1
CARACTERIZACIÓN DE LA RESPUESTA MICROGLIAL EN UN CURSO TEMPORAL POSTISQUEMIA CONSECUENTE CON EMBOLISMO ARTERIAL
INDUCIDO EXPERIMENTALMENTE EN RATA WISTAR
LUISA MARÍA ROJAS VALENCIA
Trabajo de grado presentado como requisito parcial para optar al título de Biólogo
Director
IANG SCHRONILTGEN RONDON BARRAGAN MVZ., MSc. Grupo de Investigación en Enfermedades Neurodegenerativas
Codirector
ÁNGEL ENRIQUE CÉSPEDES RUBIO MVZ., MSc., PhD.
Director Grupo de Investigación en Enfermedades Neurodegenerativas
UNIVERSIDAD DEL TOLIMA FACULTAD DE CIENCIAS PROGRAMA DE BIOLOGÍA
3
A Dios A mi mamá por el amor comprensión y apoyo constante
4
AGRADECIMIENTOS
Gracias a la Universidad del Tolima y a la Facultad de Ciencias por la formación académica. A la oficina de investigaciones y desarrollo científico por la financiación del proyecto.
Gracias a los directores, Iang Rondón por el trabajo constante, confianza exigencia motivación, responsabilidad y múltiples oportunidades que me brindo durante todo el desarrollo de la tesis. Al profesor Ángel Céspedes por mostrarme el mundo de la microglía y desafiarnos a no decir, “yo no puedo”, además de la confianza y oportunidad.
A todos los integrantes del Grupo de Enfermedades Neurodegenerativas desde la primera cohorte, Angélica por la instrucción en técnicas, a Lina y Marcela compañeras de experimentos y errores, a Sandra por ser un modelo de dedicación, a Lina por ser compañera de discusión académica y consejera. A Catalina por su colaboración en todos los asuntos del Laboratorio. A Lucia por su ayuda en el laboratorio y buena actitud y a Viviana por la motivación. A las nuevas tesistas del grupo Katerine, Stephania, Claudia y Cindy.
Al Grupo de Modelos Experimentales para las Ciencias Zoohumanas, liderado por la profesora Liliana Francis, una gran guía durante la carrera de biología, a todos los integrantes del grupo por el apoyo, colaboración y el servicio de animales para desarrollar este trabajo de investigación y algunos equipos utilizados.
5
LISTA DE ABREVIATURAS
ACA: Arteria cerebral anterior
ACC: Arteria carótida común
ACM: Arteria cerebral media
AIT: Accidente isquémico transitorio
BDNF: Factor neuronal derivado de cerebro
BHE: Barrera hematoencefálica
CI: Cápsula interna
COX 2: Ciclooxigenasa 2
DAMPs: Patrón molecular asociado al daño
ECV: Enfermedad cerebro vascular
FJ: Fluoro jade
GFAP: Proteína acídica glial fibrilar
IHC: Inmunohistoquímica
iNOS: Sintasa de óxido nítrico inducible
LFB: Luxol fast blue
NF-κβ: Factor nuclear kappa beta (κβ)
OMS: Organización mundial de la salud
SNC: Sistema nervioso central
S1BF: Corteza somatosensorial en campos de Barrel
STR: Cuerpo estriado
TNF: Factor de necrosis tumoral
TTC: Cloruro de trifenil Tetrazolio
6
CONTENIDO
Pág.
INTRODUCCIÓN 17
1. OBJETIVOS 19
1.1. OBJETIVO GENERAL 19
1.2. OBJETIVOS ESPECÍFICOS 19
2. MARCO TEÓRICO 20
2.1. ENFERMEDAD CEREBROVASCULAR 20
2.1.1. Epidemiología 20
2.1.2. Clasificación de las ECVs 21
2.1.3. Factores de Riesgo 22
2.2. ISQUEMIA CEREBRAL 22
2.2.1. Clasificación de la isquemia cerebral 24
2.2.2. Modelos de isquemia cerebral 24
2.2.2.1. De acuerdo a la extensión del infarto 25
2.2.2.2. De acuerdo al tiempo de isquemia 25
2.2.2.3. De acuerdo a la forma de oclusión 25
2.2.2.4. Isquemia embólica con coágulo autólogo 26
2.2.3. Fisiopatología de la isquemia cerebral 26
7
2.4. INFLAMACIÓN 32
2.4.1. Activadores celulares 34
2.5. RESPUESTA CELULAR ANTE LA ISQUEMIA CEREBRAL 34
2.5.1. Neuronal 34
2.6. MACROGLÍA 36
2.6.1. Astrocitos: respuesta ante la isquemia 36
2.6.2. Oligodendrocitos: Respuesta ante la isquemia 37
2.7. MICROGLÍA E ISQUEMIA 38
2.8. DESMIELINIZACIÓN 41
2.9. ALTERNATIVAS TERAPÉUTICAS 42
2.10. TECNICAS UTILIZADAS EN EXPERIMENTACIÓN PARA EVALUAR EL DAÑO CEREBRAL 43
2.10.1 El cloruro de trifenil tetrazolio (TTC) 43
2.10.2. El Luxol Fast Blue (LFB) 44
2.10.3. Cresyl Violeta 44
2.10.4. Fluoro jade (FJ) 44
3. MÉTODOS 45
3.1. ANIMALES DE EXPERIMENTACIÓN: CUIDADO Y MANEJO 45
3.2. DISEÑO EXPERIMENTAL 45
3.3. MODELO DE ISQUEMIA: EMBOLIZACIÓN ARTERIAL CON COÁGULOS AUTÓLOGOS 47
3.3.1. Preparación de los coágulos 47
3.3.2. Procedimiento quirúrgico 48
8
3.4. TIEMPOS DE SACRIFICIO – PERFUSIÓN 51
3.5. PROCESAMIENTO DE TEJIDOS: CORTE Y CRIOPRESERVACIÓN 52
3.6. DETERMINACIÓN INMUNOHISTOQUÍMICA DE ACTIVIDAD MICROGLIAL 53
3.7. HISTOLOGIA 54
3.7.1. Evaluación de la distribución del Infarto 55
3.7.2. Tinción celular y de Mielina 56
3.7.3. Neuronas en degeneración 57
3.8. MICROSCOPÍA Y REGISTRO FOTOGRÁFICO 58
3.9. DENSITOMETRÍA Y CUANTIFICACIÓN 58
3.10. ANÁLISIS ESTADÍSTICO 61
4. RESULTADOS 62
4.1. RESPUESTA MICROGLIAL 62
4.1.1. Distribución de la microglía en un curso temporal postisquemia (8, 24, 48 y 72 horas) 62
4.1.2. Microglía reactiva en diferentes zonas cerebrales 64
4.1.2.1. Giro dentado 64
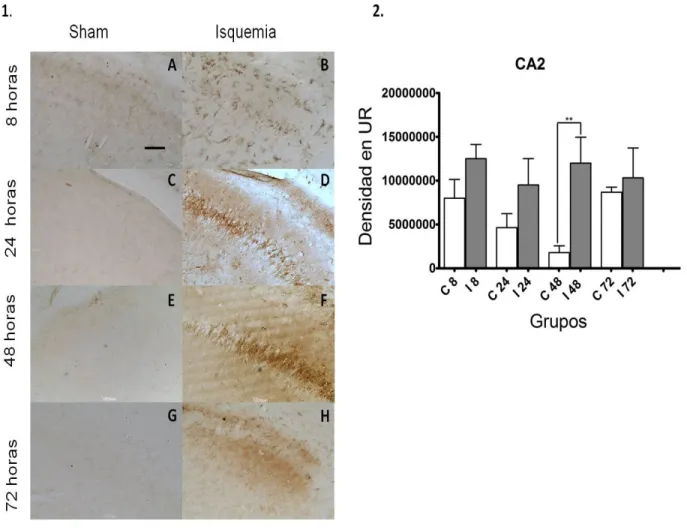
4.1.2.2. Zona CA2 65
4.1.2.3. Zona CA3 66
4.1.2.4. Zona paraventricular 67
4.1.3. Caracterización de los cambios morfológicos de la microglía 68
4.1.4. Variación de la morfología microglial en diferentes zonas cerebrales, consecuente con isquemia cerebral por embolismo arterial 73
9
4.3. DISTRIBUCIÓN DEL INFARTO CEREBRAL EN EL MODELO EMBOLICO
INDUCIDO MEDIANTE COÁGULOS AUTÓLOGOS 79
4.4. MUERTE NEURONAL Y PÉRDIDA DE MIELINA EN CORTEZA
SOMATOSENSORIAL, HIPOCAMPO Y NUCLEO ARCUATO 80
4.5 NEURODEGENERACIÓN EN HIPOCAMPO 84
5. DISCUSIÓN 86
5.1. DISTRIBUCIÓN DE MICROGLÍA REACTIVA A 24 HORAS POST ISQUEMIA 86
5.2. HIPERREACTIVIDAD MICROGLIAL POSTISQUEMIA EN ZONA
PARAVENTRICULAR (24 HORAS) E HIPOCAMPAL (48 HORAS) 88
5.3. ACTIVACIÓN MICROGLIAL Y CAMBIOS MORFOLÓGICOS 89
5.4. PREDOMINIO DE FORMAS RAMIFICADAS DE MICROGLÍA A 8 HORAS POST
ISQUEMIA Y DE MICROGLÍA AMEBOIDE A 24 HORAS POST ISQUEMIA 91
5.5. EL EMBOLISMO ARTERIAL CAROTÍDEO INDUCE ISQUEMIA CEREBRAL EN
CORTEZA MOTORA, HIPOCAMPO Y CUERPO ESTRIADO 93
5.6. PÉRDIDA DE MIELINA EN CORTEZA SOMATOSENSORIAL, HIPOCAMPO Y
ESTRIADO.POSTISQUEMIA CEREBRAL 95
5.7. NEURODEGENERACIÓN EN ZONA CA3 HIPOCAMPAL EN FASE AGUDA
POSTISQUEMIA 98
5.8. RELACIÓN MICROGLÍA-NEURONA FRENTE A ISQUEMIA CEREBRAL 99
5.9. DEGENERACIÓN NEURONAL, DESMIELINIZACIÓN Y RESPUESTA
MICROGLIAL 100
6. CONCLUSIONES 108
RECOMENDACIONES 109
10
LISTA DE FIGURAS
Pág.
Figura 1. Infarto isquémico vs hemorrágico 19
Figura 2. Fisiopatología de la isquemia cerebral 28
Figura 3. Método de extracción de sangre a partir de la vena safena interna y
formación de coágulos autólogos 46
Figura 4. Abordaje quirúrgico de la ACC derecha 47
Figura 5. Modelo de oclusión arterial mediante coágulos autólogos 48
Figura 6. Procedimiento quirúrgico para la embolización de la arteria cerebral media derecha mediante coágulos autólogos 49
Figura 7. Método de perfusión cerebral 50
Figura 8. Corte de cerebros de rata Wistar en vibrátomo semiautomático 51
Figura 9. Tinción de TTC en cortes coronales de rata Wistar 54
Figura 10. Zonas Evaluadas en el curso temporal de la respuesta microglial
consecuente a isquemia cerebral por embolismo arterial 57
11
Figura 12. Reactividad microglial en giro dentado del hipocampo, consecuente a
isquemia cerebral por embolismo arterial 62
Figura 13. Reactividad microglial en CA2 hipocampal, consecuente a isquemia cerebral por embolismo arterial 63
Figura 14. Densitometría de la reactividad microglial en CA3 del hipocampal, consecuente con isquemia cerebral por embolismo arterial 64
Figura 15. Reactividad microglial en zona paraventricular lateral, consecuente a
isquemia cerebral por embolismo arterial lateral 65
Figura 16. Morfotipos microgliales M1, M2, M3 y M4 presentes en el curso temporal de la respuesta micorglial ante isquemia cerebral 68
Figura 17. Caracterización morfológica de las células de microglía presentes en el curso temporal de la respuesta microglial ante isquemia cerebral 69
Figura 18. Proporción de las morfologías microgliales en fimbria del hipocampo ante isquemia cerebral 71
Figura 19.Proporción de las morfologías microgliales en ZPV ante isquemia cerebral 72
Figura 20. Proporción de los morfotipos de microglía en putamen caudado
consecuente con isquemia cerebral por embolismo arterial 73
12
Figura 22. Distribución del infarto cerebral, inducido mediante embolismo arterial con coágulo autólogo en ratas macho Wistar 77
Figura 23. Desmielinización y daño celular consecuente a isquemia cerebral por
embolismo arterial 78
Figura 24. Daño celular en la zona CA3 del hipocampo a 24 horas postisquemia
cerebral por embolismo arterial 80
Figura 25. Pérdida de la mielina en zona somatosensorial, consecuente con isquemia cerebral por embolismo arterial 81
Figura 26. Neurodegeneración en hipocampo, corteza somatosensorial y estriado 82
Figura 27. Gradiente de distribución de la microglía en el foco y en la periferia de un infarto isquémico 84
Figura 28. Cambios morfológicos de la microglía activa en isquemia cerebral inducida por embolismo arterial con coágulos autólogos 101
Figura 29. Modelo hipotentico fisiopatología del evento isquémico y la participación microglial en un modelo tromboembolismo arterial inducido 104
Figura 30. Distribución del infarto embolicó mediante coágulo autólogo en hembras Wistar 133
Figura 31 Medición de la densidad y área de una célula de microglía con el programa Fiji-imageJ 136
13
LISTA DE TABLAS
Pág.
Tabla 1. Tratamiento de la isquemia cerebral 40
Tabla 2. Distribución de los tratamientos 45
Tabla 3. Características de los morfotipos marcados con OX42 67
14
ANEXOS
Pág.
Anexo A. Evaluación de la distribución del infarto en el modelo de coágulo autólogo en hembras Wistar 133
Anexo B. Protocolo de densitometría global 134
15
RESUMEN
La isquemia cerebral es una de las principales causas de mortalidad y discapacidad a nivel mundial. Es una patología compleja que afecta el SNC, ha sido ampliamente estudiada pero todavía no se ha esclarecido por completo la función de grupos celulares aparte de la neurona, como lo es la glía y su función en la respuesta inflamatoria. La microglía es una célula glial, de origen mieloide que media la respuesta inmune en el CNS, incluyendo la neuroprotección y la neuroinflamación y se ha demostrado su activación ante isquemia cerebral. El objetivo del presente estudio es determinar el curso temporal de la respuesta microglial en fase aguda postisquemia. La respuesta microglial fue evaluada a 8, 24, 48 y 72 horas postisquemia en un modelo de embolismo arterial inducido experimentalmente en rata Wistar. Posteriormente se realizaron tinciones e inmunohistoquimica como marcadores de los procesos de neurodegeneración, desmielinización y la activación microglial.
En el presente estudio se observó actividad de la microglía en el foco, la penumbra y en áreas remotas al lugar de lesión (infarto), mostrando diferentes estados de activación, e.g. la transformación morfológica a células ameboides. La transformación microglial fue cercana a áreas de neurodegeneración y desmielinización. Sin embargo la activación microglial en lugares remotos no estaba asociada a procesos de neurodegeneración y desmielinización. El modelo de embolismo arterial empleado en el presente estudio ocluye áreas irrigadas por la arteria cerebral posterior como el hipocampo y mostró reproducibilidad en cuanto a la zona de lesión.
16
ABSTRACT
Stroke is a leading cause of death and disability in the world. It is a complex pathology that affects the central nervous system (CNS) in which is necessary a better understanding of cellular immune (inflammatory) response, specifically from glial cells. Microglia is a myeloid-derived cell, that mediates immune responses in CNS, including neuroprotection and neuroinflammation, and they have shown a role in stroke, but the spatio/temporal response in brain remains unclear. The aim of this study is to determinate the time course of microglial response in acute ischemic stroke. We evaluate microglial response at 8, 24, 48 and 72 hours after experimental-induced stroke with a model of embolism. Later we used cell staining and immunohistochemistry as a marker of neurodegeneration and desmielinization process and microglial activation.
Microglial activation was observed in the core, penumbra and remote areas from the lesion (infarct), showing several states of activation, e.g. morphological transformation in amoeboid cells. Microglial activation was near to areas of neurodegeneration and desmielinization. However, remote microglial activation was observed in areas without desmielinization or neurodegeneration process. The model of embolism used in this study occludes areas irrigated by posterior cerebral artery, such as hippocampus, and showed high reproducibility.
17
INTRODUCCIÓN
Las enfermedades cerebrovasculares comprenden un grupo de patologías responsables del 10 % de las muertes a nivel mundial y han sido catalogadas como las más discapacitantes. Entre estas la isquemia cerebral tromboembólica es la más frecuente, consiste en el bloqueo del flujo sanguíneo que genera un daño cerebral. Los síntomas que caracterizan el evento isquémico, son la parálisis facial y de los miembros anteriores y problemas en el habla, siendo de suma importancia su identificación y dada su corta ventana terapéutica, de apenas 3 horas para iniciar un tratamiento oportuno.
La isquemia cerebral presenta una fisiopatología compleja que ha sido ampliamente estudiada pero todavía no se ha esclarecido por completo la función de grupos celulares aparte de la neurona, como lo es la glía, encargada de brindar soporte a las neuronas y protección durante patologías como la isquemia. La glía se compone de tres grupos celulares los astrocitos, oligodendrocitos y la microglía. Esta última constituye la primera línea de defensa del sistema inmune en las patologías del SNC, y aún falta establecer sus cambios morfofisiológicos como respuesta a la isquemia en fase aguda.
18
19
1. OBJETIVOS
1.1. OBJETIVO GENERAL
Caracterizar la respuesta microglial en un curso temporal en fase aguda postisquemia por embolismo arterial con coágulo autólogo en rata Wistar.
1.2. OBJETIVOS ESPECÍFICOS
Caracterizar la distribución, la morfología y la actividad de la microglía en un curso temporal postisquemia como respuesta al embolismo arterial.
Evaluar el efecto del embolismo arterial carotideo sobre el estado de mielinización en la sustancia blanca encefálica.
20
2. MARCO TEÓRICO
2.1. ENFERMEDAD CEREBROVASCULAR
Las enfermedades cerebrovasculares (ECVs) son definidas por la Organización mundial de la Salud (OMS) como “una afección neurológica focal de aparición súbita, que perdura más de 24 horas, de presunto origen vascular y puede causar la muerte” (Frómeta, Álvarez, Sánchez, Fonseca, y Quesada, 2010), estas corresponden a un cambio patológico en la vasculatura cerebral (Yeh, Cheng, y Chen, 2011) o una alteración en el flujo sanguíneo cerebral que afecta la función de una o más regiones del cerebro de forma transitoria o permanente (Ustrell-Roig y Serena-Leal, 2007). Las ECVs representan altos costos médicos y sociales, al ocasionar la muerte o inducir a los pacientes a un estado discapacitante, constituyéndose así en un serio problema de salud pública (WHO, 2004, 2011, 2013).
21
desarrollado en la población de Armenia (Colombia), se estableció como edad promedio de presentación del ictus los 72 años, con una tasa de mortalidad de 29,9 % (Pérez Carreño, Álvarez Aristizábal, y Londoño Franco, 2011). La incidencia del ictus isquémico para dicha población se estimó en 61,9 %, siendo mayor que el hemorrágico (31,1 %), similar a la tendencia mundial (Pérez Carreño et al., 2011). Por otra parte, los factores de riesgo asociados al accidente cerebrovascular (ACV) son la hipertensión arterial (74,5 %), el tabaquismo (23,3 %), la cardiopatía isquémica previa (15,4%) y la diabetes (15,1 %) entre otros. Además se ha observado que los ACV hemorrágicos son menos comunes, aunque presentan mayor mortalidad (Pérez Carreño et al., 2011). Las ECVs se encuentran entre las enfermedades que generan mayores costos en el sistema de salud en América Latina y el Caribe (Gómez Dantés et al., 2011).
2.1.2. Clasificación de las ECVs. Las enfermedades cerebrovasculares se clasifican en isquémicas, que consisten en la deficiencia de flujo sanguíneo en un área del cerebro y el ictus hemorrágico debido a la ruptura de un vaso sanguíneo y extravasación de la sangre (Ustrell-Roig y Serena-Leal, 2007). En cuanto a los ictus hemorrágicos se clasifican en hemorragias subaracnoideas (HSA) e intracerebrales (HIC) (Caplan, Searls, y Hon, 2009) (Figura 1).
Figura 1. Infarto isquémico vs hemorrágico
22
*La isquemia cerebral representa el 85 % de los casos de accidente cerebrovascular. HSA (Hemorragia Sub Aracnoidea) HIC (hemorragia intracerebral).
2.1.3. Factores de Riesgo. Los factores de riesgo independientes que predisponen una ECV son hipercolesterolemia, hipertensión arterial, diabetes Mellitus y antecedentes de cardiopatía (Frómeta et al., 2010). Otros factores que pueden influir en el riesgo de la enfermedad son edad, sexo (siendo mayor en hombres que en mujeres en edad fértil), tabaquismo, ingestión de alcohol, sedentarismo, obesidad y antecedentes de ECV, pero estos factores no son independientes, pueden aumentar el riesgo al asociarse (Frómeta et al., 2010). También se deben tener en cuenta factores sociales y culturales para la prevención primaria, como se describe en Buergo Zuaznábar y Bembibre Taboada, (2007), además, han sido relacionados estados depresivos con la aparición de ECVs (Ramasubbu, 2000).
El estudio de factores como hipertensión, infarto al miocardio, shock cardiogénico, hiperlipemia, arritmias, obesidad-índice de masa corporal, diabetes mellitus (DM) permiten predecir el riesgo de aparición de ECV (Vigili de Kreutzenberg, Tiengo, y Avogaro, 2009; Yeh et al., 2011; Zade et al., 2010). Además estos factores de riesgo se asocian con déficits neuropsicológicos y también con demencia en Alzheimer (Henry-Feugeas, 2009).
Se han identificado como factores asociados a la mortalidad temprana postictus la edad, severidad del cuadro clínico, niveles altos de IL-6, proteína C reactiva y el sexo ya que se ha descrito que los hombres tienen mayor probabilidad de sobrevivir al primer mes post ictus que las mujeres después de la menopausia en estudios de Zurruk et al., (2007) en Colombia.
23
La isquemia cerebral es la disminución por un tiempo prolongado del adecuado suministro sanguíneo a una región del cerebro, que usualmente es causada por la obstrucción de las arterias que irrigan el cerebro desencadenando muerte celular. La disminución en el suministro sanguíneo lleva a la célula a implementar mecanismos adaptativos como la liberación de sustancias antioxidantes (McGavin y Zachary, 2007).
La OMS (2013) define infarto cerebral como aquel causado por la interrupción del suministro sanguíneo. Así mismo, Durukan y Tatlisumak (2007) la definen como una reducción en el flujo sanguíneo cerebral, en un lugar específico del cerebro, suficiente para generar daños metabólicos o déficits funcionales. El cerebro requiere del suministro de 50 ml/100g/min de sangre aproximadamente (Bárcena-Orbe et al., 2006). En un evento isquémico, se reduce hasta 15 % del volumen normal en el área infartada, conocida como foco isquémico, desencadenando muerte celular rápida (necrosis) y alrededor del foco isquémico se establece una área denominada penumbra en la cual hay compromiso del suministro energético y de oxigeno que conducen a cambios apoptóticos en las células residentes (Lipton, 1999).
En el área de penumbra se presenta isquemia sin tejido infartado (Ustrell-Roig y Serena-Leal, 2007) debido a que el flujo sanguíneo del área de penumbra proviene de arterias colaterales al foco isquémico (Arango-davila, Escobar-betancourt, Cardona-gómez, y Pimienta-jiménez, 2004). La extensión del daño depende la localización y duración del ictus (Caplan y Hacke, 2003). La reducción en el tamaño de la lesión se asocia con la reducción del tejido dañado, pero no con la recuperación de las neuronas (Nagai et al., 2010).
24
del flujo sanguíneo (AIT) en días, semanas o meses previos a la isquemia (Caplan y Hacke, 2003; Ustrell-Roig y Serena-Leal, 2007).
Diagnóstico. Inicialmente, se debe desarrollar un examen físico que incluye medición de la presión sanguínea, glicemia y el estado del corazón (para descartar murmullos) (Caplan y Hacke, 2003)., evaluar síntomas neurológicos, monitorear el cerebro con tomografía computarizada (TC), resonancia magnética (RM) (Caplan y Hacke, 2003; Ustrell-Roig y Serena-Leal, 2007) y doppler transcraneal (Caplan et al., 2009) para estudiar el lugar de oclusión.
2.2.1. Clasificación de la isquemia cerebral. La isquemia cerebral se clasifica en: trombóticas, embólicas, (Caplan et al., 2009) y lacunares (Behrouz, Malek, y Torbey, 2012).
La isquemia cerebral trombótica consiste en el bloqueo del flujo sanguíneo debido a un problema local de las paredes de los vasos sanguíneos, ocasionado por el estrechamiento del lumen, engrosamiento de músculos o por acumulación de restos lipidícos, fibrina, trombina o plaquetas (Caplan y Hacke, 2003). La isquemia cerebral embólica se origina por émbolos del sistema vascular, que viajan hasta ocluir una arteria cerebral, bloqueando el flujo sanguíneo (Caplan y Hacke, 2003; Caplan et al., 2009). Los infartos lacunares denominados también enfermedad cerebrovascular de los vasos menores, consiste en la extravasación de pequeñas cantidades de sangre en el parénquima y su origen no se ha esclarecido por completo (Behrouz et al., 2012). También puede presentarse isquemia cerebral debido a la disminución del flujo cerebral ocasionada por problemas sistémicos durante arritmias cardiacas, sangrado gastrointestinal, cirugías como la de bypass cardiaco (Caplan et al., 2009) o por falla cardiaca e infarto al miocardio (Caplan y Hacke, 2003).
25
permiten controlar variables tales como severidad, lugar de la isquemia, duración del ictus, entre otros; según estas condiciones se clasifican en:
2.2.2.1. De acuerdo a la extensión del infarto. Focales, hemisféricos o globales (Durukan y Tatlisumak, 2007).
2.2.2.2. De acuerdo al tiempo de isquemia. Permanentes o transitorios. El modelo de isquemia permanente permite el estudio de la patología isquémica, sin los efectos de la reperfusión postisquemia. Los modelos transitorios permiten evaluar los daños causados por la isquemia y el efecto de la reperfusión, considerando el daño por reperfusión del tejido (Durukan y Tatlisumak, 2007)
2.2.2.3. De acuerdo a la forma de oclusión. Embólicos y oclusivos (Lipton, 1999).
Los modelos oclusivos globales causan un infarto en los dos hemisferios, si son ligadas las arterias vertebrales de forma permanente y las carótidas de forma temporal, el modelo corresponde a 4-VO. Cuando se ligan las arterias carótida izquierda y derecha sin ocluir las arterias vertebrales, el modelo se denomina 2-VO (Lipton, 1999).
La oclusión de la arteria cerebral media (MCAO) genera infartos que afectan los ganglios basales y la corteza, si la oclusión es distal de la arteria cerebral media (ACM) solo se infarta la corteza (Stoll, Jander, y Schroeter, 1998). La oclusión de la ACM de forma transitoria (tMCAO) genera infartos más pequeños que MCAO, afectando principalmente el putamen caudado y la corteza, con una amplia zona de penumbra (Memezawa et al., 1992 citado por (Stoll et al., 1998).
Entre los modelos embólicos se encuentra la oclusión quirúrgica de MCA mediante craniectomía, la sutura intraluminal, el modelo fototrombótico (Nagai et al., 2010) y endotelina 1(Durukan y Tatlisumak, 2007).
26
modelo es ideal para el estudio de terapias trombolíticas. El modelo consiste en la inserción de coágulos autólogos en la arteria Cerebral Media (ACM) a nivel de la porción extracraneal y alcanzando la porción distal intracraneal, lo cual genera infartos de grado variable (Durukan y Tatlisumak, 2007).
Este modelo fue descrito por primera vez en perros por Hill et al., (1955) y adaptado para ratas por Kudo et al., (1982). Consiste en la formación de un coagulo a partir de sangre extraída de cada animal, el cual será inyectado en la carótida común durante la cirugía. Este modelo mostró reproducibilidad y se describieron algunos cambios comportamentales como lamer, morder y caminar en círculos (Kudo et al., 1982).
Kaneko et al., (1985) determinaron que el émbolo se puede alojar de 3 formas en el SNC; 1) de forma unilateral y proximal, en el tronco de la ACM, pero no en la porción que asciende corticalmente; 2) embolismo unilateral periférico, cuando el émbolo no se ubica en la rama principal de la ACM, no obstante varios émbolos más pequeños viajan a territorios periféricos de la ACM; 3) embolismo proximal bilateral, cuando se aloja el émbolo en la rama proximal de la MCA. En la evaluación del flujo sanguíneo regional, observaron que en las tres formas de embolización se presenta una descenso en el flujo sanguíneo, que es más fuerte en la forma 1 y 3 (Kaneko, Nakamura, y Ogawa, 1985).
Estudios recientes han demostrado que el tamaño del coágulo reviste importancia en la presentación del infarto (Rezazadeh et al., 2013), dado que si es del tamaño adecuado (1 mm de largo x 0,64 mm de ancho) este ocluye la arteria cerebral media en la parte distal, sin incluir territorios de la arteria cerebral anterior o generar infartos pequeños en ramas más profundas de la ACM (Ren et al., 2012). El tamaño del coágulo ha sido relacionado también con las tasas de recuperación (C. Wang, Yang, y Shuaib, 2001).
27
(McGavin y Zachary, 2007). En la isquemia cerebral se produce una depleción de oxígeno y glucosa (Lipton, 1999), donde se afecta el transporte de oxígeno a la mitocondria, por lo cual se detiene la fosforilación oxidativa, generando una disminución en los niveles de ATP, desencadenando la activación de la fosfofructocinasa, una de las enzimas pivote en la glucólisis anaerobia (Sugden y Newsholme, 1973), siendo el metabolismo anaerobio de la glucosa quien genera acumulación intracelular de lactato y de fosfatos inorgánicos (P. Z. Sun, Cheung, Wang, y Lo, 2011) En el tejido nervioso la depleción de ATP posee efectos que comprometen la viabilidad celular, dado que las neuronas son incapaces de obtener bajo condiciones anaerobias, energía en forma de ATP. Dicha depleción de ATP deprime la función de las bombas iónicas de la membrana, tales como Na/K ATPasa. El nivel de ATP en el foco isquémico, cae hasta valores inferiores al 25 % y en el área de penumbra entre 50-70 %, incrementando a su vez los niveles de potasio extracelular (Lipton, 1999).
Asociada a la glucólisis anaerobia y la alteración en la fosforilación oxidativa, se presenta la acumulación de radicales libres que generan daño a los fosfolípidos de la membrana celular, afectando su permeabilidad selectiva, incluyendo la función de los canales iónicos (Lipton, 1999). Durante la fase de reperfusión incrementan aún más los niveles de radicales libres (ROS) tales como el superóxido, el peroxinitrito y el peróxido de hidrógeno (Culmsee y Krieglstein, 2005). El cerebro muestra alta sensibilidad en este estado oxidativo dado que carece de catalasa, la enzima que convierte el peróxido de hidrógeno en agua, por lo cual utiliza la vía alterna del glutatión (Butterfield y Opii, 2009).
28
glutamato activa el sistema de la proteína G e induce la liberación de calcio a partir de los almacenes celulares (Platt, 2007). Los receptores ionotrópicos incrementan la permeabilidad en la membrana a iones como el sodio, potasio y calcio (Weil, Norman, DeVries, y Nelson, 2008). Además, se ha identificado un canal iónico mitocondrial llamado Ca-homeostasis acid-sensing (ASIC) que es sensible a pH bajo y participa en la mediación del influjo de calcio (Culmsee y Krieglstein, 2005). La liberación de glutamato genera hiperexcitación que aumenta la despolarización y el déficit energético, al intentar repolarizar la membrana (Arango-davila et al., 2004).
Posterior al daño, se produce un aumento en los niveles de Ca2+ el cual es secuestrado en la mitocondria y activa la sintasa de óxido nítrico. Los altos niveles de calcio también activan la fosfolipasa A que convierte fosfolípidos en ácido Araquidónico, para formar prostaglandinas y tromboxanos y de esta forma se inicia la respuesta inflamatoria (Butterfield y Opii, 2009). El nivel de Ca2+ extracelular disminuye a medida que ingresa al tejido (Lipton, 1999).
Como consecuencia del estrés oxidativo, se presenta daño mitocondrial, fallo energético y de la homeostasis del calcio; por lo cual, la membrana mitocondrial se despolariza activando el poro de transición, liberando el citocromo c y el factor inductor de apoptosis (AIF) desencadenando la muerte celular apoptótica (Culmsee y Krieglstein, 2005).
La disminución de ATP, producción de radicales libres, disminución del pH, incremento del Na y la despolarización de la membrana, llevan a la activación de mecanismos secundarios (e.g. activación de proteasas, fosfolipasas y radicales libres) que generan mayor daño celular e inician la muerte necrótica (Lipton, 1999).
29
al., 2004). Además, se establece una zona de oligemia benigna, donde disminuye el flujo sanguíneo, pero se conserva la función neuronal normal (Suwanwela y Koroshetz, 2007). En la penumbra, los niveles de ATP se mantienen entre 50 y 70 % pero se eleva el consumo de glucosa. Además se desencadenan despolarizaciones dependientes de K y activación de los receptores ionotrópicos del glutamato (Lipton, 1999).
Posterior a la restitución del flujo sanguíneo se presenta una hiperemia en la cual disminuye la viscosidad sanguínea, se presenta vasodilatación y se liberan moléculas vasoactivas. Durante este proceso hay vasoparálisis y obstrucción microvascular así como edema endotelial que impiden la reperfusión completa del tejido (Arango-davila et al., 2004), además de otros factores tales como la acción de linfocitos y neutrófilos (Price et al., 2004), la activación de las plaquetas y del complemento, daño en la barrera hematoencefálica (BHE) y disminución de la temperatura cerebral que se pueden asociar al daño (Pan, Konstas, Bateman, Ortolano, y Pile-Spellman, 2007). Durante la reperfusión se reporta un aumento en la producción de ROS (Especies Reactivas de Oxígeno) debida principalmente a la NADPH, que se suma a las ROS generadas por el daño mitocondrial y a la activación de la xantina oxidasa (Abramov, Scorziello, y Duchen, 2007).
30
Figura 2. Fisiopatología de la isquemia cerebral
Fuente: Modificado a partir de Brouns y De Deyn, (2009); Culmsee y Krieglstein, (2005); Durukan y Tatlisumak, (2007); Lipton, (1999).
2.3. RESPUESTA INMUNE DEL SNC ANTE LA ISQUEMIA.
31
Por otra parte, se ha planteado que además de la BHE en el SNC cuenta con una barrera inmunológica de tipo humoral, debido a la liberación de factores, como el FasL por parte de astrocitos y microglía (Mor et al., 1999). La presentación de antígenos se podría desarrollar de tipo humoral mediante el reconocimiento de antígenos que se encuentran en el líquido cefalorraquídeo (LCR) en una estructura similar al folículo linfoide, ubicada en las meninges, (Galea et al., 2007). Además, se ha planteado que las células dendríticas, podrían reconocer los antígenos presentes en el LCR y llevarlos hasta los nódulos linfáticos (Romo-González et al., 2012).
Se ha propuesto que éste reconocimiento de antígenos puede desencadenar una activación de la inmunidad sistémica, ya que se ha reportado la activación de linfocitos T CD4+ en sangre periférica al séptimo día post isquemia, especialmente los linfocitos T CD4 Treg encargados de regular la respuesta inmune (J. Yan et al., 2009). También se ha reportado una respuesta inflamatoria periférica posterior al AIT, similar a la observada en la isquemia focal, pero en AIT no se presenta daño en el parénquima ni en la BHE que permita el reconocimiento de antígenos (Ross et al., 2007). Posterior a la isquemia se desarrolla una inmunosupresión sistémica, lo cual podría deberse a una regulación de la noradrenalina que induce liberación de interleucina 10 (IL-10) y glucocorticoides (Macrez et al., 2011).
El parénquima cerebral cuenta con un privilegio inmunológico que regula constantemente la respuesta inmune innata y adaptativa (Galea et al., 2007), permitiendo un tipo de inmunidad local que entre otras funciones implica la activación de células residentes, las cuales median la respuesta inmune, realizando reconocimiento y eliminación de patógenos y de células en proceso de muerte (Hauwel et al., 2005).
32
Toll (TLRs), el receptor de manosa de macrófago y los Scavengers, petraxinas y receptores del complemento (Hauwel et al., 2005).
La producción de citoquinas lleva a la activación de la microglía y de los astrocitos, siendo considerada la microglía como la primera línea de defensa frente a la injuria (Streit, 2006; Vigili de Kreutzenberg et al., 2009). No obstante, se han reconocido funciones inmunes en los astrocitos (Griffiths, Gasque, y Neal, 2010)
Durante los estados patológicos, se realiza reclutamiento de leucocitos, neutrófilos, células NK (voz ingl. natural killer, asesinas naturales), células dendríticas y macrófagos (Macrez et al., 2011) al parénquima cerebral, pero también existe una regulación humoral sobre las poblaciones que ingresan por ejemplo mediante citoquinas como la CCR7 que controla el ingreso de linfocitos al parénquima así como la citoquina CCL21 que contribuye a la comunicación neurona-glía y es necesaria para la respuesta inmune durante procesos patológicos (Noor y Wilson, 2012). Además, se ha propuesto una regulación mediada por células como los linfocitos T CD4 reguladores que pueden modular la antigenicidad especifica de las células efectoras T CD8 en el parénquima del SNC reduciendo el daño neuronal (Göbel et al., 2012).
2.4. INFLAMACIÓN
33
A partir del tejido dañado se liberan señales de alerta que pueden ser los componentes celulares expuestos al medio externo posterior a una muerte necrótica, estas señales se denominan patrones moleculares asociados al daño (DAMPs) (Tizard, 2013) que actúan como señales proinflamatorias (Savage, Lopez-Castejon, Denes, y Brough, 2012).
Los DAMPs son reconocidos por receptores tales como los TLRs en las células centinelas, (e.g. microglía y astrocitos) así como neutrófilos en los vasos sanguíneos. Dichas citoquinas estimulan las cascadas de señalización intracelular que inducen la activación del factor nuclear κβ (NF- κβ) (Young et al., 2012) y el gen supresor de tumores p53 (Culmsee y Krieglstein, 2005). Ante las señales proinflamatorias los neutrófilos ingresan al parénquima mediante diapédesis, activando la iNOS y ciclooxigenasa 2 (COX2). La microglía fagocita restos celulares durante la inflamación y libera más citoquinas. Los astrocitos al activarse, también liberan citoquinas y quimioquinas al medio extracelular (Q. Wang, NanTang, y Yenari, 2007). Las citoquinas principales incluyen el TNF-α, IL-1b e IL-6 (Butterfield y Opii, 2009), IL-8 (Gopcevic et al., 2007), IL-1β (Koprich, Reske-Nielsen, Mithal, y Isacson, 2008) así como la CXCL1 (Roy, Richard, Dumas, y Vallières, 2012).
Los mediadores inflamatorios son citoquinas, quimioquinas, metabolitos derivados del ácido Araquidónico (e.g. Prostaglandinas, leucotrienos y tromboxanos), óxido nítrico (NO) (activación de nNOS e iNOS), ROS, NADPH (H. Chen, Kim, Okami, Narasimhan, y Chan, 2012) metaloproteinasas de matriz (MMP), inmunoglobulinas, moléculas de adhesión (ICAM y VCAM, selectinas P, E y L) e integrinas (CD11a-c)(Q. Wang et al., 2007).
34
Además de la producción de ROS por la Xantina oxidasa y NADPH (Abramov et al., 2007) el daño en la membrana celular permite la activación del complemento, el complejo de ataque a la membrana y la hemolisina penetran en la célula y forman un canal por el cual se intercambia libremente agua, proteínas y electrolitos, lo que lleva a la lisis celular por tumefacción (McGavin y Zachary, 2007).
Por otra parte, durante la isquemia aumenta la permeabilidad de la BHE a células como linfocitos y macrófagos, debido a daños en la BHE o mediante la estimulación con el factor de crecimiento transformante β (TGF-β) (Young Kim, Buckwalter, Soreq, Vezzani, y Kaufer, 2012).
El SNC tiene una habilidad limitada para regenerarse por lo cual una respuesta inflamatoria aguda puede generar daño celular permanente, aunque también se ha planteado que leves respuestas inflamatoria podrían ser reparadoras y benéficas (Weil et al., 2008).
2.4.1. Activadores celulares. En el foco isquémico se liberan ROS, glutamato y grandes cantidades de K al medio extracelular, que generan ondas de despolarización y daño celular en el área de penumbra (Arango-davila et al., 2004). Además, se inducen genes de expresión rápida (IEG) proteínas de choque térmico (HSP) y activación de genes proinflamatorios (Culmsee y Krieglstein, 2005) los cuales estimulan la activación de astrocitos y microglía.
2.5. RESPUESTA CELULAR ANTE LA ISQUEMIA CEREBRAL
35
Durante la isquemia se inhibe la síntesis proteica en las neuronas, lo que conduce a la muerte neuronal (Vosler et al., 2011). Uno de los mecanismos que inhibe la síntesis proteica es la estimulación de receptores NMDA que liberan calcio a nivel intracelular, el cual activa la calpaina, una proteasa, que al ser inhibida genera neuroprotección (Vosler et al., 2011). Durante la isquemia se induce estrés oxidativo, mediante la producción de anión superóxido, NO y peroxinitritos, estos últimos, inhiben las proteínas al unirse a las tirosinas, lo que causa muerte neuronal y afecta otros tipos celulares (Tajes et al., 2013).
En las neuronas sometidas a isquemia se activan factores pro inflamatorios como el NF-κβ (Pranski et al., 2012) y se desarrollan procesos de adaptación a la depleción de oxígeno y glucosa como la modificación de la proteína del citoesqueleto MAP2/τ que disminuye en el área infartada, pero se recupera su reactividad con el tiempo post isquemia, lo cual se podría relacionar con mecanismos de plasticidad neuronal. Por otra parte, un aumento en la expresión de PKC postisquemia que se relaciona con desensamblaje de microtúbulos (Arango-davila et al., 2004). La muerte neuronal se puede presentar hasta en una etapa tardía post isquemia lo que implica que las células utilizan mecanismos para adaptarse a la injuria en la fase aguda de la enfermedad (Stoll et al., 1998).
Posterior a la isquemia se dan procesos de neuroreparación como la proliferación de precursores neuronales y gliales, además de la liberación de factores que estimulan la diferenciación celular, como el factor neuronal derivado de cerebro (BDNF), factor de crecimiento de fibroblastos-2 (FGF2), factor de crecimiento epidérmico (EGF) y TGF-β (Sánchez-Mendoza et al., 2012).
36
En neuronas estimuladas con TNFα, se induce la señalización proinflamatoria mediada por el NF-kβ (Young et al., 2012) y la activación de NF-κβ, se ha identificado como un mecanismo neuroprotector posterior a la isquemia (Duckworth, Butler, Collier, Collier, y Pennypacker, 2006).
2.6. MACROGLÍA.
Posterior a la isquemia cerebral inicia la reacción glíal, caracterizada por los cambios morfológicos y funcionales de las células de macroglía y microglía los cuales se evidencian tanto en el foco isquémico como en el área de penumbra, dando como respuesta de resolución la denominada cicatriz glíal (Arango-davila et al., 2004). La microglía se activa antes que los astrocitos en el proceso de isquemia y media la respuesta inflamatoria y la reparación celular, los astrocitos presentan una activación tardía con respecto a la microglía, pero que perdura durante más tiempo (Dihné, Block, Korr, y Töpper, 2001)
2.6.1. Astrocitos: respuesta ante la isquemia. Al activarse los astrocitos aumentan su tamaño celular, número y extensión de sus ramificaciones, esta respuesta astrocitaria se caracteriza por hipertrofia e hiperplasia además del aumento en la expresión de la proteína Acídica Glíal Fibrilar (GFAP), la cual ha sido evidente en el foco isquémico y en la zona circúndate (Stoll et al., 1998). Los astrocitos alojados en la corteza cerebral se denominan protoplasmáticos tienen forma estrellada, los extremos de las prolongaciones astrocitarias envuelven los capilares y las sinapsis del neuropilo (Pascual, Gonzáles-Llanos, Cerdán, Carceller, y Roda, 2000).
37
emplean sus sistemas habituales para restituir la homeostasis, pero al no poder restituirla, sufren edema. Ya que el influjo de k, ácido láctico, radicales libres y disminución de pH es demasiado alta para los mecanismos del astrocito. Los astrocitos expanden su volumen y pueden volver a recuperarlo (Pascual et al., 2000).
Los astrocitos son muy resistentes a la deprivación de oxígeno y glucosa por separado y en asociación (OGD, voz ingl. Oxigen and glucose deprivation, deprivación de oxígeno y glucosa) mueren mucho después que las neuronas para lo cual cuentan con mecanismos como la activación de ribosomal S6K por mTOR que ayuda a reducir los niveles de ROS y afecta la síntesis de proteínas en los astrocitos (Pastor et al., 2009). El glutatión es un mecanismo antioxidante, que en isquemia global es utilizado por los astrocitos principalmente a las 24 horas y es utilizado hasta 7 post reperfusión (Sulkowski et al., 2002).
Ante los eventos excitotóxicos los astrocitos se encargan no solo de la protección de neuronas sino también de oligodendrocitos al aportarles lactato como fuente de energía (Rinholm et al., 2011).
La inflamación también es mediada por astrocitos al incrementar su expresión de TNF y COX2, la activación de COX2 se da vía MyD88, NF-κB, P38 y JNK que conduce a la producción de prostanoides (PGE2) (Font-Nieves et al., 2012) así como la liberación de citoquinas como la citoquina quimioatrayente inductora de neutrófilos (CINC-1) en respuesta al daño neuronal (Katayama, Tanaka, Yoshida, Uehara, y Minami, 2009).
2.6.2. Oligodendrocitos: Respuesta ante la isquemia. Los oligodendrocitos fueron descritos por Pio de Rio Ortega al igual que la microglía, son compuestos por mielina que está conectada o no a la vaina de mielina neuronal. Los que se encuentran de forma satélite se denominan perineuronales y regulan el microambiente (Baumann y Pham-Dinh, 2001).
38
pueden consumir lactato como fuente energética y para la síntesis de lípidos (Rinholm et al., 2011).
Los oligodendrocitos son las células más susceptibles a la isquemia después de la neurona, (Lyons y Kettenmann, 1998). En isquemia se ha reportado la disminución de oligodendrocitos en la zona infartada y que la muerte de oligodendrocitos se da previa a la perdida de las proteínas de mielina en la zona infartada. Además los oligodendrocitos que sobreviven proliferan en el borde de la lesión. El incremento de la proteína básica de mielina (MBP) se observa 5 días posteriores a la isquemia, en el cuerpo calloso y en la corteza infartada. Incluso se ha observado que la proliferación de oligodendrocitos colocaliza con astrocitos y microglía (Mandai et al., 1997). Estos resultados sugieren la invasión de oligodendrocitos sobrevivientes al tejido dañado (Mandai et al., 1997).
Durante la isquemia, el estrés oxidativo y el aumento de glutamato genera excitotoxicidad que desencadena muerte de los oligodendrocitos, (Dewar, Underhill, y Goldberg, 2003) sin embargo el lactato puede ayudar al desarrollo de los oligodendrocitos y a la mielinización durante niveles bajos de glucosa debido al transportador de monocarboxilato (Rinholm et al., 2011). Por otra parte se ha observado en estas células una expresión elevada de COX2 siendo un factor de susceptibilidad ante la excitotoxicidad (Carlson et al., 2010). Los procesos de oligogénesis y oligodendrogénesis varían según la zona del cerebro, en relación a las zonas de angiogénesis posterior a la isquemia (Jiang et al., 2012).
2.7. MICROGLÍA E ISQUEMIA
39
proinflamatorias o antiinflamatorias como interleuquinas 1, 3, 5, y 6, Factor de Crecimiento Nervioso (NGF), GTF y TNF (R. A. Taylor y Sansing, 2013). Su morfología varía de acuerdo a su función o estado de actividad y se clasifica morfológicamente en residente-ramificada, activada y ameboide, estos cambios morfológicos corresponden a cambios funcionales (Jonas et al., 2012; Stence, Waite, y Dailey, 2001). Fenotípicamente se identifica como CD68+, CD45 bajo, CD11b+, CD11c alto, MHCII+ y CD14- (Guillemin y Brew, 2004) y el fenotipo que puede presentar la microglía varía de acuerdo al estado en el que se encuentre el tejido (Dihné et al., 2001).
La microglía media una respuesta inmune plástica en el cerebro, constituyendo una interface entre sistema inmune y el parénquima (Kreutzberg, 1996). Además, cumple funciones no inmunes en el SNC, principalmente la estimulación hormonal mediada por interleuquinas como la IL-1 e IL-6 (Mor et al., 1999). También, se ha planteado una regulación hormonal de la respuesta inmune en el SNC, ya que el estrógeno puede estimular la microglía y aumentar su respuesta durante el ciclo estral (Mor et al., 1999).
La forma de la microglía varía según la zona del cerebro que ocupe, en sustancia gris es ramificada, con procesos largos, delgados y radiados, mientras que en sustancia blanca tiene un soma ovoide, con pocos procesos, ubicados a lo largo de los axones (Savchenko, Mckanna, Nikonenko, y Skibo, 2000). Durante el estado residente el soma de la microglía permanece anclado en el mismo lugar pero sus procesos se encuentran en actividad constante monitoreando el estado de la sinapsis, haciendo breves contactos con botones pre y post sinápticos (Wake, Moorhouse, Jinno, Kohsaka, y Nabekura, 2009), esta morfología ramificada consta de un soma pequeño y un gran número de ramificaciones delgadas de 2 a 6 ramificaciones principales y algunas pueden tener longitud mayor a 50 μm, con especies de espinas y se observa en zonas donde no hay daño celular (Stence et al., 2001).
40
microglíal. Las células detienen sus movimientos e inician retracción de los procesos y movimiento de lamelipodias (lo que podría indicar una estrategia para desanclarse del tejido), el movimiento de los organelos también se detiene. Estos procesos son reversibles, al volver a un pH neutro recuperan su motilidad y morfología. Al disminuir el pH a 6 o aumentar a 8 no parece afectarse la viabilidad microglial (Faff y Nolte, 2000).
La microglía residente pasa a ser activada por estimulación autocrina (Yang et al., 2010) y su rápida activación involucra citoquinas, factores de crecimiento, receptores, moléculas señalizadoras y mitógenos (Kreutzberg, 1996). El cambio morfológico se realiza en varias fases denominadas de retracción (retracción e procesos antiguos), motilidad (extensión de procesos más motiles), transicional (intermedio entre los dos tipos de procesos) y de locomoción cuando la microglía migra a través del parénquima cerebral (Jonas et al., 2012; Stence et al., 2001).
La forma activa tiene el cuerpo celular más grande y prolongaciones cortas, pero su función no es en su totalidad la de un macrófago, su activación se observa a partir de la primera hora posterior a la lesión (Stence et al., 2001). Al activarse la microglía, sufren cambios como la detirosinación y acetilación de la tubulina del citoesqueleto y se ha demostrado que cofactores de la tubulina se elevan con la activación microglial, sobretodo en células cercanas a la lesión, siendo mayor a 48 horas posterior a la lesión y que podría controlar el número y densidad de microtúbulos en la microglía en estados reactivos (Fanarraga, Villegas, Carranza, Castaño, y Zabala, 2009). Por otra parte, la acidificación y la consecuente disminución de la motilidad basal microglial, se relaciona con un rearreglo del citoesqueleto de actina (Faff y Nolte, 2000). El fenotipo reactivo actúa como un macrófago, tienen forma ameboide, esférica y pequeña, sin ramificaciones y su número aumenta entre las 6 y las 12 horas (Dihné et al., 2001; Jonas et al., 2012).
41
(LIL1CAM), RFD-7 ni alfa1-quimiotripsina y los macrófagos sí. En la microglía el MHCII es constitutivo así como el HLA DR y la diferencia más notoria es que la microglía está cubierta por espinas, mientras los macrófagos tienen forma de rosa en la superficie esta característica no varía con la edad o inmunoestimulantes. Por lo anterior es necesario utilizar varios marcadores unidos para su identificación. La microglía murina no produce ácido quinolínico y la humana si (Guillemin y Brew, 2004).
Los oligodendrocitos y la microglía son las células más susceptibles ante isquemia y el daño hipóxico después de las neuronas, pero la microglía tiene mayor porcentaje de sobrevivencia que los oligodendrocitos (Lyons y Kettenmann, 1998). La activación microglíal precede la activación de los macrófagos (Dihné et al., 2001) y en el proceso isquémico la microglía puede promover la muerte celular en las primeras fases, pero posteriormente, puede promover la supervivencia de células y reparación tisular (Wake et al., 2009), liberando factores de crecimiento (Madinier et al., 2009) y favoreciendo la recuperación de los tejidos (Stence et al., 2001).
2.8. DESMIELINIZACIÓN
42
2.9. ALTERNATIVAS TERAPÉUTICAS
Ante la isquemia cerebral se han probado diferentes terapias y clínicamente se han implementado unidades de ECV, en las cuales es monitorea la evolución de los pacientes, sus signos vitales y permiten la atención temprana de complicaciones secundarias (Caplan y Hacke, 2003) además, se miden, presión arterial, temperatura corporal y glicemia (Ustrell-Roig y Serena-Leal, 2007).
Tabla 1. Tratamientos aplicados en Isquemia Cerebral
Tratamiento Ventana terapéutica
Efecto Aprobado Fuente
rtPA 3 primeras horas
Trombolisis FDA y
EMEA
(Caplan y Hacke, 2003)
Prourocinasa 6 horas Trombolisis intraarterial
No (Ustrell-Roig y
Serena-Leal, 2007).
Aspirina 48 horas Bloquean la agregación plaquetaria
No (Ustrell-Roig y
Serena-Leal, 2007).
Heparina Profiláctico Anticoagulante No (Ustrell-Roig y Serena-Leal, 2007).
Cirugía Terapéutico Bypass arterial extracraneal
(Caplan y Hacke, 2003)
Warfarina Terapéutico Anticoagulantes (Caplan y Hacke, 2003)
Dipiridamole Terapéutico Bloquean la agregación plaquetaria
43 Fuente: autor
Tabla 1. Continuación
Tratamiento Ventana terapéutica
Efecto Aprobado Fuente
Atorvastatina Neuroprotect ora
Extender la terapia trombolítica con rtPA
(Caplan y Hacke, 2003)
Fuente autor
Entre las alternativas terapéuticas en fase de prueba se plantea el uso de la glicoproteina IIb/IIIa, las cuales bloquean los últimos pasos en la vía de la agregación plaquetaria y reducen la liberación de factor tisular (Ustrell-Roig y Serena-Leal, 2007).
2.10. TECNICAS UTILIZADAS EN EXPERIMENTACIÓN PARA EVALUAR EL DAÑO CEREBRAL
44
2.10.2. El Luxol Fast Blue (LFB). es un marcador de las fibras de mielina, se une a los lípidos de la membrana, para lo cual es necesaria la extracción de algunos fosfolípidos y gangliósidos, lípidos polares de las fibras de mielina permitiendo que la tinción se una a la fibras mielinizadas pequeñas e individuales en la capa molecular de la corteza cerebral (Geisler, Heilmann, y Veh, 2002).
2.10.3. Cresyl Violeta. marcador de neuronas y glía, debido a la interacción iónica entre la tinción básica y los grupos ácidos como el ADN/ARN (Türeyen, Vemuganti, Sailor, y Dempsey, 2004).
45
3. MÉTODOS
3.1. ANIMALES DE EXPERIMENTACIÓN: CUIDADO Y MANEJO.
Los procedimientos realizados con animales fueron evaluados y aprobados por el comité de bioética de la Universidad del Tolima, de acuerdo con las normas internacionales de protección y manejo de animales de laboratorio descritas en Guide for the care and use of laboratory animals, de acuerdo con la ley 84 de 1989 (protección animal en Colombia) y la ley 8430 de 1993 sobre investigación científica en salud.
3.2. DISEÑO EXPERIMENTAL
Se utilizaron 35 ratas, machos, línea Wistar (Rattus norvegicus) entre 8 y 12 semanas de edad, con peso promedio de 240 ± 25 g, obtenidas del Bioterio de Experimentación Animal de la Facultad de Ciencias de la Universidad del Tolima (BEAUT) donde se encontraban en condiciones controladas de temperatura (22º C), humedad relativa (60-80 %) fotoperiodo de 12 horas luz y 12 horas oscuridad, alimentados con Rodentina® extrudizada (proteína 23,5 %, Agrinal Colombia S.A.) y agua acidulada ad libitum. El tamaño muestral fue calculado con la siguiente fórmula:
46
De esta manera se estimaron 4 animales para cada grupo experimental (8, 24, 48 y 72 horas).
Para estimar las pérdidas en el n muestral por muerte, daños en los tejidos y otros errores experimentales, se ajustó a la corrección de tamaño muestral descrita por Fernández, (1996).
Lo cual indica, que para un tamaño n muestral de 16 animales se requieren 8 animales para cubrir las pérdidas por muerte o daño en el tejido, lo que sugiere que se requieren 24 animales para el grupo experimental (n= 16 + 8 de perdidas), mas 8 animales del grupo control, para un total de 32 animales.
Para la evaluación histoquímica e inmunohistoquímica se utilizaron 24 individuos asignando 4 para cada grupo experimental y 2 animales control que fueron sacrificados a 8, 24, 48 y 72 horas postisquemia. Para la prueba de TTC, se emplearon 3 unidades experimentales sacrificadas a 8, 24, y 48 horas (Tabla 2.).
47
PRUEBAS 8 horas
Grupo 1 24 horas Grupo 2 48 horas Grupo 3 72 horas Grupo 4
ISQUEMIA Inmunohistoquímica 4 4 4 4
Distribución Infarto TCC 1 1
CONTROL Sham (abordaje sin oclusión vascular)
2 2 2 2
Fuente: autor
Los animales de los grupos experimentales fueron sometidos a embolización de la arteria cerebral media derecha, bajo anestesia general, como se describe a continuación y a los controles se les realizó abordaje quirúrgico sin oclusión arterial.
3.3. MODELO DE ISQUEMIA: EMBOLIZACIÓN ARTERIAL CON COÁGULOS
AUTÓLOGOS.
48
(Figura 3 B y C) (Beeton, Garcia, y Chandy, 2007). Se colectaron aproximadamente 500 μl de sangre mediante succión con una jeringa de insulina (1 ml) y se depositaron en viales de 1,5 ml, etiquetados individualmente. La sangre colectada fue incuba a 37 ºC por 2 horas y almacenada a 4 ºC por 24 horas. Previo a la cirugía isquémica los coágulos fueron lavados con NaCl y fragmentados hasta obtener tamaños de 20 μm y un volumen 20 μl de coágulos que fueron suspendidos en 500 μl de solución salina como se observa en la figura 3 D.
Figura 3. Método de extracción de sangre a partir de la vena safena interna y formación de coágulos autólogos
Fuente autor
A. Miembro posterior extendido y rasurado. B. Punción de la vena safena interna. C. Sangre que brota de la safena interna D. Sangre colectada en viales de 1,5 ml para la formación de microcoágulos.
49
Se posicionó el animal en decúbito supino (dorsal) y se realizó una incisión de aproximadamente 3 centímetros longitudinal anterior, en sentido cráneo-caudal (cervicotomía) con bisturí.
Se disectó la piel, el tejido celular subcutáneo y la fascia del musculo cutáneo hasta visualizar las glándulas salivales submaxilares (Figura 4 A). Posteriormente se separaron los músculos esternohioideos y los músculos esternomastoideos (Figura 4 B) se replegaron con un retractor autoestático. Bajo estos se observó el músculo omohioideo. Se colocaron retractores para el músculo digástrico y se separó el omohiodeo hasta observar la arteria carótida común (ACC) y el nervio vago tal como se aprecia en la figura 4 C (Hernández, Trujillo, y Cespedes, 2011).
Figura 4. Abordaje quirúrgico de la ACC derecha
Fuente: autor y Céspedes A., 2013.
A. Se aprecia la glándula salival submaxilar (1). B. La glándula salival submaxilar es retraída lo que permite observar el músculo esternomastoideo (1) y músculo esternohiodeo (2) C. La ACC (1) es separada del nervio vago (2).
SROLHVWLUHQR1TXHVHDYDQ]ySRUOD$&,KDVWDODELIXUFDFLyQGHODDUWHULDFHUHEUDO PHGLD $&0 FRQ OD DUWHULD FHUHEUDO DQWHULRU $&$ )LJXUD % $O FDWpWHU VHDFRSOR XQD MHULQJD TXH FRQWHQtD XQ YROXPHQ GH O GH FRiJXORV DXWyORJRV SUHIRUPDGRV UHVXVSHQGLGRVHQOGH1D&O\TXHIXHURQOLEHUDGRVHQODELIXUFDFLyQKDFLD HOUDPRGHODDUWHULDFHUHEUDOPHGLDFRQHOILQGHLQGXFLUODLVTXHPLDFHUHEUDOPXOWLIRFDO )LJXUD & 3RVWHULRU D OD OLEHUDFLyQ GH ORV FRiJXORV VH UHWUDH HO FDWpWHU FXLGDGRVDPHQWH)LJXUD'\VHOLJyOD$&&HQHOSXQWRGHODLQFLVLyQ)LJXUD(\ )VHUHWLUyHOFODPSYDVFXODUUHVWDEOHFLHQGRODFLUFXODFLyQ\FRPSUREDQGRTXHQRVH SUHVHQWDUDQ KHPRUUDJLDV )LJXUD * \ + OD LQFLVLyQ IXH FHUUDGD FRQ PDWHULDO QR DEVRUELEOH/DWHPSHUDWXUDUHFWDOIXHUHJLVWUDGDGXUDQWHHOSHULRGRSUHRSHUDWRULR\XQD YH]ILQDOL]DGDODFLUXJtD'XUDQWHSRVWRSHUDWRULRVHPDQWLHQHQORVDQLPDOHVDUURSDGRV FRQPDQWDVWpUPLFDV\VHOHVSHUPLWLyOLEUHDFFHVRDODJXD\DOLPHQWR $ORVDQLPDOHV FRQWUROVHOHVUHDOL]yODFLUXJtDVLQODRFOXVLyQDUWHULDO
)LJXUD0RGHORGHRFOXVLyQDUWHULDOPHGLDQWHFRiJXORVDXWyORJRV
)XHQWH$XWRU
(Q OD LPDJHQ VH UHSUHVHQWD HO FHUHEUR GH XQD UDWD :LVWDU FRQ DPSOLILFDFLyQ GH OD HVWUXFWXUD YDVFXODU
DUWHULDO OD DUWHULD FDUyWLGD FRP~Q $&& OD ELIXUFDFLyQ HQ OD DUWHULD FDUyWLGD H[WHUQD $&( \ DUWHULD
51
representa la inyección de los coágulos autólogos a nivel de la ACM, avanzando a través de la ACC y ACI; ocasionando isquemia cerebral como se representa en el corte coronal.
Figura 6. Procedimiento quirúrgico para la embolización de la arteria cerebral media derecha mediante coágulos autólogos
Fuente Autor y Céspedes A., 2013
A. Se aprecia la ACC separada del nervio vago. B. Incisión en la ACC e introducción del catéter. C. Avance del catéter por la ACC hasta la bifurcación de la ACM con la ACA donde fueron liberados los coágulos autólogos. D. Se posicionó un hilo de nailon alrededor de ACC. E. F. Retracción del catéter y ligadura del nailon alrededor de la ACC. G y H. La ligadura de la ACC impide la reperfusión.
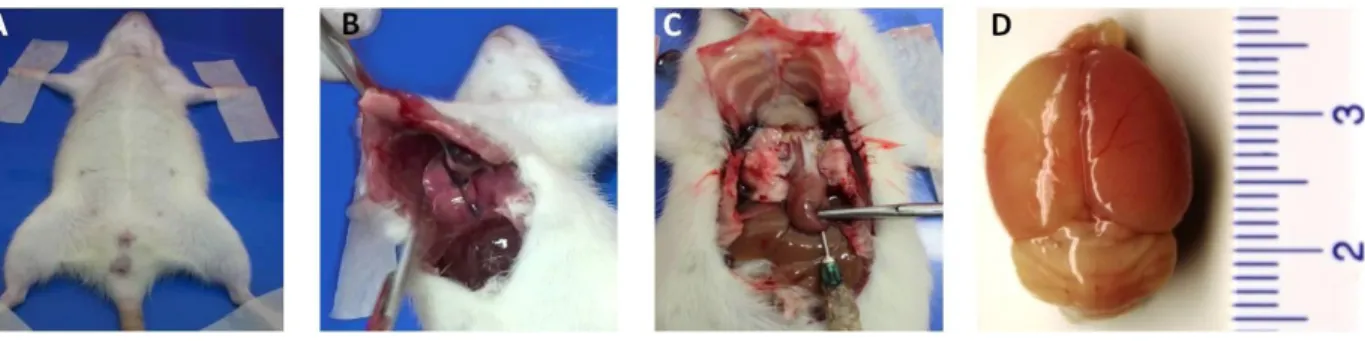
3.4. TIEMPOS DE SACRIFICIO – PERFUSIÓN
52
Los animales se sometieron a restricción alimentaria 12 horas antes de la perfusión para facilitar el efecto anestésico y se comprobaron los reflejos superficiales y profundos antes de iniciar el procedimiento quirúrgico (Figura 7 A). A continuación se incidió en la cavidad torácica (Figura 7 B) y se perfundió vía intracardiaca mediante avance aórtico (Figura 7 C), 200 ml de solución salina y posteriormente 200 ml de paraformaldehido (PFA) 4 %. Seguidamente fue extraído el cerebro de la cavidad craneana (Figura 7 D) y se sumerge en PFA a 4º por 48 horas para su fijación.
Figura 7. Método de perfusión cerebral
Fuente: autor
A. Rata Wistar bajo anestesia profunda, en decúbito supino. B. Corte en “V” realizado en la quilla esternal por el cual se accede a la cavidad abdominal y posteriormente a la toráxica. C. Punción en el ventrículo izquierdo y avance aórtico por medio del cual se perfunde solución salina y paraformaldehído. D. Cerebro perfundido de un individuo isquémico.
3.5. PROCESAMIENTO DE TEJIDOS: CORTE Y CRIOPRESERVACIÓN.
Los cerebros fueron sometidos a inmersión en gradientes de sacarosa a concentraciones de 15 %, 20 % y 30 % durante 24 horas para cada concentración y se sumergieron en solución criopreservante (30% etilenglicol, 30% Glicerol, 25% tampón fosfato 0,4 M y 15% agua desionizada) hasta el proceso de corte.
FULRSUHVHUYDQWH D & HQ FDMDV PXOWLSR]R SDUD VX SRVWHULRU PRQWDMH HQ OiPLQDV WLQFLRQHVHLQPXQRKLVWRTXtPLFD
/RV SURFHVRV GH FRUWH GH ORV FHUHEURV WLQFLRQHV LQPXQRKLVWRTXtPLFD \ DQiOLVLV PLFURVFySLFR IXHURQ UHDOL]DGRV HQ HO /DERUDWRULR GH 7R[LFRORJtD GH OD )DFXOWDG GH 0HGLFLQD9HWHULQDULD\=RRWHFQLD09=\HQ/$%,287
)LJXUD&RUWHGHFHUHEURVGHUDWD:LVWDUHQYLEUDWRPRVHPLDXWRPiWLFR
)XHQWHDXWRU
$ 6H DSUHFLD XQ FHUHEUR GH UDWD:LVWDU PRQWDGR HQ OD SODWLQD GHO YLEUiWRPR VHPLDXWRPiWLFR SDUD VHU
FRUWDGRSRUODFXFKLOODYLVWDODWHUDO%YLVWDIURQWDO
'(7(50,1$&,Ï1,10812+,67248Ë0,&$'($&7,9,'$'0,&52*/,$/
54
Este marcador fue reconocido mediante inmunohistoquímica convencional en cortes de 50 μm, de bregmas entre 2,8 y 3,6.
Los cortes fueron sometidos a lavados con PBS 0,1 M, 5 veces por 10 minutos y se depositaron en cajas multipozo y fueron sumergidos en solución de inhibición de peroxidasa endógena (1:1 metanol y PB 0,1 M + 165 μl de peróxido) por 20 minutos, se retiró la solución de peroxidasa y los cortes fueron lavados con PBS 0,1 durante 10 minutos 3 veces.
Con el objetivo de disminuir el marcaje inespecífico, se sumergieron los cortes en tampón de preincubación por 90 minutos este consiste en albumina sérica bovina (BSA) 1 % Tritón x-100 al 0,3 % en PB 0,1 M. Los cortes se incubaron en el anticuerpo primario monoclonal OX42 (Thermo) (1:100) preparado en tampón de incubación (BSA 0,3 %, Tritón 0,3 % en PB 0,1 M) durante la noche a 4 ºC.
El anticuerpo primario OX42 fue retirado de los cortes 24 horas después y fueron lavados con PBS, e incubados con el anticuerpo secundario biotinilado anti-mouse (1:250) por 2 horas a temperatura ambiente. Seguidamente se incubaron con el complejo Avidina Biotina diluido en tampón de incubación, a una concentración de 1:250 por 2 horas protegido de la luz. Al finalizar las 2 horas, los cortes fueron lavados con PBS. El marcador fue revelado con DiaminoBenzidina diluida en PBS y peróxido de hidrógeno en penumbra. Los cortes fueron montados en láminas gelatinizadas (Gelatina Porcina) y deshidratados en alcohol de 70 %, 95 %, etanol 100 % por 4 minutos y xileno durante 5 minutos 2 veces para ser sellados con Consul Mount y realizar el registro fotográfico.
55
3.7.1. Evaluación de la distribución del Infarto. Para la evaluación de la distribución del infarto se utilizó la tinción hidrocloro 3,3,5 de trifenil tetrazolio (TTC) descrita por Bederson et al., (1986).
Para la preparación de TTC (Sigma-Aldrich) se diluyeron 50 mg de TTC en 100 ml de PBS o solución salina. Se agitó en Shaker hasta homogenizar la solución que presentaba un color amarillo claro. Esta solución se cubrió con papel aluminio y se almaceno a 4 º C hasta su uso.
Para el experimento de TTC, se operaron 3 ratas Wistar con peso promedio de 212 g, según el protocolo mencionado anteriormente; las que fueron sacrificadas y perfundidas vía intracardiaca con solución salina a 4 ºC. Se extrajo rápidamente el cerebro del cráneo y se sumergió en PBS a 4 º C. A continuación se realizaron cortes coronales de 1 mm en matriz stoelting para rata, y se transfirieron a una caja multipozo que contenía TTC al 0,05 % en PBS o en NaCl. Posteriormente se incubaron durante 30 minutos a 37 º C, protegidos de la luz. Se agitó suavemente el plato multipozo cada 5 minutos; al cabo de los 30 minutos se retiró el TTC. Los cortes fueron lavados 3 veces con PBS en agitación por 1 minuto (Figura 9 A y B). Para finalizar los cortes fueron sumergidos en un plato multipozo con PFA buferado al 4% durante 30 minutos hasta 24 horas cuando se realizó el registro fotográfico (Joshi, Jain, y Murthy, 2004; Khan, Baziany, Banigesh, Hemmings, y Shuaib, 2000).
Las ratas de 8 y 24 horas sobrevivieron hasta el tiempo de sacrificio, pero la rata de 48 murió, posterior al primer día.