Cmax Maximum serum concentration Cmin Minimum serum concentration CPP Critical Process Parameter CQA Critical Quality Attribute. NCPP Non-Critical Process Parameter NF National Formulary (United States) NK Cell Natural Killer Cell. NKPP Non-key process parameter NLR Neutrophil-to-lymphocyte ratio NOR Normal operating range NSCLC Non-small cell lung cancer ORR Objective response rate.
PD-1 Programmed Cell Death-1 PDE Allowed Daily Exposure PD-L1 Programmed Cell Death Ligand-1 PFS Progression Free Survival Ph.
Background information on the procedure
- Submission of the dossier
- Legal basis, dossier content
- Information on paediatric requirements
- Information relating to orphan market exclusivity
- Similarity
- New active substance status
- Scientific advice
- Steps taken for the assessment of the product
The CHMP Rapporteur's first assessment report was circulated to all CHMP and PRAC members on . The CHMP co-rapporteur's first assessment report was circulated to all CHMP and PRAC members. The CHMP Co-Rapporteur's Critique Report was circulated to all CHMP and PRAC members on 28 March.
The CHMP rapporteurs circulated the joint assessment report of the CHMP and PRAC rapporteurs on the responses on the outstanding list.
Scientific discussion
Problem statement
- Disease or condition
- Epidemiology
- Biologic features
- Clinical presentation, diagnosis and stage/prognosis
- Management
The addition of chemotherapy to nivolumab + ipilimumab, a combination of PD-1/CTLA-4 inhibitors, showed superior efficacy compared to chemotherapy alone with early disease control at all levels of PD-L1 expression (Paz-Ares et al [Checkmate 9LA] 2021), which received a positive CHMP opinion in September 2020 (EMEA/H/C/WS1783). Unmet medical need: Immunotherapy-based treatments are the 1L standard of care in patients with advanced metastatic non-small cell lung cancer whose tumors do not harbor driver mutations (NCCN Clinical Practice Guidelines in Oncology Version 2.2021). Overall, there is therefore a need for newer treatment options that can explore the potential of immunotherapy strategies and benefit a wider patient population.
About the product
Tremelimumab AstraZeneca is for intravenous use and is administered as an intravenous infusion after dilution over 1 hour. When Tremelimumab AstraZeneca is given in combination with durvalumab and platinum-based chemotherapy, Tremelimumab AstraZeneca is given first, followed by durvalumab and then platinum-based chemotherapy on the day of administration. When Tremelimumab AstraZeneca is given as the fifth dose in combination with durvalumab and pemetrexed maintenance at week 16, Tremelimumab AstraZeneca is given first, followed by durvalumab, and then pemetrexed maintenance on the day of dosing.
During Cycle 1, Tremelimumab AstraZeneca should be followed by durvalumab starting approximately 1 hour (maximum 2 hours) after the end of the Tremelimumab AstraZeneca infusion.

General comments on compliance with GCP
Platinum-based chemotherapy infusion should start approximately 1 hour (maximum 2 hours) after completion of the durvalumab infusion.
Quality aspects
- Introduction
- Active substance
- Finished medicinal product
- Discussion on chemical, and pharmaceutical aspects
- Conclusions on the chemical, pharmaceutical and biological aspects
- Recommendation(s) for future quality development
The biological activity (potency) of the active substance is determined using a cell-based potency test. A process flow diagram for the manufacture of the final product is provided in the dossier. Most of the analytical methods used for testing the finished product are identical to those used for testing the active substance.
Information on the development, manufacture and control of the active substance and the finished product has been presented in a satisfactory manner.
Non-clinical aspects
- Introduction
- Pharmacology
- Pharmacokinetics
- Toxicology
- Ecotoxicity/environmental risk assessment
- Discussion on non-clinical aspects
- Conclusion on the non-clinical aspects
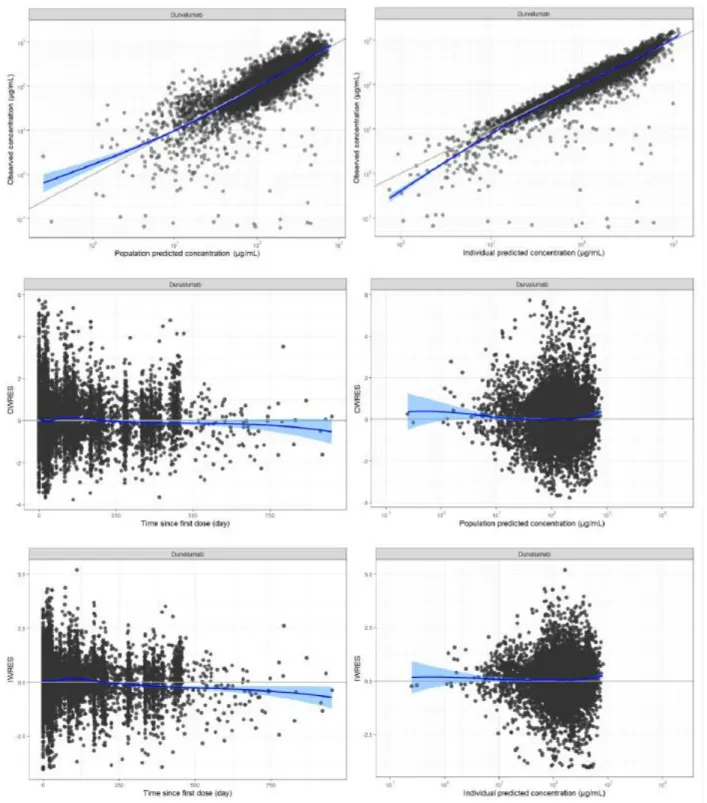
The intensity of color generated is directly proportional to the concentration of tremelimumab in the sample. In this assay, ADA could not be detected in the presence of tremelimumab above LLOQ of the bioanalytical method. It should be mentioned that the nAb assay was not functional in the presence of tremelimumab above LLOQ of the bioanalytical assay.
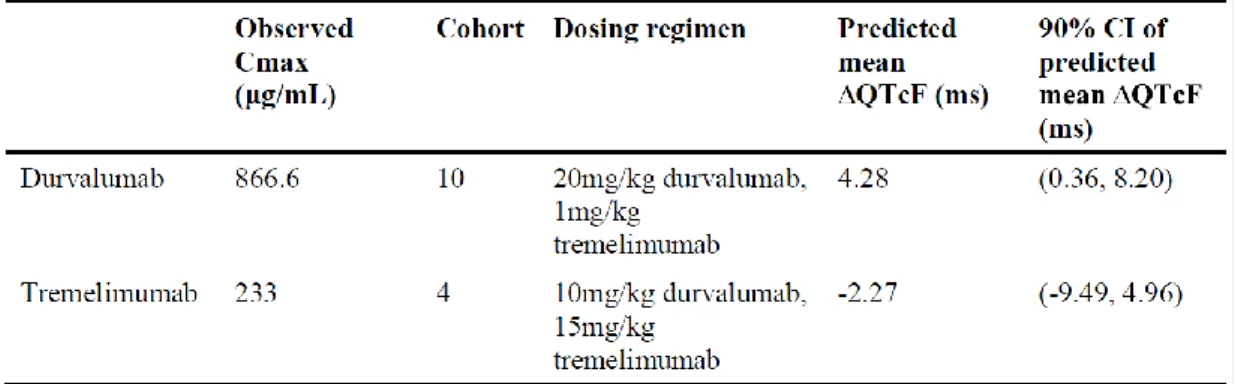
This is a very low dose compared to the highest doses used in the toxicity studies (50 and 30 mg/kg/week).
Clinical aspects
- Introduction
- Clinical pharmacology
- Discussion on clinical pharmacology
- Conclusions on clinical pharmacology
- Clinical efficacy
- Discussion on clinical efficacy
- Conclusions on the clinical efficacy
- Clinical safety
- Discussion on clinical safety
- Conclusions on the clinical safety
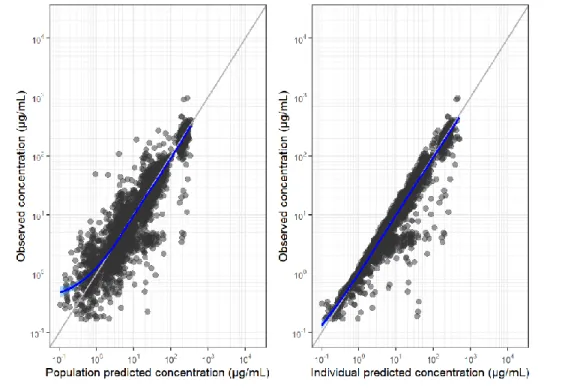
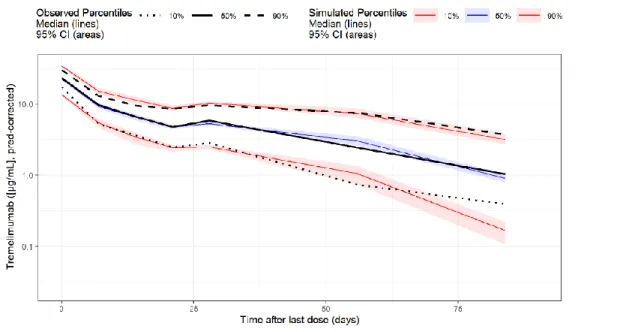
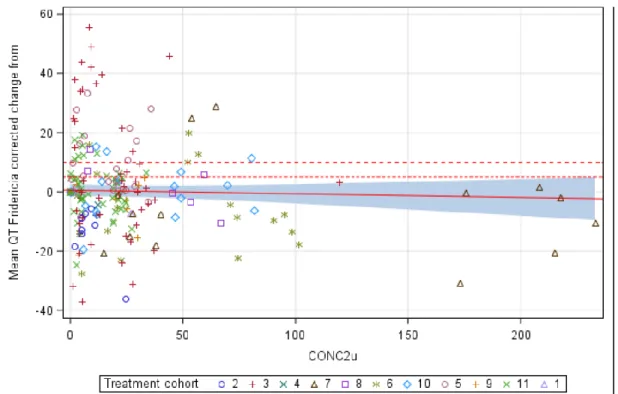
Age (range 22 to 97 years) was not identified as a significant covariate in the final PopPK model of tremelimumab. PD-L1 (PD-L1 ≥50% vs. PD-L1 <50%), histology (squamous cell vs. non-squamous cell), and disease stage (stage IVA vs. stage IVB) in the strata statement, and the CI calculated using of a profile probability approach. The dual primary endpoints of BICR-PFS and OS were analyzed in the ITT of the D+SoC vs.
SoC arms, while identical secondary endpoints were evaluated in the ITT of the T+D+SoC vs. Follow-up treatment/PFS2: A significantly higher percentage of patients received follow-up treatments in the SoC arm (60%) compared to either experimental arm (41% in T+D+SoC, 44% in D+SoC). Across the three POSEIDON arms of the PFS2 events, deaths were in the absence of second progression.
Most events were of CTCAE grade 1 or 2 severity with 1 patient (0.3%) in the T + D + SoC arm experiencing a CTCAE grade 3 event. The number of patients with events divided by the total number of patients in the age group, multiplied by 100. The number of patients with events divided by the total number of patients in the sex group, multiplied by 100.
Number of patients with events divided by the total number of patients in the racial group, multiplied by 100. Number of patients with events divided by the total number of patients in the geographic region group, multiplied by 100. POSEIDON: Of the 286 evaluable durvalumab patients in the same arm, any visit tested positive for durvalumab.
Of the 278 tremelimumab ADA evaluable patients in the T+D+SoC arm, they tested positive for tremelimumab ADA at each visit. This is consistent with the overall higher incidence of hepatobiliary disorders (8.2% patients in the T+D+SoC arm vs. 3.3% in the SoC arm). AEs by ADA status: The proportion of patients with anti-tremelimumab antibodies in the T+D+SoC arm and the T+D pan tumor pool was similar (16% and 13%, respectively), but those for anti-durvalumab antibodies were higher in POSEIDON (15% and 6%, respectively).
Risk Management Plan
- Safety concerns
- Pharmacovigilance plan
- Risk minimisation measures
- Conclusion
Undoubtedly, the addition of dual checkpoint inhibition (PD-L1 and CTLA-4) to a spinal platinum doublet results in higher overall toxicity in the target population, which should be considered in the context of frail patients, especially those of older age or more comorbidities.
Pharmacovigilance
- Pharmacovigilance system
- Periodic Safety Update Reports submission requirements
Product information
- User consultation
- Labelling exemptions
- Additional monitoring
Benefit-Risk Balance
- Therapeutic Context
- Disease or condition
- Available therapies and unmet medical need
- Main clinical studies
- Favourable effects
- Uncertainties and limitations about favourable effects
- Unfavourable effects
- Uncertainties and limitations about unfavourable effects
- Effects Table
- Benefit-risk assessment and discussion
- Importance of favourable and unfavourable effects
- Balance of benefits and risks
- Additional considerations on the benefit-risk balance
- Conclusions
However, the other primary PFS endpoint comparing the same arms showed statistical superiority and so alpha passed to the next level of testing, where OS and PFS were evaluated as key secondary endpoints in the T+D+SoC vs. a smaller effect in the subgroup of non-smoking patients has already been observed in previous immunotherapy studies. However, both subgroups were less represented in the T+D+SoC arm compared to the SoC arm.
Uncertainty about efficacy (and safety) in this subset of patients is reflected in the summary of product characteristics. Typical chemotherapy-related adverse events (anaemia, nausea, neutropenia, decreased appetite, and fatigue) were the five most common adverse events in the three arms of the trial, with a slightly higher incidence in the T+D+SoC arm compared to the SoC arm. Diarrhea and rash with potentially immune-related pathophysiology were significantly more common in the T+D+SoC arm than in the SoC arm (22% and 19%, respectively, versus 15% and 7%, respectively).
Most of these events were related to infections and cardiac events, with twice as many toxic and infectious deaths occurring in the T+D+SoC arm compared to the other two arms (15, 8, and 9, respectively ). The distribution of specific imAEs in the D+SoC arm is characteristic of PD-L1 inhibition, with a predominance of hypothyroidism (6%), hepatotoxicity (3%), pneumonitis (3%), and dermatitis/rash (2%). Pneumonia was the most common SAE in the trial and its incidence in the T+D+SoC group was double that of the SoC control group (11% vs. 5%).
Severe pneumonitis and colitis/diarrhea were more common in the T+D+SoC arm than in the other two arms. The overall percentage of patients who discontinued treatment due to AE was higher in the experimental arms (22% in T+D+SoC, 20% in D+SoC) than in the control arm (15%).
Recommendations
As detailed in the safety section, all adverse event categories have a numerically higher incidence in the trial arms, particularly in the 4-drug combination included in the therapeutic indication of tremelimumab. As expected, immune-mediated events predominated in both trial arms, and although most were low-grade and manageable (eg, hypothyroidism, rash), potentially symptomatic events (eg, diarrhea/colitis, pneumonitis) occurred primarily in the with tremelimumab. Although the combination of tremelimumab, durvalumab, and a platinum-based agent does not appear to meet an unmet medical need in the current therapeutic paradigm for advanced non-small cell lung carcinoma, it could be considered another appropriate chemoimmunotherapy regimen in this palliative setting.
The addition of tremelimumab and durvalumab to chemotherapy results in significantly increased toxicity, particularly in relation to a higher incidence of serious and grade 5 adverse events. The overall benefit-risk ratio of Tremelimumab AstraZeneca in combination with durvalumab and platinum-based chemotherapy for the first-line treatment of adults with metastatic non-small cell lung cancer without sensitizing EGFR mutations or positive ALK mutations is positive under the conditions stated in the "Recommendations" section. The requirements for the submission of periodic safety update reports for this medicinal product are set out in the list of Union Reference Dates (EURD list) provided for in Article 107c(7) of Directive 2001/83/EC and any subsequent updates published on the European Medicines Web Portal.
Conditions or restrictions regarding the safe and effective use of the medicine. The marketing authorization holder (MAH) must carry out the required pharmacovigilance activities and interventions as set out in the agreed RMP presented in Module 1.8.2 of the marketing authorization and any agreed subsequent updates of the RMP. Prior to the launch of Tremelimumab AstraZeneca in each Member State, the MAH will agree the content and format of the educational programme, including communication media, distribution modalities and any other aspects of the programme, with the National Competent Authority.
Conditions or restrictions regarding the safe and effective use of the medicinal product to be applied by the Member States. Based on the CHMP's review of the data available, the CHMP considers that tremelimumab should be classified as a new active substance in its own right as it is not a component of a medicinal product previously authorized in the European Union.
Appendix
CHMP AR on New Active Substance (NAS) dated 15 December 2022