ÍNDICE DE TABLAS
ÍNDICE DE FIGURAS
ABREVIATURAS
EMA Agencia Europea de Medicamentos (Unión Europea) Muestra procesada de estabilidad EMP en refrigeración Muestra procesada de estabilidad EMP. MCA Muestra de control alto MCB Muestra de control bajo MCD Muestra de control diluido MCM-1 Muestra de control medio 1 MeCN Acetonitrilo.
RESUMEN
INTRODUCCIÓN
La validación de métodos bianalíticos garantiza datos de alta calidad para la presentación de resultados a los organismos reguladores para el desarrollo y descubrimiento de nuevos fármacos (Kadian, Raju et al. 2016). Estas variaciones son importantes y marcan la pauta para la presentación de estudios ante las entidades reguladoras de una región o específicamente de un país (Kadian, Raju et al. 2016).
JUSTIFICACIÓN
TIPO DE INVESTIGACIÓN
ANTECEDENTES
Además, incluyen otros parámetros de evaluación como el efecto matriz, la muestra incurrida y el reanálisis. Además, analizan otros aspectos de la estabilidad para proporcionar un fácil acceso en el diseño de un método bioanalítico y su validación para cumplir con la mayoría de las directrices de las autoridades (Kadian, Raju et al. 2016).

VALIDACIÓN
- A NTECEDENTES HISTÓRICOS
- D EFINICIÓN
- L EGISLACIÓN N ACIONAL E I NTERNACIONAL PARA V ALIDACIÓN DE M ÉTODOS B IONALÍTICOS
2014 Japón, MHLW, Orientación sobre la validación de métodos bioanalíticos (ensayo de unión de ligandos) en el desarrollo farmacéutico. 2018 FDA de Estados Unidos, Inclusión de análisis de muestras planificados, incluye criterios para la validación de métodos cromatográficos y ensayos de unión de ligandos.
BIOANÁLISIS
- A NTECEDENTES HISTÓRICOS
- D EFINICIÓN
- I MPORTANCIA DEL B IOANÁLISIS
- M ÉTODOS B IOANALÍTICOS
- Métodos colorimétricos
- Métodos cromatográficos
La especificidad de los métodos colorimétricos depende del número de sustancias de la muestra que pueden formar el complejo coloreado con el reactivo elegido. Los primeros métodos bioanalíticos basados en cromatografía utilizaban cromatografía de gases (GC) para la separación ante un detector no específico, como un detector de ionización de llama (FID).
BIOEQUIVALENCIA
- D EFINICIÓN
- E STUDIOS DE BIOEQUIVALENCIA
- Regulación
- Métodos para demostrar la bioequivalencia
Para los medicamentos genéricos, los estudios de bioequivalencia confirman la equivalencia clínica entre los productos genéricos y los de referencia. La selección del tipo de estudios de bioequivalencia que se realizarán se basa en el sitio de acción del fármaco y la capacidad del diseño del estudio para comparar la administración del fármaco. (Lawrence X. Yu, 2014).
FARMACOCINÉTICA
Volumen de distribución: el volumen aparente en el cuerpo en el que se distribuye el fármaco. Dado que el volumen de sangre en humanos es de aproximadamente 5 litros, un volumen de distribución significativamente mayor que 5 litros indica que es probable que el compuesto se distribuya en el espacio extravascular.
TOXICOCINÉTICA
DESARROLLO DE MÉTODOS Y VALIDACIÓN
- P RECIPITACIÓN DE PROTEÍNAS (PP)
- E XTRACCIÓN LIQUIDO - LÍQUIDO (LL) Y EXTRACCIÓN CON LÍQUIDO ASISTIDO (ELA)
- E XTRACCIÓN EN FASE SÓLIDA (SPE) EN LÍNEA Y FUERA DE LÍNEA
- R EQUISITOS DE UNA VALIDACIÓN
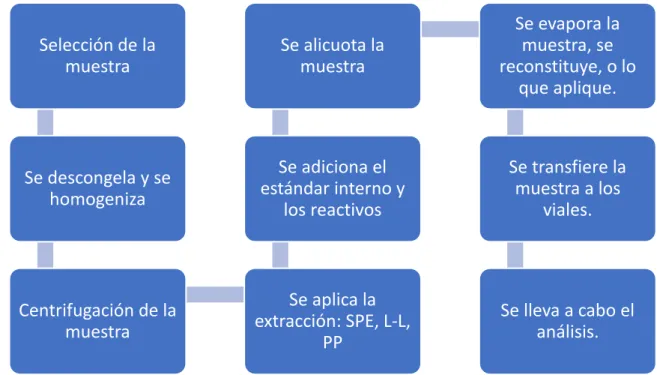
Actualmente, las técnicas de extracción más utilizadas son la extracción por precipitación de proteínas (PP), la extracción líquido-líquido (LL) y la extracción en fase sólida (SPE). En principio, el mecanismo subyacente de la precipitación de proteínas es reducir la solubilidad de los analitos mediante la adición de disolventes orgánicos como metanol (MeOH) o acetonitrilo (MeCN) o sal tampón saturada como sulfato de zinc 10 M (ZnS 4) o una alta concentración. ácido o base como ácido trifluoroacético (TFA) o ácido hidroperclórico (HClO4) al 5-10%. Otra técnica de extracción popular y selectiva ampliamente utilizada en bioanálisis es la extracción en fase sólida (SPE).
Al manipular la polaridad y el pH de la fase móvil, los analitos de interés o las impurezas no deseadas pasan a través de la fase estacionaria secuencialmente de acuerdo con sus propiedades físicas y químicas. Aunque el mecanismo de retiro es el mismo que el del SPE tradicional, el SPE en línea ofrece varias ventajas. Debido a que la preparación de la muestra se realiza durante el análisis, se elimina el tiempo necesario para la preparación de la muestra y, por lo tanto, aumenta significativamente el rendimiento.
Cuando se requiere una solución mixta para la preparación de estándares y puntos de control de calidad en la matriz, el volumen agregado debe ser menor al 5% del volumen total de la matriz (Lowes, 2017). Algunos laboratorios requieren que se establezca la estabilidad de la solución madre antes de la validación formal. Dos estándares de calibración deben estar en concentraciones inferiores a la muestra de control baja, y dos estándares de calibración deben estar en concentraciones superiores a la muestra de control alta.

INTRODUCCIÓN A LOS PARÁMETROS DE VALIDACIÓN: DEFINICIONES, MÉTODOS Y
- S ELECTIVIDAD
- E XACTITUD
- P RECISIÓN
- S ENSIBILIDAD O LÍMITE INFERIOR DE CUANTIFICACIÓN (LIC)
- C URVA DE CALIBRACIÓN (L INEALIDAD )
- E STABILIDAD
- E FECTO DE LA MATRIZ
- R ECOBRO
- R EPRODUCIBILIDAD
- E STÁNDARES DE REFERENCIA
El grado en que los resultados de la prueba del método se acercan al valor (concentración) real (o nominal). La relación entre la concentración del analito y la respuesta del instrumento (también conocida como curva estándar). La estabilidad del analito en las matrices, contenedores y condiciones utilizadas para la recolección, almacenamiento y análisis de muestras.
El cambio en la respuesta del analito debido a la presencia de otros componentes de la matriz. Eficiencia de extracción de un método analítico, expresada como porcentaje de la cantidad conocida de analito, obtenida de la comparación de los resultados analíticos de las muestras agregadas a los estándares y sometidas al proceso de extracción, con los resultados. La precisión del método en las mismas condiciones operativas durante un corto período de tiempo (varias ejecuciones o días).
Durante la validación del método y el análisis de muestras se deben utilizar estándares analíticos de identidad y pureza conocidas. Para cada lote utilizado debe estar disponible un certificado de análisis u otra documentación que confirme la identidad y pureza del estándar.

ANÁLISIS COMPARATIVO ENTRE LAS GUÍAS NACIONALES E INTERNACIONALES DE
- S ELECTIVIDAD Y E SPECIFICIDAD
- Definición
- Método
- Criterios de Aceptación
- E XACTITUD
- Definición
- Método
- Criterios de aceptación
- P RECISIÓN
- D EFINICIÓN
- M ÉTODO
- C RITERIOS DE ACEPTACIÓN
- R ECOBRO
- D EFINICIÓN
- M ÉTODO
- C RITERIO DE A CEPTACIÓN
- L ÍMITE DE CUANTIFICACIÓN
- D EFINICIÓN
- M ÉTODO
- C RITERIOS DE ACEPTACIÓN
- L INEALIDAD Y C URVA DE C ALIBRACIÓN
- Definición
- Método
- Criterios de Aceptación
- E STABILIDAD
- D EFINICIÓN
- M ÉTODO
- C RITERIOS DE A CEPTACIÓN
El patrocinador debe analizar al menos seis muestras en blanco de la matriz biológica adecuada (por ejemplo, plasma) (para métodos cromatográficos). Analizar espacios en blanco de la matriz biológica apropiada de al menos seis fuentes individuales. La NOM 177 SSA1 2013 establece que el intervalo de la curva de calibración debe determinarse con base en las concentraciones esperadas del analito a cuantificar durante el análisis de la muestra.
Se debe utilizar un mínimo de seis niveles de concentración de calibración además de la muestra en blanco (muestra de matriz procesada sin analito y sin IS) y una muestra cero (matriz procesada con IS). Las muestras en blanco y cero no deben tenerse en cuenta al calcular los parámetros de la curva de calibración. Se deben informar los parámetros de la curva de calibración (pendiente e intersección para ajuste lineal).
Las respuestas de las muestras en blanco y nulas no deben usarse en la construcción de la ecuación. La ecuación de la curva no debe incluir estándares de calibración que no cumplan con los criterios de aceptación. Al menos el 75% de las concentraciones de la curva de calibración con un mínimo de 6 puntos deben cumplir este criterio.
Los calibradores FDA 2018, distintos de la muestra cero, deben tener ± 15 % de las concentraciones nominales, excepto en LIC. La prueba de la estabilidad del fármaco en un líquido se mide en función de.
DISCUSIÓN
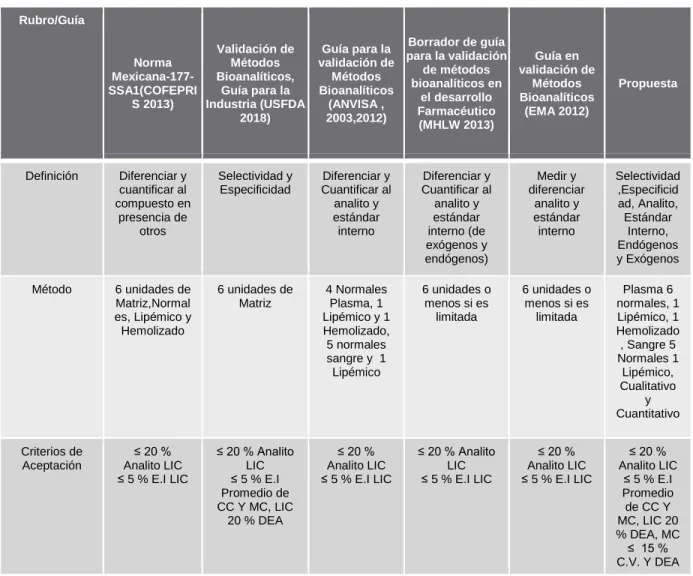
Tanto COFEPRIS 2013, FDA 2018 como EMA 2012 comentan, pero no específicamente, sobre el análisis de selectividad, incluyendo metabolitos, fármacos concomitantes, anticoagulantes especiales y todo lo que se considere que interfiere con la matriz, pero en ninguno de ellos. Establecer el método de evaluación cuantitativa de la selectividad. La ANVISA 2012, por otro lado, tiene una definición simplificada que tiene como objetivo medir la precisión en una matriz. En general, todas las guías son consistentes en el método de análisis, diferenciándose solo en el número de réplicas o niveles de concentración evaluados FDA 2018 es la única guía que establece claramente que se pueden evaluar más de 5 réplicas de cada nivel de concentración, EMA 2012. Dice que sólo se evalúan 4 niveles de concentración, tratando la integridad de la dilución como un parámetro independiente.
Este es uno de los parámetros de validación que más llama la atención, ya que no es considerado por todas las guías (EMA 2012 y NOM 177 SSA1 2013). Por otro lado, MHLW 2013 y FDA 2018 coinciden en la comparación de la señal de las muestras procesadas y en solución del analito y el estándar interno por separado, mientras que ANVISA 2012 indica que la comparación debe realizarse con la relación de área del analito. y del patrón interno por separado. En general todas las guías coinciden con el concepto, esto es de esperarse ya que es un término particularmente definido sin tendencias a variaciones en la conceptualización de la idea, la NOM 177 y MHLW 2013 no incluyen en su definición que la respuesta generada por el analito Es emitido por un instrumento, por el contrario ANVISA 2012, EMA 2012 y FDA 2018 sí incluyen que el instrumento es el que genera.
Para modelos no lineales, se debe incluir un mínimo de 8 (ocho) muestras de diferentes concentraciones en la curva de calibración y la ecuación de la curva no debe incluir estándares de calibración que no cumplan con los criterios de aceptación. Cuando un estándar de calibración cumple con los criterios de aceptación, no debe excluirse de la ecuación de la curva. Los autores manifiestan que la validación se realizó con los criterios de la FDA, sin embargo, al no evaluar la estabilidad de la molécula en sangre, no se cumplieron los criterios de la EMA 2012 y ANVISA 2012, por lo que esta prueba no cumple con los lineamientos de todas las guías mencionadas. en este papel.
CONCLUSIONES
Levonorgestrel al plasma de un implante subdérmico, evaluar la estabilidad a niveles medio, bajo y alto, seis veces en las condiciones de estabilidad de mesa, estabilidad a largo plazo, ciclos de congelación-descongelación, estabilidad de la muestra tratada.
34;Desarrollo, validación y uso de un método LC-MS/MS de alta sensibilidad para la cuantificación de levonorgestrel liberado de un implante subdérmico en plasma humano.” J Chromatogr B Analyt Technol Biomed Life Sci. NORMA Oficial Mexicana NOM-177- SSA1-2013 , Estableciendo las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos que deben cumplir los terceros autorizados, centros de investigación o instituciones hospitalarias que realicen pruebas de biocomparabilidad.
34;LC-MS/MS method for simultaneous quantification of dexmedetomidine, dezocine and midazolam in rat plasma and its application to their pharmacokinetic study." J Anal Methods Chem. Draft Guideline on Bioanalytical Method Validation in Pharmaceutical Development Japan, Ministry of Health, Work and well-being.34;A rapid LC-MS/MS assay for the quantification of piperacillin and tazobactam in human plasma and pleural fluid; application to a clinical pharmacokinetic study." J Chromatogr B Analyte Technol Biomed Life Sci.
34;Development and validation of an LC-MS/MS method for the quantification of bictegravir in human plasma and its application to an intracellular uptake study." Biomed Chromatogr: e4379.
Índice de Figuras