Síntesis de poli(imidas) aromáticas que contienen grupos benzimidazol, unidades flexibles y grupos voluminosos en a cadena principal
89
0
0
Texto completo
(2) AGRADECIMIENTOS A mi madre y padre, Ruth y Gustavo, por todas las enseñanzas, por el amor infinito y apoyo incondicional en todas mis decisiones.. A mi hermana y hermano, Pao y Tato, por brindarme una mano siempre que lo he necesitado y por el amor que siempre nos mantendrá unidos a pesar de todo.. A mis amigos, José, Cote, Gabi y Mayo por todos los buenos momentos y tiempo de calidad junto a ustedes. Panchito y Nico por las ganas incondicional de querer jugar a la pelota y escalar juntos.. A Deysma y Alain por todo lo enseñado y apoyo en esta etapa final universitaria.. A todo el equipo del laboratorio, por la ayuda y el grato recibimiento.. Finalmente, gracias a todas las personas que conocí en esta etapa de mi vida y que me brindaron un pedacito de ellos, siempre tendrán un espacio en mi corazón.. ii.
(3) LISTA DE ABREVIACIONES. Ac2O. Anhídrido acético. [Bmim] Br. Bromuro de 1-n-butil-3- metilimidazolio. CHCl3. Cloroformo. DMSO-d6. Dimetilsulfóxido deuterado. DMAc. N, N-dimetilacetamida. DMF. N,N-dimetilformamida. DSC. Calorimetría diferencial de barrido. DTGA. Análsis termogravimétrico diferencial. EDX. Fluorescencia de rayos X por energía dispersiva. FT-IR. Espectroscopía infraroja por transformada de Fourier. FT-IR-ATR. Espectroscopía infraroja por transformada de Fourier, Reflexión. total atenuada LI. Líquido iónico. LiCl. Cloruro de Litio. NMP. N-metil-2-pirrolidona. PI. Poliimida. PBI. poli(2,2´-m-(fenilen)-5,5´-bibenzimidazol). RMN. Resonancia Magnética Nuclear. SEM. Microscopía electrónica de barrido. TGA. Análisis termogravimétrico. THF. Tetrahidrofurano. Tf. Temperatura de fusión. Tg. Temperatura de Transición Vítrea. TMSCl. Cloruro de trimetilsilano. ƞinh. Viscosidad inherente. iii.
(4) ÍNDICE GENERAL AGRADECIMIENTOS ........................................................................................... ii LISTA DE ABREVIACIONES ............................................................................... iii ÍNDICE GENERAL............................................................................................... iv RESUMEN .......................................................................................................... viii ABSTRACT .......................................................................................................... ix 1. INTRODUCCIÓN ........................................................................................... 1 1.1.. Antecedentes generales .......................................................................... 1. 1.2.. Las pilas de combustible.......................................................................... 1. 1.3.. Los polibenzimidazoles en las pilas de combustible ................................ 6. 1.4.. Las poliimidas como alternativa en el transporte de protones ................. 7. 1.5.. Membranas poliméricas con líquidos iónicos........................................... 8. 2. HIPÓTESIS .................................................................................................. 10 3. OBJETIVOS ................................................................................................. 11 3.1.. Objetivos generales ............................................................................... 11. 3.2.. Objetivos específicos ............................................................................. 11. 4. METODOLOGÍA ........................................................................................... 12 4.1.. Materiales .............................................................................................. 12. 4.2.. Instrumentación y mediciones ............................................................... 12. 4.3.. Síntesis de precursores ......................................................................... 14. 4.3.1.. Síntesis de precursores tetracianos (4 – 6) ..................................... 14. 4.3.2.. Síntesis de precursores tetraácidos (7 – 9) ..................................... 16. 4.4.. Síntesis de monómeros ......................................................................... 18. 4.5.. Síntesis de poliimidas ............................................................................ 20. 4.6.. Preparación de películas ....................................................................... 22. 4.7.. Estabilidad oxidativa .............................................................................. 22. 4.8.. Absorción de agua ................................................................................. 23. 5. RESULTADOS Y DISCUSIÓN ..................................................................... 24 5.1.. Síntesis de precursores y monómeros .................................................. 24. 5.2.. Síntesis de poliimidas ............................................................................ 34. iv.
(5) 5.3.. Solubilidad y viscosidad inherente de los polímeros .............................. 42. 5.4.. Preparación de películas a partir de poliimidas con y sin líquido iónico 43. 5.5.. Propiedades térmicas de las películas con y sin líquido iónicos ............ 44. 5.6.. Análisis SEM-EDX ................................................................................. 47. 5.7.. Absorción de agua y medidas de ángulo de contacto ........................... 51. 5.8.. Estabilidad oxidativa .............................................................................. 53. 6. CONCLUSIONES ......................................................................................... 55 7. PERSPECTIVA ............................................................................................ 56 8. BIBLIOGRAFÍA ............................................................................................ 57 9. ANEXOS ...................................................................................................... 63. ÍNDICE DE TABLAS Tabla 1. Cantidades usadas para síntesis de la PI-F, rendimientos de cada experimento y visosidades de las poliimidas obtenidas.. 38. Tabla 2. Solubilidad y viscosidad inherente de poliimidas.. 42. Tabla 3. Calidad de las películas y proporción de [Bmim] Br en las películas.. 44. Tabla 4. Propiedades térmicas de películas con y sin líquido iónico bajo atmosfera de nitrógeno.. 45. Tabla 5. Valores de captación de agua y ángulo de contacto con el agua de las películas. 51. ÍNDICE DE ESQUEMAS Esquema 1. Síntesis de precursores 4, 5 y 6.. 14. Esquema 2. Síntesis de precursores tetraácidos.. 16. Esquema 3. Síntesis de precursores dianhídridos.. 18. v.
(6) Esquema 4. Síntesis de precursores y monómeros.. 24. Esquema 5. Reacción colateral debido a la presencia del ion nitrito.. 25. Esquema 6. Mecanismo aceptado para hidrólisis básica del grupo nitrilo.. 29. Esquema 7. Mecanismo de deshidratación química usando anhídrido acético como agente deshidratante. 32 Esquema 8. Síntesis de poli(imidas).. 34. Esquema 9. Mecanismo de formación de ácido poliámico.. 35. Esquema 10. Mecanismo propuesto para la imidación química usando anhídrido acético y piridina como agente deshidratante. 36. ÍNDICE DE FIGURAS Figura 1. Diagrama esquemático de cómo funciona una pila de combustible de membrana de intercambio de protones. 3 Figura 2. Estructura propuesta para Nafion®, polímero desarrollado por Dupont. 5 Figura 3. Estructura propuesta para poli(2,2´-m-(fenilen)-5,5´-bibenzimidazol) (PBI).. 6. Figura 4. Transporte de protones en membranas compuestas por PBI/LI.. 9. Figura 5. Espectros FTIR de los precursores 4, 5 y 6.. 26. Figura 6. Caracterización espectroscópica (RMN) del precursor 4.. 28. Figura 7. Espectros FTIR de los precursores 7,8 y 9.. 30. Figura 8. Comparación de espectros RMN 1H y RMN 13C entre tetraciano 4 y tetraácido 7. 31 Figura 9. Espectros FTIR de los monómeros 10, 11 y 12.. 33. Figura 10. Espectros IR con ATR de las poliimidas. 39. vi.
(7) Figura 11. Espectros 1H RMN (400 MHz, DMSO-d6) de poli(imidas).. 41. Figura 12. Curva TGA y DTGA de las PI sintetizadas (a) y películas PI/LI (b). 46 Figura 13. Micrografías SEM de las películas de PI y películas compustas de PI / IL25. Columnas izquierdas y centrales: micrografías de superficie. Columna derecha: micrografías vista lateral. 49 Figura 14. Mapeo superficial de las membranas preparadas.. 50. Figura 15. Los perfiles de una gota de agua sobre las películas.. 52. Figura 16. Estabilidad oxidativa de las PI prístinas y películas compuestas.. 53. vii.
(8) RESUMEN. En este proyecto se sintetizaron tres nuevas poli(imidas) aromáticas que contienen unidades de benzimidazol como parte de su estructura. Estas moléculas se caracterizaron mediante diferentes técnicas espectroscópicas. Para poder mejorar la solubilidad en disolventes orgánicos y su estabilización oxidativa, se incorporaron grupos voluminosos y flexibles en la estructura de las poli(imidas). Se observó que las nuevas moléculas sintetizadas fueron térmicamente estables (Td5% > 512 °C) y tuvieron la capacidad de formar películas densas y flexibles. Se prepararon películas compuestas dopadas con líquido iónico ([Bmim]+Br-) en diferentes concentraciones. Estos compositos fueron caracterizados de acuerdo a su morfología y composición elemental (SEM-EDX), capacidad de absorción de agua, ángulo de contacto y resistencia a la degradación oxidativa. Finalmente, con respeto a los resultados obtenidos se plantea que los compositos de poli(imidas)/líquidos. iónicos. serían. excelentes. candidatos. para. futuras. membranas intercambiadoras de protones.. viii.
(9) ABSTRACT. In this project, were synthesized three new aromatic poly(imides) containing benzimidazole groups as part of their structure. These molecules were characterized by different spectroscopic techniques. Bulky and flexible groups were incorporated on the structure of the poly(imides) to improve the solubility in organic solvents and the oxidative stabilization. It was observed that the new synthesized molecules were thermally stable (Td 5%> 512 °C) and had the ability to form dense and flexible films. Composite films were prepared adding ionic liquid ([Bmim] Br) to the poly (imides) in different concentrations. Morphology and elemental composition (SEM-EDX), water absorption capacity, contact angle and resistance to oxidative degradation were determined. According to the obtained results poly(imides)/ionic liquid composites would be excellent candidates for future proton exchange membranes.. ix.
(10) 1. INTRODUCCIÓN. 1.1.. Antecedentes generales. Es una preocupación global el problema que atraviesa la sociedad moderna en cuanto al suministro de combustibles fósiles se refiere, pues desde hace más de dos siglos la humanidad depende de los recursos petrolíferos. Esta dependencia y el amplio uso del petróleo en la generación de energía han llevado a una alarma sobre la escasez de este recurso natural, unido a la preocupación por el fuerte impacto ambiental que conlleva tanto su extracción como su procesamiento y utilización (agotamiento de la capa de ozono, calentamiento global, cambio climático, etc.). Estos problemas han llamado la atención de la comunidad científica, pues se ha hecho inminente la necesidad de encontrar nuevas vías para la generación de energía. Debido a esto se están haciendo grandes esfuerzos para desarrollar diversas tecnologías que permitan la generación de energía limpia. Además, se realizan numerosas investigaciones para hacer estas tecnologías. más. eficientes. y. cumplidoras. de. los. estándares. medioambientales actuales. Entre estos avances, toman ribetes de gran importancia, la energía eólica, la solar y las pilas de combustible. [1-3].. 1.2.. Las pilas de combustible. Las pilas o celdas de combustible son dispositivos electroquímicos que transforman la energía que se desprende durante la reacción química (oxidación de un combustible y reducción de un oxidante) en corriente 1.
(11) eléctrica y consta de tres partes fundamentales para formar lo que se conoce como una monocelda: un ánodo, un cátodo y un electrolito que se encuentra ubicado entre los dos primeros. Las pilas o celdas de combustible no almacenan la corriente eléctrica como lo hacen las baterías. Ellas producen corriente mientras ingrese un combustible (hidrógeno, metano, etanol, metanol, etc) y un comburente (generalmente, aire). El principio de funcionamiento de una pila de combustible consiste en el ingreso del combustible por el ánodo (en el que además se encuentra un catalizador) y el oxígeno por el cátodo. En esta reacción, a partir de hidrógeno se produce un protón y un electrón. El electrón pasa por un circuito externo en dirección al cátodo generando una corriente que es aprovechada antes de llegar a su destino y el protón atraviesa el electrolito llegando al cátodo, donde se combina con el oxígeno para formar agua (Figura 1). [4,5]. Este sistema se rige por las siguientes reacciones [6]:. En el ánodo (oxidación del hidrógeno) H2 2H+ + 2eEn el cátodo (reducción del oxígeno) ½ O2 + 2H+ H2O Teniendo lugar de manera global la reacción exotérmica: ½ O2 + H2 H2O. 2.
(12) Figura 1: Diagrama esquemático de cómo funciona una pila de combustible de membrana de intercambio de protones. Las pilas de combustibles presentan diversas ventajas con respecto a las metodologías tradicionales de generación de energía. Por ejemplo, el hecho de que al utilizar hidrógeno como combustible y oxígeno como oxidante, los productos de la reacción electroquímica son agua, electricidad y calor. No menos importante es la flexibilidad del combustible a utilizar (hidrógeno o alguna fuente de hidrógeno), aunque de ellos depende que la pila sea catalogada como una tecnología completa o parcialmente limpia. Adicionalmente, es importante destacar que algunos estudios han determinado que su rendimiento es entre un 40 a un 60% mayor que los motores de combustión, pues estos últimos poseen la limitante del ciclo de Carnot. No todo son ventajas en las pilas de combustible, porque su alto costo de fabricación hace difícil su entrada al mercado. Por otro lado, la durabilidad y confiabilidad de los sistemas que operan a temperatura altas no están demostradas completamente, y no menos relevante es que hasta el momento no se cuenta con la infraestructura adecuada para el óptimo desarrollo y masificación de esta tecnología, pues los sistemas de. 3.
(13) almacenamiento y distribución del hidrógeno combustible no son aún lo suficientemente eficientes y seguros. [7]. Las pilas de combustibles pueden ser clasificadas atendiendo a diversos criterios entre los que se encuentran la naturaleza del electrolito utilizado en las celdas, que incluyen: de membrana polimérica (PEMFC); alcalinas (AFC); de ácido fosfórico (PAFC); de carbonato fundido (MCFC) y de óxido sólido (SOFC). [8]. Las PEMFC son prometedores dispositivos de conversión de energía para aplicaciones estacionarias y de transporte debido a su eficiencia, alta densidad de energía, baja temperatura de operación y ser amigables con el medio ambiente. Es relevante saber que, para el eficiente y correcto funcionamiento de esta pila, la membrana polimérica (electrolito), que se considera como el componente clave, debe presentar ciertas propiedades como: (i) transferencia de protones eficiente desde el ánodo al cátodo, (ii) impermeabilidad a los gases (hidrógeno y oxígeno) para evitar su mezcla, y (iii) estabilidad química y mecánica [9,10]. Las celdas de combustible tipo PEMFC nacieron debido a las dificultades que se presentaron en el manejo del electrolito alcalino en las celdas AFC. La primera celda PEMFC fue desarrollada por General Electric, y fue fuente de electricidad y agua para la misión espacial Géminis. Esta celda usaba como electrolito una membrana de poliestireno con cadenas laterales de ácido sulfónico [11]. En el 1972 la empresa DuPont fabricó un polímero para las PEMFC conocido comercialmente como Nafion®, el cual poseía una estructura compuesta por una cadena principal de poli(tetrafluoroetileno) y una cadena colgante corta de perfluoroéter terminada en grupos ácido sulfónico (Figura 2). Tan importante fue su creación que hoy en día sigue siendo el material más usado para esta aplicación, pues promueve eficientemente el transporte de protones en la pila, pero con la limitación de requerir una alta humedad, por lo que la temperatura de trabajo debe ser. 4.
(14) inferior a los 80 °C y la alteración de estos parámetros provoca el mal funcionamiento de la membrana y su posterior destrucción. [12, 13, 14].. Figura 2. Estructura propuesta para Nafion®, polímero desarrollado por Dupont. El Nafion® ha sido el protagonista de numerosas investigaciones las cuales han arrojado que el transporte de protones a través de la membrana obedece a una combinación del mecanismo de Grotthuss y mecanismos vehiculares [15-17]. La estructura de este polímero permite diferenciar una cadena hidrofóbica que da resistencia al polímero y un fragmento hidrofílico (grupo ácido sulfónico) que permite la formación de agregados iónicos o clústers hidrofílicos dentro de la matriz polimérica. El modelo propuesto postula que las moléculas de agua participan como portadoras de carga protónica a través de un túnel o un camino establecido por las interacciones de tipo puente de hidrógeno. [14] Hoy en día, los polímeros perfluorsulfonados son la mejor alternativa para ser usados como membrana transportadora de protones en las PEMFC, pues en sentido general exhiben una alta resistencia al medio, tanto oxidante como reductor, presente en el interior de las celdas. Además, han demostrado experimentalmente ser estables y eficientes en ensayos de hasta 60.000 horas de duración [18]. Por otro lado, en estado de hidratación, demuestran una alta conductividad protónica. Estas membranas deben encontrarse saturadas con agua para que se establezcan las conexiones necesarias entre los fragmentos hidrofílicos que permitan el tránsito de las cargas iónicas, y que de esta forma se efectúe una adecuada conductividad. Sin embargo, de esto último se deriva una de las desventajas de este tipo de membranas, pues como el transporte de protones depende del nivel de. 5.
(15) saturación de agua, la temperatura de operación no puede sobrepasar los 100 °C y es importante tener en cuenta que con la temperatura se incrementa la velocidad de las reacciones electroquímicas que se producen en la pila. [19,20] Finalmente, otra de las desventajas de este tipo de membrana es su gran permeabilidad a metanol, lo cual limita su uso en pilas que funcionen en base a este combustible. [21, 22]. 1.3.. Los polibenzimidazoles en las pilas de combustible. Con base en los estudios realizados sobre los polímeros perfluorsulfonados, numerosos grupos de investigación se dieron a la tarea de diseñar y sintetizar nuevos polímeros que pudieran ser utilizados en la fabricación de membranas y que poseyeran iguales o mejores propiedades para el transporte de protones que los polímeros perfluorsulfonados, y que además vencieran las limitaciones térmicas de estos. En este contexto, se despierta un gran interés por la familia de los polibenzimidazoles, pues en 1995, Savinell et al. informaron el uso de PBI (poli(2,2´-m-(fenilen)-5,5´bibenzimidazol, integrante más conocido de la familia) dopado con ácido fosfórico como electrolito para pilas de combustible con prometedoras propiedades [23]. (Figura 3).. Figura 3. Estructura bibenzimidazol) (PBI).. propuesta. para. poli(2,2´-m-(fenilen)-5,5´-. El PBI posee una alta resistencia tanto química como térmica y tiene una temperatura de transición vítrea (Tg) cercana a los 450 °C. Además, la presencia de los grupos benzimidazol le proporciona a este material un cierto. 6.
(16) carácter básico (pKa = 5,5) lo que facilita su interacción con diferentes ácidos fuertes [24]. El anillo benzimidazólico de la estructura, otorga la capacidad de absorber una gran cantidad de ácido al ser dopados, favoreciendo la formación de clústers iónicos para el transporte de protones [25]. Se ha demostrado que, con un dopaje adecuado, los polibenzimidazoles permiten el transporte de protones de manera aceptable y trabajan en condiciones anhidras y a altas temperaturas [26-29]. Sin embargo, las membranas fabricadas a base de PBI arrojan deficiente resistencia mecánica a una alta carga de ácido y una baja estabilidad en el tiempo. Por otro lado, poseen baja procesabilidad, ya que la presencia de interacciones por puente de hidrógeno que se genera entre las cadenas son la causa fundamental de un empaquetamiento eficiente de las cadenas haciendo que su solubilidad se reduzca a un número limitado de solventes orgánicos y a bajas concentraciones [30]. 1.4.. Las poliimidas como alternativa en el transporte de protones. Hoy en día las poli(imidas) (PIs) han atraído la atención en estudios relacionados con membranas conductoras de protones. Los grupos imido permiten la generación de sistemas de transferencia de carga que propician las. interacciones. entre. las. cadenas. poliméricas. y. con. ello. el. empaquetamiento, resultando en materiales de alta resistencia térmica y alta Tg. Las poliimidas sulfonadas (SPIs) se han estudiado como uno de los candidatos más prometedores para esa aplicación debido a su excelente estabilidad térmica y mecánica, baja permeabilidad al metanol y buena. 7.
(17) capacidad de formación de película [31-36], pero su resistencia mecánica ha sido insuficiente frente a altas cargas de ácido [37]. Con el fin de mejorar la solubilidad de las PIs en disolventes orgánicos polares y de uso común se han desarrollado membranas poliméricas basadas en poli(imidas) y/o poli(benzimidazoles) de alto contenido aromático que contengan en su estructura unidades flexibles y grupos voluminosos como grupos CF3, uniones sulfonas y uniones éter [34,38-41]. La inclusión de CF3 y sulfonas en las membranas le da un mayor volumen, reduciendo el empaquetamiento que se produce por las interacciones entre las cadenas, mientras que las uniones éter le confieren flexibilidad los cual constituye un aporte para mejorar la solubilidad de estos materiales sin afectar significativamente la elevada temperatura de transición vítrea (Tg) y resistencia térmica que los caracterizan. 1.5.. Membranas poliméricas con líquidos iónicos. Los líquidos iónicos (LI) son sales orgánicas que se encuentran en estado líquido a temperaturas inferiores a los 100 °C. Están formados por cationes y aniones (no contienen ningún disolvente molecular), poseen una presión de vapor prácticamente nula y son muy estables tanto química como térmicamente. [42]. Gracias a sus propiedades y su versatilidad, en los últimos años los líquidos iónicos han sido usados y/o experimentados en casi todos los campos de la química, obteniéndose buenos resultados para un número considerable de aplicaciones entre las que se destacan la síntesis química [43,44], la separación de gases [ 45-47], y las pilas de combustible [48,49] Hay registros de estudios de LI con membranas como el Nafion ®, donde se han observado mejorías en cuanto a la estabilidad térmica, encontrándose,. 8.
(18) para membranas compuestas (Nafion/LI), una estabilidad térmica cercana a 350 °C y adecuados valores de conductividad protónica [50]. También se han realizado estudios con membranas que habitualmente son usadas en un rango de temperaturas de 100 - 200 °C, apreciándose un incremento de su estabilidad y siendo capaces de soportar temperaturas de operación de aproximadamente 300 °C [51-53]. Además de estos estudios, se han realizado investigaciones sobre membranas compuestas por LI y poli(imidas), generando un importante incremento en la resistencia mecánica del polímero a altas cargas de ácido, sin alterar considerablemente los valores de conductividad protónica, el principal atributo de estos materiales [54]. El mecanismo por el cual el líquido iónico en la matriz polimérica potencia la conductividad. protónica. no. está. totalmente. identificado.. Ciertas. investigaciones con membranas compuestas PBI/LI, proponen que es posible que el enlace N-H del catión de amonio del LI interactúe con el enlace C=N de PBI y que la conducción ocurra por la transferencia protónica del amonio a la unidad C=N y de esta a la amina, y así sucesivamente (Figura 4).. Figura 4. Transporte de protones en membranas compuestas por PBI/LI.. 9.
(19) 2. HIPÓTESIS. La incorporación de grupos benzimidazol, unidades flexibles (-O-; O=S=O) y grupos voluminosos (CF3) en la cadena principal de poli(imidas) aromáticas permite mejorar la solubilidad de esta familia de polímeros en disolventes orgánicos comunes, sin afectar considerablemente la resistencia térmica de estos materiales. Adicionalmente, la presencia del grupo benzimidazol en la cadena principal de los polímeros permite que estos materiales tengan posibles aplicaciones en la elaboración de membranas para pilas de combustible.. 10.
(20) 3. OBJETIVOS 3.1.. Objetivos generales. 1. Sintetizar y caracterizar espectroscópicamente poli(imidas) aromáticas que presenten grupos benzimidazol, unidades flexibles (-O-; O=S=O) y grupos voluminosos (CF3) en la cadena principal. 2. Determinar propiedades de solubilidad y resistencia térmica de los materiales sintetizados para proponer posibles aplicaciones como membranas para pilas de combustible.. 3.2.. Objetivos específicos. 1. Sintetizar y caracterizar espectroscópicamente los precursores y monómeros 4 – 12. 2. Sintetizar y caracterizar espectroscópicamente los polímeros PI-S, PIO y PI-F, así como ensayar su solubilidad en disolventes orgánicos comunes y determinar su viscosidad inherente. 3. Preparar películas de PIs y películas compuestas PIs/LI ([Bmim]Br). 4. Determinar las propiedades de cada película en cuanto a resistencia térmica (TGA y DSC), absorción de agua, morfología (SEM-EDX), ángulo de contacto y estabilidad oxidativa.. 11.
(21) 4. METODOLOGÍA 4.1.. Materiales. 4,4´-(Perfluoropropano-2,2-diil)difenol, 4,4´-sulfonildifenol, 4,4´-oxidifenol, 4nitroftalonitrilo y 1,2-diamino-4-nitrobenceno se obtuvieron de AK Scientific Inc. Company (San Francisco, EEUU). Cloruro de 4-nitrobenzoilo, reactivo de Eaton, N,N-dimetilformamida anhidra (DMF), N,N-dimetilacetamida anhidra (DMAc), piridina anhidra (Py), anhídrido acético (Ac2O) y bromuro de 1-butil-3-metilimidazol([BMIM]Br) se obtuvieron de Sigma-Aldrich-Merck Company. (Milwaukee,. WI,. EEUU).. 2-(4-aminofenil)-5-amino-1H-. benzimidazol (p-DABi). El resto de los reactivos y disolventes utilizados fueron comerciales, todos presentaron calidad analítica y se utilizaron sin purificación adicional. 4.2.. Instrumentación y mediciones. Los espectros FT-IR (pastillas de KBr) para precursores y monómeros se registraron en un espectrofotómetro de Perkin-Elmer (Fremont CA) 1310 en el rango de 4000-450 cm-1. Los espectros FT-IR-ATR (LiTaO3) para los polímeros se registraron en el espectrofotómetro Perkin Elmer Spectrum Two en el rango de 8300-350 cm-1. La temperatura de fusión de los precursores y monómeros fueron determinadas en un equipo SMP3 Stuart Scientific. Los espectros de RMN 1H y. 13C. se llevaron a cabo en un instrumento de 400. MHz (Bruker AC-200) utilizando DMSO-d6 como solvente y TMS como estándar interno. Las mediciones de viscosidad inherente se realizaron en un viscosímetro capilar del tipo Desreux-Bischof a 30 ± 0,1 °C (c= 0,5 g/dL). Los tiempos de escurrimiento fueron medidos por triplicado, empleando el promedio de estos para el cálculo de la viscosidad mediante la ecuación: 12.
(22) 𝜂𝑖𝑛ℎ =. 𝑙𝑛 (. 𝑡 𝑑𝑖𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 ) 𝑡 𝑑𝑖𝑠𝑜𝑙𝑣𝑒𝑛𝑡𝑒 𝐶. Con t = tiempo escurrimiento (s) y C = concentración (g/dL) Los valores de temperatura de transición vítrea (Tg) se obtuvieron con un sistema calorimétrico DSC 821 de Mettler-Toledo (Greifensee, Suiza) a partir de la segunda curva (10 °C/min bajo flujo de N2). Los análisis termogravimétricos se llevaron a cabo en un sistema calorimétrico TA-3000 de Mettler (Suiza) equipado con un procesador TC- 10A y una termobalanza TG-50 y con una microbalanza Mettler MT5 (rango de temperatura entre 25 °C y 900 °C a 10 °C/min bajo flujo de N2). Para medir el ángulo de contacto se utilizó un dispositivo Dataphysics OCA 20 con un goniómetro convencional y una cámara de video de alto rendimiento, controlado por el software SCA20. El análisis morfológico se realizó con un par de microscopios electrónicos de barrido ETEC Modelo U-1 a un dispositivo JeoL EDS (Universidad de Massachusetts, Worcester, MA). Las muestras se fijaron en un portamuestras y se cubrieron con una capa de oro durante 3 minutos utilizando un dispositivo de recubrimiento por pulverización catódica Edwards S150 (BOC Edwards, São Paulo, Brasil) antes del análisis.. 13.
(23) 4.3.. Síntesis de precursores. 4.3.1. Síntesis de precursores tetracianos (4 – 6). Esquema 1. Síntesis de precursores 4, 5 y 6.. En un balón de 250 mL equipado con un agitador magnético y conectado a un condensador de reflujo, se añaden 8,92 mmol de 4,4´-R-difenol, 17,8 mmol de 4-nitroftalonitrilo, 17,8 mmol carbonato de potasio y 60 mL de N,Ndimetilformamida. Posteriormente la mezcla se agita y se coloca en un baño de agua a 60 °C por 24 horas. Después de ese tiempo, la mezcla de reacción se vierte sobre 500 mL de agua con agitación vigorosa. El precipitado obtenido se filtra a presión reducida, se lava con metanol y se deja secar a 60 °C por 12 horas.. 4: Tf: 228–231 °C, Rendimiento: 96%. IR (KBr, , cm-1): 3086 (C-H arom.); 2237 (CN); 1589, 1489 (C=C); 1087 (C – F) 848 (arom. p-subst.). 1H RMN (DMSO-d6, δ, ppm): 8.11 (d, J = 8.8 Hz, 2H, (H8)); 7.79 (d, J = 2.1 Hz, 2H, (H5)); 7.43 (dd, J = 8.7, 2,2 Hz,2H, (H7); 7.27 (d, J = 8.9 Hz, 4H, (H11)); 7.21 (d, J = 9.0 Hz, 4H (H10)).. 13C. RMN (DMSO-d6, δ, ppm): 160.9 (C6); 153.6. (C9); 148.6 (C12); 135.7 (C8); 121.7 (C10); 121.6 (C7); 121.1 (C5); 120.0 (C11); 116.1 (C4); 115.3 (C1); 114.8 (C2), 107.5 (C3).. 14.
(24) 5: Tf: 206–209 °C, Rendimiento: 97%. IR (KBr, , cm-1): 3093 (C-H arom.); 2237 (CN.); 1589, 1489 (C=C); 1149 (S=O); 840 (arom. p-subst.); 732, 686 (arom. m-subst.). 1H RMN (DMSO-d6, δ, ppm): 8.18 (d, J = 8.7 Hz, 2H, (H8)); 8.08 (d, J = 8.5 Hz, 4H, (H11)); 8.01 (s, 2H, (H5); 7.62 (d, J = 8.6 Hz, 2H, (H7)); 7.38 (d, J = 8.4 Hz, 4H (H10)).. 13C. RMN (DMSO-d6, δ, ppm): 158.9. (C6); 158.6 (C9); 137.1 (C12); 136.4 (C8); 130.3 (C11); 124.4 (C7); 124.2 (C5); 120.1 (C10); 116.9 (C4); 115.6 (C2); 115.2 (C1), 109.9 (C3).. 6: Tf: 225-227 °C, Rendimiento: 98%. IR (KBr, , cm-1): 3086 (C-H arom.); 2237 (CN); 1589, 1489 (C=C); 1087 (C – F) 848 (arom. p-subst.). 1H RMN (DMSO-d6, δ, ppm): 8.17 (d, J = 8.7 Hz, 2H, (H8)); 7.96 (d, J = 1.9 Hz, 2H, (H5)); 7.56 (d, J = 8.7 Hz, 2H, (H7)); 7.51 (d, J = 8.4 Hz, 4H, (H11)); 7.33 (d, J = 8.7 Hz, 4H, (H10)). 13C RMN (DMSO-d6, δ, ppm): 159.4 (C6); 154.4 (C9); 135.9 (C11); 131.6 (C8); 128.3 (C12); 124.8, 121.9 (C14); 123.1 (C7); 122.0 (C5); 119.4 (C10); 116.4 (C4); 115.3 (C1); 114.8 (C2); 108.7 (C3); 63.9 (C13).. 15.
(25) 4.3.2. Síntesis de precursores tetraácidos (7 – 9). Esquema 2. Síntesis de precursores tetraácidos. En un balón de tres bocas, equipado con un agitador magnético y un condensador, se disuelve el precursor tetraciano (6,6 mmol) en 50 mL de etanol. Posteriormente se añaden 50 mL de una disolución acuosa de hidróxido de potasio (1,5 M) y la mezcla de reacción se calienta a reflujo durante 72 horas. Transcurrido ese tiempo se retira el calentamiento, y luego que la mezcla alcanza la temperatura ambiente se vierte sobre una mezcla de H2O/HCl 1:1. El precipitado que se obtiene se filtra a presión reducida y se seca en estufa a 60 °C durante 12 horas.. 7: Tf: > 300 °C, Rendimiento: 91%. IR (KBr, , cm-1): 3479 (OH); 1790, 1728 (C=O); 1604 (C=C); 879 (arom. p-subst.); 786 (arom. m-subst.).. 1H. RMN. (DMSO-d6, δ, ppm): 7.79 (d, J = 8.9 Hz, 2H, (H8)); 7.23-7.12 (m,, 12H, (H7, H5, H10, H11)); 13C RMN (DMSO-d6, δ, ppm): 169.0 (C1); 167.9 (C2), 160.2 (C6); 154.0 (C9); 150.9 (C12); 137.1 (C4); 131.9 (C8); 125.9 (C3); 122.3 (C10); 120.8 (C11); 118.7 (C7); 116.5 (C5).. 16.
(26) 8: Tf > 300 °C: Rendimiento: 92%. IR (KBr, , cm-1): 3587 (OH); 1743, 1738 (C=O); 1489 (C=C); 888 (arom. p-subst.). 1H RMN (DMSO-d6, δ, ppm): 8.0 (d, J = 8.8 Hz, 4H, (H11)); 7.81 (d, J = 8,5 Hz, 2H, (H8); 7.33 (d, J = 2.4 Hz, 2H, (H5)); 7.28 (dd, J = 8.5, 2,5 Hz, 2H, (H7)); 7.25 (d, J = 8.8 Hz, 4H, (H10)) 13C. RMN (DMSO-d6, δ, ppm): 169.0 (C1); 168.0 (C2), 160.6 (C9); 157.2 (C6);. 136.9 (C12); 136.6 (C4); 132.0 (C8); 130.57 (C11); 130.56 (C7);128.6 (C3); 121.7 (C5); 119.6 (C10).. 9: Tf: > 300 °C, Rendimiento: 92%. IR (KBr, , cm-1): 3510 (OH); 3078 (C-H arom.); 1789, 1697 (C=O); 1604 (C=C); 1072 (C-F); 833 (arom. p-subst.). 1H RMN (DMSO-d6, δ, ppm): 7.86 (d, J = 8.5 Hz, 2H, (H8)); 7.46 (d, J = 8.4 Hz, 4H, (H11)); 7.34 (d, J = 1.4 Hz, 2H, (H5)); 7.28 (s, 2H, (H7)); 7.23 (d, J = 8.7 Hz, 4H, (H10)).. 13C. RMN (DMSO-d6, δ, ppm): 168.7 (C1); 168.0 (C2) 158.2. (C6); 156.9 (C9); 137.0 (C4); 132.23 (C8); 132.18 (C11); 128.1 (C3); 125.9, 123.0 (C14); 120.7 (C7); 119.4 (C5); 118.8 (C10); 63.8 (C13).. 17.
(27) 4.4.. Síntesis de monómeros. Esquema 3. Síntesis de precursores dianhídridos. En un balón de 3 bocas, equipado con condensador y flujo de nitrógeno a contracorriente, se disuelven 10 mmol del precursor tetraácido en 50 mL de anhídrido acético. La mezcla de reacción de calienta a reflujo durante 12 horas. Transcurrido este tiempo la reacción se enfría a temperatura ambiente y el sólido que precipita se filtra a presión reducida y se seca a 200 °C durante 12 h.. 10: Tf: 239-241 °C, Rendimiento: 83%. IR (KBr, ʋ, cm-1): 3101 (C-H arom.); 1843, 1774 (C=O) 1597 (C=C); 887 (arom. p-subst.).. 18.
(28) 11: Tf: 245-247 °C, Rendimiento: 86%. IR (KBr, ʋ, cm-1): 3101 (C-H arom.); 1843, 1789 (C=O); 1582 (C=C); 1149 (S=O), 887 (arom. p-subst).. 12: Tf: 227-231 °C, Rendimiento: 85%. IR (KBr, ʋ, cm-1): 3101 (C-H arom.); 1843, 1782 (C=O); 1512 (C=C); 1072 (C – F), 887 (arom. p-sust).. 19.
(29) 4.5.. Síntesis de poliimidas. Todos los polímeros fueron preparados siguiendo el método general que se describe a continuación: En un balón de tres bocas, equipado con agitador mecánico y atmósfera de nitrógeno, se añade una mezcla de 2,0 mmol de la diamina, 2,0 mmol del dianhídrido correspondiente y 8 ml de DMAc y se agita a temperatura ambiente durante 6 horas. Posteriormente, se añade 1,0 mL de anhídrido acético y 0,8 mL de piridina y la mezcla se agita durante dos horas a temperatura ambiente y luego una hora a 60ºC. Transcurrido este tiempo se retira el calentamiento y cuando la mezcla de reacción alcanza la temperatura ambiente es vertida sobre 500 mL de agua destilada y desionizada, con agitación vigorosa. Las fibras obtenidas fueron lavadas con metanol y se secadas a 100 ºC durante 12 horas.. PI-O: FT-IR-ATR (film, ν, cm-1): 3080, 2973, 2924 (C-H); 1775, 1712 (C=O); 1617 (C=N); 1605, 1490, 1475 (C=C); 1364(C-N); 1273, 1226, 1077 (C-OC); 834, 808,778, 743 (C-H, fuera del plano). 1H RMN (400,13 MHz, DMSOd6, δ, ppm) 13.21 (banda ancha, 1H, H-5); 8.33 (d, J = 8.4 Hz, 2H, H-10); 7.99. 20.
(30) (banda ancha, 2H, H-18); 7.74 (banda ancha, 1H, H-3); 7.70 (s, 1H, H-8); 7.65 (d, J = 8.4 Hz, 2H, H-11); 7.46 (d, J = 7.8 Hz, 2H, H-17); 7.40 (s, 2H, H15); 7.26 (banda ancha, 4H, H-22); 7.22 (banda ancha, 5H, H-23, H-2).. 13C. RMN (100.62 MHz, DMSO-d6, δ, ppm) 166.81, 166.69 (C-20); 166.16, 166.06 (C-13); 163.24, 163.09 (C-16); 153.92, 153.88 (C-21); 151.94 (C-6); 150.22, 150.14 (C-24); 134.36 (C-7); 134.22 (C-12); 133.32 (C-4); 129.23 (C-1); 127.48 (C-10); 127.02 (C-11); 126.26 (C-9); 125.99, 125.80 (C-18); 125.38 (C-14); 125.22 (C-19); 122.66, 122.55 (C-17); 122.16 (C-2); 122.12, 122.09 (C-22); 120.52 (C-23); 118.96 (C-8); 114.72 (C-3); 111.29, 111.22 (C-15).. PI-S: FT-IR-ATR (film, ν, cm-1): 3080, 3066, 2973, 2953 (C-H); 1776, 1712 (C=O); 1618 (C=N); 1584, 1487, 1475 (C=C); 1364 (C-N); 1265, 1071 (C-OC); 1145 (S=O); 834, 810,785, 743 (C-H, fuera del plano). 1H RMN (400,13 MHz, DMSO-d6, δ, ppm) 13.27 (banda ancha, 1H, H-5); 8.34 (d, J = 7.9 Hz, 2H, H-10); 8.07 (d, J = 8.5 Hz, 4H, H-23); 8.04 (banda ancha, 2H, H-18); 7.75 (banda ancha, 1H, H-3); 7.71 (s, 1H, H-8); 7.66 (d, 4H, H-11, H-15); 7.59 (d, J = 8.0 Hz, 2H, H-17); 7.34 (d, J = 8.3 Hz, 4H, H-22); 7.27 (d, J = 8.2 Hz, 1H, H-2).. 13C. RMN (100.62 MHz, DMSO-d6, δ, ppm) 166.68, 166.48 (C-20);. 166.03, 165.85 (C-14); 160.40, 160.22 (C-21); 159.94, 159.84 (C-16); 151.96 (C-6); 136.60, 136.53 (C-24); 134.49 (C-7); 134.36 (C-12); 133.27 (C-4); 130.26 (C-23); 129.31 (C-1); 127.52 (C-10); 127.38 (C-14); 127.18 (C-19); 127.05 (C-11); 126.20 (C-9); 126.13, 125.94 (C-18); 125.26 (C-17); 125.15 (C-17); 122.06 (C-2); 119.55, 119.47 (C-22); 119.07 (C-8); 114.83 (C-3); 114.42, 114.36 (C-15).. PI-F: FT-IR-ATR (film, ν, cm-1): 3072, 2975, 2924 (C-H); 1775, 1712 (C=O); 1618 (C=N); 1602, 1508, 1477 (C=C); 1365 (C-N); 1258, 1076 (C-O-C); 1235, 1170 (C-F); 834, 810,780, 742 (C-H, fuera del plano). 1H RMN (400,13. 21.
(31) MHz, DMSO-d6, δ, ppm) 13.33 (banda ancha, 1H, H-5); 8.34 (d, J = 8.0 Hz, 2H, H-10); 8.03 (banda ancha, 2H, H-18); 7.75 (banda ancha, 1H, H-3), 7.71(s, 1H, H-8); 7.67 (d, J = 8.0 Hz, 2H, H-11); 7.58 (s, 2H, H-15); 7.56 (banda ancha, 2H, H-17); 7.52 (d, J = 8.3 Hz, 4H, H-23); 7.32 (d, J = 8.4 Hz, 4H, H-22); 7.27 (d, J = 8.5 Hz, 1H, H-2).. 13C. RMN (100.62 MHz, DMSO-d6,. δ, ppm) 166.75, 166.59 (C-20); 166.10, 165.96 (C-13); 161.46, 161.28 (C16); 156.13, 156.03 (C-21); 151.96 (C-6); 134.46 (C-7); 134.33 (C-12); 133.31 (C-4); 131.99 (C-23); 129.28 (C-1); 128.30, 128.22 C-24); 127.51 (C10); 127.05 (C-11); 126.57 (C-14); 126.36 (C-19); 126.24 (C-9); 126.10, 125.91 (C-18); 124.21, 124.09 (C-19); 124.00 (q, J = 287.5 Hz, C-26); 122.07 (C-2); 119.57, 119.50 (C-22); 118.97 (C-8); 114.87 (C-3); 113.23, 113.15 (C15); 63.41 (hept, J = 25.0 Hz, C-25). 4.6.. Preparación de películas. Las películas fueron preparadas por método de casting a partir de una solución de 500 mg de polímero en 10 mL de DMAc. La solución fue filtrada sobre una placa de teflón de 5 cm de diámetro que fue posteriormente colocada sobre una placa de calentamiento a 50 °C durante 12 horas. Posteriormente, la película fue retirada y colocada en una malla de acero inoxidable para ser tratada a 200 °C por 24 horas. Las membranas compuestas con LI fueron preparadas siguiendo la misma metodología anterior y añadiendo a la disolución un 25 % en peso de líquido iónico. 4.7.. Estabilidad oxidativa. Se sumergieron 100 mg de cada película en una solución de Fenton, que consiste en una disolución acuosa de H2O2 al 3% m/m y 3 ppm de FeSO4 a. 22.
(32) 80 ° C. El método consiste en obtener la estabilidad oxidativa a partir del tiempo transcurrido antes que las películas se vuelvan quebradizas.[55]. 4.8.. Absorción de agua. Para cada película formada se midió la propiedad de absorción de agua, para ello fue necesario pesar las películas previamente secas, luego se sumergieron en agua desionizada por 24 horas a 30 ºC y a 80 ºC. Las películas se retiraron, se limpiaron suavemente con papel y se pesaron nuevamente. Este procedimiento se realizó cinco veces para asegurar una medición promedio. [56]. 23.
(33) 5. RESULTADOS Y DISCUSIÓN 5.1.. Síntesis de precursores y monómeros. La ruta sintética de los precursores y monómeros se expresa en el siguiente esquema:. Esquema 4. Síntesis de precursores y monómeros. La obtención de los monómeros dianhídridos (10 - 12) se llevó a cabo en tres etapas [57] (esquema 4). La primera etapa consistió en la formación de los precursores tetracianos (4 - 6) mediante una reacción de sustitución nucleofílica aromática entre el difenol (1 - 3) y 4-nitroftalonitrilo en medio básico. Donde la base (carbonato de potasio) extrae el hidrógeno ácido del difenol, formando difenolato de potasio, nucleófilo capaz de atacar y sustituir el grupo nitro en una reacción de sustitución nucleofílica aromática. Para esta reacción, es importante señalar que la formación del enlace C-nucleófilo determina la velocidad de esta y no la ruptura del enlace C-NO2 [58,59]. El átomo de carbono al cual se encuentra enlazado el grupo nitro se encuentra deficiente de electrones debido al efecto atractor inductivo (-I) tan fuerte de dicho grupo, favoreciendo que el nucleófilo ataque más rápidamente.. 24.
(34) Además, el grupo nitro al ser un fuerte atractor de electrones se convierte en un buen grupo saliente siendo desplazado rápidamente. Una condición experimental importante para considerar en esta reacción es que la temperatura no debe superar los 60 °C. De lo contrario la reacción pasa de un color rojo a un color negro que indica la presencia de reacciones colaterales [60]. Estas reacciones colaterales no deseadas fueron estudiadas por Parker y colaboradores [61], los que plantearon que el ion nitrito en el medio puede atacar a 4-nitroftalonitrilo, desplazando al grupo nitro para formar un nitritoderivado, el cual reacciona nuevamente con el ion nitrito para formar un fenóxido que puede competir con el difenol y atacar al 4-nitroftalonitrilo para formar un derivado diariléter (Esquema 5). Es sabido que el grupo nitro no es desplazado fácilmente por el ion nitrito, sin embargo, altas temperaturas pueden favorecer esta reacción. [62].. Esquema 5. Reacción colateral debido a la presencia del ion nitrito. Los tres precursores tetracianos (4 – 6), al ser caracterizados por FT-IR, mostraron una señal intensa que se corresponde con el estiramiento del triple enlace del grupo ciano a 2237 cm-1, la cual se puede observar claramente en la figura 5.. 25.
(35) ν C-N N. N. N. O. O. O. N. ν C-N N. N. N. O S O. O. O. N. ν C-N N. F F C C C F F F. N. F N. 4000. O. 3500. 3000. O. 2500. N. 2000. 1500. 1000. 500. -1. N° de onda ( cm ). Figura 5. Espectros FTIR de los precursores 4, 5 y 6. En el espectro de RMN 1H del compuesto 4 (Figura 6) es posible asignar cada una de las señales de los hidrógenos correspondientes, resaltando el. 26.
(36) patrón de sustitución para de los hidrógenos H10 y H11. Así mismo es posible apreciar que el grupo de señales a 7,43 ppm con multiplicidad doble doblete del hidrógeno H7 se corresponde con un acoplamiento a corta distancia con el hidrógeno H8 y uno a larga distancia con el hidrógeno H5, con constantes de acoplamiento de 8,7 y 2,2 Hz respectivamente. En los espectros RMN. 13C. fueron asignadas todas las señales, entre las. cuales se observa que el carbono unido al grupo ciano (C3) aparece alrededor de los 107 ppm, resultado que era esperado gracias al fuerte efecto apantallante del grupo nitrilo sobre su carbono . Además, es posible observar que las señales de los carbonos C6, C9 y C12 se encuentran desplazadas a campo bajo gracias al efecto desapantallante de los átomos de oxígeno a que se encuentran enlazados cada uno de ellos. Teniendo en cuenta las similitudes estructurales de los compuestos 4, 5 y 6, las observaciones descritas son válidas para los tres compuestos, a excepción, fundamentalmente del carbono C12, cuyo corrimiento estuvo condicionado a la naturaleza del grupo central, desplazándose a campo alto, 148,65 ppm en el compuesto 4, 137,12 ppm en el compuesto 5 y 128,35 ppm en el compuesto 6.. 27.
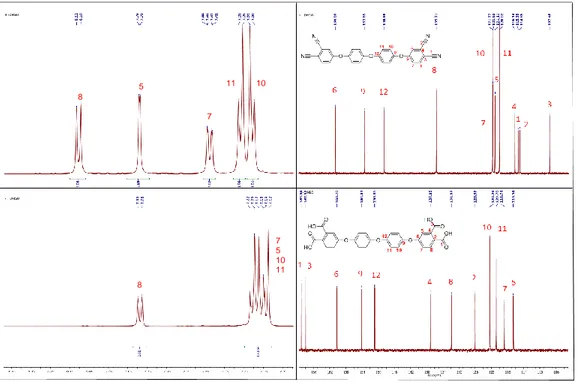
(37) 8. 11. 5. 10. 7. Figura 6. Caracterización espectroscópica (RMN) del precursor 4. La síntesis de los compuestos tetraácidos carboxílicos (7 - 9) a partir de los precursores 4, 5 y 6 se realizó mediante una hidrólisis alcalina, usando hidróxido de potasio en una mezcla de etanol-agua (1:1 vol/vol). Esta reacción fue monitoreada detectando la presencia de amoniaco gaseoso a la salida del refrigerante, usando para ello un papel indicador de acidez. Una vez concluida la emanación de amoniaco, la reacción se detuvo y fue llevada a pH 3 con la adición de una solución de HCl acuoso al 20 %. El mecanismo que explica esta reacción [63], propone una primera etapa donde el ion hidroxilo es adicionado nucleofílicamente al carbono del grupo nitrilo. Posteriormente la hidroxi-imina que se forma, isomeriza a una amida por acción del agua. Al persistir las condiciones de hidrólisis, el ion hidroxilo. 28.
(38) se adiciona al carbono carbonílico de la amida, seguido por una desprotonación del grupo OH y una eliminación del ion amiduro (Esquema 6).. Esquema 6. Mecanismo aceptado para hidrólisis básica del grupo nitrilo. La formación de amoniaco proviene de la acción del calor sobre el hidróxido de amonio, el que a su vez se forma rápidamente debido a la interacción entre el agua presente en el medio y el ion amiduro. Finalmente, la sal formada pasa a ser un ácido tetracarboxílico a través de la acidificación del medio, utilizando ácido clorhídrico diluido. En el espectro de FT-IR de estos compuestos (7 – 9) se comprueba la desaparición de la intensa señal del estiramiento C-N del grupo ciano de los respectivos precursores y la aparición de una banda ancha de gran intensidad entre 3479 cm-1 y 3587 cm-1 que se corresponde con el. 29.
(39) estiramiento O-H y que constituye la señal más característica de los ácidos carboxílicos. Además, se observa el estiramiento del grupo carbonilo entre los 1697 cm-1 y 1789 cm-1. (Figura 7).. ν O-H O. ν C=O. HO. HO O. O OH O. O. O. HO. O. ν O-H O. ν C=O. HO. HO O. O OH. O S O. O HO. O O. ν O-H O HO O O HO. 4000. 3500. 3000. HO. F F C F C F C F F. O OH O O. 2500. 2000. ν C=O 1500. 1000. 500. -1. N° de onda (cm ). Figura 7. Espectros FTIR de los precursores 7,8 y 9.. 30.
(40) La caracterización espectroscópica de 7, 8 y 9 fue realizada con éxito, siendo posible asignar las señales correspondientes a todos los carbonos y los protones de los tres compuestos. Comparando las señales de 7, 8 y 9 con la de sus respectivos precursores tetraciano, es posible apreciar en los espectros RMN 1H el significativo desplazamiento a campo bajo de los hidrógenos H5 y H8 condicionado por el reemplazo del grupo ciano por el grupo ácido carboxílico. En el caso de los espectros RMN. 13C. es posible apreciar el desplazamiento. del carbono C1 y C2 a campo bajo, alrededor de los 168 ppm, región clásica en la que aparecen los carbonos carbonílicos de los ácidos, gracias al efecto desapantallante del oxígeno. (Figura 8). 11. 5. 10. 8 7. 7 5 10 11 8. Figura 8. Comparación de espectros RMN 1H y RMN 13C entre tetraciano 4 y tetraácido 7. El último paso de la síntesis consistió en la obtención de los monómeros del tipo anhídridos de ácidos (10 – 12) a partir de una ciclación química de los. 31.
(41) tetraácidos (7 – 9) utilizando como agente deshidratante anhídrido acético. Esta reacción se encuentra favorecida debido a la posición orto en la que se encuentran los grupos ácidos de la molécula. El mecanismo propuesto para esta reacción se observa a continuación (Esquema 7).. Esquema 7. Mecanismo de deshidratación química usando anhídrido acético como agente deshidratante. El producto obtenido fue aislado por filtración a presión reducida, pues al enfriar la mezcla de reacción el compuesto precipitaba en forma de cristales puros. En los espectros FT-IR obtenidos para los dianhídridos (10 - 12) desaparece la señal correspondiente al estiramiento O-H de los ácidos carboxílicos y aparecen las señales de estiramiento simétrico y asimétrico de los carbonilos a 1843, 1782; 1843, 1774 y 1843, 1789 cm-1, respectivamente. Sin embargo, para el caso de estas moléculas no fue posible la caracterización por RMN, pues la presencia de agua en el disolvente DMSO-d6 provocó la hidrólisis de las moléculas anhídridas obteniendo así señales que no resultaban fidedignas.. 32.
(42) O. O. O. O O. O. O. O. O. O. O O. O. O. 4000. O S O. O O. O. 3500. F C F C F C F F. 3000. O. O. F. O O. O. O O. 2500. O. 2000. 1500. 1000. 500. -1. N° de onda (cm ). Figura 9. Espectros FTIR de los monómeros 10, 11 y 12.. 33.
(43) 5.2.. Síntesis de poliimidas. El método implementado para la preparación de las poliimidas aromáticas consiste en dos etapas, la primera de ellas es la formación del ácido poliámico a partir de la reacción entre la diamina y el dianhídrido de ácido a temperatura ambiente y en disolución, empleando para ello un disolvente polar aprótico (DMAc). La segunda etapa consiste en la ciclodeshidratación intramolecular del ácido poliámico para generar la poliimida final. Esta ciclodeshidratación puede realizarse química o térmicamente, siendo la primera de ella la utilizada en este trabajo. (Esquema 8).. Esquema 8. Síntesis de poli(imidas). 34.
(44) La formación del ácido poliámico, comienza con el ataque nucleofílico del par de electrones no enlazantes del nitrógeno de la amina monomérica, sobre el carbono carbonílico del dianhídrido de ácido (10 – 12), ocasionando la. apertura. del. dianhídrido. y. dando. origen. al. ácido. poliámico. correspondiente. (Esquema 9).. Esquema 9. Mecanismo de formación de ácido poliámico. Es importante tener en cuenta que el peso molecular de la poliimida final se define por el peso molecular que se logre del ácido poliámico intermedio y si bien, la síntesis del ácido poliámico es una reacción bastante simple a nivel procedimental, existen algunos factores que pueden afectar la obtención de altos pesos moleculares. Un factor importante para tener en cuenta es la pureza de los monómeros, la cual debe estar cercana al 99 %. Además, la estequiometría de los reactantes debe ser exactamente uno es a uno, el tiempo de reacción debe ser suficientemente largo para lograr una mayor conversión y no deben existir impurezas provenientes del disolvente utilizado.[64] El mecanismo propuesto para la ciclación química se muestra en el esquema 10. El primer paso consiste en la N-acetilación de la piridina, lo cual cataliza la reacción de imidación. Para esto, la piridina reacciona con el anhídrido acético formando el catión acilpiridinio y el anión acetato (reacción 1). Como el ácido poliámico (pKa = 8,8 en DMF) es un ácido más fuerte que el ácido acético (pKa = 13,5 en DMF), el ion acetato puede ser protonado rápidamente desplazando el equilibrio de la reacción 1 hacia la derecha (reacción 2). Por lo tanto, la alta concentración del ion acilpiridinio promueve la reacción con el ion conjugado del ácido poliámico generando el. 35.
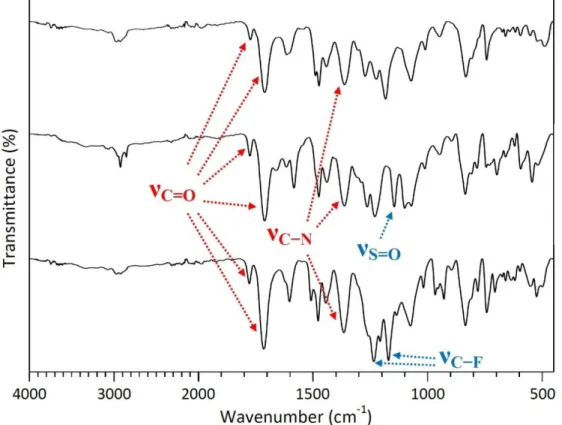
(45) intermediario anhídrido-amida y reconstruyendo la piridina (reacción 3). Como es esperado, el equilibrio de esta reacción se ve favorecido con una alta concentración de ácido poliámico.. Esquema 10. Mecanismo propuesto para la imidación química usando anhídrido acético y piridina como agente deshidratante. Posteriormente, el intermediario anhídrido-amida es desprotonado por la piridina para formar dos estructuras aniónicas resonantes diferentes (reacción 4). El ataque nucleofílico intramolecular del nitrógeno cargado negativamente produce la imida correspondiente, mientras que el ataque del oxígeno cargado negativamente produce el isómero isoimida. Sin embargo, como el nitrógeno cargado negativamente es mejor nucleófilo que el oxígeno cargado negativamente, la formación de la imida se ve favorecida.. 36.
(46) El ion acetato que se genera de la reacción 4 es protonado rápidamente por el ácido poliámico presente en el medio de la reacción, lo que desplaza el equilibrio hacia la derecha e impide la reacción inversa. Una vez que se consume el ácido poliámico, el ion acetato se genera nuevamente al reaccionar el ácido acético formado con la piridina (k-b, reacción 5). Este ion acetato puede reaccionar con la isoimida formada para generar nuevamente el intermediario anhídrido-amida (k5, reacción 4), el cual se cicla a la forma imida más estable. [65,66] En este trabajo tuvimos el inconveniente que la diamina monomérica no era soluble en DMAc, ni en los disolventes polares apróticos que se ensayaron. Por esta razón nos dimos a la tarea de realizar un estudio de la reacción en el que se modificaran algunas variables. Los ensayos fueron realizados en la síntesis de PI-F con el objetivo de aplicar al resto de los polímeros la alternativa con mejores resultados en cuanto a peso molecular se refiere. Los ensayos realizados estuvieron encaminados a mejorar la solubilidad de la diamina en el disolvente (DMAc). Para ello se realizó un primer ensayo en el cual se añadió cloruro de litio a la mezcla de reacción, con el fin de disminuir las interacciones intermoleculares entre las moléculas de la diamina. Otra alternativa ensayada fue la de adicionar clorotrimetilsilano (TMSCl) para mejorar la nucleofilia del monómero y a la vez también mejorar la solubilidad [67]. Finalmente se ensayó la reacción sin aditivos y con el monómero diamina suspendido en la mezcla de reacción. Los resultados de rendimiento y viscosidad al modificar las variables se observan en la Tabla 1.. 37.
(47) Tabla 1. Cantidades usadas para síntesis de la PI-F, rendimientos de cada experimento y visosidades de las poliimidas obtenidas. Experimentoa. TMSCl (mmol). LiCl (mmol). DMAc (mL). inh (dL/g) b. 1. -. 2. 4. 0.32. 2. -. 2. 8. 0.35. 3. 2. -. 4. 0.43. 4. 2. -. 8. 0.50. 5. -. -. 8. 0.72. a. Para cada experimento se ocuparon 2 mmol de cada uno de los monómeros b. Viscosidad inherente (0,5 g/dL of NMP, at 30 °C).. Tanto la adición de LiCl como de TMSCl propiciaron la solubilidad del monómero diamina, permitiendo que el medio de reacción fuera homogéneo. Sin embargo, en ninguno de los dos casos los resultados fueron satisfactorios (experimentos 1 y 3), a pesar de que se realizaron dos experimentos adicionales donde se incrementó el volumen de disolvente, lo cual no modificó significativamente la viscosidad del polímero resultante (experimentos 2 y 4). Finalmente, fue realizado un experimento sin aditivos y con 8 mL de disolvente. En esta oportunidad la mezcla diamina-disolvente era un medio heterogéneo y a él se añadió el monómero dianhídrido. A medida que la reacción avanzaba la mezcla se iba haciendo traslúcida y más viscosa. Después de dos horas se obtuvo una solución de ácido poliámico, completamente homogénea y muy densa. Finalmente se procedió a la ciclación química según se describió con anterioridad y los valores de viscosidad fueron de hasta 0,72 dL/g. Utilizando esta última metodología experimental fue posible obtener poliimidas con un alto peso molecular.. 38.
(48) Para lograr confirmar las estructuras químicas de las poliimidas sintetizadas se utilizó la espectroscopía FT-IR y RMN. Las bandas de absorción observadas en los espectros FT-IR, correspondientes a las vibraciones de estiramiento C=O del grupo imida, con valores alrededor de 1775 y 1712 cm 1 (simétricas. y asimétricas) en los productos finales (Figura 10), confirmaron. el paso de ácido poliámico a poliimida. Otra característica que permitió comprobar las estructuras fue la ausencia de las bandas anchas entre 3600 y 3200 cm-1 que corresponden a O-H o N-H. Además, para el compuesto PIS se observaron las bandas de las vibraciones de estiramiento S=O (aproximadamente 1145 cm-1) y para el compuesto PI-F las bandas de las vibraciones de estiramiento C-F (alrededor de 1235 y 1170 cm-1).. Figura 10. Espectros IR con ATR de las poliimidas Por otro lado, la caracterización mediante 1H RMN y. 13C. RMN también. permitió confirmar las estructuras de las poliimidas sintetizadas. En la Figura. 39.
(49) 11 se observan regiones específicas de los espectros 1H RMN con las señales de hidrógeno correspondientes para cada poliimida. Los desplazamientos químicos de los protones del resto de diamina (H2, H3, H5, H8, H10 y H11) son prácticamente iguales en todos los polímeros, ya que estos protones están ubicados lejos del grupo R, proveniente del fragmento proporcionado por el monómero de dianhídrido, que diferencia entre sí a las poli(imidas). La diferencia principal obtenida en los cambios químicos en los espectros de 1H RMN fue entre los protones H23 de cada polímero, debido a los diferentes efectos de blindaje causados por los grupos R en la posición orto con respecto a estos núcleos. Se observó un comportamiento similar en los espectros de. 13C. RMN (ver la. sección de Materiales y métodos), donde la mayoría de las diferencias en cuanto a entorno magnético químico es, entre los núcleos de carbono cercanos al grupo R (C21, C22, C23 y C24) y en el caso de PI-F, la presencia de las señales correspondientes a los núcleos de carbono C26 (q, J = 287,5 Hz) y C25 (hept, J = 25,0 Hz) respectivamente. La detección de señales duplicadas, para los átomos de carbono de los fragmentos proporcionados por los monómeros de dianhídrido, podría deberse a los dos entornos químicos posibles generados por la asimetría de la diamina.. 40.
(50) PI-O. PI-S. PI-F. Figura 11. Espectros 1H RMN (400 MHz, DMSO-d6) de poli(imidas).. 41.
(51) 5.3.. Solubilidad y viscosidad inherente de los polímeros. La solubilidad de las poliimidas fue determinada a temperatura ambiente y calentando la muestra hasta ebullición. El ensayo se realizó para diferentes disolventes orgánicos de uso común (Tabla 2). Se logró observar que todos los polímeros fueron solubles a temperatura ambiente en solventes polares apróticos como DMSO, DMF, DMAc, y NMP y además, fueron solubles en m-cresol cuando la muestra fue calentada. Es importante agregar que la PIF fue soluble además en THF. Este fenómeno puede estar asociado a la presencia de grupos CF3, cuyo volumen puede generar espacios entre las cadenas, facilitando la entrada del disolvente, la solvatación, el hinchamiento y posterior solubilización del polímero, a pesar del alto contenido aromático en su estructura. Tabla 2. Solubilidad y viscosidad inherente de poliimidas. PIs PI-O PI-S PI-F aViscosidad. inh (dL/g)a 0.68 0.73 0.72. DMSO (+) (+) (+). Solvente utilizado/Solubilidad DMF DMAc NMP m-Cresol CHCl3 (+) (+) (+) (+)* (-) (+) (+) (+) (+)* (-) (+) (+) (+) (+)* (-). inherente (c = 0.5 g/dL in NMP at 30 °C). Solubilidad: (+) Soluble a temperatura. ambiente; (+) * Soluble bajo calentamiento (temperatura de ebullición del disolvente); (-) insoluble incluso después de calentar.. La viscosidad de una disolución polimérica es una herramienta muy útil para determinar el peso molecular de este. Para los polímeros lineales, la viscosidad de una disolución se relaciona con el tamaño de las cadenas poliméricas y por lo tanto con el peso molecular. En este sentido, se han definido distintos tipos de viscosidad: viscosidad relativa (ηr), específica (ηsp), reducida (ηred), inherente (ηinh) e intrínseca ([η]).. 42. THF (-) (-) (+).
(52) La viscosidad intrínseca no depende de la concentración, debido a que se determina extrapolando los datos obtenidos de viscosidad inherente o reducida, a concentración cero (dilución infinita). Si bien este parámetro se independiza de la concentración, es función de la estructura del polímero, de la temperatura y del disolvente empleado. Los pesos moleculares. de los. polímeros. sintetizados no fueron. determinados, no obstante, los elevados valores de viscosidad inherente obtenidos nos permiten estimar que son altos. Esta estimación fue respaldada por el hecho de que las tres poliimidas sintetizadas fueron usadas para preparar películas densas de buena calidad y suficientemente resistentes a la manipulación.. 5.4.. Preparación de películas a partir de poliimidas con y sin líquido iónico. Las películas formadas por poliimidas con y sin líquido iónico fueron preparadas de acuerdo con lo descrito en el apartado 4.6. Las películas formadas sin líquido iónico demostraron ser resistentes al tacto y manipulación. Para poder realizar las películas con líquido iónico fue necesario encontrar la relación adecuada entre este y la poliimida. De esta forma se analizaron las calidades de las películas con diferentes concentraciones de LI (10, 15, 20, 25, 30, 35 y 40 % en peso). El polímero PI-F fue elegido para preparar las películas compuestas junto a [Bmim]Br. Este último se escogió debido a su bajo costo y a la similitud de la estructura catiónica (imidazolio) con la estructura diamina (benzimidazol). Después del tratamiento térmico final (200 °C durante 24 h), las películas se testearon manualmente en relación a su resistencia mecánica (Tabla 3).. 43.
(53) Tabla 3. Calidad de las películas y proporción de [Bmim]Br en las películas. Muestra. [Bmim]Br (% m/m) *. Calidad de la película. PI-O PI-S PI-F PI-F/LI10 PI-F/LI15 PI-F/LI20 PI-F/LI25 PI-F/LI30 PI-F/LI35 PI-F/LI40. 0 0 0 10 15 20 25 30 35 40. (+) (+) (+) (+) (+) (+) (+) (-) a (-) b (-) c. *500 mg de polímero en cada intento. Calidad: (+) Buena, resistente a la manipulación; (-) Frágil; a Frágil al girar la película. b Frágil al remover la película. c Frágil en la placa de teflón.. Puesto que el LI posee una naturaleza no volátil y una elevada estabilidad térmica, se ocupa como plastificantes y además como proveedores de portadores de cargas libres para la preparación de la nueva película de electrolito de polímero sólido [68]. Es importante mencionar que la carga máxima de LI que permitió obtener una película flexible y densa fue 25 % en masa (PI-F/LI25), confirmando así el efecto plastificante del líquido iónico a baja concentración. Las cargas superiores a 25% en masa produjeron películas frágiles, probablemente debido a la posible reducción de las interacciones de la cadena del polímero. 5.5.. Propiedades térmicas de las películas con y sin líquido iónicos. Un importante conjunto de propiedad que se midieron para las películas formadas, son las propiedades térmicas, la cuales se evaluaron por medio de análisis termogravimétrico (TGA y DTGA) y calorimetría diferencial de barrido (DSC). Cabe mencionar que para las películas con LI (PI/LI) solo se analizaron por TGA y DTGA. Los resultados obtenidos se pueden observar en la Tabla 4.. 44.
(54) Tabla 4. Propiedades térmicas de películas con y sin líquido iónico bajo atmósfera de nitrógeno. Muestra. Td5% (°C)a. Tg (°C)b. Rc (%)c. PI-O. 545. 225. 57. PI-S. 512. 230. 62. PI-F. 549. 260. 58. PI-F/LI25. 281. n.d. 60. PI-F/LI25. 298. n.d. 47. PI-F/LI25. 298. n.d. 59. aTemperatura. a 5% en Pérdida de peso (velocidad de calentamiento de 10 °C/min).. bTemperatura. de transición vítrea a partir del segundo ciclo de calentamiento (velocidad de. calentamiento de 10 °C/min).. c. Peso residual después de calentar a 900°C. n.d: No. determinado.. De acuerdo con los resultados obtenidos, todas las poliimidas fueron térmicamente estables y con temperaturas de descomposición (T d5%) superiores a 512 ° C. Estas propiedades se podrían atribuir al alto contenido aromático en la cadena principal de los polímeros. Se observa que la curva DTGA indica dos pérdidas significativas de peso para cada polímero. La primera ocurre entre 520-570 °C, y es asociada a la reacción de despolimerización, ruptura y volatilización simultáneas de fragmentos asociados con el grupo central en cada segmento de anhídrido y otros enlaces simples lábiles. La segunda perdida ocurre alrededor de 620-650 °C, y es asociada a la descomposición completa de las cadenas aromáticas de cada polímero.. 45.
(55) a). b). Figura 12. Curva TGA y DTGA de las PI sintetizadas (a) y películas PI/LI (b). Para todas las PIs, la temperatura de descomposición al 5 % fue cercana a 300 ° C y más baja que la de las poliimidas sin LI. Este fenómeno puede estar relacionado con la presencia del LI, el cual se descompone a 273 °C [69]. El gráfico DTGA de las PI/LI arrojó una primera perdida en peso alrededor de 250-350 °C. Una observación importante fue que el líquido iónico mostró valores de temperatura de descomposición más elevados, posiblemente por estar protegido por las cadenas poliméricas. Todas las películas de poliimidas con líquidos iónicos arrojaron excelentes propiedades térmicas en comparación con el sistema PBI/H3PO4, que se utiliza en PEMFC y se degrada a 160 °C. [38]. Como se mencionó anteriormente solo se midieron valores de Tg para las películas sin LI, donde se obtuvieron resultados que oscilaban alrededor de 225-260 °C con una tendencia a disminuir la Tg a medida que aumenta la flexibilidad del grupo central. Por esta razón el compuesto PI-F tiene el valor más alto debido a la rigidez de su centro C(CF3)2 en comparación con las otras moléculas que contienen O y SO2 como centro. Este comportamiento se ha reportado para poliimidas que contienen dianhídridos con grupos C(CF3)2 en sus estructuras [70-74].. 46.
(56) Teniendo en consideración los valores de estabilidad térmica y de Tg obtenidos para todas las poliimidas, los nuevos materiales serían adecuados para la aplicación a altas temperaturas, incluyendo la membrana de intercambio de protones. 5.6.. Análisis SEM-EDX. Las PIs demostraron tener una morfología de superficie fibrosa, mientras que PI-F /LI25 y PI/LI25 arrojaron una superficie continua con presencia de algunos aglomerados. Esta diferencia se atribuye posiblemente a la presencia de [Bmim]Br, que puede interactuar con los grupos polares de las cadenas de polímero, produciendo agregados que podrían extenderse en la superficie durante el proceso de fabricación de las membranas. Para PI-O/LI25 se observó una morfología de superficie de aglomerados libres, un indicio de una mejor compatibilidad entre PI-O y [Bmim]Br. La vista lateral de las películas mostró la estructura interna de las películas de criofracturas y los compuestos. Esto podría mostrar la presencia de una superficie fibrosa con una disposición de fibra transversal en forma de capas, a excepción de PI-O que se mantuvo inalterada, mostrando un aspecto globular irregular. En la Figura 13 se pueden observar las micrografías SEM para las diferentes películas.. 47.
(57) PI-F. PI-F/IL25. PI-S. PI-S/IL25. PI-O. 48 PI-O/IL25.
(58) PI-O. PI-O/IL25. Figura 13. Micrografías SEM de las películas de PI y películas compustas de PI / IL25. Columnas izquierdas y centrales: micrografías de superficie. Columna derecha: micrografías vista lateral. El análisis EDX y las asignaciones elementales mostraron que la distribución de [Bmim]Br fue homogénea en la película. En Figura 14 se observa la presencia de átomos de bromo en la superficie y a través de las películas en una distribución aleatoria. Los puntos brillantes en cada imagen confirman la presencia de bromo distribuido uniformemente en la superficie y a través de la película. Esto indica buena compatibilidad entre el LI y la matriz del polímero.. 49.
(59) Vista superficial PI-F/IL25. PI-S/IL25. PI-O/IL25. Vista lateral PI-F/IL25. PI-S/IL25. PI-O/IL25. Figura 14. Mapeo superficial de las membranas preparadas.. 50.
(60) 5.7.. Absorción de agua y medidas de ángulo de contacto. Los valores para la absorción de agua de PI y PI/LI25 se obtuvieron a partir de la diferencia de masa y se expresaron como un porcentaje en la Tabla 5. Tabla 5. Valores de captación de agua y ángulo de contacto con el agua de las películas. Película*. PI-O PI-O/LI25 PI-S PI-S/LI25 PI-F PI-F/LI25. Captación de agua a 30 °C (% en peso) 5.3 9.0 8.0 12.0 2.4 4.4. Captación de agua a 80 °C (% en peso) 6.4 10.1 9.1 13.2 3.2 5.3. Ángulo de contacto con el agua (°) 88.2±1.3 90.7±0.8 84.4±1.1 84.8±0.8 87.6±0.5 96.0±1.8. *El espesor fue de alrededor de 64-68 μm.. En los resultados se observó que la cantidad de átomos de oxígeno en la unidad repitente desempeña un importante factor en la absorción de agua ya que las películas que contienen oxígeno en su estructura (PI-O y PI-S) pueden absorber dos a tres veces mayor cantidad de agua que las películas con grupos C(CF3) (PI-F). De esta manera se propone que el mecanismo de absorción de agua podría estar relacionado a la presencia de enlaces de hidrógeno entre los átomos de oxígeno de la PI y el agua del medio. A su vez la presencia de LI en la matriz polimérica aumenta aún más la capacidad para absorber agua en todas las muestras. La muestra PI-S/LI25 mostró tener la mayor absorción de agua (12% en peso). Además, se observó que la absorción de agua fue mayor en condiciones de 80 ° C como condición de trabajo. Sin embargo, este aumento observado no fue suficiente para indicar que la cantidad de agua absorbida por las películas fue alta. Este fenómeno. 51.
Figure



+7




Outline
Documento similar
que hasta que llegue el tiempo en que su regia planta ; | pise el hispano suelo... que hasta que el
dente: algunas decían que doña Leonor, "con muy grand rescelo e miedo que avía del rey don Pedro que nueva- mente regnaba, e de la reyna doña María, su madre del dicho rey,
Luis Miguel Utrera Navarrete ha presentado la relación de Bienes y Actividades siguientes para la legislatura de 2015-2019, según constan inscritos en el
Tras establecer un programa de trabajo (en el que se fijaban pre- visiones para las reuniones que se pretendían celebrar los posteriores 10 de julio —actual papel de los
En cuarto lugar, se establecen unos medios para la actuación de re- fuerzo de la Cohesión (conducción y coordinación de las políticas eco- nómicas nacionales, políticas y acciones
La campaña ha consistido en la revisión del etiquetado e instrucciones de uso de todos los ter- mómetros digitales comunicados, así como de la documentación técnica adicional de
b) El Tribunal Constitucional se encuadra dentro de una organiza- ción jurídico constitucional que asume la supremacía de los dere- chos fundamentales y que reconoce la separación
La recuperación histórica de la terciaria dominica sor María de Santo Domingo en los últimos años viene dada, principalmente, por causa de su posible influjo sobre personajes
