METODOLOGÍA SINTÉTICA
APLICADA A LA SÍNTESIS DE
FÁRMACOS
TEMA 8
Inhibidores de quinasas como agentes
anticáncer
Índice 1. Introducción
2. Quinasas como enzimas clave en el proceso de señalización celular 2.1. Estructura de las quinasas
2.2. Colocación del ATP en el bolsillo de unión de la quinasa 2.3. Mecanismo de fosforilación
2.4. Unión de la quinasa al sustrato diana 2.5. Estados activado e inactivado
3. Tipos de quinasas 3.1. Quinasas RTKs
3.2. Transducción de señal por activación de RTKs: vía RAS-RAF 3.3. Transducción de señal por activación de RTKs: vía JAK-STAT 4. Quinasas como dianas moleculares en el tratamiento del cáncer
4.1. Desregulación de la actividad quinasa 4.2. Inhibidores de quinasas
4.2.1. Fármacos inhibidores de quinasas hiperactivas
4.2.2. Fármacos inhibidores de Receptores Tirosina-Quinasa 4.3. Tipos de fármacos inhibidores de quinasas
5. Imatinib
5.1. Translocación cromosómica y formación de la quinasa híbrida bcr-abl 5.2. Quinasa abl
5.3. Modo de acción del imatinib
5.4. Unión del imatinib al centro activo de la quinasa abl 5.5. Síntesis de imatinib
5.5.a.1. Análisis retrosintético de imatinib 5.5.b.1. Síntesis de imatinib
5.5.c.1. Cuestiones
5.5.b.2. Síntesis alternativas de imatinib 5.5.c.2. Cuestiones
5.6. Resistencia a imatinib 6. Nilotinib
6.1. Modo de acción del nilotinib 6.2. Síntesis de nilotinib
6.2.a.1. Análisis retrosintético de nilotinib 6.2.b.1. Síntesis de nilotinib
6.2.c.1. Cuestiones
6.2.a.2. Análisis retrosintético alternativo de nilotinib 6.2.b.2. Síntesis
7. Dasatinib
7.1. Unión del dasatinib a la quinasa abl 7.2. Síntesis de dasatinib
7.2.c. Cuestiones 8. Sorafenib
8.1. Síntesis de sorafenib
8.1.a. Análisis retrosintético de sorafenib 8.1.b. Síntesis
8.1.c. Cuestiones 9. Gefitinib
9.1. Síntesis de gefitinib
9.1.a. Análisis retrosintético de gefitinib 9.1.b. Síntesis
9.1.c. Cuestiones 10. Erlotinib
10.1. Síntesis de erlotinib 10.1.c. Análisis 10.1.b. Síntesis 10.1.c. Cuestiones 11. Sunitinib
11.1. Síntesis de sunitinib
11.1.a. Análisis retrosintético de sunitinib 11.1.b. Síntesis
11.1.c. Cuestiones 12. Lapatinib
12.1. Síntesis de lapatinib
12.1.a. Análisis retrosintético de lapatinib 12.1.b. Síntesis
13. Pazopanib
13.1. Angiogénesis
13.2. Estructura del VEGF 13.3 Tipos de VEGF
13.4. Receptores del VEGF
13.4.1. Receptor VEGF-1 13.4.2. Receptor VEGF-2 13.4.3. Receptor VEGF-3 13.5. Uniones VEGF-VEGFR 13.6. Desarrollo del pazopanib 13.7. Síntesis del pazopanib
13.7.a. Análisis retrosintético del pazopanib 13.7.b. Síntesis
Inhibidores de quinasas como compuestos anticáncer
1. Introducción
La gran mayoría de fármacos antitumorales centran su acción en todas las células de crecimiento rápido, incluyendo las normales, de ahí los graves efectos secundarios de la quimioterapia. Desde hace algunos años se han empezado a aplicar nuevas terapias anticáncer denominadas terapias dirigidas. En estas terapias se emplean fármacos que bloquean el crecimiento y la diseminación del cáncer interfiriendo con biomoléculas implicadas en los procesos de señalización celular, consiguiendo de esta manera que las células cancerosas detengan su crecimiento y su división incontrolada.1
2. Quinasas como enzimas clave en el proceso de señalización celular
Las quinasas son enzimas clave en el proceso de señalización celular encargándose de la transferencia de grupos fosfato desde ATP a un sustrato diana. Este proceso es, desde el punto de vista químico, una fosforilación, por lo que a las quinasas también se las denomina como ATP(x)fosfotransferasas, donde (x)= molécula a la que se transfiere el grupo fosfato (véase el esquema 1). El proyecto del genoma humano ha permitido la identificación de 520 quinasas que constituyen lo que se denomina como el «quinoma» humano.2
Esquema 1
La fosforilación de las proteínas implica la adición de un grupo fosfato a un aminoácido de la cadena peptídica y es un proceso clave en las vías de señalización celular, ya que cada grupo fosfato lleva consigo dos cargas negativas, y la fosforilación de un aminoácido de la cadena peptídica provoca un cambio conformacional en la proteína que es fosforilada. El cambio conformacional en la proteína fosforilada permite un control regulatorio sobre la actividad de ésta.
La fosforilación por las quinasas es un proceso reversible. Las proteínas pueden ser desfosforiladas por enzimas denominadas proteína-fosfatasas. Estos dos grupos de enzimas, las quinasas y las fosfatasas, a menudo trabajan juntas para apagar y encender las señales celulares. Las quinasas juegan un papel muy importante en varios procesos de señalización intracelular, incluyendo aquellos que controlan el crecimiento y la división celular.
2.1. Estructura de las quinasas
Las quinasas unen covalentemente un grupo fosfato, a partir de un ATP donador, a residuos de serina, treonina y/o tirosina de otras proteínas o sustratos. La reacción de fosfotransferencia requiere la presencia de tres sitios específicos:
a) Un sitio de unión de ATP.
b) Un dominio que cataliza la transferencia de un grupo fosfato del ATP.
1 http://www.cancer.gov/espanol/recursos/hojas-informativas/tratamiento/terapias-dirigidas.
c) Un sitio de unión del sustrato.
Las quinasas constan de una estructura bilobular con un lóbulo N-terminal y uno C-terminal.
El lóbulo N-terminal, denominado dominio regulatorio, está constituido principalmente de láminas β y de una hélice α (hélice αC) que es parte del sistema de activación. El lóbulo N-terminal también contiene un bucle altamente flexible rico en glicina que cubre y ancla los fosfatos del ATP no transferibles(véase la figura 1).
El lóbulo C-terminal está constituido fundamentalmente de hélices α y se une al lóbulo N-terminal mediante un bucle denominado bisagra. En el lóbulo C-N-terminal se encuentra el bucle de activación. El sitio de unión del ATP está situado en la interfaz entre los dos lóbulos.
Figura 1. Estructura de una quinasa unida a ATP
En la figura 2 se muestra el complejo formado por una tirosina-quinasa receptora de insulina unida a ATP.3 En la parte a de la figura 2 se aprecia que el bolsillo de unión de ATP a la quinasa reside en un profundo surco colocado entre el lóbulo N-terminal y el lóbulo C-terminal.
En la parte b de la figura 2 se muestra con más detalle el centro activo de la enzima. Los enlaces de hidrógeno que mantienen unido al ATP al bolsillo están indicados con líneas de trazos en color amarillo y son los que se establecen entre N1 y N6 de la parte de adenina con aminoácidos del dominio bisagra. El fosfato γ del ATP está colocado hacia el exterior y muy cerca del hidroxilo fenólico de un residuo de tirosina. Obsérvese también la presencia de dos cationes Mg2+ en el centro activo de la enzima. Estos cationes juegan un papel clave en el proceso de fosforilación.
Bis ag
ra
Figura 2. ATP unido a una tirosina-quinasa
Todas las quinasas contienen 12 motivos o sub-dominios altamente conservados, y 12 residuos invariantes. En la figura 3 se representa esquemáticamente la estructura de una quinasa con indicación de los diferentes subdominios.
Espe cific
idad
del s ustr
ato
D5
Bucle Gly
Segm ento
de
activ ación
Figura 3. Sub-dominios de una quinasa
D1 representa el sub-dominio de glicina que actúa como tapadera flexible coordinándose con los grupos fosfato no hidrolizables del ATP.
D5 indica el bucle bisagra, que une la región N-terminal con la C-terminal. El bucle bisagra se coordina con la parte de adenina del ATP.
D6 indica una región invariante de aspártico. Esta parte es la zona que aporta la funcionalidad básica encargada de la abstracción de un protón del grupo hidroxilo que va a ser fosforilado.
D7 marca una región invariante de aspártico denominada DFG, que contiene la secuencia Asp-Phe-Gly. En esta zona se coordina el catión Mg2+ que se unirá y orientará a los fosfatos β y
γ del ATP.
D7.5 indica el lóbulo de activación, que abarca las regiones DFG y APE.
D8 marca la hélice αEF (región APE), que determina la especificidad de la quinasa hacia la fosforilación de serina/treonina o hacia la fosforilación de tirosina.
Los elementos estructurales que son clave en la actividad catalítica de la quinasa se indican en la figura 4 y son:
a) El bucle rico en glicina, que se ubica encima del ATP (en azul en la figura 4). b) La hélice αC (en magenta en la figura 4).
c) La bisagra, en amarillo, que se une a la parte de adenosina del ATP formando un puente de hidrógeno bidentado.
d) El bucle de activación (en azul oscuro), que contiene una secuencia muy conservada de DFG y termina con APE.
…GxGxxG…
Helix-C
DFG……APE loop
Hinge ATP …GxGxxG…
Helix-C
DFG……APE loop
Hinge ATP
Figura 4. Elementos clave en la actividad catalítica de la quinasa
En la figura 5 se esquematizan las regiones del bolsillo de unión de ATP y la colocación de esta molécula en dicho bolsillo. Como se verá más adelante, el bolsillo de unión de ATP es la diana farmacológica de la gran mayoría de los fármacos antitumorales inhibidores de quinasas. En este bolsillo se distinguen las siguientes zonas:
1) La zona hidrofóbica de unión de la parte de adenina. En esta zona los N-1 y N-6 del anillo de adenina de la molécula de ATP se coordinan mediante enlaces de hidrógeno con aminoácidos del dominio bisagra.
3) La zona hidrofílica de unión de la parte de fosfato. Esta región parece ser la menos importante en términos de unión de inhibidores debido a su elevada exposición al disolvente. El grupo trifosfato se encuentra constreñido por el bucle de glicina y está colocado en una región de aminoácidos básicos, los cuales, junto con el aspartato del dominio D6, que desprotona al OH fosfoaceptor, están implicados en el proceso catalítico.4
4) El bolsillo hidrofóbico o bolsillo de selectividad, denominado bolsillo adyacente en la figura 5. Este bolsillo no está ocupado por ATP, pero juega un papel clave en el proceso de fosforilación. Su tamaño está controlado por dos residuos de aminoácido en las posiciones 129 y 183. En esta zona se coloca el dominio DFG, cuyo residuo de aspartato es el encargado de orientar a los fosfatos β y γ del ATP mediante coordinación con un catión Mg2+.
Figura 5. Bolsillo de unión de ATP en una quinasa
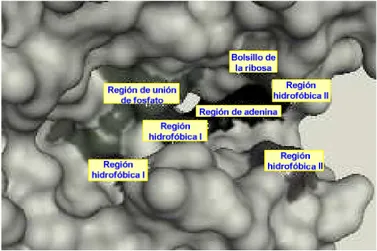
En la figura 6 se representa la superficie molecular del bolsillo de unión de ATP. Se indican las regiones de unión de adenina, la zona de unión de la ribosa, la zona de unión de fosfato y los bolsillos hidrofóbicos.
Figura 6. Representación de la superficie molecular del bolsillo de unión de ATP
2.2. Colocación del ATP en el bolsillo de unión de la quinasa
En la figura 7 se representan tres visiones consecutivas de una proteína quinasa tipo A (PKA), generadas por rotación de 90º alrededor del eje vertical. El lóbulo N-terminal es la región coloreada en gris, la región C-terminal está coloreada en ocre, la hélice αC en rojo, el bucle de activación en azul claro, la extensión N-terminal en púrpura, la extensión C-terminal en rosa, las proteínas asociadas en verde y la bisagra en azul oscuro.5
Figura 7. Representaciones de una proteína quinasa A
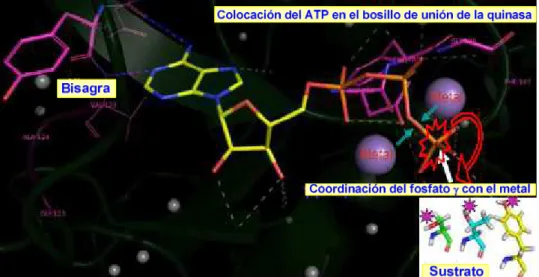
En la figura 8 se representa la estructura del complejo ATP-proteína quinasa A. La parte de adenina del ATP se coloca dentro del bolsillo de unión formando enlaces de hidrógeno entre los nitrógenos N1 y N6 con los aminoácidos de la zona bisagra. Los otros grupos no polares de la parte de purina establecen interacciones de van der Waals con el bolsillo.
Figura 8. Complejo ATP-proteína quinasa A
Los grupos hidroxilo de la ribosa se unen mediante enlaces de hidrógeno a un residuo de glutamato de la cadena lateral y a un grupo carbonilo de E170 (glutamato-170, numeración de los aminoácidos de la quinasa A).6
El grupo trifosfato apunta hacia afuera del bolsillo a fin de poder transferir el grupo fosfato
γ en la reacción de fosforilación. En el lóbulo N-terminal, y desde la hélice αC, un residuo de glutamato (E91) y una lisina (K72), colocada en la lámina β3, se coordinan con los fosfatos α y
β (véase la figura 8). Los fosfatos α y γ se coordinan con un catión Mg2+ que, a su vez, se coordina con el aspartato D184 del dominio DFG y con una asparagina N171. Los fosfatos β y
γ se coordinan con un segundo catión Mg2+ que, a su vez, se coordina con D184. Una tercera serie de interacciones adicionales con el bucle de glicina estabilizan aun más la conformación del ATP mediante unión a los fosfatos β y γ.
En la figura 9 se representa con más detalle la colocación del ATP en el bolsillo de unión de la quinasa. Los enlaces de hidrógeno que unen a la adenina con la zona bisagra se indican con líneas de trazos de color azul. Los enlaces de hidrógeno que une a los grupos hidroxilo de la ribosa con el bolsillo de unión se indican con líneas de trazos de color amarillo. Los cationes Mg2+ se representan mediante esferas de color púrpura. En el recuadro inferior de la figura 9 se indican las estructuras de los aminoácidos serina, treonina y tirosina. El grupo hidroxilo que contienen estos aminoácidos, que es el que experimentará el proceso de fosforilación, se remarca mediante una estrella de color purpura.
Figura 9. Colocación del ATP en el bolsillo de unión de la quinasa
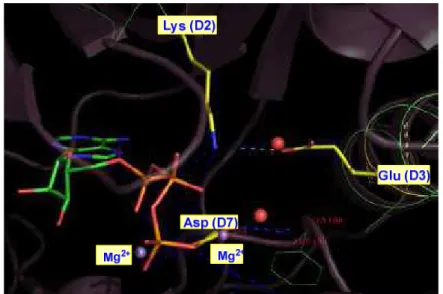
En la figura 10 se representa de nuevo el ATP en el bolsillo de unión de la quinasa. En la conformación activa de la quinasa un residuo de lisina del dominio D2 forma un puente salino con el glutámico del dominio D3. Esta interacción asegura la coordinación del aminoácido aspartato (parte DFG del dominio D7) con el fosfato en γ y con el catión Mg2+.
Figura 10. Coordinación de los fosfatos β y γ de ATP en el bolsillo de unión
2.3. Mecanismo de fosforilación
En la parte superior de la figura 11 se indican las interacciones de la parte de trifosfato de ATP con los aminoácidos del bolsillo de unión de una serina-treonina quinasa.
Parte de Ser o Thr que se va a fosforilar
Transferencia de fosfato
Estado de transición
ATP
ATP
ADP
Lys-72 (D2-D3)
Lys-168
Asp-184 (D7-DFG)
Asp-166 (D6)
Mg2+
Mg2+ Mg2+
Mg2+
Figura 11. Mecanismo de fosforilación de una serina/treonina quinasa
Los grupos fosfato en β y γ del ATP se coordinan con un catión Mg2+, coordinado a su vez a la parte de carboxilato de un aspartato (parte DFG del dominio D7).
Una lisina, Lys-72, protonada en el grupo amino, se coordina con los grupos fosfato en α y
β de ATP (dominio D2 y D3).
Otra lisina, Lys-168, se coordina con el fosfato en γ, aumentando la reactividad electrofílica de esta parte del ATP. En tirosina-quinasas el residuo estabilizante de los fosfatos α y γ es arginina (véase más adelante la figura 12).
El hidroxilo nucleofílico del sustrato que se va a fosforilar se ioniza por cesión del protón al carboxilato de Asp-166 (dominio D6).
El ataque nucleofílico del anión alcóxido, que tiene lugar mediante la intervención del estado de transición indicado en la figura 11, provoca la transferencia de fosfato.
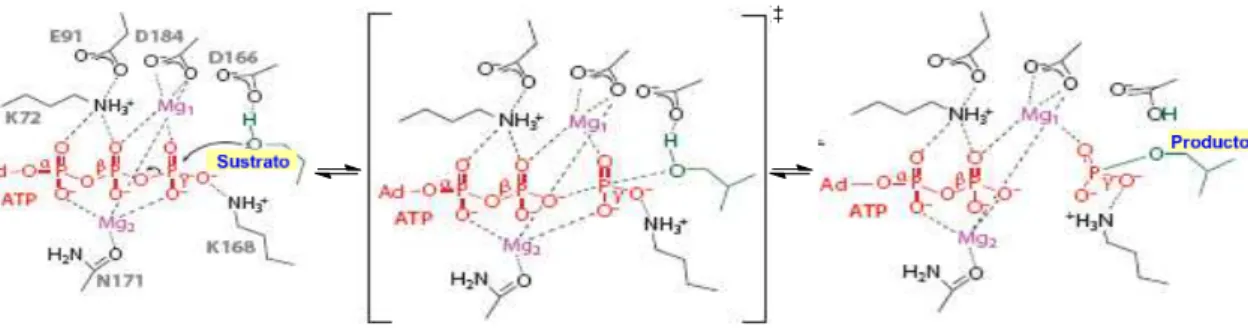
En la figura 12 se indica el mecanismo de fosforilación en la proteína kinasa A. El grupo hidroxilo del sustrato que se va a fosforilar se alinea de forma que el par solitario de electrones del oxígeno se dirige al átomo de fósforo del fosfato γ. El estado de transición es de tipo disociativo e implica un intermedio metafosfato en el cual la ruptura del enlace P-O entre los fosfatos β y γ está muy avanzada, mientras que el enlace con el nucleófilo entrante está empezando a formarse.5
Figura 12. Mecanismo disociativo de fosforilación en la proteína quinasa A (PKA)
La carga negativa que va creciendo sobre el fosfato γ se compensa con los iones Mg2+ y sobre el residuo de lisina. A medida que la reacción progresa, la acidez del hidroxilo del sustrato se incrementa y su pKa va disminuyendo en relación con el pKa de aspartato, permitiendo de esta forma la transferencia del protón desde el hidroxilo del sustrato (pKa =12) al aspartato (pKa = 4.5). Este protón es, probablemente, transferido al fosfato dianiónico del producto fosforilado, restableciéndose de esta forma la función carboxilato en el sitio catalítico de aspartato.5
Algunas quinasas también pueden llevar a cabo la fosforilación mediante un mecanismo asociativo, generándose en el estado de transición un fosforano pentavalente. En este mecanismo la formación del enlace entre el oxigeno del sustrato y el fosfato γ se produce simultáneamente con la ruptura del enlace entre los fosfatos β y γ, o incluso puede que la formación del enlace entre el sustato y el fosfato γ esté mas avanzada que la ruptura del enlace entre los fosfatos β y γ.7
En el esquema 2 se representa la quinasa con la letra E, el sustrato que se va a fosforilar con la letra S y el producto de la reacción con la letra P. K3 es la constante de velocidad para la etapa de fosforilación y k4 la constante de velocidad para la etapa de disociación de los productos. Se ha demostrado que la etapa catalítica es rápida (k3 ~300-500 s−1) y la etapa de disociación es lenta (k4 ~20-30 s−1), de lo que se deduce que, una vez que los sustratos se han orientado
correctamente, la etapa de fosforilación es fácil y rápida y que la etapa limitante de la velocidad es la liberación de los productos (ADP y la proteína fosforilada).8
Esquema 2
La eficiencia catalítica de las quinasas es enorme puesto que la reacción catalizada por la enzima es 1014 veces más rápida que la reacción no catalizada.
2.4. Unión de la quinasa al sustrato diana
La afinidad de las quinasas por los sustratos peptídicos que van a ser fosforilados es débil, del orden de 2 × 10−4 M, por lo que es necesario utilizar concentraciones milimolares del mismo para saturar la quinasa y conseguir la cristalización del complejo. Hasta el momento sólo se han conseguido cristalizar tres complejos de la quinasa con el sustrato proteínico. Uno de ellos es el formado por la quinasa PKR (proteína quinasa dependiente de RNA) con el factor de iniciación de la translación elF2α (véase la figura 13).9
En la figura 13 se observa que la lámina β de elF2α se acopla con la hélice G de la quinasa PKR. El sitio de fosforilación sobre elF2α está colocado en una región desordenada entre los residuos 47-59.
Figura 13. Complejo PKR/ elF2α
8 (a) Skamnaki, V. T.; Owen, D. J.; Noble, M. E.; Lowe, E. D.; et al. Biochemistry 1999, 38,
14718-14730. (b). Adams, J. A.; Taylor, S. S. Biochemistry 1992, 31, 8516-8522.
2.5. Estados activado e inactivado
Las quinasas pueden adoptar dos estados conformacionales: el estado activado o el inactivado. El estado activado tiene una conformación abierta, con el bucle de activación colocado lejos del surco catalítico.
El estado inactivado se adquiere cuando el bucle de activación se desplaza hacia la zona de unión de ATP, bloqueando así la entrada de esta biomolécula.
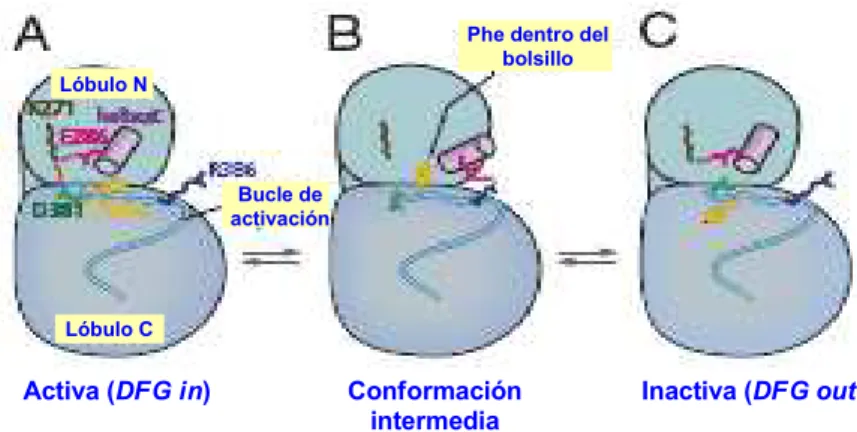
En las quinasas el bucle de activación, que contiene el fragmento DFG, puede adquirir diferentes conformaciones denominadas DFG-in, DFG-out y DFG-like. En la conformación inactiva la posición de la fenilalanina del dominio DFG ocupa parcialmente el bolsillo de unión del ATP (véase la figura 14). La zona catalítica está desconectada debido a la ausencia de ATP en el bolsillo de unión. El aspartato del dominio D7 tampoco se dirige hacia el bolsillo de unión, lo que impide la coordinación del magnesio que se requiere para la catálisis. En la conformación inactiva DFG-out la hélice αC mantiene su orientación hacia dentro del bolsillo.
Figura 14. Conformación inactiva de la quinasa
El cambio conformacional desde DFG-out (quinasa inactiva) a DFG-in (quinasa activa) provoca que el residuo aspartato del dominio DFG ocupe la zona del bolsillo de unión, que en la conformación DGF-out está ocupada por el anillo de fenilo de la fenilalanina, disponiendo a la quinasa para llevar a cabo su acción catalítica (véase la figura 15).10
Figura 15. Conformación activa de la quinasa
En la figura 16 se indican con más detalle las conformaciones que adopta el bolsillo de unión de la quinasa en sus estados activado e inactivado.
Figura 16. Conformaciones activa (parte A) e inactiva (parte B) de una quinasa
En el estado activado, parte A de la figura 16, la hélice αC (en rojo en la figura) se empaqueta contra el lóbulo N-terminal y el aspartato del dominio DGF se coordina con el catión Mg2+, orientando al fosfato γ hacia el sustrato que se va a fosforilar.
En la parte B de la figura 16 se representa el estado inactivado de la quinasa abl unida a Gleevec®, un fármaco inhibidor. En este estado inactivado, la interacción del aspartato con el catión Mg2+ está interrumpida y la fenilalanina del dominio DFG apunta hacia dentro del bolsillo de unión del ATP.
Se ha propuesto un mecanismo para explicar el cambio conformacional entre el estado activado (DFG-in) e inactivado (DFG-out) de las quinasas.11 Las estructuras marcadas como A y C en la figura 17 representan las conformaciones activa e inactiva, respectivamente. Estas dos conformaciones se diferencian en la conformación del dominio DFG.
En la conformación A (estado activado) la hélice αC se encuentra próxima al bolsillo de unión de ATP, de forma que la lisina del dominio D2 y el glutamato del dominio D3 pueden formar el puente salino.
Phe dentro del bolsillo
Bucle de activación
Lóbulo C Lóbulo N
Activa (DFG in) Conformación Inactiva (DFG out) intermedia
Figura 17. Cambio conformacional DFG-in/DFG-out
11 Shan, Y.; Seeligerb, M. A.; Eastwood, M. P.; Frank, F.; Xua, H.; Jensen, M.; Drora, R. O.; Kuriyanb,
En la parte B de la figura 17 se representa la conformación de transición entre el estado activado y el inactivado. La conformación B difiere de la A y de la C en dos aspectos significativos. El primero es que la hélice αC se coloca lejos del bolsillo de unión de ATP. El segundo es que el desplazamiento hacia fuera de la hélice αC crea un bolsillo en la base del lóbulo N-terminal que es ocupado por el grupo fenilo de la fenilalanina del dominio DFG.
El movimiento del dominio DFG promueve la liberación de ADP, que es la etapa limitante de la actividad catalítica de la quinasa.12 Cuando la quinasa está unida a ATP el aspartato del dominio DFG interacciona con un catión magnesio colocado a 2.3 Å que, a su vez, se coordina con los fosfatos β y γ del ATP (véase el apartado 2.3). Este catión magnesio puede ser liberado después de la transferencia de fosfato, reduciendo la carga positiva en las inmediaciones del aspartato del DFG y provocando la protonación de este residuo. La transferencia del protón puede venir del disolvente o del aspartato que ha inducido la ionización del hidroxilo de la tirosina (o serina o treonina). Esta protonación es la que provoca el giro desde la conformación DFG-in a la DFG-out y facilita la liberación del ADP.
3. Tipos de quinasas
Existe un sistema de clasificación sistemático de las enzimas basado en los números EC (en inglés Enzyme Commission numbers). Cada número EC está asociado a las reacciones catalizadas por las enzimas de manera que enzimas diferentes, que proceden, por ejemplo, de organismos diferentes pero que catalizan la misma reacción, reciben el mismo número EC.
Cada código de enzimas consiste en las dos letras EC seguidas por 4 números separados por puntos. Estos números representan una clasificación progresivamente más específica. Por ejemplo, la enzima tripéptido aminopeptidasa tiene el código EC 3.4.11.4, construido así:
3 por hidrolasa (enzima que usa el agua como reactivo en el proceso químico). 4 por hidrolasa que actúa sobre los enlaces peptídicos.
11 porque actúa sobre el terminal amino del aminoácido del péptido. 4 por que actúa sobre el amino terminal de un tripéptido.
Las quinasas se clasifican según el código EC como EC 2, ya que son transferasas que trasfieren un grupo funcional desde una sustancia a otra.
Otra clasificación menos sistemática divide a las quinasas en dos superfamilias: la familia de las denominadas quinasas convencionales (ePKs) y la familia que engloba a las denominadas quinasas atípicas (aPKs). En la figura 18 se representa de forma gráfica el árbol taxonómico del quinoma humano (quinasas convencionales).
Como se aprecia en la figura 18, las quinasas convencionales se dividen en las familias: 1.- Familia AGC: quinasas enlazadas a proteínas G.
2.- Familia CAMK: quinasas reguladas por calmodulina. 3.- Familia CMGC: quinasas dependientes de ciclinas. 4.- Familia CK1: caseína-quinasas.
5.- Familia GYC: quinasas receptoras asociadas a guanilato-ciclasa.
12 Para un estudio computacional sobre el mecanismo de liberación de ADP del bolsillo de unión de la
6.- Familia STE: compuesta de tres subfamilias que se activan entre si y que luego activan a las MAP-quinasas (del inglés Mitogen Activaten Protein Kinases).
7.- Familia TK: tirosina-quinasas.
8.- Familia TKL (del inglés Tyrosine Kinase-Like). En este grupo se encuadran las quinasas similares a las TK pero que carecen del motivo específico TK.
Figura 18. Arbol taxonómico del quinoma humano
Las quinasas atípicas no tienen una secuencia muy similar a la de las quinasas convencionales, pero tienen actividad enzimática de quinasa. La familia está formada por cuatro subfamilias:
1.- Familia alfa
3.- Familia PDHK (del inglés Pyruvate Dehydrogenase Kinases)
4.- Familia RIO
De las 520 quinasas humanas identificadas, 90 son quinasas, 43 son tirosina-quinasaslike y el resto, son serina/treonina quinasas.13
Las tirosina-quinasas se dividen en dos grupos:14
a) Tirosina quinasas que funcionan como receptoras (RTKs). Estas enzimas son proteínas transmembrana que contienen un dominio extracelularal que se unen diferentes ligandos, y un dominio intracelular, con actividad tirosina-quinasa.15 Se conocen 58 RTKs humanas (denominación EC 2.7.10.1) divididas en 20 subfamilias.
b) Tirosina quinasas citoplasmáticas no-receptoras (no-RTKs). Estas enzimas son proteínas intracelulares que funcionan por debajo de las RTKs en rutas de transducción de señal. Se conocen 32 no-RTKs humanas (denominación EC 2.7.10.2) divididas en 10 subfamilias.
3.1. Quinasas RTKs
Las tirosina-quinasas receptoras (RTKs) están unidas a receptores transmembrana. Existen seis grandes clases de receptores transmembrana y son los siguientes:
1- Canales Iónicos Dependientes de Ligando. 2- Receptores Tirosina-Quinasa (RTKs). 3- Receptores Serina/Treonina-Quinasa. 4- Receptores Guanilato Ciclasa.
5- Receptores Acoplados a Proteína G (GPCR, del inglés G Protein Coupled Receptor). 6- Receptores de Citoquinas.
Los receptores de la clases 1 a 4, entre los que se encuentran las tirosina-quinasas receptoras, son moléculas bifuncionales que unen hormona y sirven a su vez como efectores al actuar como canales iónicos o enzimas. En cambio, los receptores 5 y 6 ligan hormona pero deben reclutar otra molécula para catalizar su función.
En la figura 19 se indican de forma esquemática las diferentes estructuras de los receptores con actividad tirosina-quinasa (RTKs).16
Como se explicará más adelante, muchos de los fármacos anticáncer inhibidores de quinasas tienen su diana terapéutica en los receptores del factor de crecimiento epidérmico, denominados con el acrónimo EGFR del inglés Epidermal Growth Factor Receptor. La familia de receptores EGFR está compuesta por cuatro miembros: EGFR (ErbB1), HER2 (ErbB2, HER2/neu), HER3 (ErbB3) y HER4 (ErbB4), que pueden formar tanto homodímeros como heterodímeros.
13 Schlessinger, J. Cell. 2000, 103, 211-225.
Figura 19. Familia de receptores tirosina-quinasa
En la figura 20 se indica esquemáticamente la estructura de un receptor del factor de crecimiento epidérmico (EGFR).
Figura 20. Estructura de un EGFR
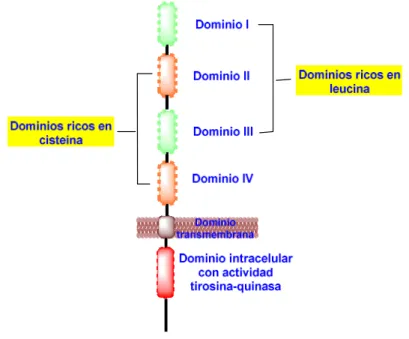
Los receptores EGFR están compuestos por:
3) De un dominio intracelular que contiene la región con actividad tirosina-quinasa (véase la figura 20).
La región extracelular contiene cuatro dominios (I-IV).
a) Los dominios I y III tienen una longitud de, aproximadamente, 160 aminoácidos y contienen unos dominios solenoide formados por hélices β de tipo LRR (del inglés Leucine Rich Repeat). Ambos dominios se unen a los ligandos.
b) Los dominios II y IV tienen una longitud de, aproximadamente, 150 aminoácidos y son dominios ricos en cisteina.
Cuando el ligando EGF se une al receptor pone en contacto dos sitios distintos en los dominios I y III de un único receptor EGFR (véase la parte 2 de la figura 21).
Esta unión bivalente promueve un cambio conformacional en la región extracelular que desenmascara un brazo de dimerización en el dominio II. Antes de la unión del ligando, este brazo de dimerización está completamente sepultado mediante interacciones intramoleculares con el dominio IV.
La unión del ligando permite que el brazo de dimerización del dominio II interaccione con un segundo complejo ligando-receptor povocándose la dimerización de las dos unidades receptoras (véase la parte 3 de la figura 21).
Figura 21. Proceso de dimerización de EGFR
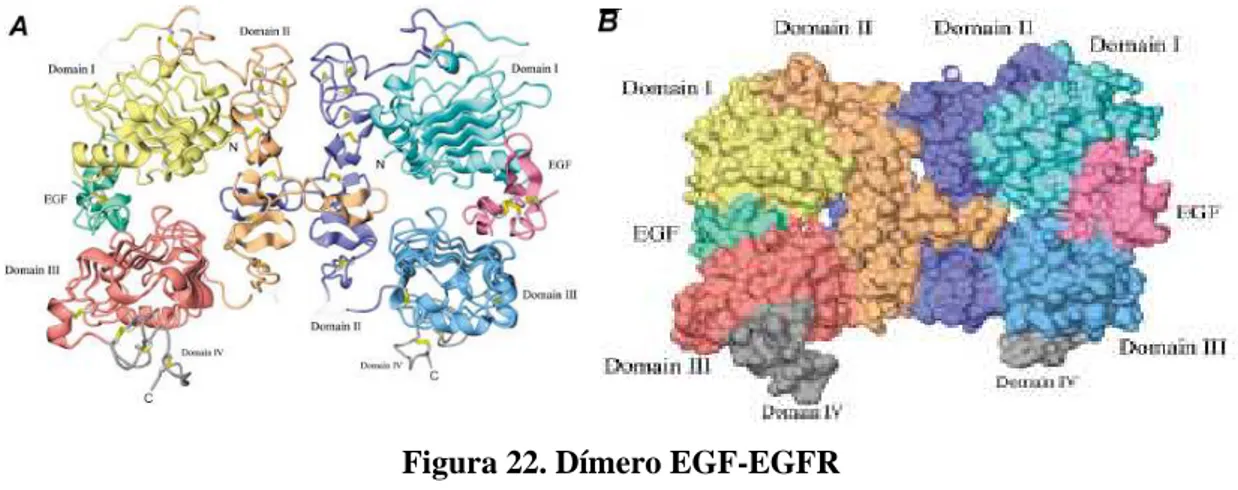
En la figura 22 se indica una representación del dímero formado por la unión de EGF a la región extracelular de EGFR. El EGF está coloreado en verde en la parte izquierda del dímero y en magenta en la parte derecha. En la parte A de la figura se representa el dímero en diagrama de cintas y en la parte B en diagrama de superficie.17
Figura 22. Dímero EGF-EGFR
17 Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J-H.; Saito, K.; Sakamoto, S.; Inoue,
El proceso de dimerización del receptor se puede llevar a cabo mediante otros mecanismos, que se indican a continuación.
1) Mediante ligando dimérico. Una subunidad del ligando se une a un receptor por un lado y otra subunidad del ligando se une al otro receptor por el lado opuesto. En total se ligan dos subunidades receptoras (véase la figura 23).
Figura 23. Dimerización del RTK mediante unión a ligando dimérico
2) Mediante ligando que contiene dos sitios de unión. El ligando posee en su estructura dos sitios de unión al receptor, cada uno de los cuales se une a una subunidad receptora (figura 24).
Figura 24. Dimerización del RTK mediante unión dual
3) Mediante unión de ligando a dímero receptor preexistente. El receptor en este caso ya está dimerizado, aún en ausencia de ligando, pero dispuesto y orientado de tal forma que no puede activarse antes de su unión con el ligando, como ocurre con el receptor de la insulina (véase la figura 25).
Tyr Tyr Tyr Tyr
Insulina
P P
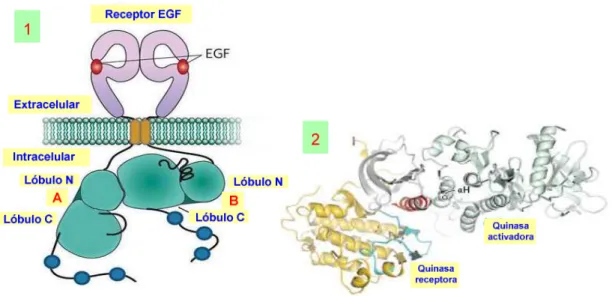
La dimerización extracelular del EGFR provoca un proceso de dimerización asimétrica en los dominios intracelulares tirosina-quinasa. Así, el lóbulo N-terminal de la parte intracelular quinasa del EGFR (véase la parte A de la figura 26-1) entra en contacto con el lóbulo C-terminal de la otra quinasa (véase la parte B de la figura 26-1). En la parte 2 de la figura 26 se aprecia cómo la dimerización asimétrica de los dominios quinasa se produce cuando la quinasa receptora, colocada a la izquierda, es activada mediante interacción de su hélice αC con la hélice B de la quinasa activadora.18 Este contacto entre los dominios quinasa los activa hacia el proceso de trans-fosforilación.
Figura 26. Dímerización asimétrica de los dominios quinasa en un EGFR
Como se acaba de explicar, la dimerización extracelular del EGFR provoca la dimerización asimétrica de los dominios intracelulares del receptor, que tienen actividad tirosina-quinasa. Esta dimerización intracelular provoca la autofosforilación de zonas proteínicas del dominio citosólico. Los dominios fosforilados interaccionan con proteínas citosólicas iniciando el proceso de transducción de la señal (véase la figura 27).
Figura 27. Efectos de la unión del EGF al receptor EGFR
En la figura 28 se indica esquemáticamente una tirosina-quinasa receptora tipo EGFR. Se aprecia la región extracelular receptora de la molécula señalizadora, la región dentro de la
membrana plasmática y la región intracelular que es la que tiene actividad tirosina-quinasa. Cuando la quinasa está inactiva existe en la membrana como monómero.
Figura 28. Proceso de dimerización y señalización mediado por una RTK
En la figura 28 se representa también la dimerización del receptor tirosonina-quinasa como consecuencia de la unión de una molécula señalizadora. Después de la unión del ligando, los monómeros se asocian en dímeros y los residuos de tirosina de la región intracelular se fosforilan. Este proceso promueve la fosforilación de otras proteínas que, a continuación, inician el proceso de señalización celular.
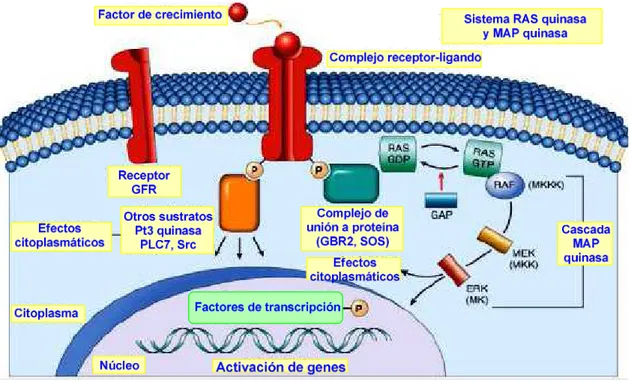
3.2. Transducción de señal por activación de RTKs: vía RAS-RAF
La cascada de acontecimientos que tiene lugar después de la dimerización del receptor es la siguiente:
1) La dimerización activa el dominio intracelular del receptor que se autofosforila en los residuos de tirosina.
2) Los residuos de tirosina fosforilados forman sitios de unión para proteínas que contienen dominios SH2, como la fosfolipasa C o la proteína adaptadora GRB2 (del inglés Growth factor Receptor-BoundProtein2, véase la figura 29).
3) Por un lado, la unión de la fosfolipasa C provoca su activación y la subsiguiente ruptura de PIP2 (fosfatidilinositol 3,5-bifosfato) en InsP3 (inositol 1,4,5-trifosfato) y DAG (diacilglicerol).
4) Por otro lado, la unión del GRB2 al receptor fosforilado provoca la activación de la proteína Sos (del inglés Son Of Sevenless,19 véase la figura 29).
5) SOS causa la activación de la proteína RAS mediante liberación en ésta de GDP y adquisición de GTP (véase la figura 29). Las proteínas RAS forman una familia de unos 50
19 Con el nombre de Son of Sevenless (SOS) se agrupan una serie de genes que codifican factores de
miembros.20 Son proteínas pequeñas que unen nucleótidos de guanina y son parte del sistema de transmisión de señales relacionado con la multiplicación y desarrollo celular. A la familia de enzimas RAS también pertenecen las proteínas ARF y RAB, vinculadas a la regulación del tránsito vesicular de proteínas, las RAN, relacionadas con la importación de proteínas en el núcleo y las RHO involucradas en la organización del citoesqueleto.
Figura 29. Transducción de señal por activación de RTKs: vía RAS-RAF
6) La RAS-GTP activa a la serina/treonina quinasa RAF21 que, a su vez, fosforila y estimula a MEK22 y ésta a ERK (del inglés Extracellular signal-Regulated Kinase, véase la figura 29).
7) La cascada de sucesos desemboca en la formación de AP-1, un factor de transcripción del núcleo celular que estimula la expresión de genes implicados en el crecimiento celular (véase la figura 29).
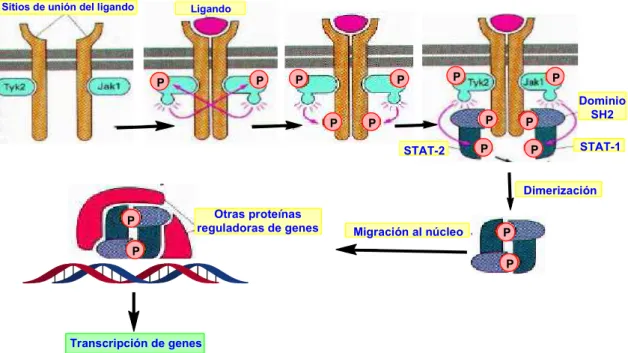
3.3. Transducción de señal por activación de RTKs: vía JAK-STAT
Existen más vías de señalización en las que participan también receptores tirosina-quinasa. Una de las más importantes es la cascada de señalización que se inicia con un proceso de heterodimerización provocado por la unión de un monómero tirosina-quinasa con otro, también con actividad tirosina-quinasa, denominado JAK (del inglés Janus Kinase), de los cuales existen cuatro tipos: JAK-1, JAK-2, JAK-3 y JAK-4.
La cascada de acontecimientos que tiene lugar después de la heterodimerización del receptor es la siguiente.
20 Con el monbre de Ras (del inglés Retrovirus Associated Sequences) se denomina un oncogén aislado de
sarcomas murinos inducidos por virus. Aunque inicialmente los genes Ras fueron aislados en virus de sarcomas murinos altamente oncogénicos, posteriormente fueron detectados también en tumores humanos y en células normales.
21 El acrónico Raf deriva del ingés Rapidly Accelerated Fibrosarcoma.
22 La quinasa MEK (del inglés MAPK kinase/ERK kinase) es una quinasa de doble especificidad (Ser/Thr
1) La heterodimerización provoca la fosforilación cruzada de ambos monómeros receptores (véase la figura 30).
2) Las proteínas STAT (del inglés Signal Transducers and Activators of Transcription), que contienen un dominio SH2, se unen al receptor fosforilado y se activan por fosforilación.
3) Las STAT se disocian del receptor y se dimerizan mediante intervención de sus dominios SH2.
4) Las STAT diméricas migran al núcleo y se unen al ADN y a otras proteínas reguladoras de genes activando la transcripción génica (véase la figura 30).
P
STAT-1 STAT-2
Sitios de unión del ligando Ligando
Dominio SH2
Migración al núcleo
Dimerización
Otras proteínas reguladoras de genes
P P P P
P P P P P P P P P P P
Transcripción de genes
Figura 30. Transducción de señal por activación de RTKs: vía JAK-STAT
4. Quinasas como dianas moleculares en el tratamiento del cáncer
Las quinasas son capaces de fosforilar una amplia variedad de sustratos, como proteínas estructurales, enzimas metabólicas, reguladores del ciclo celular, factores de transcripción, etc. Muchos de los sustratos fosforilados por las quinasas participan en las rutas de transducción de señal regulando procesos celulares fundamentales como el metabolismo, la proliferación celular, la diferenciación celular, la supervivencia, la migración o la angiogénesis.23
La perturbación de la actividad de las quinasas resulta en una transmisión de señal anormal que produce una desregulación de los procesos celulares que puede conducir al desarrollo de fenotipos malignos.24 De hecho, las quinasas son reguladores clave en las neoplasias, haciendo que la biología del cáncer y de otras enfermedades se caracterice por una situación molecular de transmisión de señal y actividad proteína quinasa aberrante.25
4.1. Desregulación de la actividad quinasa
La desregulación de la actividad proteína-quinasa puede tener lugar por:
23 Pawson, T. Cell. 2004, 116, 191-203.
24 Cohen, P. Nat. Rev. Drug. Discov. 2002, 1, 309-315.
a) Una reordenación genética, que genera una proteína híbrida que contiene el dominio catalítico de la quinasa, como en el caso de la tirosina-quinasa c-abl híbrida responsable de la leucemia mieloide crónica.26 Esta quinasa se produce por la translocación del material genético entre los cromosomas 9 y 22. Así, el gen abl situado en el cromosoma 9, que codifica una tirosina-quinasa de la familia Src,27 se inserta en el gen bcr (del inglés Breakpoint Cluster Region) del cromosoma 22 (véase la figura 31).28 El gen resultante de la translocación es el encargado de codificar a la proteina quinasa aberrante.
Figura 31. Formación del gen bcr-abl por translocación
Esta translocación génica fue descubierta en 1960 por científicos que trabajaban en aquel entonces en centros de investigación de la ciudad de Filadelfia. Estos científicos eran Peter Nowell, de la Escuela de Medicina de la Universidad de Pensilvania y David Hungerford del instituto Fox Chase Cancer Center. Desde entonces se conoce al cromosoma resultante de esta translocación como cromosoma Filadelfia, por el nombre de la ciudad donde se ubican ambos centros de investigación.
El gen de fusión, denominado gen bcr-abl, contiene secuencias de la c-Abl quinasa y se expresa a niveles muy superiores a los del gen que codifica la quinasa c-Abl normal. El resultado es la expresión de una vía que estimula el ciclo de división celular e inhibe la reparación del ADN, causando la inestabilidad del genoma y siendo una causa potencial de la temida «crisis en cadena» de la leucemia mieloide crónica, patología con una elevada tasa de mortalidad.
b) Disrupción de la función normal, que se produce cuando una mutación convierte a una quinasa en constitutivamente activa, como es el caso del oncogen c-Kit en tumores gastrointestinales.29
c) Expresión aumentada o aberrante de la quinasa o del ligando para la quinasa receptora transmembrana (RTK), como ocurre en la amplificación de la tirosina-quinasa receptora HER-2 (una variante de EGFR) en el cáncer de mama (véase la figura 32), o la expresión aberrante del
26 Hantschel, O.; Superti-Furga, G. Nat. Rev. Mol. Cell. Biol. 2004, 5, 33-44.
27 Las quinasas Src son tirosina-quinasas citoplasmáticas no receptoras que están codificadas por el
proto-oncogen Src (abreviatura de sarcoma).
28 El nombre de gen abl deriva de Abelson, que es la denominación que se da a una proteína producida
por un virus causante de leucemias.
factor de crecimiento vascular (VEGF) y su receptor (VEGFR), que aumentan la angiogénesis y el crecimiento tumoral.
Figura 32. Amplificación del gen Her2 y aumento de la producción de la quinasa HER-2
d) Desregulación de la actividad quinasa por activación de oncogenes o pérdida de genes supresores, lo que puede contribuir también a la tumorogénesis, como ocurre en la mutación de la GTPasa Ras.30 La proteína Ras es clave en la proliferación celular y se encuentra mutada en el 30% de los tumores sólidos. Ras se activa mediante el reclutamiento de las proteínas señalizadoras Grb2 y Sos, como consecuencia de la activación del receptor con actividad tirosina-quinasa (RTK).
Figura 33. Señalización celular mediada por Ras
En la figura 33 se esquematiza el proceso de activación de Ras. Así, la activación del RTK activa a su vez a la proteína efectora Sos que activa a Ras abriendo el bolsillo de unión a GTP, lo cual favorece la salida de GDP y la entrada de GTP. La proteína Ras participa en una vía de señalización que gobierna el proceso de división celular. Ras se inactiva con el tiempo debido a la hidrólisis del GTP por la actividad GTPasa intrínseca de Ras. Las mutaciones que afectan a la
actividad GTPasa de Ras producen proteína Ras constitutivamente activada y las células que la expresan proliferan sin control dando lugar a un cáncer.
4.2. Inhibidores de quinasas
Las estrategias convencionales en el tratamiento del cáncer se basan en una combinación de cirugía, radioterapia y quimioterapia. Sin embargo, dado que la base molecular que subyace en muchos tipos de cáncer es una disfunción de la actividad de las quinasas, estos enzimas constituyen dianas moleculares del mayor interés en el diseño de inhibidores específicos.
Para llevar a cabo su función, cualquier quinasa debe ser capaz de unir la proteína-sustrato y el ATP, antes de que la actividad catalítica pueda transferir el grupo fosfato. De acuerdo con ello, el diseño racional de inhibidores de quinasas se basa en impedir la unión de ATP, o del sustrato, o de ambos a la quinasa, de modo que quede bloqueada la función de la enzima.
Aunque la estructura 3D se conoce solamente para menos del 10% de las quinasas humanas, la información estructural de este reducido grupo de proteínas facilita la predicción y comparación de los sitios de unión a ATP de moléculas relacionadas con los sustratos.31
Hay que señalar que el desarrollo de inhibidores de quinasas como agentes terapéuticos estuvo durante algún tiempo ralentizado debido a dos factores:
1) La elevada concentración intracelular de ATP (2-10 mM), que se pensaba que competiría con el inhibidor, haciendo imposible la inhibición de la quinasa.
2) El origen común de todas las quinasas, que hace que todas tengan un mecanismo catalítico similar, por lo que se consideró improbable el desarrollo de inhibidores específicos, ya que el bloqueo de la actividad de una determinada quinasa inhibiría, simultáneamente, procesos fisiológicos no relacionados, lo que podría acarrear graves efectos secundarios.32
El procedimiento más usual en la búsqueda de inhibidores de quinasas ha sido el rastreo de grandes colecciones de sustancias químicas, compuestas por cientos de miles de productos naturales o sintéticos. La composición de una librería de estos compuestos suele estar basada en: a) La estructura del bolsillo de unión del ATP de una quinasa (librería basada en la estructura).
b) La estructura de compuestos conocidos que se unen al bolsillo de ATP de la quinasa (librería basada en el ligando).
El grado de conservación de los sitios de unión de ATP en las diferentes quinasas no es absoluto y es posible desarrollar sustancias con elevada selectividad. Además de elevada selectividad, los inhibidores de quinasas deben ser capaces de unirse con una gran afinidad. Idealmente, ésta debe ser de varios órdenes de magnitud mayor que la afinidad de la quinasa por ATP, ya que el inhibidor solo estará presente en la célula en concentraciones de rango micromolar-nanomolar.
4.2.1. Fármacos inhibidores de quinasas hiperactivas
Anteriormente se ha indicado que la desregulación de la actividad quinasa puede tener lugar por una reordenación cromosómica que origina un oncogen capaz de codificar una quinasa híbrida, como la quinasa bcr-abl. Las quinasas anómalas se caracterizan por su hiperactividad, lo que provoca procesos de fosforilación descontrolada que desembocan en la formación del cáncer. La inhibición de este tipo de quinasas aberrantes es una de las nuevas dianas farmacológicas en los tratamientos anticáncer.
En la parte A de la figura 34 se indica esquemáticamente el modo de acción de la quinasa bcr-abl. La unión del sustrato a la quinasa provoca la fosforilación de aquél y la subsiguiente activación del efector.
En la parte B de la figura 34 se indica la unión del inhibidor (imatinib) en el bolsillo de unión de ATP. Este hecho impide la actividad fosforilante de la enzima. El sustrato no fosforilado no puede unirse al efector y la vía de señalización celular se interrumpe.
Figura 34. Bloqueo de la acción quinasa con un inhibidor (imatinib)
El primer fármaco inhibidor de quinasas empleado en terapias anticáncer fue el imatinib, que se comercializó por Novartis en 2001. Este producto es un inibidor de tirosina-quinasas y se emplea en el tratamiento de la leucemia mieloide crónica (LMC).
Figura 35. Estructura del imatinib
Figura 36. Estructuras del nilotinib y del dasatinib
El sorafenib es un inhibidor multiquinasa ya que es capaz de inhibir a tirosina-quinasas y a serina/treonina quinasas. Se emplea en el tratamiento del cáncer renal primario avanzado y del cáncer hepático primario (hepatocarcinoma).
Figura 37. Estructura del sorafenib
4.2.2. Fármacos inhibidores de Receptores Tirosina-Quinasa (RTKs)
Los factores de crecimiento GF (del inglés Growth Factor) son proteínas que regulan la proliferación celular, se encargan de la supervivencia celular, estimulan la migración y la diferenciación celular y controlan la apoptosis.
El Factor de Crecimiento Epidérmico o EGF (del inglés Epidermal Growth Factor) es una pequeña proteína que contiene 53 aminoácidos y tres puentes disulfuro (véase la figura 38). Los puentes disulfuro del EGF se representan en amarillo en la figura 38 y son el Cys6-Cys20, Cys14-Cys31 y el Cys33-Cys42. Las cadenas laterales de los residuos 13 (Tyr), 41 (Arg) y 47 (Leu) juegan un importante papel en la funcionalidad del EGF.
Figura 38. Estructura del Factor de Crecimiento Epidérmico (EGF)
Figura 39. Representación de un receptor del factor de crecimiento epidérmico (EGFR)
El proceso de transducción de la señal se lleva a cabo mediante vías como:
a) La cascada de las MAPK quinasas (del inglés Mitogen-Activated Protein Kinase), que es activada por la proteína Ras, estimulando la síntesis y activación de factores de transcripción como FOS y JUN. Estos factores inducen la producción de nuevos factores de crecimiento, de receptores para dichos factores y de proteínas que controlan la entrada de la célula en el ciclo celular.
b) La cascada de la PI3K (fosfoinositol 3-quinasa), que activa la quinasa Akt, implicada en la proliferación celular y la supervivencia celular por inhibición de la apoptosis.
Los receptores del factor de crecimiento epidérmico (EGFR) tienen actividad tirosina-quinasa intrínseca y están sobreexpresados en las células tumorales de origen epitelial. La desregulación de los EGFR activa la proliferación, invasión y supervivencia celular. También activa la metástasis y la angiogénesis y desactiva los mecanismos de apoptosis (véase la figura 40).
En la figura 41 se representa esquemáticamente las consecuencias positivas del bloqueo del receptor EGFR por parte de un inhibidor de tirosina-quinasa (TKI).
Figura 41. Consecuencias de la inhibición de la actividad EGFR
Los fármacos como gefitinib, erlotinib, sunitinib, lapatinib, vandetanib y pazopanib, basan su acción anticáncer en el bloqueo del proceso de autofosforilación de receptores tirosina-quinasa, como los receptores EGFR, VEGFR, etc.
O MeO N N N HN F Cl O Gefitinib O O MeO N N HN Erlotinib MeO N H N H O O N H N Sunitinib Cl O F HN N N O H N S O O Lapatinib Br HN N N MeO Vandetanib N N N N N NH S O O NH2 Pazopanib F
4.3. Tipos de fármacos inhibidores de quinasas
La actividad inhibidora de quinasas se puede conseguir mediante tres tipos de inhibidores. a) Inhibidores tipo I. Estos inhibidores ocupan la parte de adenosina del ATP en el bolsillo de unión induciendo una conformación del residuo DGF denominada DGF-in. El residuo DGF contiene la secuencia Asp-Phe-Gly, muy conservada en todas las quinasas. Esta secuencia se encuentra localizada muy cerca del bolsillo de unión de ATP. El residuo de aspartato de DGF es muy importante para la acción catalítica de la quinasa y apunta hacia el sitio de unión de ATP en la conformación DGF-in, donde se coordina con un ión magnesio unido a ATP y activa al fosfato γ para que tenga lugar la fosforilación del sustrato receptor.
Cuando la quinasa está activa, y dispuesta a su acción fosforilante, la conformación DFG es DFG-in.
En la figura 43 se indica la coordinación de un fármaco inhibidor tipo I en el bolsillo de unión de ATP.
Figura 43. Coordinación de un inhibidor tipo I al bolsillo de unión de ATP (HCK = del inglés Hemopoietic Cell Kinase, PP1= pirazolo pirimidina tipo I)
b) Inhibidores tipo II. Estos inhibidores, al igual que los de tipo I, también ocupan la parte de adenosina del bolsillo de unión pero, al contrario que éstos, inducen una conformación del residuo DFG denominado DGF-out (véase la figura 44). Cuando la conformación es DGF-out la quinasa está inactiva y no puede fosforilar porque el aspartato del dominio DFG se aleja del fosfato γ.
c) Inhibidores tipo III. Estos inhibidores bloquean la actividad quinasa sin desplazar el ATP de su bolsillo de unión a la enzima (véase la figura 45).
Hélic e
Tipo III
Parte de ATP
Inhibidor tipo III
Bisa gra Inhibición no competitiva de ATP H N F I Br F F H N O O OH HO
Figura 45. Coordinación de un inhibidor tipo III a la quinasa
5. Imatinib
En los últimos años de la decada de 1980, investigadores de la entonces empresa farmacéutica Ciba-Geigy (hoy Novartis) iniciaron, bajo la dirección de N. Lydon, A. Matter y B. J. Druker, oncólogo del Oregon Health and Sciences University e investigador del Howard Hughes Medical Institute,33 un proyecto enfocado a la identificación de compuestos con actividad inhibitoria de quinasas. Uno de los compuestos que mostraban esta clase de actividad era la 2-fenilaminopirimidina (compuesto 7.88 del esquema 3). Este compuesto mostraba baja potencia y poca especificidad en la inhibición de quinasas pero fue el punto de partida en el desarrollo de la estructura de otros inhibidores más potentes.34
Un aumento de la actividad se consiguió con la adición de un grupo 3´-piridilo en la posición 4 del anillo pirimidínico (estructura 7.89).
La introducción de un grupo benzamido en el anillo fenílico aumentó aun más la actividad inhibidora (estructura 7.90).
Un paso clave en el desarrollo del imatinib fue observar que las sustituciones en la posición 6 del anillo fenílico disminuían la actividad inhibidora (estructuras 7.91). Por el contrario, la introducción de un grupo metilo en esta posición 6 del anillo fenílico retenía o incluso aumentaba la actividad contra tirosina-quinasas (estructura 7.91, R=Me, véase la interacción de este inhibidor tipo II con el bolsillo de unión de ATP en la figura 42).
La primera serie de compuestos con estructura 7.91 tenían poca biodisponibilidad oral y poca solubilidad en agua. La introducción de una cadena lateral altamente polar, como la de N-metilpiperazina, aumentaba la solubilidad y la biodisponibilidad oral. Al nuevo compuesto se le nombró primeramente como STI571 y después imatinib.
El imatinib inhibía la actividad de la quinasa c-abl con un IC50 entre 0.1-0.35 μM. En pacientes con leucemia linfoblástica aguda el valor de IC50 estaba en el rango 0.1-0.5 μM, lo que demostraba que el fármaco penetraba la membrana celular. Novartis comercializó el imatinib, en forma de sal de mesilato, con el nombre de Gleevec® en Estados Unidos y como Glivec® en Europa y Australia.
33 Druker, B. J. Nat. Med.2009, 15,15-28.
34 (a) Zimmermann, J.; Buchdunger, E.; Mett, H.; et al. Bioorg. Med. Chem. Lett. 1996, 6, 1221-1226. (b)
Esquema 3. Desarrollo de la estructura del imatinib como inhibidor de quinasas
5.1. Translocación cromosómica y formación de la quinasa híbrida bcr-abl
El imatinib es un inhibidor específico de la quinasa bcr-abl y ha demostrado ser muy eficaz en pacientes con leucemia mieloide crónica, tumores del estroma gastrointestinal y otros cánceres.35 La leucemia mieloide crónica (LMC) es una enfermedad maligna de la sangre y la médula ósea de carácter progresivo y lento que se presenta en la edad madura, con mayor incidencia entre los 35 y 55 años de edad. Se caracteriza por una formación descontrolada de granulocitos inmaduros de forma que el tejido sanguíneo se vuelve tan espeso, por la acumulación de estas células sanguíneas anómalas, que las células sanas, como los glóbulos rojos y las plaquetas, no pueden llevar a cabo sus funciones. El resultado es anemia, disfunción del sistema inmune, infecciones y, en última instancia, leucemia.
La leucemia mieloide crónica resulta de una anomalía cromosómica adquirida, originada por el intercambio del gen abl del cromosoma 22 con el gen bcr del cromosoma 9 (véase la figura 46). El resultado de esta translocación cromosómica es la formación de cromosoma 22 alterado, denominado cromosoma Filadelfia, que contiene el gen bcr-abl. Este gen codifica una proteína quinasa alterada denominada quinasa bcr-abl que posee una actividad quinasa aumentada e incontrolada, lo que provoca la desregulación del ciclo celular y la inhibición de la apoptosis. Por otra parte, la quinasa bcr-abl disminuye la adhesión de las células leucémicas a
las células del estroma de la médula ósea, permitiendo la salida de aquéllas al torrente sanguíneo.
Figura 46. Formación del cromosoma Filadelfia
5.2. Quinasa abl
La quinasa abl es una tirosina quinasa no receptora con un peso molecular de 145 KDa. Existen dos variantes de esta quinasa: la abl-a y la abl-b. La diferencia entre estas dos quinasas es que la abl-b ha sufrido una modificación post-translacional mientra que la abl-a no tiene esta modificación. La quinasa abl-b está miristoilada en el lóbulo N-terminal. Por razones que se desconocen, la quinasa abl-b se encuentra en mayor cantidad en las células que la abl-a.
En la estructura de la quinasa abl se distinguen tres dominios denominados SH1, SH2 y SH3 (véase la figura 47). En el dominio SH1, en el que se encuentra la región catalítica, abarca desde el aminoácido 235 al 509.
Los dominios SH2 y SH3 están implicados en procesos de regulación. Ayudan a mediar las interacciones proteína-proteína y a regular el proceso de transducción de señales.
El dominio SH3 es el que controla y regula la actividad de la quinasa. Este dominio es el que se une a la proteína Grb236 y es el que pierde su función de control en la proteína híbrida bcr-abl, lo que resulta en una actividad quinasa incrementada.
El dominio SH2 tiene una elevada afinidad por los residuos de tirosina fosforilada y se sabe que identifica una secuencia de 3 a 6 aminoácidos en el motivo peptídico de la proteína diana.
La quinasa abl-a se localiza tanto en el citoplasma como en el núcleo celular. En el citoplasma participa remodelando el citoesqueleto durante el proceso de división, diferenciación y adhesión celular. Se cree que la quinasa abl-a utiliza su dominio de unión a la actina para acoplarse a los filamentos de esta proteína y controlar sus movimientos durante las transformaciones citoesqueletales.
Figura 47. Estructura de la quinasa abl
La quinasa abl-a que se encuentra en el núcleo participa regulando el ciclo celular, en especial en el punto de control (checkpoint) de la fase G1/S. Así, si una célula es expuesta a una radiación dañina, como la de los rayos X o la de los rayos γ, la quinasa abl se activa y fosforila a la proteína p73 en el sitio Y99. La proteína p73 pertenece a la familia de proteínas p53, que son supresoras de tumores. La proteína p73 activada actúa sobre el gen p21 que codifica la proteína p21, que inhibe la actividad de la kinasa dependiente de ciclina. El resultado de esta vía de señalización es la parada del ciclo celular en el punto de control G1/S.
La quinasa abl-a se puede transferir del núcleo al citoplasma y viceversa, dependiendo de las funciones que la célula requiera en un momento dado.
5.3. Modo de acción del imatinib
El imatinib ocupa el bolsillo ATP de la quinasa bcr-abl impidiendo la fosforilación del sustrato, que ya no puede unirse al efector. De este modo, la vía específica de transducción de señales, activada anormalmente en el proceso de transformación leucémica, resulta inactivada por el imatinib interrumpiéndose la transducción de la señal (véase la figura 48).
Figura 48. Modo de acción del imatinib
5.4. Unión del imatinib al centro activo de la quinasa abl
El imatinib se une a la quinasa-abl mediante interacciones del nitrógeno piridílico con el NH de metionina-318 (Met-318, CH3SCH2CH2CH(NH2)COOH), del NH anilínico con el OH de la treonina-315 (Thr-315, CH3CH(OH)CH(NH2)COOH), del NH del carbonilo amídico con el carboxilato de ácido glutámico-286 (Glu-286, (-)OOCCH2CH2CH(NH2)COOH), del oxígeno carbonílico con el NH del ácido aspártico-381 (Asp-381, HOOCCH2CH(NH2)COOH) y del nitrógeno piperazínico con los carboxilatos de isoleucina-369 (Ile-369, CH3CH2CH(CH3)CH(NH2)COOH) y de histidina-362 (His-362, Imid-CH2CH(NH2)COOH) Además de estas interacciones, otras de tipo van der Waals también son importantes en el reconocimiento del imatinib por la quinasa (véase la figura 49).
Ile-369(COO-) N
N N
N
H3C
N H
O
N
N CH3
H H
Met-318(NH)
Thr-315(OH)
Glu-286(COO-)
Asp-381(NH)
His-362(COO-)
Figura 49. Interacciones del imatinib con el sitio de unión de ATP a la quinasa
En la figura 50 se indica la colocación del imatinib en el bolsillo de ATP de la quinasa abl. El resultado de la unión del imatinib a la quinasa es el solapamiento de los anillos de piridina y pirimidina con el sitio de unión del ATP, lo que impide la actividad fosforilante de la enzima.37
37 (a) Nagar, B.; Bornmann, W. G.; Pellicena,P.; Schindler, T.; R. Veach, D. R.; Miller, W. T.; Clarkson,
B
is
a
g
ra
Figura 50. Colocación de imatinib en el bolsillo ATP de la quinasa-abl
El imatinib (Gleevec®) es un inhibidor tipo II y se une a la conformación inactiva de la quinasa abl, que es la que presenta el dominio DFG-out. En la figura 51 se representan las moléculas de imatinib (Gleevec®) y de ATP en el bolsillo de unión de la quinasa. Cuando la conformación del bucle activador es DFG-out (color verde en la figura) el ATP no puede ocupar el bolsillo de unión porque su grupo fosfato colisionaría con el resto fenilo del dominio DFG. En esta conformación DFG-out la hélice αC se coloca lejos del bolsillo (en color verde en la figura 51), dejando el suficiente hueco para que sea ocupado por el imatinib.
Figura 51. Conformaciones DFG out/DFG in en la unión de imatinib a la quinasa-abl
5.5. Síntesis de imatinib
5.5.a.1. Análisis retrosintético de imatinib
El análisis retrosintético del imatinib se inicia con la escisión del enlace amídico. Esta operación genera dos fragmentos. Uno de de los fragmentos es el cloruro de ácido 7.92 y el otro es la pirimidina 7.93 (véase el esquema 4).
Esquema 4
La desconexión del anillo pirimidínico conduce a la arilguanidina 7.95 y a la piridina 7.96. Estos compuestos se prepararán a partir de la orto-toluidina 7.97 y de la 2-acetilpiridina 7.98, respectivamente.
5.5.b.1. Síntesis de imatinib38
1) Síntesis de la arilguanidina 7.95
La síntesis de la 1-(metil-5-nitrofenil)guanidina se inicia con la nitración de la 2-metilanilina 7.97 que se lleva a cabo con ácido nítrico en etanol (esquema 5). Esta reacción proporciona la 2-metil-5-nitroanilina 7.99 que se transforma en la guanidina 7.95 mediante reacción con la cianamida.
Esquema 5
2) Síntesis de la piridina 7.93
Para la síntesis de la piridina 7.96 se elige como compuesto de partida la 3-acetilpiridina 7.98 (esquema 6). La enolización de este compuesto con metóxido sódico, seguida de reacción de formilación con formiato de etilo, conduce a la cetoaldehídopiridina 7.100. El tratamiento de
38 (a) Zimmermann, J. EP Patente 564,409, 1993. (b) Zimmermann, J. U.S. Patente 5,521,184, 1996. (c)
este compuesto con dimetilamina y ácido acético, en tolueno a reflujo, proporciona la piridina 7.96.
Esquema 6
3) Síntesis del cloruro de ácido 7.92
El cloruro de ácido 7.92 se obtiene mediante aminación reductiva de la N-metilpiperidina con ácido p-formilbenzoico,39 seguida de conversión del correspondiente ácido en cloruro de ácido (esquema 7).
Esquema 7
4) Construcción del anillo de pirimidina y pasos finales
Los pasos finales en la síntesis del mesilato de imatinib se inician con la condensación entre la guanidina 7.95 y la piridina 7.96 (esquema 8).
O
N N
7.96
O2N N
H NH
NH2
7.95
+ NaOH,iPrOH
reflujo, 12 h O2N N
H N N N 7.94 7.92, piridina luego
CH3SO3H
N N N H O N H N N N
Mesilato de Imatinib
· CH3SO3H
H2, Pd/C
THF, 21 h
H2N N
H N N N 7.93 Esquema 8
La condensación se lleva a cabo en isopropanol a reflujo en presencia de NaOH y proporciona la piperazina 7.94. La reducción del grupo nitro mediante hidrogenación molecular proporciona 7.93, que por amidación con el cloruro de ácido 7.92 forma el imatinib. Este compuesto se convierte en el mesilato por adición de 1 equivalente de ácido metanosulfónico.
39 Koroleva, E. V.; Kadutskii, A. P.; Farina, A. V.; Ignatovich, J. V.; Ermolinskaya A. L.; Gusak, K. N.;