A todo el personal de espectroscopia del Instituto de Química de la UNAM, en especial a Kimi Rocío Patiño, quien siempre tiene una sonrisa amigable. El presente trabajo se realizó en los laboratorios 1-1 y 1-5 del Instituto de Química de la UNAM, bajo la dirección del Dr. Acoplamiento del dipéptido 79 con depsitripéptido 28 y pruebas de ciclación mediante el método de la azida………..… …….
INTRODUCCIÓN
MARCO TEÓRICO
CIANOBACTERIAS
- Clasificación. 8
La clasificación de las cianobacterias aún no ha sido completamente validada por el Código Internacional de Nomenclatura Bacteriana. La gran mayoría de las cianobacterias tienen una estructura distintiva, que se muestra esquemáticamente en la Figura 1. 2.1.3.3.4 Carboxisomas (C).18 Todas las cianobacterias contienen carboxisomas (en promedio de 3 a 12 carboxisomas), que son agregados pseudocristalinos de la enzima principal. . para la fijación de CO2, ribulosa 1,5-bisfosfato-carboxilasa (RuBisCOase).

Gramicidina S 6
- Depsipéptidos
- Síntesis Química de Depsipéptidos
- PLANTEAMIENTO DEL PROBLEMA
- OBJETIVOS
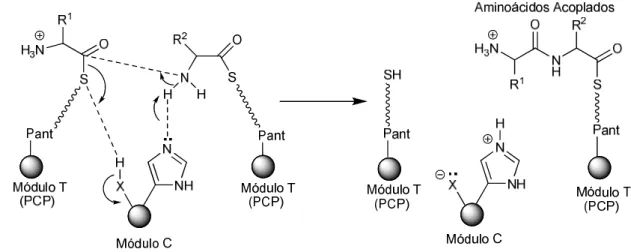
Cuando se inhibe la hidrólisis de GTP, también se inhibe el intercambio de tubulinas y como resultado hay una despolimerización de los microtúbulos ya formados y una inhibición de la formación de nuevos microtúbulos. La síntesis de depsipéptidos ha ayudado enormemente al desarrollo de la química de los agentes de acoplamiento porque muchas de las moléculas constituyentes son grandes y muchos de los aminoácidos constituyentes están N-metilados, lo que provoca un alto impedimento estérico en el momento del acoplamiento. Luesch, et al.4 informaron en 2002 de la adquisición de 6 nuevos depsipéptidos a partir de extractos lipófilos de la cianobacteria Lyngbia sp, a los que denominaron ulongamidas A-F.

GENERAL
PARTICULARES
DISCUSIÓN DE RESULTADOS
- Acoplamiento del dipéptido 80 con el depsitripéptido 28 y obtención de la ulongamida A (1)
39 Otra fragmentación de la molécula a través del sitio b produce el depsitripéptido objetivo 28 y el dipéptido 29. El paso final de la síntesis fue la protección del extremo N-terminal de 33 con Boc, que se llevó a cabo a temperatura ambiente usando dioxano-agua (50 :50) como solvente. . La segunda ruta utilizó L-alanina 69, que es un aminoácido disponible comercialmente.
Además, el grupo protector (Boc) se introduce en la molécula desde el inicio de la síntesis. El primer paso en la síntesis fue la protección del grupo amino de L-alanina 69 con Boc, produciendo N-Boc-L-alanina 72 con un rendimiento del 87%. Durante el desarrollo de la síntesis para realizar los enlaces de los fragmentos constituyentes de la ulongamida A (1), se utilizaron dos agentes de acoplamiento: diciclohexilcarbodiimida (DCC, 17) y benzotriazol-1-il-oxi-tris-(dimetilamino)hexafluorofosfato). -fosfonio (BOP, 21), y el aditivo hidroxibenzotriazol (HOBt, 18).
En el esquema de síntesis original de ulongamida A (1), se propuso sintetizar y utilizar el dipéptido 29; Sin embargo, debido a dificultades experimentales durante el desarrollo de la síntesis, hubo que sintetizar otros dos dipéptidos 79 y 80 (Figura 26). El dipéptido 80 se sintetizó mediante dos rutas diferentes (Esquema 16), para las cuales se prepararon ésteres de N-metil-terc-butilo. La síntesis de 31d se realizó de la misma manera que la de 31c, con la única diferencia de que se inició con Z-L-valina 86.
En la ruta I de la síntesis del dipéptido 80 (Esquema 16), los aminoácidos N-metil-Z-L-fenilalanina 32b y el éster terc-butílico de N-metil-L-valina 31c se unieron mediante BOP, dando el dipéptido diprotegido 80a. . con una eficiencia del 58%. En la ruta II, el compuesto 32b y el éster terc-butílico de L-valina 31d se acoplaron mediante DCC/HOBt para producir el dipéptido 94 con un rendimiento del 87%. La señal en δ (ppm) se observa como un multiplete que integra cuatro átomos de hidrógeno y corresponde a los dos grupos metileno de la cadena hidrocarbonada.

NOESY
CONCLUSIONES
Aunque durante el trabajo experimental se realizaron varios cambios en el diseño de la síntesis, ésta tuvo éxito, ya que se obtuvieron los fragmentos planificados, el depsitripéptido 28 y un derivado del dipéptido 29, el dipéptido 80, que se acoplaron convergentemente para producir el depsipentapéptido lineal 97. que al degradarse produjo el también planificado depsipentapéptido 30, que logró la ciclación y ulongamida A (1). El éxito en la ciclación también indicó que el sitio situado entre los fragmentos de valina y el aminoácido tiazol elegido para llevarla a cabo era el adecuado. Los cambios más importantes en el patrón de síntesis se realizaron en el dipéptido 29, el primero de los cuales fue la N-metilación de sus fragmentos constituyentes Val y Phe, que inicialmente se llevó a cabo mediante el método de Quitt con pérdida de pureza quiral. ;.
Es importante señalar que el método de Benoiton se ha convertido actualmente en la forma clásica de N-metilación de aminoácidos. Se realizó otro cambio en los grupos protectores: el dipéptido 29, el segundo fragmento convergente, no pudo acoplarse al 28 debido a su alta reactividad, lo que indica que la elección del bencilo como grupo protector no era apropiada. En general, la síntesis del depsitripéptido 28 no causó muchos problemas, a excepción de la síntesis del fragmento correspondiente al aminoácido tiazol 34, que se simplificó sustituyendo el ácido propiólico por L-alanina como material de partida para su síntesis; mientras que para la síntesis del β-aminoácido 33, la reacción de Mitsunobu permitió la introducción de un grupo azido por inversión de configuración y mejoró la síntesis descrita.
La elección del método de ciclación es importante: la ciclación con el método de la azida dio malos resultados, conduciendo principalmente a la reducción del carbonilo en el extremo carboxilo del depsipentapéptido 90 al aldehído 91. La ciclación y la obtención de ulongamida A (1) fueron exitosas mediante disolvente para DMF, aumentando la temperatura (80-90 °C) y utilizando un agente de acoplamiento, BOP (23), lo que sugirió la necesidad de proporcionar a la molécula mayor energía y fuerza nucleofílica para poder ser ciclada. Se encontró que el agente de acoplamiento BOP era adecuado para realizar la ciclación final y obtener ulongamida A (1).
Los métodos elegidos para la determinación de la pureza quiral: rotación óptica y preparación de derivados fueron una herramienta muy útil e importante, ya que permitieron identificar la ulongamida A (1) con sus centros quirales en la configuración deseada.
PERSPECTIVAS
PARTE EXPERIMENTAL
- Métodos Generales
Se secó sobre sulfato de sodio anhidro y se destiló a presión reducida, combinando la fracción con p. Se seca con sulfato de sodio anhidro y se evapora el disolvente para dar 27,42 g (93%) de 49 en forma de un aceite amarillo. Después de secar con sulfato de sodio anhidro y evaporar el disolvente, el residuo se purificó mediante cromatografía ultrarrápida en columna usando una mezcla de hexano-acetato de etilo (80:20) como eluyente.
Se separaron las fases, la fase orgánica se secó con sulfato de sodio anhidro, se tituló con solución de hidróxido de sodio 0,5 N y se almacenó en una botella de color ámbar herméticamente cerrada. Después de secar con sulfato de sodio anhidro y evaporar el diclorometano, el residuo se purificó mediante cromatografía en columna (gel de sílice con una mezcla de hexano-acetato de etilo (80:20) como eluyente). La fase orgánica se secó con sulfato de sodio. el disolvente se evaporó y el residuo se purificó mediante cromatografía ultrarrápida en columna, usando una mezcla de hexano-acetato de etilo (80:20) como eluyente.
Se acidificó con solución de ácido clorhídrico 1 N, se extrajo con diclorometano (3 x 50 ml), se secó con sulfato de sodio anhidro y se filtró. La fase orgánica se secó con sulfato de sodio anhidro, el disolvente se evaporó y el residuo se purificó mediante cromatografía en columna (gel de sílice 230-400), usando una mezcla de acetato de etilo-hexano (90:10) como eluyente. La fase orgánica se secó con sulfato de sodio anhidro, el disolvente se evaporó y el residuo se purificó mediante cromatografía ultrarrápida en columna, usando una mezcla de hexano-acetato de etilo-trietilamina como eluyente.
Después de secar con sulfato de sodio anhidro y evaporar el disolvente, el residuo se purificó mediante cromatografía en columna (gel de sílice 230-400) usando una mezcla de hexano-acetato de etilo (70:30) como eluyente.
Metil- L -valinato de bencilo (31a)
Los extractos orgánicos ácidos se combinaron, se secaron con sulfato de sodio anhidro y el disolvente se evaporó, produciendo 19 g (89,0%) de N-metil-Boc-L-valina 83 en forma de un aceite amarillo. El producto se purificó mediante cromatografía ultrarrápida en columna usando una mezcla de hexano-acetato de etilo (90:10) como eluyente para producir 23,34 g (84%) de 84 en forma de un aceite amarillo pálido. Se evaporaron el disolvente y el exceso de ácido trifluoroacético, se añadió agua helada (250 ml) y se basificó con una solución saturada de bicarbonato.
Metil-Boc- L -fenilalanina (32a)
Los extractos orgánicos ácidos se combinaron, se secaron con sulfato de sodio anhidro y se evaporó el disolvente. La purificación se llevó a cabo mediante cristalización en una mezcla de acetato de etilo-hexano, dando 37,04 g (87,7%) de N-metil-Boc-L-fenilalanina 32a en forma de un sólido blanco, p.f.
Metil-Z- L -fenilalanina (31b)
Metil- L -valin [N´-(Boc)]-hidrazida (31b)
- REFERENCIAS
99 El residuo se purificó por cristalización en acetato de etilo-hexano, proporcionando 29,2 g (93%) de N-metil-Z-L-fenilalanina 32b como un aceite amarillo pálido. Los extractos orgánicos se combinaron, se secaron sobre sulfato de sodio anhidro y se evaporó el disolvente. Se filtró a través de celite y el disolvente se evaporó hasta sequedad, dejando el residuo obtenido durante al menos una hora en la bomba de alto vacío antes de usarlo en la siguiente reacción.
Filtrar a través de celite y evaporar el disolvente a una temperatura no superior a 30 °C y una presión no inferior a 20 mmHg durante 20 minutos, porque el éster es muy volátil. Filtrar a través de celite y evaporar el disolvente a una temperatura no superior a 30 °C y una presión no inferior a 20 mmHg durante 20 minutos, porque el éster es muy volátil. Se enfrió a entre 0 - 5ºC y se añadió diciclohexilcarbodiimida 17 (4,3 g, 20 mmol) disuelta en diclorometano (15 ml) en tres porciones, con intervalos de 10 minutos entre cada adición.
La fase orgánica se secó sobre sulfato de sodio anhidro y el disolvente se evaporó para dar 8,14 g (87%) de 94 en forma de un aceite amarillo pálido. La fase orgánica se secó sobre sulfato de sodio anhidro y el disolvente se evaporó para dar 4,1 g (85%) de 80a en forma de un aceite amarillo pálido. La fase orgánica se secó sobre sulfato de sodio anhidro, el disolvente se evaporó y el compuesto se purificó mediante cromatografía en placa preparativa usando una mezcla de hexano-acetato de etilo (70:30) como eluyente.
El disolvente se evaporó hasta sequedad, se diluyó con acetato de etilo (20 ml) y se lavó sucesivamente con solución de HCl 1 N (2 x 5 ml), con solución saturada de bicarbonato de sodio (2 x 5 ml) y con salmuera (5 ml). La fase orgánica se secó con sulfato de sodio anhidro, el disolvente se evaporó y el producto se purificó mediante cromatografía en placa preparativa. La fase orgánica se secó con sulfato de sodio anhidro, el disolvente se evaporó y el producto se purificó mediante cromatografía en placa preparativa.