Suspensión coloidal de polímeros estrella : propiedades estructurales y dinámicas con simulaciones computacionales
80
0
0
Texto completo
(2) Universidad de Sonora Repositorio Institucional UNISON. Excepto si se señala otra cosa, la licencia del ítem se describe como openAccess.
(3) 11. Agradecimientos. A mis padres: María Enriqueta y Francisco y mis hermanos: Lucía Guadalupe, Francisco Javier, Saúl Omar; Ustedes que me han permitido entrar a su vida como lo han hecho en la mia, apoyándome en el camino que he decidido tomar, por estar conmigo incluso cuando yo no quería estar con nadie; gracias a ustedes soy la persona que soy ahora.. No cabe dudo-que cuando uno se para en los hombros de gigantes alcanza metas que jamás pensó en acercárseles, por eso (y más) quiero agradecer a mi directora de tesis a la profesora Laura Yeomans. Desde que nos conocimos me ha apoyado con su consejo, su experiencia y ha escuchado mis inquietudes, dudas y loqueras,_,aconsejándome a ser un mejor físico y una mejor persona; de aquí para adelante.. A mis sinodales el profesor Amir y el profesor Ramón, por haberme ayudado en esta última etapa de este trabajo y brindarme sus consejos que fueron de gran ayuda.. Éste a sido un camino duro, pero no lo hubiese logrado sin el apoyo de mis maestros de la Licenciatura: Angelina, Rosas, Corella, Rogelio, Horacio, entre muchos más; que gracias a ellos pude lograrlo. En particular quiero agradecer a dos maestros, al profesor Carlos Calcáneo y la profesora Gabriela Robles quienes me apoyaron desde el inicio y cuando yo estaba en el dilema de seguir o no este carrera me conoencieron de quedarme, siempre les agradeceré por eso y por estar aún al pendiente de mí. También al Departamento de Física de la Universidad de Sonora.. A el profesor Acuña, por apoyarme en los últimos años y estar al pendiente de mí y mis estudios, por abrir mi mente con sus preguntas que no tiene una respuesta concreta; yo sé que al final de todo le caigo bien.. Y por último a tí que te tomas el tiempo en leer este trabajo, espero te sirva..
(4) ,. Indice 1. Sistema. coloidal de partículas. suaves. 1. 1.1 Modelos de suspensiones coloidales. 2. 1.1.1. Esferas duras. ... 3. 1.1.2. Esferas cargadas. 4. 1.2 Polímeros estrella . . .. 5. 2 Elementos. 12. de Simulación. 2.1 Configuraciones iniciales. 12. 2.2 Celda principal de simulación y condiciones periódicas .. 13. Convención de imagen mínima y radio de corte. 2.2.1 2.3. Termalización. .. 2.4. Método de simulación de Monte Carlo. 17. 19. 2.5 Método de simulación de dinámina Browniana 2.6. Parámetros importantes. 22 23. 2.6.1. Parámetros del sistema. 23. 2.6.2. Parámetros generales de la simulación .. 24. 2.6.3. Parámetros particulares de la simulación. 25. 3 Sistema. monodisperso. de polímeros. estrella. 3.1 Funciones de distribución ...... 26 26. 3.2. Desplazamiento cuadrático medio. 32. 3.3. Resultados selectos de sistemas fluidos. 38. 3.3.1 4. 16. Sistema. Criterio de Lówen. polidisperso. de polímeros. .. 42. estrella. 46. 4.1. Concepto de polidispersidad. .. 4.2. Efecto de la polidispersidad en las fases fluidas . iii. 46. 51.
(5) ÍNDICE. iv. 4.3 Diagrama de fase de polímeros estrella con polidispersidad . . . . . . . . . . . . . . 55 Bibiografía. 60. Apéndice A Barras de Error. 63. Apéndice B Programas de simulación. 64.
(6) Resumen En este trabajo de tesis se presentan los resultados obtenido del estudio de una suspensión de polímeros estrella que constituye un modelo de partículas ultrasuaves.. Este estudio se realiza. haciendo uso de simulaciones por computadora con las técnicas de Monte Carlo y dinámica Browniana. Se calculan las propiedades estructurales y dinámicas para suspensiones coloidales monodispersas y polidispersas. En el capítulo 1 se expone una breve descripción de modelos de interacción típicos para suspensiones coloidales de partículas duras y suaves. Se presenta el modelo de potencial de los polímeros estrella basado en el modelo de Daoud-Cotton y Witten-Pincus; que está definido en dos regiones: una corresponde a crecimiento logarítmico para distancias menores al diametro de corona a y un decaimiento exponencial tipo Yukawa para distancias mayores que a. En este capítulo también se presenta los resultados obtenido de la literatura para el diagrama de fases correspondiente a esta suspensión. Los métodos de simulación con los que se abordo la exploración en la suspensión coloidal se describen en el capículo 2, lo cual incluye la presentación de los algoritmos de Ermak para la dinámica Browniana y metropolis para Monte Carlo; así como también los elementos necesarios para la implementación de los códigos en lenguaje Fortran necesarios con los cuales se presentaron las propiedades estrucuturales y dinámicas en este trabajo ele tesis. En el capítulo 3 se expone sobre la implementación del cálculo de la función de distribución radial, el desplazamiento cuadrático medio y el coeficiente de difusión dependiente del tiempo para una suspensión coloidal monodispersa.. Se muestran los resultados obtenido con las simulaciones. para diferentes condiciones de funcionalidad y fracción en volumen. Específicamente se explora. las regiones fluidas de baja y alta concentración y se presenta el diagrama de fase sólido-líquido obtenido mediante la aplicación del criterio fenomenológico de Lówen. Las suspensiones coloidales reales son sistemas de naturaleza polidispersa, es por ello que es de nuestro interes la exploración del impacto que pudiese tener la polidispersidad en el tamaño V.
(7) ÍNDICE. vi. en los polímeros estrella. El capítulo 4 se dedica para plantear los resultados obtenidos con las simulaciones para sistemas con polidispersidad (en tamaño) menores o iguales al 15%, tomando como modelo de polidispersidad una función de distribución uniforme para los diámetros de las partículas. En la parte final de este trabajo de tesis se presenta las conclusiones y se incluyen como anexos los códigosde los programas desarrollados para las simulacionesde Monte Carlo y dinámica Browniana para sistema de polidispersidad p arbitraria.
(8) Capítulo Sistema. 1 coloidal de partículas. Una suspensión coloidal está formada por partículas con un diámetro CT :::::;. suaves CT. aproximado entre 1 Onm :::::;. IOµm las cuales se encuentran en un solvente continuo, es decir las partículas que conforman. al solvente son mucho más pequeñas, del orden de diámetros de Angstrom. Experimentalmente lo anterior-implica para la exploración de las estructuras de los sistemas atómicos se requiera técnicas de difracción de Rayos-X, mientras que para los sistemas coloidales la técnica de dispersión estática y dinámica de luz (láser visible). El tiempo característico de las partículas coloidales Te :::::;. 10 µs mientras que las partículas del solvente. Te. T8. está comprendido en el intervalo 1 ns : : :;. -. es del orden de 0.1 ps esto es debido a. que la masa de las partículas coloidales me es mucho mayor que la masa de las partículas del solvente tn;. Lo anterior es la razón fundamental por la cual la trayectoría de las partículas. coloidales es errático debido a las multiples interacciones de las partículas del solvente sobre las partículas coloidales. Debido a la diferencia en los tiempos característicos de los sistemas atómicos y coloidales los fenómenos dinámicos de los sistemas coloidales son mucho más lentos que los atómicos, por ello es más accesible el estudio dinámico de las partículas coloidales desde el punto de vista experimental; por ejemplo la cinética del crecimiento cristales se puede observar haciendo uso de microscopia en tiempo real. Dentro de las técnicas experimentales utilizadas para el estudio de las propiedades dinámicas de una suspensión coloidal se encuentra la dispersión dinámica de luz y la videomicroscopía, entre otras. En 1827 Robert Brown al examinar granos de polen suspendidos en agua através del microscopio, notó el movimiento caótico de ellas, pensando que se trataba de organismos vivos. Es por eso que a este movimiento de se llama Movimiento Browniano. Un aspecto que caracteriza a las suspensiones coloidales es la polidispersidad, es decir, no todas 1.
(9) las partículas. coloidales tiene el mismo tamaño ( o propiedades. etc). Existen técnicas para sintetisar punto la polidispersidad esto puede introducir. como la carga, la funcionalidad,. partículas coloidales y que permiten controlar. hasta cierto. en tamaño del sistema, sin embargo no se puede eliminar por completo,. dificultades al momento de establecer comparaciones. entre modelos teóricos. sencillos basados en la suposición de que todas las partículas son del mismo tamaño (monodispersa) y resultados experimentales.. Para poder observar el efecto de la polidispersidad. de la suspensión una opción es obtener de un sistema experimental los tamaños de las partticulas, otra opción es incorporar de las partículas.. en las propiedades. una función de distribución. algún modelo de distribución. en. de tamaño. En este trabajo de tesis, se explora este aspecto incluyendo la polidispersidad. mediante un modelo de función de distribusión. uniforme[l. J.. La estructura y las propiedades dinámicas de una suspensión coloidal dependen fuertemente de la fracción en volumen (o concentración reducida), que es un parámetro que brinda información de la razón entre el volumen que ocupan las partículas coloidales y el volumen total de la suspensión. La fracción en volumen la fija quien prepara la suspensión, experimentalmente la fracción en volumen resultante en una suspensión dependerá de la cantidad de partículas colidales y del volumen del solvente; mientras que en las simulaciones se coloca un número de partículas coloidales en una celda de forma tal que la fracción en volumen sea la deseada. En este trabajo de tesis nos interesa explorar el comportamiento de una suspensión coloidal de polímeros estrella. Es decir, nos interesa estudiar un material blando conformado por partículas blandas.. 1.1. Modelos. de suspensiones. coloidales. En los laboratorio de investigación se han usando o sintetizado diferentes partículas coloidales con ras cuales. se preparan suspensiones coloidales con diferentes características. Para cada uno de estos. sistemas colidales se estudian mediantes diferentes técnicas experimentales o esquemas teóricos, se proponen modelos de interacciónentre las partículas, es decir, se construyen propuestas para el potencial de interación par entre ellas. Dependiendo de las características físicas y fisicoquímicas de las partículas coloidales y del solvente se puede proponer diferentes tipos de modelos de potenciales de interacción par entre las partículas y existen diferentes tipos de clasificaciones de ellos, dentro de estas se puede mencionar: modelo de potencial atractivo, modelo de potenciales repulsivos, modelo de potencial duros, modelo de potenciales suaves, modelos de potenciales ultrasuaves. En esta sección se ilustraran tres tipos de modelos de potenciales repulsivos de tipo duro, suave y 2.
(10) ultrasuave.. 1.1.1. Esferas duras. El modelo de potencial de esferas duras de una suspensión coloidal monodispersa se caracteriza por un solo parámetro, este parámetro es el diámetro de las partículas coloidales. Este modelo de potencial es de tipo discontinuo:. u(r) =. oo sir< { O. -. sir>. a. (1.1). a. u(r). /. r. a. \. -. e.e. Figura 1.1: Potencial de esferas duras. Con este potencial se modelan partículas duras; uno de los modelos experimentales que pueden ser descritos con un modelo de esferas duras es el de partículas de polimetilmetacrilato, estabilizados estéricamente (partículas de PMMA). En 1986 Pusey y Van Megen, haciendo uso de suspensiones coloidales de PMMA mostraron su comportamiento como esferas duras. Su trabajo llevo a una gran cantidad de estudios experimentales de la física estadística usando coloides como modelos y a partir de este importante trabajo se ha determinado la ecuación de estado de coloides de esferas duras, se ha observado la cinética de cristales y la transición vitria [2]. En la figura 1.2 se ilustra una serie de muestras experimentales de suspensiones coloidales de esferas duras obtenidas por P. N. Pusey y Van Megen. En éstas se observa el efecto de la concentración en la fase de la suspensión. La muestra más diluida se ilustra en el extremo derecho y que corresponde a una fase fluida, es decir no presenta efectos de cristalización, mientras que las muestras intermedias más concentradas, presentan una cristalización de la suspensión coloidal 3.
(11) Figura 1.2: Muestras experimentales de las fases de una suspensión de esferas duras.. en diferentes grados. A concentraciones mayores se observa que en todo el volumen se encuentra cristales homogéneo ( en forma de pequeños cristales regulares) o heterogéneo ( en forma de grandes cristales irregulares).. En la muestra más concentrada. ( extremo izquierdo) la suspensión coloidal. de esferas duras se manifiesta en un estado amorfo y permanece así por varios meses.. Uquid. /. Uquid·solid. FCC crystal. coexístence. A94. "Glass". .545. ~. ,56 Ranálm. Volume Iracnon. esoee packing. cp-. Figura 1.3: Diagrama de fase de una suspensión coloidal obtenido por simulasión de esferas duras.. En la figura 1.3 se ilustra el esquema del diagrama ele fase obtenido con sumulaciones por computadoras para una suspensión ele esferas duras en la cual se indican los valores ele fracción en volumen que corresponden a las fases fluidas, coexistencia líquido-sólido, cristalina (fcc), vítrea, así como también los valores ele la máxima fracción de empaquetamiento máxima ele empaquetamiento. 1.1.2. aleatorio (RCP) y la. cristalino (CCP).. Esferas cargadas. Un modelo muy utilizado para describir el potencial ele interacción par en una suspensión coloidal de partículas cargadas es la teoría DLVO (propuesta por Derjaguin, Landau, Verwey y Over4.
(12) beek). Este potencial consiste en dos tipos de interacciones relevantes: La fuerza electrostática de naturaleza Coulombiano y la fuerza de Van der Waals la cual es inversamente proporcional a r6.. Es posible modelar adecuadamente a estas suspensiones incluyendo sólo la parte de repulsión. electrostática:. ¡3u(r) = A e-zr r. (1.2). Figura 1.4: Potencial de Yukawa.. Este potencial Coulombicoapantallado por el efecto del solvente que posee iones de carga opuesta sobre la superficie del coloide; es de tipo repulsivo, continuo y depende de sólo dos parámetros: A denominada contante de interacción y z identificada como parámetro de apantallamiento. Este. potencial es conocido como potencial de Yukawa[13]. En la figura 1.5 se muestra un diagrama de fases de partículas coloidales cargadas obtenido mediante simulación de Monte Carlo, dicho sistema es descrito usando el modelo primitivo que consiste de coloides y contraiones en un solvente continuo de constante dieléctrica uniforme.. 1.2. Polímeros. estrella. En esta sección se presenta una breve revisión sobre las suspensiones de polímeros estrella, que es el sistema modelo sobre el cual se trabajó con simulaciones de computadora en esta tesis. Las características de los modelos de potenciales ilustrados anteriormente corresponden a los casos de potenciales duros (modelo de esferas duras) y a los casos de potenciales suaves (modelo de 5.
(13) colloid charge Q=20e. /:.. 1.4 so lid. fluid. colloid packing fraction. Figura 1.5: Diagrama de fase de una suspensión coloidal de partículas cargadas obtenido por simulación.. esferas cargadas); como veremos en esta sección, el potencial de interacción de polímeros estrella corresponde al tipo de modelos de potencial ultrasuave. Los polímeros estrella son macromuléculas compuestas por f cadenas poliméricas ancladas químicamente a un centro común. Al número de brazos f conectados se le llama funcionalidad de la estrella. Cuando las cadenas poliméricas ancladas son del mismo tamaño se habla de estrellas regulares, la longitud de las cadenas se polimerizan de forma que a nivel microscópico su longitud sea tal que la dimensión del núcleo central sea irrelevante en comparación con la dimensión característica de la estrella. En la figura 1.6 se ilustran configuraciones de polímeros estrella para dos funcionalidades diferentes. La síntesis de los polímeros estrella da inicio en la década de 1990 y como lo señala C. Likos las razones del interés de la comunidad científica por estos sistemas se pueden agrupar en diferentes ambitos. Primeramente. desde el punto de vista tecnológico los polímeros estrella son importantes en aplicaciones industriales como por ejemplo su uso como modificadores de viscosidad en la industria petrolera; también son usados como materiales de recubrimiento y en aplicaciones farmacéuticas y médicas. Desde el punto de vista experimental la sintesis de polímeros estrella regulares hizo posible explorar la física de sistemas monodispersos tanto por el número de brazos como por su longitud; se señala que los ejemplos importantes de polímeros estrella sintetizados corresponden a funcionalidades de valores entre 8 ~. f~. 128. Las técnicas experimentales utilizadas. para estudiar propiedades estáticas de estos sistemas son la dispersión de neutrones de ángulos pequeños (SANS) y dispersión de Rayos-X de ángulos pequeños (SAXS), por otra parte la técnica 6.
(14) (a). (b). Figura 1.6: Polímeros estrella obtenidos por simulación a) 10 de funcionalidad b) 50 de funcionalidad.. de dispersión dinámica de luz (DLS) para estudiar sus propiedades dinámicas y propiedades colectivas. Finalmente se indica que los polímeros estrella constituyen un sistema importante dentro de la materia condensada blanda ya que relaciona dos grandes campos de ella, por una parte la física.de polímeros y por otra parte la física coloidal. travendo como consecuencia el interés desde el punto de vista teórico. En la figura l. 7 se ilustra las diferentes escalas de longitud en las cuales se puede considerar a los sistemas de polímeros estrella que va desde lo macroscópico ( 1 cm) hasta lo microscópico (1 A.). Los polímeros estrella para f pequeñas (J = 1 y 2) se asemejan a una estructura lineal, mientras que al incrementar el valor de f va adquiriendo una estructura cada vez más rígida y esférica, de forma tal que para el caso f > > 1 el polímero adquiere el comportamiento de una esfera dura, en cuyo caso el modelo adecuado pudiese ser el modelo de potencial de esferas duras ya ilustrado. El modelo de potencial de interacción propuesto para los polímeros estrella es una combinación 7.
(15) IÁ. i---t. ,___. lnm. mit-r<~.!opic bond. 1eagtb. persistence leneJb of i\. ~ii1gfo diáb. t·~xüün;iün uf -;1. ~iliglt'.. star. (e). (.J. ·a o (.J "'or.r;. a,·eTi:it,~e-intt'tsüu-. E. distauce. (U. (d). Figura l. 7: Ilustración de las escalas de longitud importante en los polímeros estrella.. entre un potencial tipo Yukawa para r r. <. CJ.. >. CJ. y un potencial con comportamiento logarítmico para. Estos dos modelos de potencial son unidos adecuadamente para que el potencial y su. primer derivada sea continua en r =. CJ.. La longitud característica. CJ. es identificada como diámetro. de corona dentro del modelo de Daoucl-Cotton[3] quien visualiza la parte interior de los polímeros estrella como conformada por cascarones esféricos de "grumos" de tamaño creciente de forma tal que. CJ. se identifica con la distancia del centro de la esfera hasta la distancia media de la última. capa de grumos, como se ilustra en la figura 1.8.. La dependencia logarítmica del modelo del potencial fue propuesta por Witten y Pincus en 1986. El decaimiento exponencial tipo Yukawa, por otro lado, es incluido de forma empírica y ajustado para que coincida con los resultados experimentales obtenidos con dispersión SANS; de forma tal que la expresión matemática del potencial de interacción efectivo es de la forma: 8.
(16) Figura 1.8: Modelo de Daoud-Cotton. de un polímero estrella.. 1. + -·J]. -ln(r/a). 1+-. 2. (1.3) o lr. v1 r J]exp[-2(;;: - 1)] sir> a. 1+-. 2 donde kB es la constante de Boltzman y T es la temperatura absoluta. En la figura 1.9 se ilustra el comportamiento de este modelo de potencial para 18 ::; f ::; 256. Se observa lo señalado anteriormente, en el sentido de que conforme se aumenta la funcionalidad el comportamiento del potencial se a próxima al de esfera dura. En este sentido la funcionalidad f es interpretable como un parámetro de dureza (o suavidad) de las polímeros estrella. 140. 120 100. '--. f. \\ \\. 80. :;:;:. =. ; 1. 60. '1. 40 20. o o.o. 0.5. 1.0. 1.5. 2.0. 2.5. 3.0. r/cr. Figura 1.9: Potencial par de polímeros estrella, línea del extremo izquierdo corresponde a f = 18 y extremo derecho f = 256.. En 1999 Watzlawek, Likos y Lówen presentaron el diagrama de fases de un sistema monodis9.
(17) perso de polímeros estrella, obtenido mediante el método de cálculo de energías libres e integración termodinámica. apartir de Monte Carlo. En la figura 1.10 se incluye el diagrama de fases obenido. por ellos, en el que se observa una fase fluida y 4 diferentes estructuras cristalinas. Para fracciones en volumen menores a O. 7 y funcionalidades mayores a 34 se predice estructuras. cristalinas fcc. (cúbica centrada en la cara) y bcc (cúbica centrada en la base); para fracciones en volumen mayores y. f > 44 se predicen estructuras cristalinas más complejas como bco ( ortorombicos centrado. en las caras) y diamante[3].. f. -1. f. 0.03. 34. 0.02. 40 48. 64 96. 0.01 0.00. O.O. 0.2. 0.4. 0.6. 0.8. 1.0. 1.4. 1.2. TJ Figura 1.10: Diagrama de fase de polímeros estrella obtenido por simulación de computadora. En esta figura f representa la funcionalidad y r¡ es la fracción en volumen.. ----,~ºº QM. l.,. izo!. o. 1. ;.. 1. 100(. !º. 80~. -. 1. •. f. - - cquíl~bri;1~1 phasc_di~_gram [ MCI - lü grass liuc. \. - e. o. 1. o 9. ,. o o o o\.... Eo. : 60E-º. 00000. \. o. o. 0000. -. fluid. ..........i... _. 0.1. fcc. bit..- • -~-. 0011,••ll~-. -. \. .1. 0.2. ,bcc. •. _/.,,. -----. <; ..•....... _. ..........i. :. º·~. 1- -. : ... 0.4. =:.:::i-:.::. 0.5. c... --~..J .1 .. 0.6. Figura 1.11: Diagrama de fase de polímeros estrella obtenido experimentalmente.. En el 2007 M. Laurati y colaboradores; obtuvieron resultados experimentales con la técnica 10.
(18) de dispersión SANS en un sistema micellar PEP-PEO de polímeros estrella, en la región de funcionalidades. intermedias. y fracciones en volumen menores a 0.5. En la figura 1.11 se muestra el. diagrama de fase obtenido por ellos, en donde se observan las fases fluidas y s cristalinas (fcc y bcc) predichos previamente por Likos y colaboradores [4]-[5]. Uno de los objetivos de este trabajo de tesis es explorar el diagrama de fases en la transición fluido-sólido haciendo uso de simulaciones con dinámica Browniana y el criterio fenomenológico de Lówen, así también se explora la modificación del diagrama por efecto de polidispersidad en tamaño de los polímeros estrella.. 11.
(19) Capítulo 2. Elementos de Simulación En este capítulo expondremos los elementos básicos necesarios para implementar simulaciones computacionales de un sistema coloidal modelo. Específicamente, presentaremos lo relativo a los métodos de Monte Carlo y de Dinámica Browniana. El método de Monte Carlo nos permitirá calcular propiedades estáticas como la función de distribución radial y propiedades termodinámicas de los sistemas, mientras que las simulaciones de dinámica Browniana será permiten calcular propiedades dinámicas como el desplazamiento cuadrático medio y el coeficiente de difusión dependiente del tiempo. Las propiedades estructurales y dinámicas son propiedades estadísticas de muchos cuerpo, y para obtenerlas se procede a calcular promedios relativos al tipo de propiedad.. Por ejemplo,. para calcular la función de distribución radial habremos de calcular distancias promedios entre partículas y para calcular desplazamientos cuadráticos medios seguiremos a cada partícula en su movimiento y obtendremos el promedio del cuadrado de su distancia en el tiempo. En simulaciones de Monte Carlo y Dinámica Browniana se determinará las posiciones de todas las partículas de un sistema, consistente con un modelo de potencial de interacción entre ellas y respetando los principios fundamentales de física clásica. En particular, nos enfocaremos en. visualizar a nuestro sistema de estudio como formado por un número N fijo de partículas dentro de un volumen V a una temperatura T.. 2 .1. Configuraciones. iniciales. En las simulaciones, se necesita partir de una configuración inicial para las N partículas, es decir se debe asignar la posición de cada partícula coloidal del nuestro sistema en el volumen V de la simulación. Como las propiedades estructurales y dinámicas que interesan son de equilibrio, sus 12.
(20) valores no deben depender de cual sea la configuración inicial con la que se debe dar inicio al movimiento de las partículas.. En nuestro caso partiremos de configuraciones iniciales aleatorias,. es decir, cada una de las posiciones de las partículas se asignarán al azar mediante el uso de un generador de números aleatorios. En la figura 2.1, se muestra un ejemplo.. N. Figura 2 .1: Configuración inicial aleatoria.. La configuración inicial se puede incluir al programa dando lectura a un archivo externo o bien se puede incluir a través de una subrutina que genera la configuracióninicial. En un sistema modelo como el de esferas duras, es común poner como condición que las partículas no se traslapen desde la (lJ. configuracióninicial, pero en un sistema modelo de partículas suaves, como el de polímeros estrella, en donde se pueden traslapar o no dependiendo de factores como la concentración o suavidad de las partículas, esta condición pueda ser innecesaria para lograr que el sistema alcance el equilibrio en un tiempo adecuado de ejecución.. 2.2. Celda principal de simulación y condiciones periódicas. En los sistemas reales ya sean fluidos, gases, fluido-gas, fluido-sólido, etc. el número de partículas que interactuan entre si es muy grande, es decir, en una escala microscópica es del orden de miles de millones de partículas (del orden del número de Avogadro, NA "'"' 1023 partículas). Este es un número tan grande que no se puede utilizar al simular un sistema, de forma que en los programas de simulación se considera un número muchísimo más paqueño, de cientos o miles de partículas. En nuestros programas utilizamos usualmente 500 partículas y en casos específicoshasta 2000, debido 13.
(21) a que entre mayor es el número de partículas es mayor el tiempo que tarda el programa en realizar los cálculos ( decimos, es mas costosa la ejecución del programa) Ante este problema los simuladores. pensaron. y con resultados. equivalentes.. en una herramienta para facilitar el trabajo de. emular los sistemas reales, haciendo uso de una celda principal la cual se repite "infinitamente". y en donde las celdas vecinas son réplicas exactas de la celda principal y que acostumbramos a llamar celdas imagen de la celda principal (ver figura 2.2). Cabe destacar que en la celda principal es donde se encuentran las N partículas con las que se realizarán los cálculos estadísticos de los que hablamos previamente.. Figura 2.2: Celda principal y celdas imagen.. La celda principal puede ser de diferentes formas, dependiendo del sistema a simular y de las condiciones externas, como por ejemplo la forma de las paredes en un sistema confinado. En simulaciones de sistemas en bulto generalmente se utiliza la celda cúbica que es la más sencilla. Como este es nuestro caso, se escoge como celda principal un cubo de arista L, en donde las partículas se pueden desplazar en :3 dimensiones. El tamaño de la arista de la celda principal no es un parámetro de entrada en nuestro programa de simulación, sino que L se ajustará. dependiendo de la concentración. ti. de partículas y del número. de partículas N que se usen en la simulación, es decir:. ti. =. i-+. L. = ( :). i/3. (2.1). De esta forma se puede observar como el ancho L de la celda depende directamente del número de partículas N involucradas en la simlucación. Para una concentración n dada, la celda será mas grande conforme se aumente el número de partículas. Este aspecto aparentemente trivial resulta 14.
(22) ser determinante para el caso de sistemas modelo cuyo potencial de interacción sea de largo alcance, en estos casos se requieren celdas de tamaño suficientemente grande. En nuestro caso N = 500. partículas nos permite observar satisfactoriamente la estructura de los sistemas a mediano alcance.. (). C) (). C) Figura 2.3: Condiciones periódicas.. Las partículas no están atrapadas en la celda principal, simplemente cuando una partícula sale de ella por uno de los lados de la celda, otra partícula entra por el lado opuesto. De esta forma se mantiene el número de partículas constante dentro de la celda principal y de las celdas imagen también. Para obtener estos movimientos se debe incluir en el programa lo que se conoce como condiciones periódicas. Una forma de hacerlo es escribiendo el siguiente código, cada vez que se mueve a las partículas: X(I)=X(I)-BOXL*NINT(X(I)/BOXL). en donde X(I) es la posición en x de la i-ésima partícula dentro del volumen de la celda principal, BOXL es la longitud de la arista de la celda (L) y con el comando NINT(X(I)/BOXL) FORTRAN se obtiene el entero más cercano al argumento. Esta instrucción se hace también para las coordenadas y y z de las partículas. 15.
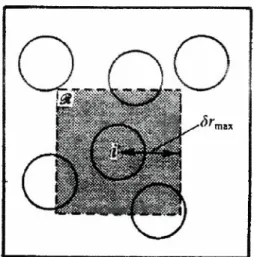
(23) 2.2.1. Convención de imagen mínima y radio de corte. En los programas de simulación un punto muy importante es el cálculo del potencial de interacción entre las partículas, así como también las condiciones periódicas en la frontera. Es decir, cuando se calcula la energía potencial de la i-ésima partícula se suman los términos de la energía potencial por pares de ésta con las otras N - 1 partículas restantes en la celda principal, así también podríamos incluir su interacción con las partículas que se encuentrán en las celdas imágenes vecinas necesarias para representar la condición en que se encuentra nuestro sistema de polímeros estrella. Hacer lo anterior llevaría a considerar un número infinito de términos en la energía potencial, y esto es prácticamente imposible de calcular en la práctica.. Para muchos de los modelos de potencial de interacción usados en las simulaciones, el incluir a las partículas de las celdas imagen resulta innecesario, debido a que el alcance del potencial es finito y limitado ( corto alcance); ésto nos permite incluir una aproximación conocida como convención. de imagen mínima. Esta condición consiste en considerar a la i-ésima partícula en el centro de una región cuyas dimensiones son similares a las de la celda principal, de forma tal que al calcular la interacción con las demás partículas sólo se tiene que considerar a las partículas restantes que se encuentran en esta región. En la figura 2.4 se ilustra gráficamente lo que implica utilizar la convención de imagen mínima.. C) \J (). \J Figura 2.4: Condición de imagen mínima.. 16.
(24) En lenguaje de programación, la condición de imagen mínima para la distancia en. Xij. se escribe. de la siguiente manera: XIJ=XIJ-BOXL*NINT(XJJ/BOXL). en donde XJJ es la distancia en x entre la i-ésima y la j-ésima partícula. Adicionalmente, para los sistemas de partículas cuyo potencial de interacción es de corto alcance, en las simulaciones se incluye la aproximación del radio de corte (re)· Esta consiste en despreciar el valor del potencial de interacción u( r) entre aquellas partículas cuya distancia de separación sea mayor a re, de forma tal que al calcular el potencial de interacción de la i-ésima partícula, sólo se incluyen los términos del potencial par de aquellas partículas cuya distancia con la partícula i sea menor a re· Por ejemplo, en la figura 2.4, el radio de corte relativo a la partícula 1 está representado por el círculo a trazos, de forma tal que en lugar de calcular los cuatro términos de potencial relativos a la interacción (1- 2 y 1- 3', 1 - 4' y 1 - 5'), sólo calcularán dos términos (1 - 2 y 1 - 4') con la aplicación del radio de corte. Esto permite optimizar el_tiempode ejecución en las simulaciones. No existe un criterio general para seleccionar el valor del radio de corte, ya que en principio dependería de la forma específica del potencial de interacción entre las partículas. Sin embargo observemos que el valor más grande que puede tomar es re= L/2, es decir, la mitad de la longitud de la arista de la celda de simulación. Un criterio conservador sería entonces tomar este valor para re y en nuestras simulaciones este es el criterio que hemos incluido.. 2.3. Termalización. Como se señaló previamente, la simulación da inicio con la. construcción de la configuración inicial, arbitraria. Es a. partir de esta configuración que se empieza a mover a las N partículas haciendo uso de los algoritmos de Monte Carlo o bien de dinámica. Browniana ( entre otros, como por ejemplo el de Dinámica Molecular) para determinar la posición de las N partículas en cualquier otra configuración de interés. Es en ese momento que las partículas empiezan a interactuar entre sí en una simulación. En palabras más simples, es cuando cada una de las partículas se percatan de la presencia de las otras y se empiezan a "sentir" ( colisionando, uniendose, repeliéndose, etc) y en consecuencia.la energía potencial del sistema va cambiando. El sistema tiende finalmente al equilibrio promedio. Cuando esto ocurre se dice que el sistema ha alcanzado el equilibrio término y al proceso que va desde el inicio hasta el estado en equilibrio, 17.
(25) se le llama termalización. Para saber cuando las configuraciones seguimiento. constantemente a la energía potencial intermolecular. corresponden. al equilibrio, se da. promedio del sistema.. Cuando. la energía potencial promedio fluctúa alrededor de un valor constante es cuando se puede decir que se ha llegado al equilibrio o bien decimos que el sistema se ha termalizado acorde al modelo de interacción par. En la figura 2.5 mostramos como ejemplo el comportamiento de la energía potencial promedio de un sistemas de polímeros estrella. Hacemos notar que independientemente de la configuración inicial el mismo estado termalizado se alcanzará, modinámicos. siempre y cuando todos los parámetros ter-. y del potencial de interacción tambien sean los mismos. Curva de Termalización Caso con: $ = 0.45 ro ·¡¡ e. =. 51.3. f 30 POLJ = 0%. w oo.. ro. 'ªw. 50,4. e w. 100000. 200000. 300000. 400000. Ulo. Figura 2.5: Termalización.. De lo anterior se desprende que otro parámetro importante en la simluación es el número de configuraciones. necesarias para termalizar el sistema.. En el programa. se utiliza. un parámetro. llama.do NENER el cual se utiliza para representar el número de configuraciones construirse. en el programa antes de que alcance la termaliza.ción.. muy pequeño ( centenas o miles de configuraciones), de miles o millones de configuraciones),. que deben de. Este parámetro en ocaciones es. pero en otras puede ser muy grande ( cientos. dependerá del tipo particular del sistema modelo y debe. de estarse verificando del valor adecuado de este parámetro. Algo curioso que obtuvimos en algunas simulaciones de sistemas muy interactuantes (sistemas muy concentrados,. entre otros) es que la termalización. pero al seguir evolucionando. llegaba a un aparente estado de equilibrio,. el sistema, la energía potencial. promedio presentaba un cambio y. fluctuaba alrededor de otro valor. A este fenómeno lo identificamos 18. como estados metaestables,.
(26) debido a que llegan a un estado en equilibrio por lapso de tiempo para después llegar a otro estado, aparentemente más favorable, de equilibrio. A pesar de que en este trabajo de tesis no nos enfocamos a situaciones metaestables, en la figura 2.6 ilustramos un ejemplo de como se manifiesta este tipo de fenómeno[6]. Curva de Termalización de un Sistema Meta-Estable 337,5~-------------~ 337.0. -¡¡; -¡:;. =1. e:. 335,5. o Q.. 335.0. a,. -~"'. 334,5. w. 333,5. a, e:. Caso de: ~=0.8 f= 100 POLI= 0%. 334,0. 333.0 332.5 332.0 100000. 200000. 300000. 400000. Figura 2.6: Estados metaestables.. Una vez que el sistema se ha termalizado, se esta en condiciones de empezar a almacenar las configuraciones necesarias con las cuales se calcularán las propiedades de estadísticas de equilibrio del sistema. Estas configuraciones de equilibrio serán el conjunto de posiciones x, y y z de cada una de las N partículas del sistema. Estas configuraciones se almacenan en tres matrices ( una para cada tipo de coordenada), de forma tal que el número de renglones es igual al número N de partículas y el número de columnas K I será igual al número de configuraciones de equilibrio por almacenar. En los programas actuales de simulación no se almacenan estas matrices inmensas, lo que se hace es incluir subrutinas adicionales para el cálculo de las propiedades de interés. Estas subrutinas hacen uso de las matrices de configuración y concluida la ejecución del programa de simulación se eliminan. En el capítulo siguiente abordaremos lo relativo a las subrutinas para el cálculo de las propiedades estructurales y dinámicas del sistema de interés en esta tesis.. 2.4. Método de simulación de Monte Carlo. Como se señaló previamente, el método de simulación de Monte Carlo nos permite calcular propiedades estructurales de equilibrio, es decir, propiedades que dependen exclusivamente de las posiciones de las N partículas del sistema modelo. En términos mecánico estadísticos, lo que esto. 19.
(27) significa es que las propiedades. dependerán. unicamente. del espacio de configuración del sistema.. Haciendo uso del ensemble canónico clásico, se sabe que la probabilidad. PN de que un sis-. tema se encuentre en una configuración alrededor de rN es proporcional al factor de Boltzmann correspondiente,. (2.2) donde U ( rN) corresponde a la energía potencial del sistema en la configuración rN y rN es una notación compacta para referirnos al conjunto de posiciones de las N partículas del sistema que caracterizan a la configuración. Como se describe a continuación esta probabilidad será básica para entender la esencia del algorítmo de Metropolis utilizado en una simulación convencional de Monte Garlo ( N V T). Lo primero que se necesita para aplicar el método es una configuración inicial del sistema la cuál tendrá asociada una energía potencial U0(rN), así mismo, cada partícula de esta configuración tendrá asociada una energía U;0(rN) (con i. =. 1, 2, ... , N). Para generar las subsiguientes. configuraciones del sistema, es decir, para mover las partículas, se procede de la siguiente forma: 1. Se selecciona una de las partículas y se le desplaza aleatoriamente en las direcciones x, y y z una distancia proporcional a un desplazamiento máximo Sr max previamente seleccionado ( en. nuestro caso lo hemos seleccionado igual a 0.1 o-). La constante de proporcionalidad corresponde a un número aleatorio o, entre O. y. 1 con distribución uniforme (ver figura (2.7)).. Figura 2.7: Desplazamientos en Monte Garlo.. 20.
(28) 2. Si la energía potencial de la partícula es menor en la nueva posición que en la que se tenía antes del movimiento descrito en el punto 1, entonces de la ec. (2.2) implicará que PF. > p1. En. este caso, como la nueva posición es más probable, simplemente se acepta este movimiento y como consecuencia la nueva posición de la partícula queda definida. 3. Por el contrario, si la energía potencial de la partícula es mucho mayor en esta nueva posición que en la que se tenía antes del movimiento descrito en el punto 1 ( esto lo referimos a un valor f3b.Ui(rN) > 75, tomado de Tildesley), entonces este movimiento tentativo se rechaza. En consecuencia la partícula se deja con la posición previa. exp(-PW). Always aeeept Accept. '. ,¡. o Figura 2.8: Criterio para aceptar o rechazar movimientos en Monte Garlo.. 4.. Si la energía potencial de la partícula es mayor en esta nueva posición que en la que. tenía antes del movimiento descrito en el punto 1, pero ¡3b,.Ui(rN) < 75, entonces implicaría que PF. < Pt , pero puede o no ocurrir que la partícula pudiese ocupar esta nueva posición. Para decidir. si se acepta o se rechaza, lo que se hace es seleccionar otro número aleatorio 'Y entre O y l. Sí 'Y. <. e-/3b.Ui(rN),. la posición se acepta, por el contrario se rechaza. En el primer caso tendremos una. nueva posición para la partícula, en el segunde).la partícula se deja. con la posición que tenía en el punto l. En la figura 2.8 de ilustra el esquema de aceptación o rechazo de este criterio [7]. Ésto se hace una y otra vez, es decir, se repite el procedimiento anterior para todas las N partículas obteniendo con ello una nueva configuración del sistema. Si luego se repite de nuevo esta rutina se obtiene otra configuración y así sucesivamente hasta conseguir un número de configuraciones suficientemente grande del sistema termalizado. Cabe señalar que es posible ajustar el valor del desplazamiento máximo a partir de cuantificar el número acomulado de movimientos aceptados y rechazados cada número de configuracionesgeneradas. Si es mayor el número de movimientos aceptados, entonces se permite que los movimientos 21.
(29) tentativos correspondan a un desplazamiento mayor. Si por el contrario el número de movimientos rechazados es mayor que los aceptados, entonces se mueve a las partículas con movimientos tentativos de menor longitud. Ello se implementa imponiendo una taza de aumento o disminución de Sr max del 5%.. 2.5. Método de simulación de dinámina Browniana. Para las simulaciones con dinámica Browniana básicamente se considera el algoritmo de Ermok. Este algoritmo está basado en la ecuación de Langevin siguiente:. (2.3) donde F¡ representa la fuerza que ejercen las N - 1 partículas coloidales sobre la i-ésima partícula; fa es una fuerza aleatoria debido a la interacción de las partículas brownianas con el medio solvente; 0:. es el coeficiente de fricción y V¡ es la velocidad con la que se mueve la i-ésima partícula. Como. podemos observar el último término corresponde a la fuerza de Stokes producto del carácter viscoso del medio solvente ( 0:. =. 61rr¡r para partículas esféricas).. Ahora bien, para calcular la posición de la partículas de una manera relativamente simple se utiliza el algoritmo de Ermak. Este consiste en considerar el régimen sobreamortiguado del sistema, es decir, omitir o despreciar el término inercial, m dit¡. '.:::::. O de las partículas. Esto es. similar a considerar la cinemática del régimen terminal de un balín en caída a través de un fluido. Lo anterior permite obtener la siguiente expresión básica para calcular las posiciones de las N partículas en la simulación:. r¡(t. + ~t). =. r¡(t). + R(~t) +. D. k8~Fij(t)~t. (2.4). en donde r, es la posición de la partícula i, Res un desplazamiento aleatorio Gaussiano, F¡j es la fuerza de interacción entre la partícula i-ésima con las otras N -1 partículas restantes en el sistema, Do es el coeficiente de difusión libre de la partícula y b.t el tiempo de paso entre configuraciones, este último debe de ser suficientemente pequeño para que las partículas se muevan muy poco. Al igual que en el caso de las simulaciones de Monte Carlo, en dinámica Browniana se parte de una configuración inicial arbitraria y para construir las configuracionessiguientes se debe mover a cada una de las N partículas de acuerdo a la ecuación 2.3. Esto se repite sistemáticamente para generar tantas configuracionescomo sea necesario[7]. 22.
(30) 2.6. Parámetros. importantes. Para implementar las simulaciones se quiere de la espeficicación de algunos parámetros utilizados en los programa de Monte Garlo y de dinámica Browniana, mismos que podemos clasificar en dos grupos: parámetros propios del sistema modelo y aquellos parámetros necesarios en la simulación. A continuación se describen brevemente.. 2.6.1. Parámetros. del sistema. ·• Fracción de Volumen (PHI): Es el parámetro que se usa para indicar la razón del volumen que ocupan las partículas de nuestro sistema en el volumen de la celda principal. En programas de simulación usado en este trabajo de tesis fue uno de los parámetros que se varió para estudiar al sistema.. Nv. (2.5). </J=v. donde N es el número de partículas utilizadas en la simulación, v corresponde al volumen de una de las partículas y V es el volumen total disponible en la celda unitaria. Como puede observarse <p es un parámetro sin dimensiones.. •. La concentración. (DENS): Es el número de partículas (NP) por unidad de volumen, es. decir, cuando la concentración es grande las partículas interaccionan más entre si que cuando la concentración es baja.. Por ejemplo, en un sistema de esferas duras esperamos que a bajas. concentraciones las partículas se muevan con mayor facilidad que en un caso más concentrado. Como se desprende de su definición la concentración es un parámetro con unidades inversas de volumen.. En el caso del programa que se uso se calculó la concentración. la concentración adimensionalizada con a3.. reducida que es. Esto nos permite escribirla en términos de </> de la. siguiente manera:. p* = 6</> 7r. •. Funcionalidad. (2.6). ( f): Este es un parámetro del potencial de interacción entre las partículas. y representa el número de brazos en los polímeros estrella.. Como se señaló previamente en el. capítulo 1 para funcionalidades grandes los polímeros estrellas tienden a comportarse como esferas. 23.
(31) duras, mientras que para funcionalidades pequeñas o moderadas éstos se comportan como esferas suaves o ultrasuaves.. Este es un parámetro el cual tambien variamos para estudiar al sistema. dependiendo de la suavidad o rigidez de las partículas. ·• Polidispersidad. (POLI):. También denotada como p, nos da información de la distribución. de tamaños de las partículas del sistema. Si la polidispersidad p = O, indicará que el sistema es monodisperso, es decir, todas las partículas serán del mismo tamaño. Su valor nos indica la dispersión porcentual en los diámetros de las partículas respecto a su valor promedio . Los valores de p usados en nuestros programas fueron: O, 5, 10 y 15 %.. 2.6.2. Parámetros. generales de la simulación. • Número de partículas (NP): Corresponde al número total de partículas que se utilizan para simular al modelo de polímeros estrella. En la mayoría de las ejecucionesdel programa se consideró N = 500 partículas y sólo en casos excepcionalesse usaron 1000 o 2000 partículas. • Número total de configuraciones (NS): Este parámetro es incluye para establecer el número total de configuracionesgeneradas al mover a las N partículas del sistema. En los programas utilizamos de dinámica Browniana se utilizó en general un valor de NS = 401000, mientras que en Monte Carlo fue sucifiente NS= 25000. •. Número de configuraciones necesarias para llegar a la termalización. (NENER):. Utilizado para indicar el número de configuracionesnecesarias para que la energía promedio del sistema alcance un valor estable, es decir cuando el sistema ya se haya termalizado. Un valor típico NEN ER = 1000 fue suficiente en la gran mayoría de los casos de dinámica Browniana y IVIonte Carlo. •. Frecuencia de Guardado. (NFREC): Cómo el nombre lo dice, este parámetro es uti-. lizado para indicar la frecuencia con la que se guardarán configuracionesde equilibrio del sistema. Recordemos que con estas configuraciones después se realizaran los cálculos de propiedades de interés. En nuestro caso utilizamos NEN ER = 100. Sabiendo que el número total de configuracioneses NS, que dejamos pasar NENER de noequilibrio y que almacenaremos configuracionescon una frecuencia de N F REC, podemos calcular el número total K I de configuracionesde equilibrio con el que se calcularan las propiedades estadísticas: 24.
(32) KI = NS-NENER NFREC. (2.7). Haciendo uso de los valores típicos utilizados en nuestras simulaciones, se concluye que las propiedades de equilibrio se calcularon con 1000 - 4000 configuraciones.. 2.6.3. Parámetros. particulares. de la simulación. Dependiento del tipo de simulación ( dinámica Browniana o Monte Carlo) se deberá especificar: el desplazamiento máximo inicial o el tiempo de paso, respectivamente. • Desplazamiento máximo inicial (DRMAX): Se refiere al valor de entrada del desplazamiento máximo con el que podrán moverse aleatoriamente las partículas en la simulación de Monte Carlo, es decir, corresponde al parámetro óTmax que describimos en la presentación del algorítmo de Metrópolis. Recordemos que dependiendo del valor del número aleatorio ( entre O y 1) que se genere, el desplazamiento de las partículas será en general un submultiplo de ór max, más aún, este valor de ór max en realidad se irá ajustando conforme transcurra la simulación, dependiendo de la proporción de movimientos aceptados y rechazados que incluye el algorítmo. Cabe señalar que esto último no es necesario, sin embargo se sugiere por algunos autores (Tyldesdey) para optimizar la ejecución del programa. El valor del parámetro que usamos en nuestras simulaciones fue de 8r:n_ax = 0.1 (la décima parte del diámetro de las partículas). • Tiempo de paso (DT): Corresponde al valor del intervalo de tiempo f:!...t que media entre movimientos consecutivos de las partículas, mismas que se desplazan de acuerdo al algorítmo de Ermack en las simulaciones de Dinámica Browniana. Como consecuencia, !:::,.t es el intervalo de tiempo que hay entre cada configuración o "fotografía" que se genera en la simulación. A anterior, a diferencia la simulación de Monte Carlo, lo anterior nos permite seguir a las partículas en el tiempo, construir sus trayectorias y por sobre todo, nos permite calcular propiedades dinámicas, como por ejemplo, los desplazamientos cuadráticos medios y los coeficientes de difusión dependientes del tiempo. En nuestro programa se uso en general un valor de !:::,.t* = 0.0001 (la diezmilésima parte del tiempo que le lleva a una partícula difundirse libremente una distancia igual a su diámetro).. 25.
(33) Capftulo 3 Sistema rnonodisporso estrella. de polímeros. En este capítulo se mostrarán algunos resultados importantes sobre la elaboración de esta tesis basados en cálculos y teorías propias de la descripción de una suspensión coloidal. En el capítulo 1 se estableció el sistema de polímeros estrella mientras que el en capítulo 2 se plantearon los elementos básicos de la simulación. En este capítulo se presentaran otros factores involucrados en la simulación, que nos permitiran estudiar el comportamiento de nuestro sistema cuando hay cambios en sus variables tanto la funcionalidad (!), densidad, etc. Esta explorará se hará mediante el cálculo de la función de distribución radial, el desplazamiento cuadrático medio, coeficiente de difusión, etc. También se determinará el diagrama de fase de los polímeros estrella explorando casos en los que la funcionalidad permanecerá constante variando la densidad o al revés, para así tener más claro como es el comportamiento de los cambios de fase desde el punto de vista de la simulación.. 3.1. Funciones de distribución. En el capítulo anterior se han presentado los aspectos de simulación necesarios que nos permiten describir el movimiento de las partículas del sistema modelo, es decir, de como interaccionan unas con otras adquiriendo nuevas configuraciones consistentes con los algorítmos de Monte Carlo y Dinámica Browniana.. Consistente con las condiciones y parámetros propios del sistema, las. partículas se destribuyen en promedio unas respecto a otras, de forma tal que en equilibrio es posible hablar de sus propiedades estructurales. En esta sección se presenta los elementos básicos que nos permiten obtener la función de distribución radial, misma que constituye una de las propiedades estructurales más importantes que nos permiten describir termodinámicamente a las. 26.
(34) suspensiones coloidales en bulto. se parte de considerar al sistema compuesto de. Para empezar a explicar este comportamiento. N partículas coloidales en un volumen V a una temperatura T, constantes. Para describir la. distribución de las partículas alrededor de un pequeño grupo n nos basaremos en la descripción mecánico estadística del ensamble canónico, la cual nos dice que la probabilidad de que el sistema se encuentre en un conjunto de microestados df es dw = pdI' con la densidad de probabilidad definida como[9] e-f3H. p(V, T). =. (3.1). donde (3 = (kBT)-1 y ZN(V, T) es la función de partición canónica:. j ···j. ZN(V, T) = N!~3N. e-f3U. (3.2). dI'. estas integrales son sobre todas las posiciones y todos los momentos de las partículas del sistema de tal forma que el elemento de volumen en el espacio fase es df = d7 1 d7 2. · · ·. d7 Ndp 1 dp 2. · · ·. dp N. y H es la energía mecánica total del sistema:. (3.3) sustituyendo las ecs. (3.1 )-(3.3) en la probabilidad df e integrando en los momentos se obtiene la probabilidad de encontrar a la partícula 1 en d71, a la partícula 2 en d7 2, etc., independientemente de los momentos que tenga cada una de ellas: dP(71 72. . . . '. --+r. '. N. ) _ -. d71d7 ') · .. d71\, e f3Ud----3 d=-+ 7 1 r ')- · · · d -=+ T ji¡ .. e-f3U. J. .. (3.4). La integral que aparece en el denominador de la ecuación anterior es conocida como integral de configuración IN por lo que es posible reescribir la ecuación (3.1) como:. (3.5) Ahora, si se desea obtener la probabilidad de que la partícula 1 se encuentre entre. d7 1,. la partícula 2 entre. 72. y. d7 2. y así sucesivamente hasta la partícula. ti. ( con. 71. y. n < N),. independientemente de las posiciones de las N - n partículas restantes, se debe de integrar dP -sobrelas posiciones de estas últimas, para obtener: 27.
(35) J. dPd7 n+1d7 n+2 · · · d7 N =. L (j · · · J. e-f3U d7 n+l· · · d7 N )d71 · · · d7 n. (3.6). definiendo a esta densidad de probabilidad p(n) como: p(n)(¿1¿2,. •.•. ,-:;tn)= J···Je-f3Ud7n+l"""d7N. (3.7). con la ecuación anterior se puede reescribir la ec. (3.6) como. (3.8) esta probabilidad nos brinda información acerca de la forma en la que se distribuye un grupo de n partículas en un sistema de N partículas en total. Para encontrar la probabilidad pn(7172, en las posiciones 7 1, 7. 2, • • • ,. · • · ,. 7 n) de encontrar a cualquiera de las N partículas. 7 n, se parte de dPn, pero como ahora sólo nos interesa encontrar N!. a cualquiera de las ti partículas, las posibilidades se incrementen en un factor de ( N _ n) ! y sólo basta multiplicar a d.P¿ para obtener la probabilidad deseada.. P. (n) N! p(n)(--:-:t -4 - (N _ n ) · T 1, r. 2, · · ·. 1. '. -4 T N). NI. =. (7\T. ·. _\I. J ... J -fJUd--:-:t e r n+ 1 T. · · ·. d. -4 T N. (3.9). Para el caso de ti = 1 la función de distrubición toma la forma:. (3.10) De la ec. (3.10) y la definición de IN se observa que. J p(ll(71)d71. = N.. También sabemos. que la concentración total pes la razón del número de partículas Nen el volumen 1/ en el que esta están contenidas, entonces. p. _ = N V. J. = V1. /1l(7)d71. (3.11). Para el caso de un fluido homogenéneo, p(l)(7 i) es constante, luego entonces se satisface que _ p{l) V. p--. J. 4. _. dr1---p. p(l) _ (1) V. (3.12). Por otra parte, si las partículas no están correlacionadas, es decir que sus posiciones son independientes, se puede utilizar separación de variable en la ec. (3.9) obteniendo lo siguiente 28.
(36) /nl(71, 72, · · · , 7 n) = /1l(71)/1l(7 2) · · · /1)(7 n). (3.13). que para un fluido homogenéo se reduce a. (3.14). En caso de que las partículas sí estén correlacionadas, lo cual es el caso más general, se introduce una función de correlación. g(n) (71,. 7 2, · · · , 7. n),. de esta forma se puede escribir la función de. distribución como p(nl(71, 72, · · · , 7 n) = png(n)(71,. 72, · · · , 7 n)· De lo anterior y considerando. la ec. (3.9) se obtiene:. ..,.,+ g (n) (-+ r1 T 2 ' '. · · ·. '. ..,.,+ ) = vN! T n JVn (N - n)!JN. J···J. e. -f3U. d. -+ T' +1 n. · · ·. -+ drs. (3.15). El caso más importante de las funciones de correlación es g(2l(71, 72), debido a que la función de correlación de dos partéulas se puede obtener experimentalmente.. Por otra parte, es posible. mostrar que las propiedades termodinámicas de un sistema ( energía interna, presión, potencial químico, etc.) se pueden expresar en términos de esta función de distribución.. Ello permite. establecer un puente entre propiedades macroscópicas y microscópicas de un sistema. Se puede observar que, considerando n.. =. 2. en la ec. (3.15) e integrando. en. 71. y. 7 2,. se. obtiene:. (3.16). para el caso del potenciales de interacción centrales, es decir, aquellos que dependen solamente de la distancia relativa entre las partículas, es posible expresar a la función de distribución par como 9(2)(71, 72) = g(2l(712) = g(r), entonces. ¡ -+/ dr. finalmente, como. 1. V g(r)d -+ r = -¡;(N - 1). J d71 = V y en coordenadas. fo. 00. esféricas. d7 = 41rr2dr,. pg(r)41rr2dr = (N - 1) ~ N 29. (3.17). se obtiene. (3.18).
(37) A la función g(r) se le llama función. de distrtibución. radial y la ecuación anterior indica que. pg(r)41rr2 nos proporciona el número de partículas N(r) que hay entre r y r-s-d« alrededor de una partícula,. pg(r)41rr2dr ~ N(r). (3.19). de tal forma que esto permite expresar pg(r) como la densidad local alrededor de una partícula. Por otra parte, esta ecuación nos permite escribir:. g(r)"' N(r) "' p41rr2dr. (3.20). y como el denominador corresponde al número de partículas en bulto y sin interacción, es decir. Nid = p41rr2dr, se puede escribir la ec. (3.20) como. g(r) ~. ~i:). (3.21). Como se ve mas delante, la función de distribución radial es una función oscilante y cada uno de sus máximos nos da información de los primeros vecinos, segundos vecinos, etc. alrededor de cada partícula. Independientemente de la altura de dichos máximos y de sus posiciones, es decir, independientemente del potencial de interacción entre las partículas, todas las g ( r) satisfacen los siguientes dos límites: g(r) ---+ 1 cuando r ---+ oc, ello corresponde al hecho de que cuando las partículas estan muy separadas realmente se encuentran descorrelacionadas, consistente con la ec. (3.14); por otra parte si consideramos que las partículas poseen un tamaño impenetrable (ej., esferas duras), implicará que no podrá encontrarse ninguna partícula a distancias menores a dicha longitud característica, lo que permite escribir que en el límite r ---+ O necesariamente g ( r) ---+ O. En lo que sigue, se expondrá la forma en que se obtiene la función de distribución radial mediante simulaciones. Como podrá observarse, su cálculo parte de la ec. (3.21) y del conocimiento de un conjunto de configuraciones de las partículas obtenidas ya sea con Monte Carla o dinámica Browniana (o cualquier otro algoritmo, como por ejemplo dinámica Molecular). Como se indicó en el capítulo 2, el cálculo de la g(r) se incluye en el programa de simulación como una subrutina a la que se accede luego de concluir con los movimientos necesarios de las N partículas consideradas. Para adaptar lo que se ha dicho de teoría en el programa de simluación, ya sea con método de Monte Carla o de dinámica Browniana se procedió de la siguiente manera: Se debe fijar en una partícula y dar un radio ri con el cual se formará un cascarón alrededor de la partícula en la se 30.
(38) encuentra parado, después se cuenta el número de partículas que se encuentren en el cascarón; al contar las parículas en el cascarón se guarda el número de partículas en un histograma. Se vuelve a hacer todo lo anterior, sólo que ahora es para un cascarón diferente, en donde el radio del nuevo cascarón será de r2i = 2ri lo cual nos dará otro número de partículas en el nuevo cascarón, el cual se guarda en el histograma. De la misma manera se elige una nueva partícula en la cual se repetirá el procedimiento anterior; estos resultado se guardan en el histograma con los cuales se harán los calculas más adelante. El radio del cascarón donde se realizan los conteos, en el programa, se fija de la siguiente maneta:. DELTAR=O.OIDO MAXBIN=DINT(BOXL/2.DO/DELTAR) en donde M AX BI N es el número máximos de elementos en el vector que representa el histograma consistente con BOX L/2 y DELT AR es la distancia radial entre cascarones.. Mientras que el. conteo de distribuciones de partículas alrededor de una se debe de hacer por medio de las matrices de posición que ya se obtuvieron por medio de la sub rutina generadora de posiciones, con las cuales se mide la distancia que hay de una partícula a las otras. XLO=CX(L,J) XLT=CX(M,J) XLOT=XLO-XLT YLO=CY(L,J) YLT=CY(M,J) YLOT= YLO-YLT ZLO=CZ(L,J) ZLT=CZ(M,J) ZLOT=ZLO-ZLT con estas componentes en x, y y z se puede calcular su componenete de imagen mínima para después calcular la magnitud del radio r que hay entre una y otra partículas. XLOT=XLOT-BOXL*NINT(XLOT/BOXL) YLOT= YLOT-BOXL *NINT(YLOT /BOXL) ZLOT=ZLOT-BOXL*NINT(ZLOT/BOXL) ROT=SQRT(XLOT**2+ YLOT**2+ZLOT**2) 31.
(39) y para finalizar los siclos se ve si ya termina de hacer todo el muestro de una partícula con el resto de las partículas en 'el sistema, para concluir la subrutina. NBIN=INT(ROT/DELTAR)+l IF(NBIN.LE.MAXBIN). THEN. NHIST(NBIN)=NHIST(NBIN)+l. ENDIF. 40 CONTINUE 25 CONTINUE y esto se hace con todas las N partículas del sistema. A continuación se muestra un resultado de esta subrutina, de un caso específico tanto en dinámica Browniana como en Monte Carla, viendo como coninciden lo cual nos da seguridad de que los programas están bien. Función de Distribución. A. 1.8. / \ ~ \. 1.6. '. 1 '. 1,4. 1~~ ,...~. 1.2 ~. --DB " MC. ' /. 1,0. f. 0.8. '. \./. Caso. 0.6 0.4. 0.2. con:. $ = 0.1 f= 100 POLI= 5%. /. 1 J. o.o+-_,......,....._,~~~~~-~~~~-~--J. Figura 3.1: g(r)-Función de distribución para un caso específico.. 3.2. Desplazamiento. cuadrático. medio. Cuando una partícula coloidal se encuentra en suspención en un sistema compuesto por otras partículas coloidalesy el medio solvente, el movimiento que describe cada partícula coloidal es un movimiento aleatorio. Este comportamiento se debe fundamentalmente por las múltiples interacciones de las partículas del solvente sobre las macropartículas ( o partículas coloidales). Debido al carácter errático del desplazamiento de ellas, para su descripción se requiere de herramientas estadísticas dentro de las cuales se construye el concepto de desplazamiento cuadrático medio. 32.
(40) En palabras similares a las de Eliezer Braun [9], imaginemos un recipiente con una suspensión coloidal ( o en nuestro caso, imaginemos la celda central con las N partículas coloidales), en la que cada partícula está en constante movimiento azaroso y en todas direcciones. Centrando ahora nuestra mirada en una sola partícula a la que se desea medir que tanto se mueve en pequeños lapsos de tiempo. Primero se coloca el cronómetro en un tiempo inicial. t. = O y se dice que a ese. tiempo la partícula se encuentra en la posición O. Se deja correr el tiempo hasta que el cronómetro indica un tiempo t. =. T. en el cual la partícula se movió a otra posición x, y y z. Para tener una. idea de que tanto se movió la partícula, se calcula d1,r. =. (x2. + y2 + z2)112.. Después se coloca de. nuevo a la partícula en el origen de coordenadas, haciendo ahora que vuelva a correr el tiempo hasta. t =. T. y medimos lo distancia, que ahora llamaremos d2,n que se movió la partícula.. Al. comparar las distancias vemos que d1,r f- d2,r· Se puede repetir sucesivamente este procedimiento y se observará que a pesar de que el intervalo de tiempo es el mismo, las distancias recorridas son en general diferentes a las anteriores. Si se toman los cuadrados de las distancias encontradas en una sucesión muy grande de experimentos como este, podemos calcular precisamente su promedio. Al promedio de la distancia cuadrática recorrida por las partículas la denotaremos como. < d2 > y. lo llamaremos desplazamiento cuadrático medio. Ahora bien, si se hace muchas veces lo anterior para las N partículas de la suspensión coloidal y vamos almacenando los diferentes intervalos de tiempos y las distancias correspondientes recorridas, obtendremos el comportamiento del desplazamiento cuadrático medio como función del tiempo. Como se señaló en la introducción,. al movimiento azaroso que manifiestan las partículas. coloidales se le refiere como movimiento broumiano. En una suspensión coloidal muy diluida, las partículas coloidales no interaccionan entre sí y solamente lo hacen con el solvente, de forma tal que cada una de ellas se mueve en difusión libre. El estudio del movimiento browniano de una partícula, fue estudiado ampliamente por científicos como Einstein, Langevin, Smoluchowski y Perrin desde los primeros años del siglo XX[9]. La ecuación de Langevin que se presentó en el capítulo anterior ( ecuación 2.3), incluye la fuerza aleatoria. fa(t) debida al golpeteo azaroso de las partículas del solvente sobre la macropartícula.. Este elemento caótico ( o estocástico) trae como consecuencia que la posición y velocidad sean ahora variables aleatorias.. Para obtener la función de distribución de probabilidad asociada a. estas variables, se necesita incluir hipótesis factibles con el fenómeno descrito del movimiento browniano. 33.
(41) Es posible mostrar[8] que si se asume que, el promedio de la fuerza aleatoria es nulo,. < fa(t) >=. O, y si la correlación de la fuerza aleatoria a dos tiempos diferentes no es relevante, < fa(t)fa(t'). >=. Gó(t - t') (donde Ges constante y b(t - t') es la función delta de Dirac), entonces la función de distribución de probabilidad es una función gaussiana ( con media µ y desviación estandard <7). Conocida la función de distribución de probabilidad se pueden calcular todos los momentos o propiedades estadísticas asociadas con la posición y velocidad de la partícula browniana[15]. Específicamente, es posible mostrar que el desplazamiento cuadrático medio en una dimensión toma la siguiente forma:. (b.x2) = ([x(t) - x0])2 =. P!x +. 2 [1 - exp (- :) { od: ª2m 2 f3 m - 1 2. ]. at)] }. [ exp ( - 2at) · (- m m - 1 ] - 2 [ 1 - exp. (3.22). donde m es la masa de la partícula browniana, a = 67rr¡r es el coeficientede fricción definido en la fuerza de Stokes y /3 =. k~T.. Observando que para t < <. es decir, para tiempos muy cortos:. !I!'., a. (3.23). y ele aquí que para este régimen ele tiempos tan cortos, se señale que la partícula browniana se mueve balísticamente; esto es, no siente el efecto caótico de los choques moleculares del solvente.. Por otra parte, para tiempos muy grandes, esto es t > > ~, el comportamiento del desplazamiento cuadrático medio es lineal: 2t ( (b.x )2) = (3a. (3.24). En síntesis, para este caso de difusión libre, podemos identificar dos regímenes de tiempos importantes.. t. <<. TB. régimen balístico. t. >>. TB. régimen difusivo 34. (3.25).
Figure




+7




Documento similar