensure protection against HSV-2. 1.4.- Dendritic cells and HSV infection Dendritic cells (DCs) are immune cells operating at the interface of innate and adaptive immunity, with the unique ability to activate naïve T cells (Steinman & Banchereau, 2007). Under homeostatic conditions, DCs reside immobilized in an immature state in peripheral tissues, possessing high phagocytic capability, but low T cells priming faculty (Banchereau et al., 2000). Upon activation by stimulation via, e.g., pattern recognition receptors (PRRs), antigen uptake, combined or not with specific pro-inflammatory cytokines, immature DCs (iDCs) undergo a maturation process (Wilson & Villadangos, 2004). Mature DCs (mDCs) enhance their capacity to process antigen and the ability to present them, which is accompanied by increased major histocompatibility complex (MHC) class-I and class-II expression levels, together with upregulation of co-stimulatory molecules belonging to the B7 family (CD80 and CD86), as well as CD40 and CD83, among others (Banchereau & Steinman, 1998; Bueno, Tobar, Iruretagoyena, & Kalergis, 2005). Furthermore, changes in the type and amount of surface molecules is important for interactions with other immune cells. mDCs upregulate molecules, such as intercellular adhesion molecule 1 (ICAM-1, CD54) and CD83 (Banchereau & Steinman, 1998; L. J. Zhou & Tedder, 1996). In addition, upon maturation DCs undergo a switch in their chemokine receptor expression profile, which amongst other changes in cell.
(26) 25. phenotype are important for cell migration to areas containing high concentrations of T lymphocyte in secondary lymphoid organs. This process is mediated via the CCR7-CCL19 chemokine-chemokine receptor axis (Banchereau et al., 2000; Ohl et al., 2004). Considering that DCs are essential inducers of antiviral immune responses, by activating T cell mediated immunity, they represent key immune evasion targets for invading pathogenic microorganisms, such as viruses that persistently infect the host, such as herpesviruses. DCs generated from PBMCs express the HSV entry receptors HVEM (Hve-A) and nectin-2 (Hve-B), and hence are potential targets for HSV infection. Indeed, HSV infects these cells and impairs the function of DCs. Noteworthy, HSV-2 can interfere with DC function, particularly by blocking their maturation, migration to lymph nodes, promote the secretion of pro-inflammatory cytokines by these cells and inhibit the function of their autophagosome (Figure 3) (Elboim, Grodzovski, Djian, Wolf, & Mandelboim, 2013; Gobeil & Leib, 2012; Puttur et al., 2010; Raftery, Winau, Kaufmann, Schaible, & Schonrich, 2006; Stefanidou et al., 2013). HSV can also block the activity of inducible nitric oxide synthase (iNOS) and NO production, by interacting with caveolin-1 (Cav-1) in DCs (Figure 3) (B. Wu et al., 2015). Furthermore, HSV-2 can limit DC presentation of viral antigens on MHC-I molecules by interfering with the transport of antigenic peptides (TAP) from the cytoplasm to the endoplasmic reticulum and decrease the expression of T cell costimulatory molecules on the DC surface, thus hampering effective T cell activation (Figure 3) (Bosnjak et al., 2005; Elboim et al., 2013; Gobeil & Leib, 2012; Hill et al., 1995). HSV uses the ɣ34.5 protein (ICP34.5) to block autophagy in infected cells. Infection of MEFs with ICP34.5-null HSV-1 induces autophagy, while infection of PKR-null MEFs with the same virus did not, these findings suggest that autophagic degradation of herpes simplex virions depend of antiviral eIF2α kinase signaling.
(27) 26. pathway (including PKR and eIF2α) (Talloczy et al., 2002). Moreover, ICP34.5-null strain are attenuated in MEFs, infection with wild-type HSV-1 yields 1,000-fold-more progeny than an ICP34.5-null strain within 72 hours of infection (Talloczy, Virgin, & Levine, 2006). Additionally, and most importantly, HSV-2 elicits DC apoptosis early after infection, further limiting the chances of the host to mount an effective antiviral T cell response (Figure 2) (Bosnjak et al., 2005; C. A. Jones et al., 2003; Stefanidou et al., 2013).. 1.5.- The adaptive immune response elicited to HSV-2 Dendritic cells are professional antigen presenting cells (APCs) that play key roles at initiating and modulating immune responses at the interface of innate and adaptive immunity (Gonzalez, Carreno, Figueroa, & Kalergis, 2007). DCs and macrophages are the major APCs in mucosal organs, such as the genital tract and mouth, where they sense and capture microbes and particles that are potentially harmful to the host (Duluc et al., 2013; Iijima, Thompson, & Iwasaki, 2008; Soloff & Barratt-Boyes, 2010). Once captured, antigens derived from these elements are processed and presented to antigen-specific T cells (Iijima, Thompson, et al., 2008). T cells interacting with DCs presenting antigens they recognize will acquire functional attributes, based on the membrane-bound and soluble molecules presented by the DCs. These attributes include cytotoxic capacities, the secretion of soluble cytokines and regulatory functions that modulate other immune cells, among others. Importantly, these functions can ultimately define the overall immune response to an antigen (Gonzalez et al., 2007). There is evidence both, in the mouse model and humans that effector CD8+ T cells are important in clearing HSV-2 infection within the genital mucosa (Koelle et al., 1998; Parr & Parr, 1998)..
(28) 27. The effector functions of CD8+ T cells primarily depend on the production of IFN-γ and supported by perforin release, as well as Fas-mediated signaling (Dobbs, Strasser, Chu, Chalk, & Milligan, 2005). In humans, the infiltration of cytotoxic CD8+ T cells within genital lesions correlates with viral clearance (Koelle et al., 1998). Interestingly, CD8+ T cells have the ability to shape the severity and frequency of HSV-2 reactivations (Schiffer et al., 2010). However, CD8+ T cells alone are not sufficient for viral clearance, as several studies suggest that CD4+ T cell help is also required for virus control (Koelle, Huang, Hensel, & McClurkan, 2006). Indeed, earlier studies have established that depletion of CD4+ T cells in mice leads to increased HSV2 viral burden and decreased cell-mediated lysis, as well as diminished IFN-γ production, as compared to CD8+ T cell depletion (Milligan & Bernstein, 1997; Milligan, Bernstein, & Bourne, 1998). On the other hand, HSV-2 has been shown to elicit antiviral IgG and IgA antibodies upon natural infection (Morrison, Zhu, & Thebeau, 2001). Although antibodies elicited after natural infection display neutralizing capacity in vitro, they only seem to confer limited viral clearance capacity or prevent infection with this virus. This notion is supported by the results of a recent clinical trial based on a subunit formulation considered as a potential vaccine (Belshe et al., 2012). Although the formulation produced neutralizing antibodies in the vaccinated, the elicited antibodies where not protective (Belshe et al., 2012; Cohen, 2010). However, a protective role for antibodies against HSV-2 has been evidenced in animal models for HSV-2 infection upon vaccination suggesting that particular antibodies may be indeed protective, while others not. Furthermore, these results suggest that HSV-2 may dampen the antiviral antibody response upon natural infection by eliciting poor titers of protective antibodies. Whether poor titers of protective antiviral antibodies are a consequence of poor B cell stimulation or activity.
(29) 28. of these cells, these questions remain to be determined. Furthermore, the viral surface glycoprotein gE has been described to interfere with antibody function by binding the Fc portion of these molecules (Lubinski et al., 2002)..
(30) 29. Figure 3. Herpes simplex viruses negatively modulate dendritic cell function. (1) HSV viral proteins bind caveolin-1 and sequester iNOS synthase, dampening NO production within the cell, which is involved in cellular antiviral responses. (2) ICP0 interferes with TLR-IRF3 signaling, thus reducing type-I interferon (IFN-I) production by DCs. (3) The HSV ICP47 viral protein interferes with peptide translocation from the cytoplasm to the endoplasmic reticulum, which is mediated by TAP (Transport Associated with Antigen Processing), thus decreasing antigen presentation to CD8+ T cells on MHC-I molecules. (4) Viral protein γ34.5 blocks autophagosome maturation, thus reducing the capacity of DCs to process viral antigens. (5) DC infection with HSV interferes with the migration of these cells to draining lymph nodes, where T cell activation takes place. (6) Glycoprotein D (gD) has been previously described to decrease T cell activation by negatively modulating TCR signaling. Such inhibition likely affects both, CD4+ and CD8+ T cell activation. (7) Ultimately, DC infection with HSV elicits considerable apoptosis. Reproduced from (Retamal-Diaz, Kalergis, Bueno, & Gonzalez, 2017)..
(31) 30. Thus, if HSV-2 is impaired from interfering with DC function and B cell antibody antiviral activity, these cells might contribute altogether to the stimulation of CD4+ and CD8+ T cells that secrete IFN-γ, which would limit HSV-2 infection (Iijima, Linehan, et al., 2008). In addition, a balance between CD4+ and CD8+ T cells subsets, within the vaginal mucosa is critical for producing an effective adaptive response against HSV-2 (Fernandez et al., 2008). Thus, assessing the role of immune mediators elicited upon infection with HSV-2 that modulate the function of these cells may contribute to the design and identification of novel strategies to counteract this virus.. 1.6.- Vaccine strategies against HSV-2 Development of a vaccine against HSV-2 would be a major step forward in public health, mainly because of its high prevalence, worldwide distribution and both, epidemiological and molecular association with HIV-1 infection (Barnabas et al., 2011; Corey et al., 2004; Freeman et al., 2006; Masson et al., 2014; Wasserheit, 1992). In spite of many formulations based on subunits or live-attenuated vaccines against HSV-2 advanced to clinical trials but these approaches have failed. Most efforts to date have concentrated on subunit vaccine mainly based on glycoprotein D from HSV-2 (gD-2), alone or in combination with other HSV envelope glycoproteins, other viral proteins, as well as different adjuvants (Mertz et al., 1990). Glycoprotein gD is highly conserved within HSV serotypes (Cheshenko et al., 2013) and is essential for the entry of the virus into target cells both, immune and non-immune (as describe above) (D. C. Johnson & Baines, 2011; Salameh, Sheth, & Shukla, 2012). Furthermore, this immunodominant viral protein harbors epitopes for CD4+ T cells (Kim et al., 2008), CD8+ T cells (Chentoufi et al.,.
(32) 31. 2008) and neutralizing antibodies (A. V. Nicola, Willis, Naidoo, Eisenberg, & Cohen, 1996). Although clinical data show that natural exposure to HSV induces antibodies directed against the viral protein gD with neutralizing activity against the virus in vitro, their effectivity in vivo seems to be overestimated as individuals having such antibodies nevertheless display symptomatic infection (Cairns et al., 2014). Concomitantly, the predominant vaccine strategy based on the viral gD protein plus adjuvants against HSV failed in a phase 3 clinical trial; the formulation did not reduce HSV-2 infection or minimize the shedding of the virus (RetamalDíaz et al., 2015; X. P. Zhu et al., 2014). However, the magnitude of these responses may have been too weak for protecting against HSV-2 upon exposure (Bernstein et al., 2005; Cairns et al., 2014; Chentoufi, Kritzer, Yu, Nesburn, & Benmohamed, 2012; Halford, 2014). Although the vaccine was not efficacious against HSV-2 infection and genital disease, it protected 35% against HSV-1 infection (with or without disease) and showed 58% cross-protection against HSV-1 genital disease (Belshe et al., 2012). However, a recent study that re-evaluated of epitope-specific antibody responses in the gD-2 Herpevac trial formulation both humans and guinea pigs, the antibody responses shown to significantly fewer epitopes in immunized humans than guinea pigs. This data suggests that neutralizing antibodies and antibodies to multiple gD2 epitopes involved in virus entry and cell-to-cell spread represent important immune correlates of protection (Hook et al., 2018). Because subunit vaccine candidates have failed so far at eliciting protective immunity against HSV-2 in clinical trials, other more traditional approaches, such as those based on attenuated mutant viruses have re-emerged as prophylactic alternatives for eliciting immunity against this virus (Angello R. Retamal-Díaz, 2016; Retamal-Diaz, Suazo, Garrido, Kalergis, & Gonzalez, 2015). The notion that an attenuated HSV may achieve protective immunity against.
(33) 32. HSV-2 is somewhat based on the fact that a weakened herpesvirus, namely the varicella zoster Oka strain is currently used as a protective and therapeutic vaccine against varicella and shingles (Quinlivan & Breuer, 2014; Quinlivan, Breuer, & Schmid, 2011). At present, several attenuated viruses have shown to be safe and confer protective immunity against HSV-2 in animal models. For instance, an HSV-2 mutant that has the nuclear localization sequence of the viral protein ICP0 (0ΔNLS) deleted has been shown to be safe in vivo and elicit antibodies against numerous virus infected cell proteins (ICP) and gB, within others (Geltz, Gershburg, & Halford, 2015; Halford, Geltz, Messer, & Hasenkrug, 2015; Halford et al., 2011). Other attenuated HSV mutants have also provided promising results in numerous animal models and should move on onto clinical trials, such as the HSV-2 dl5-29 strain, which is has UL5 and UL29 gene deleted from its genome (X. Da Costa, Kramer, Zhu, Brockman, & Knipe, 2000; Diaz & Knipe, 2016; Mundle et al., 2013). The deletion of UL39 gene and designated ICP10ΔPK, because it has the protein kinase domain of the large subunit of HSV-2 ribonucleotide reductase (ICP10) deleted (Aurelian, Kokuba, & Smith, 1999; Casanova, Cancela, Alonzo, & Benuto…, 2002; Gyotoku, Ono, & Aurelian, 2002; Wachsman, Kulka, Smith, & Aurelian, 2001), an HSV-2 virus that has mutations in gD (K. Wang et al., 2015), which limits neuron infection, and an HSV mutant that has glycoprotein E (gE) deleted (S. Awasthi et al., 2012). Additional mutant viruses tested as vaccines in animal models are HSV1 VC2, which is a glycoprotein K (UL53 gene) and envelope protein UL20 (UL20 gene) deficient virus (Stanfield, Pahar, Chouljenko, Veazey, & Kousoulas, 2017), AD 472 which has UL55-56 (𝛾134.5 gene), UL34.5 and the US10-12 region deleted (Prichard et al., 2005) and finally RAV 9395, a mutant virus with UL55 and UL56 genes deleted (Spector et al., 1998). Importantly, all.
(34) 33. these viruses elicited humoral and cellular immune responses. Noteworthy, HSV-1 VC2 was recently tested in macaques with promissory results (Stanfield et al., 2017). All these mutants have shown to be safe in animals, elicit either HSV-specific both cellular and humoral response, and confer protection against HSV-2 infection, this strategy constitute a new insight into development an effective HSV-2-vaccine.. 1.6.1.- HSV-2 deleted for Us6 (glycoprotein D) a vaccine candidate The glycoprotein D of HSV-2 (gD-2, encoding US6 gene) is essential for virus entry into target cells, deletion of this gene should likely result in an attenuated virus that is impaired at infecting cells (∆gD-2). Yet, if such virus is phenotypically complemented by growing it on Vero cell that expresses gD-1 from HSV-1 (VD60 cells) on the surface of the virion, it would be capable of infecting cells, although its replication and progeny could be hampered. A virus with such characteristics (∆gD-/+gD1, ∆gD-2) was recently created and tested in animals and shown to be safe, highly immunogenic and confer protection against later challenges with high doses of clinical isolates of HSV-1 and HSV-2 in the skin and genital tissue (C. Petro et al., 2015; C. D. Petro et al., 2016). These unexpected results may be partially explained by the fact that gD has been previously described to inhibit T cell proliferation and induce their death (La, Kim, Kwon, & Kwon, 2002; Sloan et al., 2006; Vanden Oever & Han, 2010; Yang et al., 2015). Additionally, the surface expression of gD on HEK-293 cells has been reported to decrease the cytotoxic activity of NK cells (Grauwet et al., 2014). Together, these findings suggest that gD may negatively modulate the induction of an effective antiviral response and thus, its deletion from the virion may induce more favorable immune responses than the wild-type virus. We postulated that live attenuated HSV-2 on.
(35) 34. dendritic cells would provide more HSV antigens for induction of virus-specific antibodies and cellular immunity than would subunits vaccines. Therefore, in the first chapter of this thesis we assessed the ∆gD-2-DCs interaction by modulating their activation, maturation and capacity to activate T cells.. 1.7.- The unfolded protein response The endoplasmic reticulum (ER) is the first compartment in an ordered membranous network called the secretory pathway. This pathway is responsible for the synthesis, modification and delivery of biologically active proteins to different sites within the cell or their secretion to the extracellular milieu. Similar to many other biochemical pathways, protein flux through the secretory pathway is controlled at its early steps. While the ER is the entry site for the vast majority of proteins processed in the secretory pathway, transit from the ER to the Golgi complex is considered the rate-limiting step for the secretion of many glycoproteins(RojasRivera, Rodriguez, Sepulveda, & Hetz, 2018). Importantly, early steps in the maturation of secretory proteins takes place in the ER, such as the folding of nascent polypeptide chains and post-translational modifications that are important for proper folding and function of the protein. Importantly, if the influx of nascent unfolded polypeptides into the ER exceeds the folding and/or processing capacity of this organelle, the normal physiological state of the ER will be perturbed (Ron & Walter, 2007). Additionally, changes in redox state of the cells (Eletto, Chevet, Argon, & Appenzeller-Herzog, 2014), variations in calcium levels, glucose deprivation (included starvation) (de la Cadena, Hernandez-Fonseca, Camacho-Arroyo, & Massieu, 2014), iron imbalance (Oliveira, de Sousa, & Pinto, 2011) and hypoxia (Pereira, Frudd, Awad, & Hendershot, 2014) also induce significant changes in the ER that may limit its function..
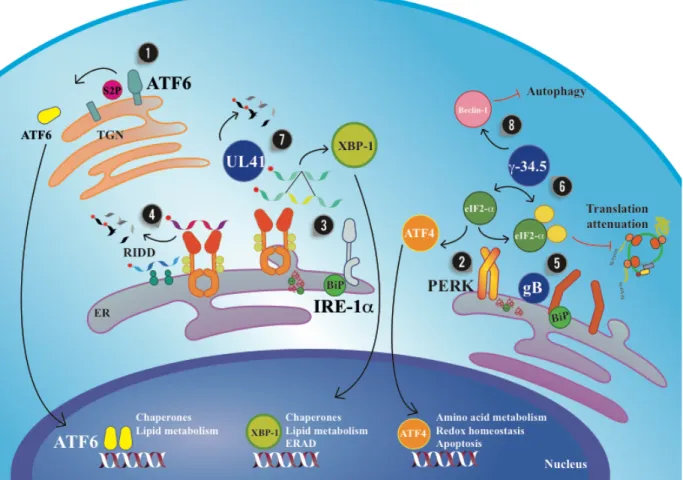
(36) 35. Excessive traffic of proteins into the ER during viral infection (i.e. HSV infection) will also likely stress this organelle and alter the normal functioning of the cells. Remarkably, cell harbor sensors and effectors for situations in which ER is stressed either to resolve the inconvenient or determine cell death if irreparable. Conditions that lead to a response to stress of the ER, can be sensed and transduced in the cells by signaling pathways and is termed the unfolded protein response (UPR). As indicated above, the UPR can trigger different signaling pathways with different outcomes: either to limit the rate of mRNA translation, altogether increasing the folding capacity of the ER by transcriptional activation of molecular chaperones to promote protein folding, as well as promoting the clearance of the misfolded or unfolded proteins through ER-associated protein degradation (ERAD) and autophagy, which has been reported to have cytoprotective effects (Schroder & Kaufman, 2005). Three main branches have been identified in unfolded protein responses. These branches operate in parallel and each possess unique mechanism for signal transduction. One branch is mediated by the activating transcription factor 6 (ATF6), another by the double-stranded RNAactivated protein kinase (PKR)-like ER kinase (PERK), and a third one by the inositol-requiring enzyme 1 (IRE1) (Figure 4). Under homeostatic conditions, the luminal domains of these ER stress sensors are retained in an inactive state through their association with binding immunoglobulin protein BiP (a chaperone of the heat shock protein HSP70 family, also known as GRP78 and HSPA5). However, owing to the higher affinity of BiP for misfolded proteins, BiP dissociates from the ER stress sensor as misfolded proteins accumulate in the ER lumen, thereby releasing the stress sensors and allowing it to activate receptors of the different branches. In turn, the dimerization of the receptors PERK, ATF6 and IRE1 lead to the production of b-.
(37) 36. ZIP transcription factors, which work alone or combined with each other to activate UPR target genes (Pincus et al., 2010; Schindler & Schekman, 2009). The first branch of the UPR activation is mediated by ATF6, which is a transcription factor that is initially synthesized as an ER-resident transmembrane protein bearing a large ERluminal domain (Haze, Yoshida, Yanagi, Yura, & Mori, 1999). Upon accumulation of unfolded proteins, ATF6 it is packaged into transport vesicles that pinch off the ER and deliver it to the Golgi apparatus (Schindler & Schekman, 2009). There, it encounters two proteases, S1P and S2P (site-1 and site-2 protease), that sequentially remove the luminal domain and the transmembrane anchor, respectively (Ye et al., 2000). The released N-terminal fragment, which is cytosolic ATF6(N), then moves into the nucleus to induce the transcription of UPR target genes. Among ATF6’s effectors are prominent ER-resident proteins involved in protein folding, such as BiP, protein disulfide isomerase, and glucose-regulated protein 94 (GRP94; a chaperone of the Hsp90 family). The processing mechanism of ATF6 resembles that of sterol response element binding protein (SREBP), a transcription factor that controls sterol and fatty acids biosynthesis and is regulated in mammalian cells by the same proteases (M. S. Brown & Goldstein, 1999; Ye et al., 2000). Whereas the mechanism by which SREBP exits the ER is well understood (see below XBP-1 and lipid metabolism), little is known about how ATF6 responds to ER stress. The ER-luminal domain shows no sequence homology with that of other proteins. What is known is that ATF6 associates with BiP, and it’s release under conditions of ER stress may contribute to ATF6 activation. The ATF6 luminal domain also contains intra- and intermolecular disulfide bonds that may monitor the ER environment as redox sensors (see Fig. 3)..
Figure



+7





Documento similar
Because immune cells in the nasal mucosa can control and regulate viral infections via killing infected res- piratory epithelial cells, low numbers of NK and or T cells in
Functional analysis through Rab27a depletion showed a significant decrease in the number of infected cells and viral production in Rab27a-silenced cells.. Conclusions: Altogether,
± SEM, n=6) of healthy cells. C) Control and SCaMC-1-KD COS-7 and 143B cells are equally sensitive to 1 μM staurosporine induced cell death. Cells were treated with the drug for 0,
Chemokines and Chemokine Receptors Critical to Host Resistance following Genital Herpes Simplex Virus Type 2 (HSV-2) Infection.. Resistance to experimental
Given that the main disadvantage of polyene macrolides is their host toxicity, because of their haemolytic action on mammalian cells, we studied the haemolytic activity of the
Supporting this idea, immunoprecipitation assays proved the interaction between proteins pVII and IVa2 in infected cells (Zhang and Arcos, 2005). 2) The second
Here we report that infection of human cells with HIV-1 conveys the proteolytic cleavage of GCN2 and that purified HIV-1 and HIV-2 proteases produce direct proteolysis of GCN2 in
cruzi infection and ET-1 cooperatively activated the Ca 2+ / calcineurin (Cn)/nuclear factor of activated T cells (NFAT) signaling pathway in atrial myocytes, leading to COX-2
