Connective tissue growth factor induction by lysophosphatidic acid requires transactivation of transforming growth factor type beta receptors and the JNK pathway
9
0
0
Texto completo
(2) 450. C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. target genes. Smad-7, on the other hand, is a strong inhibitor of this pathway [13,14]. Besides Smad-mediated transcription, TGF-β activates other signaling cascades, including MAPK pathways [15]. Some of these pathways regulate Smad activation, but others might induce non-transcriptional responses [15]. Additionally, TGF-β can activate the ERK [16], JNK [17], p38 MAPK kinase [18] and phosphatidylinositol-3-kinase (PI3K)-mediated pathways [17,19]. Recent data show significant involvement of phospholipids in wound healing and the development of fibrosis. This group includes LPA, sphingosine-1-phosphate (S1P), lysophosphatidylcholine, sphingosylphosphorylcholine, and ceramide-1-phosphate. LPA functions as a serum-derived growth factor, and also exhibits multiple pleiotropic effects as an inter- and intra-cellular lipid mediator of cellular functions [8,20] such as proliferation [21], migration [22], and survival [23]. LPA and S1P are novel, potent regulators of wound healing and fibrosis that have diverse effects on many types of cells involved in the wound-healing process. The effects of LPA and S1P are principally mediated via specific cell surface receptors. Some of the biological responses mediated by LPA and S1P are coincident but others differ [24]. Most of the biological effects of S1P and LPA have been attributed to high-affinity ligand binding to two closely related subfamilies of specific G-protein-coupled receptors (GPCRs), namely S1P1-5 and LPA1-4 [24,25]. Several lines of investigation in a variety of cell types have shown that many of the growth-promoting effects of the GPCR agonists LPA, S1P, angiotensin, PGE2, and others are mediated through activation of receptor tyrosine kinases, a phenomenon termed as transactivation [26]. The transactivation of receptor tyrosine kinases by GPCR ligands has been demonstrated in various cell types, and this cross-talk provides additional non-canonical signaling pathways which regulate cellular function. There is experimental evidence to support cross-talk between LPA and epidermal growth factor receptors (EGF-R) [27], c-Met receptors [28] and platelet derived growth factor receptors (PDGF-R) [29]. On the contrary, cross-talk between S1P and TGF-β has been proposed to regulate metalloproteinase expression, whereby TGF-β reduced S1P phosphatase activity [30]. On the other hand, S1P activation of its described receptors can stimulate TGF-βRI kinase activity, resulting in phosphorylation of Smad2 and Smad3 and their subsequent nuclear translocation [31,32]. In this paper, we have evaluated if the expression of CTGF mediated by two important pro-fibrotic agents, LPA and TGF-β, is related. We have shown that treatment of myoblasts with LPA together with TGF-β increases CTGF expression. The augmented expression of CTGF by LPA required active TGF-βR activity and the presence of Smad-2/3. Nevertheless, LPA itself did not induce either Smad-2/3 phosphorylation or Smad-4 translocation to the nucleus. Interestingly, LPA mediated the induction of stress fibers required for TGF-βRI serine/threonine kinase activity, but did not induce cell proliferation. Finally, we have shown that LPA induces CTGF expression by activating JNK, but not ERK pathways, and this activation was independent of TGF-βR activity. These results are important for the understanding of the roles of pro-fibrotic growth factors and phospholipids involved in wound healing and fibrotic diseases. 2. Materials and methods 2.1. Cell cultures The skeletal muscle cell line C2C12 was obtained from adult mouse leg muscle (American Type Culture Collection, USA), and was grown as described [11]. Myoblasts were treated with TGF-β1 (R&D, USA) and/or LPA (Sigma Chemicals, USA) at the indicated concentrations. Inhibitors were used to suppress TGF-βRI kinase activity (SB431542, Sigma-Aldrich, USA), MEK1/2 activity (U0126, Alomone, Jerusalem, Israel), or JNK activity (SP-600125, Enzo Life Sciences NY, USA). Cells were serum-starved for 18 h and then treated for the indicated times.. 2.2. Transient plasmid transfection The TGF-β responsive constructs used were p3TP-lux [33] and pCTGF-Luc a plasmid containing part of murine promoter for CTGF, pDN-TGF-βRII-HA (kindly donated by Dr. J. Massagué, Sloan Kettering, HHMI, New York, USA) was used to overexpress a dominant negative mutant of TGF-βRII (DN-TGFβRII-HA). Briefly, cells were plated in 24-well plates until they reached 60% confluence. Cells were then incubated in Opti-MEM I containing 1 μg of each plasmid, 0.02 μg of pRL-SV40, 2 μl of PLUS reagent and 1 μl of LipofectAMINE. After 6 h, FBS was added to the medium, and the cells were cultured for a further 12 h. Medium was changed to fresh growth medium and the following reagents were added: TGF-β1 dissolved in 0.5% FBS; LPA or the indicated inhibitors. Dual luciferase activity assays (Promega, USA) were performed after 24 h. 2.3. Immunoblot analysis For immunoblot analyses, cell extracts obtained from myoblasts were prepared in 50 mM Tris–HCl, pH 7.4, 0.1 M NaCl, 0.5% Triton X100 with a cocktail of protease inhibitors and 1 mM PMSF. For analysis of phosphorylated proteins, cell extracts were prepared in RIPA buffer as previously described [34]. Aliquots were subjected to SDS gel electrophoresis in 5% or 10% polyacrylamide gels, electrophoretically transferred onto nitrocellulose membranes (Schleicher & Schuell) and probed with rabbit anti-phosphoSmad-2 (1:1000, Calbiochem, USA), anti-phosphoERK-1/2 (1:1000), anti-phosphoJNK (1:1000), antiSmad2 (1:1000), anti-Smad3 (1:500) (Calbiochem, USA), mouse anti-α-tubulin (1:5000, Sigma-Aldrich, USA), mouse anti-GAPDH (1:2000, Chemicon, USA) or with rabbit anti-HA (1:2000, Santa Cruz Biotechnology, USA). All immunoreactions were visualized by enhanced chemiluminescence (Pierce, USA). 2.4. RNA isolation and Northern blot analysis Total RNA was isolated from cultures as described previously [35,36]. RNA samples (20 μg) were electrophoresed on 1.2% agarose/ formaldehyde gels, transferred onto Nytran membranes (Schleicher & Shuell, Dassel, Germany) and hybridized with random primed [32P] dCTP-labeled cDNA probes for mouse CTGF or tubulin in hybridization buffer overnight at 42 °C or 65 °C, respectively [33]. Hybridized membranes were then washed at 42 °C and exposed on a Phosphor Imager. 2.5. Immunofluorescence microscopy Smad-4 intracellular localization in C2C12 myoblasts was analyzed by indirect immunofluorescence. Cells were grown on glass coverslips and then fixed in 3% paraformaldehyde, permeabilized with 0.05% Triton X-100 and incubated for 1 h with mouse anti-Smad-4 (1:100, Santa Cruz Biotechnology, USA). The incubation buffer used contained 50 mM Tris–HCl, pH 7.7,0.1 M NaCl, and 2% bovine serum albumin. After buffer removal and several washes with the above buffer, bound antibodies were detected by incubating the cells for 30 min with affinity-purified rhodamine-conjugated anti-mouse antibodies (1:100, Pierce Biotechnology, USA). For actin filament staining, fixed cells were incubated with phalloidin conjugated with FITC (0.1 μM, Sigma Chemical, USA) for 40 min and rinsed with Blotto reagent. For nuclear staining, sections were incubated with 1 μg/ml Hoechst 33258 in PBS for 10 min. After rinsing, the coverslips were mounted and viewed under a Nikon Diaphot inverted microscope, equipped for epifluorescence. 2.6. Short interfering RNA (siRNA) transfection The specific siRNAs for both Smad-2/3 (Santa Cruz Biotechnology, USA) were used as well as control siRNA (Ambion, USA). Briefly, for.
(3) C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. transfection, myoblasts were seeded into six-well plates until they reached 60% confluence. Subsequently, cells were incubated for 6 h in 800 μl of Opti-MEM I medium containing siRNAs for Smad-2/3 or control siRNA plus 8 μl of Lipofectamine 2000 (Invitrogen, USA) [37]. For experiments with pMyo-Luc plasmid reporters, these were cotransfected with siRNA. Following transfection, FBS was added to the medium, and the cells were cultured for a further 48 h. 2.7. Growth stimulation assay Growth stimulation was demonstrated by measuring [3 H]thymidine incorporation into C2C12 cells [38]. Briefly, cells were seeded into individual wells of a 24-well culture dish and allowed to attach overnight. The next day, LPA or TGF-β was added to the wells. After a 24-h incubation, the cells were pulsed with [3 H]thymidine (64 Ci/mmol in 50 μl of culture medium/well; ICN, USA) for 2 h. The radioactive tissue culture medium was aspirated from the wells and the cells were washed 3×10 min with 10% trichloroacetic acid at room temperature. The acid-extracted cells were solubilized for 20 min at room temperature with 300 μl of 0.2 N NaOH. A 100 μl aliquot from each well was taken for scintillation counting.. 451. 3. Results 3.1. LPA and TGF-β1 present an additive effect on CTGF expression Previously, we determined that both TGF-β and LPA alone have the ability to induce the expression of CTGF [11], so we wanted to study the effect of both together. CTGF mRNA expression was induced by LPA (20 μg/ml) and TGF-β1 (5 ng/ml) (Fig. 1A). LPA caused a transient increase in CTGF mRNA after 1 h of incubation, whereas TGFβ induced CTGF expression after 3 h of incubation. When both CTGF inducers were added together, an additive effect was observed with a maximal effect after 3 h incubation. Quantitative analyses of two independent experiments were also performed (Fig. 1A). Next, we evaluated the induction of a specific reporter that contained the promoter region of mouse CTGF (pCTGF-Luc activity) transfected into myoblasts together with pRL-SV40 in order to normalize results (Córdova, manuscript in preparation). As seen in Fig. 1B, in myoblasts transfected with CTGF reporter, luciferase activity was higher when myoblasts were incubated with both LPA and TGF-β1, compared to when myoblasts were treated with LPA or TGF-β1 separately. These results indicated that LPA and TGF-β1 can induce CTGF expression in an additive manner.. 2.8. Protein determination 3.2. LPA induction of CTGF requires active TGF-βRs Protein levels were determined in aliquots of cell extracts using the bicinchoninic acid protein assay kit (Pierce) with BSA as a standard.. To gain insight in the molecular requirements involved on CTGF induction by LPA, we decided to evaluate whether TGF-βRs are. Fig. 1. LPA and TGF-β have an additive effect on CTGF expression. (A) Left. C2C12 myoblasts were incubated with LPA, TGF-β or both at 37 °C during 1 and 3 h.. Then total RNA was isolated and blotted with a specific probe for CTGF. Right. Ratio CTGF/tubulin determined by densitometric analysis. The values shown are the results obtained from three independent experiments and correspond to the mean and standard deviation. *Pb 0.05, in one way ANOVA test. (B) C2C12 cells were transiently transfected with plasmid containing sequence for luciferase under the control of the promoter of CTGF (pCTGF-Luc) together with a plasmid used as a transfection control containing pRL-SV40 sequence. TGF-β, LPA or both was added to the cells and after 24 h, cells were processed, and luciferase activity was determined. The values correspond to the mean and standard deviation of three independent experiments, *P b 0.001, in one-way ANOVA test..
(4) 452. C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. necessary for CTGF induction. We used a dominant negative form of TGF-βRII lacking the cytoplasmic domain (DN-TGF-βRII-HA), which is required, upon binding of TGF-β, to recruit, bind and transphosphorylate TGF-βRI receptors, thereby stimulating their serine/threonine kinase activity [13]. Myoblasts were transiently co-transfected with cDNAs for pCTGF-Luc and for DN-TGF-βRII-HA and incubated with TGFβ1 and LPA. As shown in Fig. 2A (left), a strong inhibition on the. induction of pCTGF-Luc activity in response to LPA was observed. As expected, the induction of pCTGF-Luc activity in response to TGF-β was also negatively affected by the presence of DN-TGF-βRII-HA. Further analysis (Fig. 2A, right) showed the expression of DN-TGF-βRII-HA determined by an immunoblot using anti-HA antibodies. Next, we analyzed the direct role of the serine/threonine kinase activity of TGF-βRI, using the specific inhibitor SB 431542 [37,39]. As. Fig. 2. LPA-induced CTGF expression requires active TGF-β receptors. (A) Left. Myoblasts were co-transfected with plasmids containing sequence for luciferase under the control of the promoter of CTGF (pCTGF-Luc) together with the plasmid pRL-SV40 and a plasmid containing the sequence for a dominant negative form of TGF-βRII containing a HA epitope (DN-RII- HA). The activities in response to TGF-β or LPA were determined after 24 h. Values correspond to the mean and standard deviation of three independent experiments. *Pb 0.005, in one-way ANOVA test. Right. Immunoblots to determine the levels of DN-RII-HA were performed for HA epitope. Levels of tubulin are shown as loading control. (B) C2C12 myoblasts were preincubated for 1 h at 37 °C with DMSO (Control) or an inhibitor of TGF-βRI kinase activity (SB-431542). Then cells were incubated with LPA, TGF-β or both at 37 °C during 3 h. After that, total RNA was isolated and blotted with a specific probe for CTGF or tubulin as a loading control. (C) Left. C2C12 cells were transiently transfected with a plasmid containing the sequence for luciferase under the control of the promoter of CTGF (pCTGF-Luc) together with a plasmid used as a transfection control containing the pRL-SV40 sequence. Then cells were preincubated for 1 h at 37 °C with DMSO (Control) or an inhibitor of TGF-β RI kinase activity (SB-431542). TGF-β or LPA was added, and after 24 h, cells were processed, and luciferase activity was determined. The values correspond to the mean and standard deviation of four independent experiments. *Pb 0.001, in one-way ANOVA test. Right. C2C12 cells were transiently transfected with plasmid containing the sequence for luciferase under the control of the promoter of PAI-1 (p3TP-lux) together with a plasmid to control the transfection (pRL-SV40). Then cells were preincubated with SB-431542 for 1 h at 37 °C, TGF-β was added subsequently for 24 h after which the cells were processed, and luciferase activity was determined. The values correspond to the mean and standard deviation of three independent experiments. *Pb 0.005, in t-test..
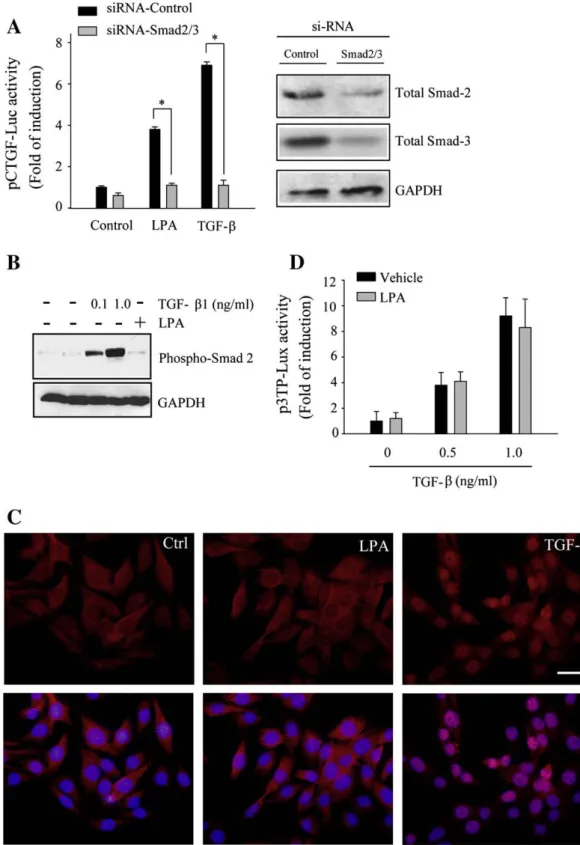
(5) C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. seen in Fig. 2B, SB 431542 strongly prevented CTGF mRNA induction in response to LPA. As expected, the kinase inhibitor also inhibited the induction of CTGF in response to TGF-β1. A similar experiment was performed to determine the effects of TGF-βRI inhibition by evaluating the induction of pCTGF-Luc activity (Fig. 2C, left). The serine/threonine kinase activity inhibitor totally prevented the inductive effect of LPA. Similarly, an inhibitory effect on the induction of luciferase activity in response to TGF-β was also demonstrated (Fig. 2C, left). Fig. 2C (right) shows a positive control for the inhibitory effect of SB 431542 on the induction of a reporter construct in which the promoter of plasminogen activation inhibitor-1 was coupled to luciferase cDNA. This reporter construct, p3TP-lux, was activated upon the binding of TGF-β to its transducing receptors [33]. Myoblasts were transfected with p3TP-lux together with pRL-SV40 (in order to normalize results) and then incubated with TGF-β1 in the presence or absence of SB 431542 [33,40,41]. The inhibitor completely reduced the induction of p3TP-lux activity in response to TGF-β1. These results strongly suggest that the induction of CTGF in response to LPA requires the activity of TGF-βRs, in the absence of ligand. 3.3. LPA requires Smad-2/3 proteins for the induction of CTGF expression, but not their phosphorylation or their nuclear translocation To further determine if the Smad pathway was a requisite for CTGF induction in response to LPA, we tested whether pCTGF-Luc induction required Smad-2/3 expression. To test this hypothesis, we silenced Smad-2/3 via siRNA knockdown. Fig. 3A (left) shows that in myoblasts where Smad-2/3 were silenced, the induction of pCTGF-Luc activity in response to LPA was completely inhibited. A similar result was observed for the induction of pCTGF-Luc activity in response to TGFβ1. This was expected, since the induction of CTGF in response to TGFβ1 is known to be at least partially mediated by Smad proteins which are also involved in the basal expression of CTGF [42]. The results of Smad-2/3 knockdown using siRNA are shown in Fig. 3A (right), and protein levels were determined by Western blot analysis. These results suggest that the expression of CTGF in response to LPA requires the presence of Smad-2/3 proteins. Next, we studied if the downstream steps of the Smad-dependent pathway were required; i.e. Smad-2/3 phosphorylation and Smad-4 nuclear translocation. Myoblasts were incubated with TGF-β and LPA and the levels of phospho-Smad-2 and -3 were determined by Western blot analysis. Fig. 3B shows that, as expected, TGF-β1 stimulated the phosphorylation of Smad-2, but in contrast, LPA did not have any effect on the phosphorylation of Smad-2 or -3. Then, we determined if LPA had the ability to induce the translocation of Smads to the nucleus. Fig. 3C shows that LPA did not induce the nuclear translocation of Smad-4, as expected, since nuclear translocation of Smad-4 associated to Smad-2/3 requires the latter to be phosphorylated [43]. In contrast, as expected, TGF-β1 induced the nuclear translocation of Smad-4. Finally, we studied if LPA was able to induce the specific TGF-β1 reporter p3TP-lux. Fig. 3D shows that LPA did not induce any reporter activity, nor did it have any effect on TGF-β1 induced activity, when both molecules were added. These results strongly indicate that although LPA requires the presence of Smad-2/3 in order to induce CTGF, LPA has no effect on Smad phosphorylation or on their nuclear translocation. 3.4. LPA requires TGF-βR activity to induce actin stress fiber formation but not to induce proliferation We have shown above that LPA induces CTGF by a mechanism that requires the transactivation of TGF-βRs, therefore we analyzed if this requirement was a general feature involved in LPA biological responses. It is known that in myoblasts, LPA is able to induce actin stress fiber formation [44,45] and cell proliferation [46]. Fig. 4A shows that in myoblasts, LPA induced stress fiber formation; however, this induction was significantly abolished by the TGF-βRI serine/threonine. 453. kinase activity inhibitor SB 431542. In contrast, the inhibitor did not have any significant effect on cell proliferation induced by LPA. Fig. 4B shows quantitative analyses of the results on the induction proliferation as determined by stained nuclei counting. Similar results were observed when 3 H-thymidine incorporation was used as a measure of cell proliferation, as shown in Fig. 4C. These results indicate that LPA requires TGF-βRs for stress fiber formation but not for mitogenic activity. 3.5. CTGF induction in response to LPA requires the activation of JNK but not ERK pathways. JNK requirement is independent of TGF-βRI-mediated activity In addition to Smad-mediated transcription, TGF-β activates other signaling cascades, including MAPK pathways [15,47]. Among these, TGF-β can activate the ERK and JNK pathways [17,48]. Above, we showed that the induction of CTGF in response to LPA required active TGF-β-mediated signaling; therefore, we decided to evaluate if other non-Smad-dependent pathways were required for the induction of CTGF. Myoblasts were incubated for varying times with LPA and the levels of phosphorylated-JNK (phospho-JNK) and phosphorylatedERK (phospho-ERK-1/2) were determined. Fig. 5A shows that a transient induction of phospho-JNK and phospho-ERK-1/2 was observed after a 5 min incubation with LPA, which decreased with time. To study if this transient induction of phospho-JNK and -ERK induced by LPA required an active TGF-βRI, myoblasts were incubated with LPA in the presence or absence of SB 431542 and the amount of phospho-JNK and phospho-ERK-1/2 was determined. Fig. 5B shows that the serine/threonine kinase activity of TGF-βRI was not required for induction of phospho-JNK or phospho-ERK-1/2 in response to LPA. Next, we evaluated the direct role of the JNK and ERK pathways on the expression of CTGF in response to LPA. Myoblasts were transfected with the reporter pCTGF-Luc, and the effects of JNK and ERK inhibitors were determined on the induction of CTGF in response to TGF-β1 and LPA. Fig. 5C shows that the specific inhibitor of phospho-JNK (SP 600125) prevented the induction of CTGF reporter activity in response to LPA. In contrast, the MEK inhibitor (UO 126) did not have any effect on CTGF induction in response to LPA. Additionally, we could demonstrate that inhibition of TGF-β1-dependent pathways by SB 431542 affected the expression of the specific CTGF reporter in response to LPA. Finally we evaluated the effect of the JNK inhibitor on the expression of CTGF in response to LPA and TGF-β. Fig. 5D shows that under both experimental situations the expression of CTGF was diminished by the inhibitor. These results suggest that the induction of CTGF by LPA requires the JNK pathway and this induction is independent of serine/threonine kinase activity associated with TGFβRI. 4. Discussion In this paper, we have shown that the expression of CTGF, a key growth factor involved in wound healing and fibrotic diseases, is mediated by both TGF-β and LPA signaling pathways. The latter requires the transactivation of TGF-βRs. To our knowledge, these results are novel, since this is the first study to show that LPA requires the presence of active TGF-βRs to exert at least a part of its biological effects. Previously, it has been reported that S1P was able to transactivate TGFβRs. Prior evidence showed that GPCR ligands are able to activate tyrosine kinase receptors in various cell types. Experimental evidence supports a cross-talk between LPA and tyrosine kinase activity associated with receptors for EGF [27], c-Met ligand [28] and PDGF [29]. This cross-talk provides additional non-canonical signaling pathways which have been shown to regulate cellular function. The described requirement TGF-βRs in the LPA-mediated induction of CTGF expression and stress fiber formation, but not myoblast proliferation, might be an essential step during the fibrotic process..
(6) 454. C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. Fig. 3. CTGF expression induced by LPA requires Smad-2/3 expression but not its phosphorylation or nuclear translocation. (A) Left. Myoblasts were transfected with siRNA control or for Smad-2/3 together with the plasmid containing the sequence for luciferase under the control of the promoter of CTGF (pCTGF-Luc) and the plasmid pRL-SV40 as transfection control. The cells were incubated a 37 °C with TGF-β or LPA, and after 24 h, the luciferase activity was determined. The values correspond to the mean and standard deviation of three independent experiments. *P b 0.001, in one-way ANOVA test. Right. Immunoblots to determine the Smad2, Smad3 and GAPDH protein levels in cells transfected with a siRNA control or siRNA for Smad-2/3. (B) C2C12 cells were incubated with TGF-β or LPA at 37 °C during 30 min. After that, cells were lysed and immunoblotted to detect protein levels of phospho-Smad-2 and GADPH. (C) Myoblasts were incubated with TGF-β or LPA for 60 min and processed for indirect immunofluorescence for Smad-4. The bottom panel shows the merger of Smad-4 with nuclei labeled with Hoechst 33258. The scale bar corresponds to 100 μm. (D) C2C12 cells were transiently transfected with the plasmid containing the sequence for luciferase under the control of the promoter of PAI-1 (p3TP-lux) together with a plasmid to control the transfection (pRL-SV40). Then cells were incubated with the indicated amount of TGF-β alone or together with LPA. After 24 h, cells were processed, and luciferase activity was determined. The values correspond to the mean and standard deviation of three independent experiments. No significant differences were observed with treatment with LPA (one-way ANOVA test)..
(7) C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. 455. Fig. 4. LPA requires TGF-β receptors activity to induce actin stress fiber formation but not to induce myoblasts proliferation. (A) Myoblasts were serum starved for 18 h and then incubated with LPA (LPA), the inhibitor of kinase activity of TGF-βRI SB-431542 (SB), or both (LPA + SB) during 48 h. Then, the cells were processed for indirect immunofluorescence, using phalloidin coupled to fluorescein. Nuclei were labeled with Hoechst 33258 as indicated in Material and methods. The scale bar corresponds to 100 μm. (B) Number of cells incubated with LPA, the inhibitor of kinase activity of TGF-βRI (SB-431542), or both (LPA + SB-431542) during 48 h. The quantification corresponds to the number of Hoechst-labeled nuclei per mm2 (mean and standard deviation of three independent experiments; *P b 0.001, in one-way ANOVA test). (C) Myoblasts were serum starved for 18 h and incubated with LPA, the inhibitor of kinase activity of TGF-βRI (SB-431542), or both (LPA + SB-431542) during 24 h. Growth stimulatory effect was measured by [3 H] thymidine incorporation as described in Materials and methods. Treatment with TGF-β is shown as a positive control of [3 H] thymidine incorporation. The values correspond to cpm mean and standard deviation of three independent experiments; *P b 0.001, in one way ANOVA test.. Elevated levels of TGF-β and LPA have been reported in several fibrotic tissues [1,8,49]. Likely as a consequence of these elevated levels, increased amounts of CTGF have also been described in several fibrotic tissues [50–55]. At the molecular level, we showed that the induction of CTGF by LPA was completely inhibited by the use of a dominant-negative form of the TGF-βRII, a receptor form that is not able to recruit TGF-βRI and is consequently unable to trigger the signaling pathway. We also showed that the inhibition of serine/ threonine kinase activity associated with TGF-βRI inhibited CTGF expression induced by LPA. These experiments were done in the absence of TGF-β, reinforcing the idea that LPA might be transactivating TGF-βRs to exert part of its effect on CTGF expression. Another interesting result on the induction of CTGF mediated by LPA was the fact that the presence of Smad-2/3 was required, although neither their phosphorylation nor their translocation to the nucleus was observed in response to LPA. A similar observation has been previously described for apoptosis induced by TGF-β which is regulated by cross-talk between. the Akt/PKB serine/threonine kinase and Smad-3 through a mechanism that is independent of Akt kinase activity. Akt interacts directly with unphosphorylated Smad-3 to sequester it outside the nucleus, preventing its phosphorylation and nuclear translocation. This results in the inhibition of Smad-3-mediated transcription and apoptosis [56]. On the other hand, it has been shown that, independent of the presence of TGF-β, S1P induces not only the phosphorylation of Smad proteins but also their partnering with Smad-4 and translocation into the nucleus [31,32,57]. We found that the serine/threonine kinase activity of TGF-βRI was not required for the mitogenic effect of LPA. It has been demonstrated that LPA induces phospho-JNK without affecting total JNK levels [46]. We found that LPA-induced phosphorylation of JNK did not require transactivation of TGF-βRI. However, inhibition of JNK phosphorylation had a strong inhibitory effect on the expression of CTGF. Thus, the expression of CTGF can be stimulated by at least two LPA-dependent mechanisms; one dependent on the TGF-β signaling pathway and another independent of TGF-βRs but dependent on JNK phosphorylation..
(8) 456. C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457. Fig. 5. CTGF expression induced by LPA requires the activation of JNK in an TGF-βRI activity independent manner. (A) C2C12 cells were incubated with LPA at 37 °C during the indicated times. After that, cells were lysed and immunoblotted for phospho-JNK and phosphor-ERK. Levels of tubulin are shown as a loading control. (B) Myoblasts were preincubated with the inhibitor of kinase activity of TGF-βRI (SB-431542) and then incubated with LPA at 37 °C. After that, cells were lysed and immunoblotted for phospho-JNK and phosphor-ERK. Levels of tubulin are shown as a loading control. (C) C2C12 cells were transiently transfected with a plasmid containing the sequence for luciferase under the control of the promoter of CTGF (pCTGFLuc) together with a plasmid used as a transfection control containing pRL-SV40 sequence. Cells were preincubated for 1 h with DMSO (Vehicle) or inhibitors for TGF-βRI kinase activity (SB-431542), for JNK activity (SP-600125) and for ERK activity (UO-126). TGF-β or LPA was added, and after 24 h, cells were processed, and luciferase activity was determined. The values correspond to the mean standard deviation of four independent experiments. *P b 0.005, **P b 0.001, in one-way ANOVA test. (D) C2C12 myoblasts were preincubated for 1 h at 37 °C with DMSO (Vehicle) or an inhibitor of JNK activity (SP-600125). Then cells were incubated with LPA (left) or TGF-β (right) at 37 °C during 3 h. After that, total RNA was isolated and blotted with a specific probe for CTGF or tubulin as a loading control.. The role of CTGF in fibrosis has gained attention in recent years [58–61]. CTGF overexpression is known to occur in a variety of fibrotic skin disorders [50,62], in renal fibrosis [51], hepatic fibrosis [52] and pulmonary fibrosis [53]. An increase in CTGF mRNA and protein levels has been reported in dystrophic dogs [54,55], and microarray analyses have indicated increased levels of CTGF in mdx dystrophic mice [63]. In Duchenne muscular dystrophy, TGF-β and fibrosis are augmented [64–67], with similar results found in the diaphragm muscles of dystrophic mdx mice [68,69]. It would be interesting to evaluate the amount of LPA and its receptors in dystrophic muscle to determine its role on CTGF production and its deleterious fibrotic effects. Acknowledgements This study was supported by research grants from FONDAPBiomedicine # 13980001, CARE PFB12/2007, FONDECYT 11080212 and MDA 89419. The Millennium Institute for Fundamental and Applied. Biology (MIFAB) is financed in part by the Ministerio de Planificación y Cooperación (Chile). References [1] D. Pohlers, J. Brenmoehl, I. Löffler, C.K. Müller, C. Leipner, S. Schultze-Mosgau, A. Stallmach, R.W. Kinne, G. Wolf, Biochim. Biophys. Acta 1792 (2009) 746. [2] A.B. Roberts, B.K. McCune, M.B. Sporn, Kidney Int. 41 (1992) 557. [3] C.P. Denton, D.J. Abraham, Curr. Opin. Rheumatol. 13 (2001) 505. [4] A. Holmes, D.J. Abraham, S. Sa, X. Shiwen, C.M. Black, A. Leask, J. Biol. Chem. 276 (2001) 10594. [5] G.R. Grotendorst, Cytokine Growth Factor Rev. 8 (1997) 171. [6] A. Leask, D.J. Abraham, FASEB J. 18 (2004) 816. [7] A.M. Tager, P. LaCamera, B.S. Shea, G.S. Campanella, M. Selman, Z. Zhao, V. Polosukhin, J. Wain, B.A. Karimi-Shah, N.D. Kim, W.K. Hart, A. Pardo, T.S. Blackwell, Y. Xu, J. Chun, A.D. Luster, Nat. Med. 14 (2008) 45. [8] J.P. Pradère, J. Gonzalez, J. Klein, P. Valet, S. Grès, D. Salant, J.L. Bascands, J.S. Saulnier-Blache, J.P. Schanstra, Biochim. Biophys. Acta 1781 (2008) 582. [9] S. Muehlich, N. Schneider, F. Hinkmann, C.D. Garlichs, M. Goppelt-Struebe, Atherosclerosis 75 (2004) 261. [10] M. Goppelt-Struebe, S. Fickel, C.O. Reiser, Biochem. J. 45 (Pt 2) (2000) 217..
(9) C. Cabello-Verrugio et al. / Cellular Signalling 23 (2011) 449–457 [11] C. Vial, L.M. Zúñiga, C. Cabello-Verrugio, P. Cañón, R. Fadic, E. Brandan, J. Cell. Physiol. 215 (2008) 410. [12] F. Lopez-Casillas, C. Riquelme, Y. Perez-Kato, M.V. Ponce-Castaneda, N. Osses, J. Esparza-Lopez, G. Gonzalez-Nunez, C. Cabello-Verrugio, V. Mendoza, V. Troncoso, E. Brandan, J. Biol. Chem. 278 (2003) 382. [13] J. Massagué, Nat. Rev. Mol. Cell Biol. 1 (2000) 169. [14] E. Piek, C.H. Heldin, P. Ten Dijke, FASEB J. 13 (1999) 2105. [15] R. Derynck, Y.E. Zhang, Nature 425 (2003) 577. [16] Y. Imamichi, O. Waidmann, R. Hein, P. Eleftheriou, K. Giehl, A. Menke, Biol. Chem. 386 (2005) 225. [17] S.C. Lien, S. Usami, S. Chien, J.J. Chiu, Cell. Signal. 18 (2006) 1270. [18] L. Yu, M.C. Hébert, Y.E. Zhang, EMBO J. 21 (2002) 3749. [19] A. Rodríguez-Barbero, F. Dorado, S. Velasco, A. Pandiella, B. Banas, J.M. LopezNovoa, Kidney Int. 70 (2006) 901. [20] D.E. Gerrard, A.L. Grant, Domest. Anim. Endocrinol. 11 (1994) 339. [21] E.J. van Corven, A. van Rijswijk, K. Jalink, R.L. van der Bend, W.J. van Blitterswijk, W.H. Moolenaar, Biochem. J. 281 (Pt 1) (1992) 163. [22] F. Hao, M. Tan, X. Xu, J. Han, D.D. Miller, G. Tigyi, M.Z. Cui, Biochim. Biophys. Acta 1771 (2007) 883. [23] W.H. Moolenaar, L.A. van Meeteren, B.N. Giepmans, Bioessays 26 (2004) 870. [24] K.R. Watterson, D.A. Lanning, R.F. Diegelmann, S. Spiegel, Wound Repair Regen. 15 (2007) 607. [25] J.W. Choi, D.R. Herr, K. Noguchi, Y.C. Yung, C.W. Lee, T. Mutoh, M.E. Lin, S.T. Teo, K.E. Park, A.N. Mosley, J. Chun, Annu. Rev. Pharmacol. Toxicol. 50 (2010) 157. [26] N.J. Pyne, C.M. Waters, J.S. Long, N.A. Moughal, G. Tigyi, S. Pyne, Adv. Enzyme Regul. 47 (2007) 271. [27] M. Laffargue, P. Raynal, A. Yart, C. Peres, R. Wetzker, S. Roche, B. Payrastre, H. Chap, J. Biol. Chem. 274 (1999) 32835. [28] D. Shida, J. Kitayama, H. Yamaguchi, K. Hama, J. Aoki, H. Arai, H. Yamashita, K. Mori, A. Sako, T. Konishi, T. Watanabe, T. Sakai, R. Suzuki, H. Ohta, Y. Takuwa, H. Nagawa, Exp. Cell Res. 301 (2004) 168. [29] D.R. Cerutis, A.C. Dreyer, M.J. Vierra, J.P. King, D.J. Wagner, J.L. Fimple, F. Cordini, T.P. McVaney, L.C. Parrish, T.M. Wilwerding, J.S. Mattson, J. Periodontol. 78 (2007) 1136. [30] M. Yamanaka, D. Shegogue, H. Pei, S. Bu, A. Bielawska, J. Bielawski, B. Pettus, Y.A. Hannun, L. Obeid, M. Trojanowska, J. Biol. Chem. 279 (2004) 53994. [31] B. Sauer, R. Vogler, H. von Wenckstern, M. Fujii, M.B. Anzano, A.B. Glick, M. Schäfer-Korting, A.B. Roberts, B. Kleuser, J. Biol. Chem. 279 (2004) 38471. [32] C. Xin, S. Ren, B. Kleuser, S. Shabahang, W. Eberhardt, H. Radeke, M. SchäferKorting, J. Pfeilschifter, A. Huwiler, J. Biol. Chem. 279 (2004) 35255. [33] C. Cabello-Verrugio, E. Brandan, J. Biol. Chem. 282 (2007) 18842. [34] N. Osses, E. Brandan, Am. J. Physiol. Cell Physiol. 282 (2002) C383. [35] E. Brandan, M.E. Fuentes, W. Andrade, J. Neurosci. Res. 32 (1992) 51. [36] P. Chomczynski, N. Sacchi, Anal. Biochem. 162 (1987) 156. [37] R. Droguett, C. Cabello-Verrugio, C. Santander, E. Brandan, Exp. Cell Res. 316 (2010) 2487. [38] J. Villena, E. Brandan, J. Cell. Physiol. 198 (2004) 169. [39] N.J. Laping, E. Grygielko, A. Mathur, S. Butter, J. Bomberger, C. Tweed, W. Martin, J. Fornwald, R. Lehr, J. Harling, L. Gaster, J.F. Callahan, B.A. Olson, Mol. Pharmacol. 62 (2002) 58.. 457. [40] J. Massagué, J. Andres, L. Attisano, S. Cheifetz, F. López-Casillas, M. Ohtsuki, J.L. Wrana, Mol. Reprod. Dev. 32 (1992) 99. [41] C. Santander, E. Brandan, Cell. Signal. 18 (2006) 1492. [42] Y. Chen, I.E. Blom, S. Sa, R. Goldschmeding, D.J. Abraham, A. Leask, Kidney Int. 62 (2002) 1149. [43] J. Massagué, R.R. Gomis, FEBS Lett. 580 (2006) 2811. [44] W.H. Moolenaar, Trends Cell Biol. 4 (1994) 213. [45] A. Graness, K. Giehl, M. Goppelt-Struebe, Cell. Signal. 18 (2006) 433. [46] G. Jean-Baptiste, Z. Yang, C. Khoury, M.T. Greenwood, Biochem. Biophys. Res. Commun. 335 (2005) 1155. [47] J.S. Kang, C. Liu, R. Derynck, Trends Cell Biol. 19 (2009) 385. [48] H. Watanabe, M.P. de Caestecker, Y. Yamada, J. Biol. Chem. 276 (2001) 14466. [49] T.A. Wynn, J. Pathol. 214 (2008) 199. [50] A. Igarashi, K. Nashiro, K. Kikuchi, S. Sato, H. Ihn, M. Fujimoto, G.R. Grotendorst, K. Takehara, J. Invest. Dermatol. 106 (1996) 729. [51] Y. Ito, J. Aten, R.J. Bende, B.S. Oemar, T.J. Rabelink, J.J. Weening, R. Goldschmeding, Kidney Int. 53 (1998) 853. [52] V. Paradis, D. Dargere, M. Vidaud, A.C. De Gouville, S. Huet, V. Martinez, J.M. Gauthier, N. Ba, R. Sobesky, V. Ratziu, P. Bedossa, Hepatology 30 (1999) 968. [53] J.A. Lasky, L.A. Ortiz, B. Tonthat, G.W. Hoyle, M. Corti, G. Athas, G. Lungarella, A. Brody, M. Friedman, Am. J. Physiol. 275 (1998) L365. [54] L. Passerini, P. Bernasconi, F. Baggi, P. Confalonieri, F. Cozzi, F. Cornelio, R. Mantegazza, Neuromuscul. Disord. 12 (2002) 828. [55] G. Sun, K. Haginoya, Y. Wu, Y. Chiba, T. Nakanishi, A. Onuma, Y. Sato, M. Takigawa, K. Iinuma, S. Tsuchiya, J. Neurol. Sci. 267 (2008) 48. [56] A.R. Conery, Y. Cao, E.A. Thompson, C.M. Townsend Jr., T.C. Ko, K. Luo, Nat. Cell Biol. 6 (2004) 366. [57] H.H. Radeke, H. von Wenckstern, K. Stoidtner, B. Sauer, S. Hammer, B. Kleuser, J. Immunol. 174 (2005) 2778. [58] B. Perbal, Lancet 363 (2004) 62. [59] A. Leask, Front. Biosci. 1 (2009) 115 (Elite Ed). [60] D. Abraham, Rheumatology 47 (Suppl 5) (2008) v8 (Oxford). [61] A. Leask, Cell. Signal. 20 (2008) 1409. [62] A. Igarashi, K. Nashiro, K. Kikuchi, S. Sato, H. Ihn, G.R. Grotendorst, K. Takehara, J. Invest. Dermatol. 105 (1995) 280. [63] J.D. Porter, A.P. Merriam, P. Leahy, B. Gong, S. Khanna, Hum. Mol. Genet. 12 (2003) 1813. [64] K. Alvarez, R. Fadic, E. Brandan, J. Cell. Biochem. 85 (2002) 703. [65] P. Bernasconi, C. Di Blasi, M. Mora, L. Morandi, S. Galbiati, P. Confalonieri, F. Cornelio, R. Mantegazza, Neuromuscul. Disord. 9 (1999) 28. [66] R. Fadic, V. Mezzano, K. Alvarez, D. Cabrera, J. Holmgren, E. Brandan, J. Cell. Mol. Med. 10 (2006) 758. [67] V. Mezzano, D. Cabrera, C. Vial, E. Brandan, J. Cell Commun. Signal 1 (2007) 205. [68] J.V. Hartel, J.A. Granchelli, M.S. Hudecki, C.M. Pollina, L.E. Gosselin, Muscle Nerve 24 (2001) 428. [69] S. Cáceres, C. Cuellar, J.C. Casar, J. Garrido, L. Schaefer, H. Kresse, E. Brandan, Eur. J. Cell Biol. 79 (2000) 173..
(10)
Figure



+2

Documento similar
The purpose of the research project presented below is to analyze the financial management of a small municipality in the province of Teruel. The data under study has
“ CLIL describes a pedagogic approach in which language and subject area content are learnt in combination, where the language is used as a tool to develop new learning from a
Serum levels of vascular endothelial growth factor-A 165 , soluble vascular endothelial growth factor receptor 1, and hepatocyte growth factor were assessed by
Podoplanina / antígeno PA2.26 como promotor de la migración e invasión tumoral en carcinomas humanos
To analyze the effects of PA2.26 expression on the phenotype of human epithelial cells, we transiently transfected HeLa carcinoma cells and HaCaT immortalized keratinocytes with
High copy number plasmids, generally smaller than 10kbp that have 10 to 100 copies per cell, appear not to code for partition systems and their stable inheritance must be assured
What is perhaps most striking from a historical point of view is the university’s lengthy history as an exclusively male community.. The question of gender obviously has a major role
Conversely, the evaluation of mRNA levels of genes encoding pro-in flammatory cytokines [interleukin-1 beta (IL-1 b ), tumor necrosis factor alpha (TNF a ), and transforming
Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D 3 is mediated by mutual modulation of receptor expression. The down-regulation
