A todos los empleados de CECYPE y CIATEJ por crear esta Maestría y darme la oportunidad de ser parte de ella. Benigno Figueroa Núñez por todo el apoyo que me brindó para poder estudiar mi maestría y por la información que pude adquirir gracias a su guía.
RESUMEN
2 bioequivalentes al producto de referencia; Sin embargo, al analizar las áreas parciales, se observó una absorción más rápida durante los primeros tiempos de muestreo con el producto de prueba en comparación con el producto de referencia, lo que puede representar un efecto más rápido.
INTRODUCCIÓN
MARCO TEÓRICO
Fundamentos de la investigación clínica
- Fundamentos éticos
- Fundamentos científicos
- Fundamentos normativos/regulatorios
Todo ensayo clínico realizado en México deberá cumplir con lo establecido en la Ley General de Salud, el Reglamento de la Ley General de Salud en Materia de Investigaciones en Salud y las Normas Oficiales Mexicanas (NOM-177-SSA1-2013, NOM-012-SSA3-2012). y NOM-220-SSA1-2012), en Buenas Prácticas Clínicas, y debe realizarse en estricto apego a los Principios Éticos para la Investigación Médica en Seres Humanos de la Declaración de Helsinki, emitida por la 64 Asamblea General de la Asociación Médica Mundial. , Fortaleza, Brasil, octubre de 2013 y las Directrices Éticas Internacionales para la investigación biomédica en seres humanos, elaboradas por el Consejo de Organizaciones Internacionales de Ciencias Médicas (CIOMS), en colaboración con la Organización Mundial de la Salud, Ginebra 2002. En la ética de Investigación en sujetos humanos El principio se refiere principalmente a la justicia distributiva, que establece la distribución justa de cargas y beneficios al participar en una investigación (CIOMS, 2002).
Interacción de fármacos
Requisitos que deben cumplir los terceros autorizados, centros de investigación o instituciones hospitalarias que realicen pruebas de biocomparabilidad. El aumento del efecto de un fármaco como resultado de la administración de otro fármaco se denomina sinergia farmacológica.
Descripción farmacológica del paracetamol
- Estructura de paracetamol
- Parámetros farmacocinéticos del Paracetamol
- Eventos adversos
- Experiencia post-comercialización
- Efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad 14
- Estructura química de cafeína
- Parámetros farmacocinéticos de Cafeína
- Eventos adversos
Su metabolismo hepático produce metabolitos activos (paraxantina, teofilina y teobromina) que contribuyen a la acción farmacológica de la cafeína (Shariq et al., 2005). Además de depender de la dosis, la incidencia de eventos adversos puede variar entre consumidores habituales y no consumidores de cafeína (Nawrot et al., 2003).

ANTECEDENTES
La relación entre las concentraciones plasmáticas de paracetamol y los efectos estimados por los potenciales evocados se evaluó mediante un modelo compartimental farmacocinético-farmacodinámico. Se realizaron pruebas discriminantes de disolución y deconvolución in vitro de las concentraciones plasmáticas in vivo de las formulaciones de prueba y de referencia para explorar las diferencias en la absorción.
PLANTEAMIENTO DEL PROBLEMA
22 Este estudio se basa en los resultados del estudio de Renner et al., (2007), en el que se observó que la Cmax de la combinación de paracetamol y cafeína es mayor que la observada para el paracetamol simple, manteniéndose por encima del rango de bioequivalencia. . de 80-125% (IC al. Con base en este estudio, se espera que la cafeína modifique la farmacocinética del paracetamol, aumentando la tasa de absorción.
JUSTIFICACIÓN
24 Para ello se propone un estudio clínico controlado, el diseño experimental seleccionado es uno de los recomendados por la NOM-177-SSA1-2013, cruzado, tipo abierto, con dos secuencias y dos periodos de tratamiento (esquema A – B, B – A), en dosis única, con un tiempo de muestreo de veinticuatro horas, que permita determinar al menos el 80% del AUC, con un total de 14 muestras y un período de lavado de siete días, en 26 sujetos sanos de ambos sexos, en ayunas. Se sugiere un diseño cruzado donde cada sujeto esté bajo su propio control para reducir la variabilidad y caracterizar adecuadamente la farmacocinética de los fármacos en estudio, teniendo en cuenta además que se trata de fármacos seguros en las dosis a administrar en el estudio. y que tienen una vida media de eliminación corta y, por tanto, no requieren un período de lavado prolongado.
HIPÓTESIS
OBJETIVOS
Objetivo general
Objetivos secundarios
METODOLOGÍA
Etapa clínica
- Diseño experimental y plan del estudio
- Tamaño de muestra
- Selección de población de estudio
- Criterios de inclusión
- Criterios de no inclusión
- Criterios de exclusión
- Método de asignación de sujetos a la secuencia de tratamiento
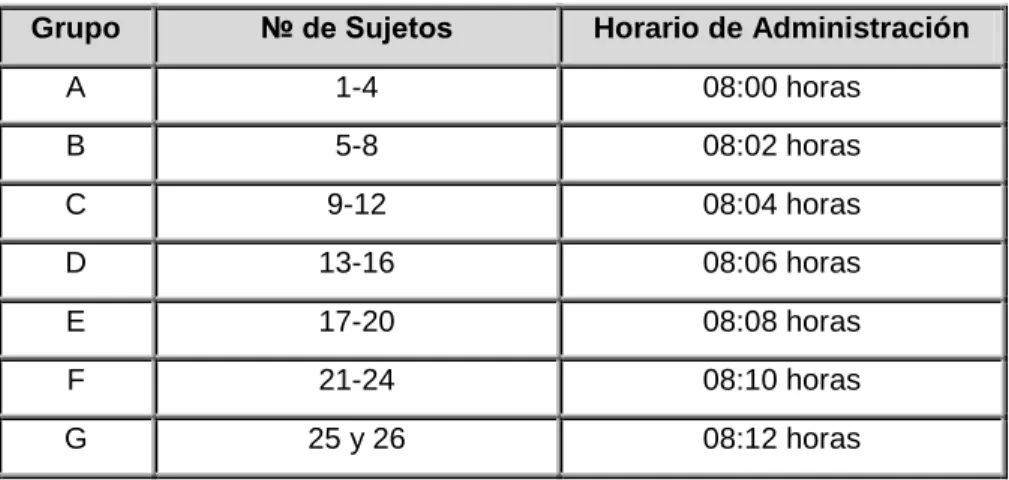
- Selección y horario de administración de dosis para cada sujeto
- Medicamentos que se administraron
- Almacenamiento
- Criterios de aceptación de medicamentos de prueba y referencia
- Criterios de rechazo de medicamentos de prueba y referencia
- Muestras de retención
- Disposición final
- Métodos o procedimientos del estudio
- Selección de sujetos
- Periodos del estudio
- Eventos Adversos
- Definiciones
- Relación del Evento Adverso con el producto de investigación
- Severidad y gravedad del Evento Adverso
- Documentación y reporte de los Eventos Adversos
- Aspectos éticos y legales
- Gestión de calidad
Esta lista de aleatorización se generó el día antes de la dosificación en el primer período del estudio. El período de lavado duró siete días desde la administración del medicamento en el período anterior. El reporte de eventos adversos se realizó desde la administración de la primera dosis del fármaco en el primer período del estudio y hasta un período de siete días después de la última ingesta del fármaco del estudio.
La información sobre la suspensión de la administración del fármaco sospechoso no está disponible o no está clara.
Etapa analítica
- Envío de muestras
- Manejo de las muestras biológicas por la Unidad Analítica
- Desecho de las muestras biológicas
- Etiquetado de las muestras ultra congeladas
- Análisis químico de las muestras biológicas
- Condiciones cromatográficas e instrumentales
- Condiciones del espectrómetro de masas
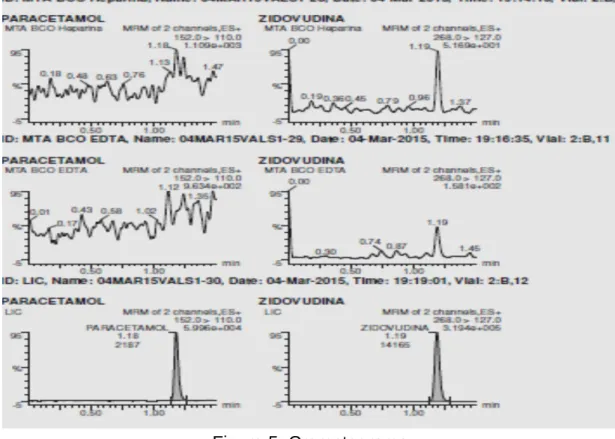
Según acuerdo con el patrocinador, las muestras fueron retiradas de la ultracongelación un año después de realizado el estudio y colocadas en el almacén temporal de Residuos Biológico-Infecciosos Peligrosos, tras lo cual fueron recogidas por la empresa contratada para tal fin. Para la determinación de la cantidad de paracetamol, se ha desarrollado un método analítico para su medición mediante cromatografía líquida acoplada a un detector masa/masa (UPLC MS/MS), en polaridad positiva, según sus propiedades físico-químicas y lo que está reportado en la literatura científica internacional. adecuándolo a las condiciones de laboratorio y validado para cumplir con los criterios de la NOM-177-SSA1-2013 y el Sistema de Calidad de la unidad analítica de la Clínica de Enfermedades Crónicas y Procedimientos Especiales, S.C. Se realizó un método UPLC MS/MS para cuantificar el paracetamol en plasma humano, siguiendo las condiciones que se describen a continuación.
Extracción líquido-líquido: para el análisis de las muestras de plasma se utilizó una alícuota de los microtubos, la cual se transfirió a un microtubo de procesamiento, se añadió estándar interno (Zidovudina), luego se añadió solución de extracción, se agitó vigorosamente y se Se centrifugó la mezcla, se congeló y decantó la muestra, la fase orgánica, luego se evaporó, se reconstituyó y finalmente se tomó una alícuota del sobrenadante para inyectar en el sistema cromatográfico.
Etapa estadística
- Análisis farmacocinético
- Estadística descriptiva
- Análisis estadístico para biodisponibilidad comparativa
- Análisis de varianza
- Análisis estadístico para la evaluación de la interacción farmacocinética
- Desviación al Plan Estadístico
Análisis estadístico para evaluar la interacción farmacocinética Para evaluar la interacción farmacocinética se construyó el intervalo de confianza. Para evaluar la interacción farmacocinética se construyó el clásico intervalo de confianza del 90% para la relación entre la media de los fármacos de prueba y de referencia. Si la concentración previa a la dosis es igual o inferior al 5 % del valor de Cmax en ese sujeto, los datos del sujeto se pueden incluir sin ningún ajuste en todas las mediciones y cálculos farmacocinéticos. En los casos en que el valor previo a la dosis sea superior al 5% de la Cmax, el sujeto debe ser eliminado de todas las evaluaciones del estudio.
Se considera que un sujeto de investigación tiene concentraciones muy bajas si el AUC es inferior al 5% de la media geométrica del AUC del fármaco de referencia (debe calcularse sin incluir valores atípicos).

RESULTADOS Y DISCUSIÓN
Resultados etapa clínica
No hubo cambios relevantes en los signos vitales de los sujetos participantes durante el estudio. No hubo eventos adversos adicionales en este período de seguimiento, finalizando así la fase clínica del estudio. 60 Durante el estudio se tomaron 14 muestras de sangre por período, en los momentos definidos en el protocolo clínico.
Para la recolección de muestras se asumió una tolerancia de ± 1 minuto durante el período de absorción y dispersión (hasta la muestra 07 inclusive) y ± 3 minutos durante el período de eliminación.

Resultados etapa analítica
- Descripción del análisis de muestras
- Criterios de aceptación de las corridas analíticas
Asimismo, se demostró que las muestras de plasma son estables a temperatura ambiente durante al menos 27 horas y al menos 3 ciclos de congelación-descongelación. Las muestras procesadas son estables en condiciones de muestreador automático (6°C) durante al menos 27 horas, en refrigeración durante al menos 23 horas y las muestras son estables en congelación (-70°C ± 10°C) durante al menos 62 días. . Individualmente, al menos dos tercios del total de muestras de control deben cumplir el criterio de precisión.
Las muestras deberán cumplir un CV(%) ≤15, un Dev(%)≤15 y al menos dos tercios del total.
Etapa estadística
- Estadística descriptiva de la población de estudio
- Datos individuales de Concentración plasmática (Cp) Vs. Tiempo (t)
- Análisis de Varianza
También se demuestra que el sujeto con el caso número 10 no fue incluido en el análisis. La Tabla 19 muestra los datos individuales, así como las estadísticas descriptivas de los parámetros farmacocinéticos del paracetamol, que se utilizan normativamente para determinar la equivalencia entre dos productos (Cmax, AUC0-t y AUC0-∞). 78 En la Tabla 20 se muestran los resultados promedio de los parámetros farmacocinéticos, teniendo en cuenta su media aritmética, se observa una alta variabilidad la cual se puede atribuir a la sensibilidad de la media aritmética a valores extremos, para lo cual el análisis se realizó teniendo en cuenta la media geométrica y la media armónica de los parámetros farmacocinéticos utilizados para determinar la equivalencia, que se muestran en la tabla 21.
79 La Tabla 21 muestra una ligera diferencia en el parámetro Cmax, que es mayor en el fármaco analizado que contiene cafeína.

Estadística de bioequivalencia
- Análisis de áreas parciales
Al igual que en el estudio de Renner et al. (2007), en nuestro estudio se observó una absorción más rápida del producto que contiene cafeína, que en este caso es el medicamento de prueba (Sedalmerck Max®). Se puede concluir que la modificación en la tasa de absorción del paracetamol no depende de la dosis, ya que en el estudio de Renner et al. utilizaron dosis dos veces superiores a las utilizadas en este estudio (1000 mg de paracetamol y 130 mg de cafeína. ) .. En el estudio de Renner et al., también se demostró que la cafeína en sí no tiene un efecto antinociceptivo o analgésico, pero potencia el efecto del paracetamol.
En nuestro estudio, el producto de prueba (en combinación con cafeína) y el producto de referencia (sin cafeína) también fueron bioequivalentes considerando la biodisponibilidad general (AUC0-t y AUC0-∞), y considerando Cmax, la diferencia en la tasa de absorción a través de la análisis de áreas parciales, los cuales se realizaron tomando como antecedentes el estudio de Renner et al., así como las recomendaciones de la FDA.

CONCLUSIONES
88 Para evaluar la eficacia del paracetamol y demostrar que su efecto analgésico es más rápido cuando se administra en combinación con cafeína, se necesitan más estudios para evaluar su efecto analgésico solo y en combinación con cafeína en pacientes con dolor agudo. Se demostró que el paracetamol es seguro y bien tolerado en la población del estudio, tanto solo como en combinación con cafeína, y solo ocurrieron dos eventos adversos, ninguno de los cuales estuvo relacionado con el medicamento.
PERSPECTIVAS
NORMA Oficial Mexicana NOM-012-SSA3-2012, que define los criterios para la ejecución de proyectos de investigación sobre la salud de los seres humanos, publicada en el DOF. La cafeína acelera la absorción y aumenta el efecto analgésico del paracetamol; Revista de farmacología clínica.
ANEXOS
Anexo 1. Cronograma de periodo total de estudio