edigraphic.com
Artículo de revisión
Bioquímica especial
* Laboratorio de Neurobiología y Bioingeniería Celular de la Facultad de Medicina de la Universidad Autónoma de Querétaro.
Correspondencia:
MD, PhD Hebert Luis Hernández-Montiel. Laboratorio de Neurobiología y Bioingeniería Celular de la Facultad de Medicina de la Universidad Autónoma de Querétaro, Clavel Núm. 200, Prados de la Capilla, Santiago de Querétaro, 76170, México. Fax: 2161087. e-mail: [email protected]
Recibido: 08-09-2005 Aceptado: 16-10-2006
Aspectos moleculares y prospectos de terapias en
la enfermedad de Parkinson
Hebert Luis Hernández-Montiel*
RESUMEN
La enfermedad de Parkinson es el trastorno neurodegene-rativo del movimiento más común y está caracterizado por la pérdida progresiva de las neuronas dopaminérgicas en la sustancia nigra, pars compacta. En el pasado, el principal obstáculo para el desarrollo de terapias de neuroprotección y restauración había sido el limitado conocimiento de los eventos moleculares que provocan esta neurodegeneración. En los últimos años y con el advenimiento de modernas téc-nicas de investigación se han podido conocer con mayor de-talle la fisiopatología de esta enfermedad. Esto ha favorecido que se hayan desarrollado y perfeccionado, a través de dis-tintas líneas de investigación, nuevas terapias que preven-gan o protejan de la enfermedad de Parkinson. El presente trabajo hace una revisión de los aspectos fisiopatológicos involucrados en la enfermedad de Parkinson, desde los as-pectos genéticos hasta los ambientales, que provocan la disfunción y muerte de las neuronas dopaminérgicas. Hace referencia también a las nuevas terapias que están en desa-rrollo y que podrían disminuir la progresión de la enferme-dad en aquellos pacientes portadores y con medidas neuro-protectoras, evitar que aparezcan nuevos casos.
Palabras clave: Enfermedad de Parkinson, disfunción mi-tocondrial, neurodegenerativo, apoptosis, terapias restaura-tivas y de neuroprotección.
ABSTRACT
Parkinson’s disease is the most common neurodegenerati-ve moneurodegenerati-vement disorder and it is characterized by a progres-sive loss of dopaminergic neurons located in the substan-tia nigra pars compacta. The main obstacle to develop neuroprotective and restorative therapies has been a limit-ed understanding of the key molecular events causing neu-rodegeneration. In the past few years with the development of new methodological techniques, the knowledge about the physiology of this disease has increased, favoring the development of new preventive therapies. This review sum-marizes physiopathological aspects in Parkinson´s di-sease, including genetics to environmental factors that might promote dopaminergic dysfunction and death. In the same way, it focuses in the development of new thera-pies to reduce the evolution of the disease in carrier pa-tients avoiding arise of new cases, through neuroprotec-tive measures.
Key words: Parkinson´s disease, mitochondrial dysfunc-tion, neurodegenerative, apoptosis, neuroprotective and res-torative therapies.
INTRODUCCIÓN
La enfermedad de Parkinson (EP) es un padeci-miento neurodegenerativo que aumenta su
inciden-cia con la edad, en el cual las neuronas dopaminér-gicas nigroestriatales mueren de forma progresiva, provocando los síntomas clínicos clásicos de la en-fermedad. Se desconoce la causa exacta de la muer-te de las neuronas dopaminérgicas, pero evidencias recientes sugieren que el estrés oxidativo forma parte de la fisiopatología de esta enfermedad, en este sentido las neuronas dopaminérgicas de pa-cientes con la EP presentan un incremento de mar-cadores de estrés oxidativo en proteínas, lípidos y ADN. Se ha reportado la reducción de la actividad del complejo I de la cadena respiratoria mitocon-drial, la presencia de la microglia reactiva en esta zona del cerebro y un incremento en los niveles de citocinas proinflamatorias en el líquido cerebroes-pinal de pacientes con EP.
edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
Herbicidas como el paraquat y la rotenona, el com-puesto metil-fenil-tetrahidropiridina (MPTP) y los metales pesados han sido involucrados en esta enfer-medad. Alteraciones genéticas que afectan genes como el gen Parkin, α sinucleína y componentes del sistema proteosomal, se encuentran en algunos casos de EP. La evidencia reciente sugiere que los procesos fisiopatológicos de la EP podrían ser diversos y mul-tifactoriales, por lo que el desarrollo de nuevos trata-mientos tendrá que tomar en cuenta todos estos posi-bles factores involucrados. En esta revisión se tratarán al inicio los diversos factores asociados al desarrollo de la enfermedad y después, se hablará de las nuevas terapias de neuroprotección y restaura-ción que a partir de los últimos conocimientos de esta enfermedad, se han desarrollado y que podrían disminuir la progresión de la enfermedad en aquellos pacientes portadores y con medidas neuroprotecto-ras, evitar que aparezcan nuevos casos.
Antecedentes
La enfermedad de Parkinson es el segundo padeci-miento neurodegenerativo, detrás de la enfermedad de Alzheimer (EA), y es el primero con desórdenes de movimiento. Con una prevalencia que aumenta con la edad y afecta alrededor del 1% de la población ma-yor de 65 años, los pacientes con EP exhiben los sín-tomas clínicos clásicos que incluyen bradicinesia, temblor en reposo, rigidez e inestabilidad postural.1 Mientras que la población normal tiene una pérdida de alrededor del 4.4% de células de la sustancia ni-gra, los pacientes con la EP tienen una pérdida 10 veces mayor,2 la pérdida del 70-90% de neuronas dopaminérgicas (DA) de la sustancia nigra pars compacta (SNc) y la presencia de inclusiones intra-citoplásmicas conocidas como cuerpos de Lewy en algunas de las neuronas dopaminérgicas restantes son patognomónicos de EP.3 El 90% de la EP es de tipo esporádico y en el 10% restante se encuentran mutaciones en diferentes genes que causan la EP fa-miliar. Al inicio de los síntomas, la dopamina en el estriado ha disminuido un 80% y el 60% de las neu-ronas dopaminérgicas mesencefálicas de la pars compacta se han perdido (Figura 1). La neurodege-neración y los cuerpos de Lewy también se encuen-tran en las neuronas noradrenérgicas del locus ceruleus, serotoninérgicas del rafe y colinérgicas del núcleo basal de Meynert y del motor dorsal del vago, así como en la corteza cerebral (especialmente en el cíngulo y la corteza entorrinal), bulbo olfato-rio y el sistema nervioso autónomo.4,5
Daño oxidativo y disfunción mitocondrial
Todos los organismos aeróbicos se encuentran conti-nuamente expuestos al estrés oxidativo.6 La fosforila-ción oxidativa conlleva la transferencia de electrones, la cual puede provocar la generación de radicales li-bres (RL), independientemente de las especies exis-tentes que tienen uno o más electrones no apareados.
Snpc
a) Cerebro normal Putamen
Caudado
Tracto nigroestriado
Snpc
Putamen Caudado
b) Cerebro con Enf. de Parkinson
Figura 1. Muerte neuronal en la enfermedad de Parkinson. a)
edigraphic.com
Muchos RL son especies reactivas inestables que pue-den extraer un electrón de las moléculas vecinas para completar su propio orbital. Esto provoca la oxida-ción de las moléculas vecinas, moléculas biológicas críticas que incluyen al ADN, proteínas y lípidos de la membrana, son sujetos de daño oxidativo.
La mitocondria es la fuente más importante de RL, generando radicales superóxido a partir de ubi-quinona y NADH deshidrogenasa (complejo I).7 Las mitocondrias son organelos cuya membrana interna y matriz contienen una multitud de enzimas y esta última contiene copias de un genoma mitocondrial distinto, el mtADN. El superóxido producido en las células por las reacciones oxidativas es normalmente convertido a H2O2, por la superóxido dismutasa (SOD). Este superóxido puede reaccionar con el óxido nítrico para formar peroxinitrito (ONOO-),8 reacción que ocurre con una velocidad tres veces mayor al rango de superóxido dismutación por la peróxido dis-mutasa. La generación de peroxinitrito depende de la concentración de superóxido y de óxido nítrico en la célula y puede existir en una forma activa parecida al radical hidróxilo. A pH fisiológico puede difundir a través de muchos diámetros celulares, causando daño por oxidación de lípidos, proteínas y ADN. También puede reaccionar con Cu/Zn-SOD para for-mar el ion nitronium, el cual puede nitrar residuos
de tirosina.9 La 3-nitrotirosina así generada es un ex-celente marcador bioquímico de daño oxidativo me-diado por peroxinitrito.
Se han encontrado de forma consistente en los pa-cientes con EP un decremento en los niveles del an-tioxidante glutatión. Se ha podido determinar que esta baja posiblemente es debida a su sobreutilización en las reacciones de estrés oxidativo y que podría te-ner una gran participación en la degete-neración nigra
de todos los desórdenes con deficiencia de dopamina nigroestriatal, tales como la parálisis supranuclear progresiva y la atrofia sistémica múltiple.10
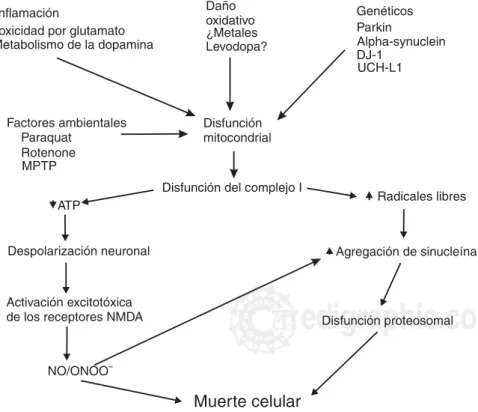
Evidencia reciente sugiere que la inhibición del complejo I mitocondrial puede ser la causa central de los casos de EP esporádica y que esta inhibición cau-sa la agregación de la α sinucleína, lo cual contribu-ye a la muerte de las neuronas DA. Esta anormalidad enzimática sólo se encuentra en la sustancia nigra pars compacta que provoca un decremento de la acti-vidad de la cadena respiratoria. Hasta el momento no se ha identificado una anormalidad genética del geno-ma nuclear, mitocondrial, una endo o exotoxina que explique la incidencia de los casos en la EP esporádi-ca (Figura 2). A pesar de esto, el incremento de la in-cidencia de EP con la edad se liga al hecho de que el envejecimiento provoca deleciones por encima del 5% de todas las moléculas genómicas mitocondriales en
Disfunción del complejo I ATP
Despolarización neuronal
Activación excitotóxica de los receptores NMDA
NO/ONOO¯
Muerte celular
Disfunción proteosomal Agregación de sinucleína
Radicales libres Disfunción
mitocondrial Daño oxidativo ¿Metales Levodopa? Inflamación
Metabolismo de la dopamina Toxicidad por glutamato
Factores ambientales
MPTP Paraquat Rotenone
Genéticos Parkin
Alpha-synuclein
UCH-L1 DJ-1
Figura 2. La alteración del complejo I como causa central de la enfermedad de Parkinson esporádica. La disfunción del complejo I incrementa el estrés oxi-dativo, la formación de radicales libres y la reducción de la formación del ATP. Esta disfunción puede ser multifactorial y puede presentarse en otras enfermeda-des que contribuyen a la disfunción. Esto permite la agregación de sinucleína con la alteración de los proteosomas y contribuye a la muerte neuronal. Este mismo estado puede ser inducido por factores ambientales e inflamación cró-nica. La disminución de ATP produce despolarización de la membrana y con-tribuye al daño excitotóxico y por radi-cales libres como el peroxinitrito (ONOO-)
edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
el cerebro.11 Muchas de las rutas conocidas de muer-te neuronal se encuentran involucradas con el com-plejo I: excitotoxicidad, especies reactivas de oxígeno, apoptosis dependiente e independiente de caspasas, necrosis y daño por inflamación. Las observaciones sugieren que la inhibición del complejo I crea un
me-dio ambiente oxidativo que provoca la agregación de la α sinucleína, con la subsecuente muerte de neuro-nas DA. La evidencia bioquímica de los defectos en el complejo I de la cadena respiratoria en la sustancia
nigra de pacientes con EP,12,13 junto con los hallazgos de que la potente toxina
1-fenil-4-metil-1,2,3,4-tetrahi-P P
P P
P P P14-3-3P
Bad
Bcl-2
Bcl-XL
PI3K/Akt MAPK
Cit c
Bax Calcineur
ina
Sobrevida
Ser 112 Ser 136
Receptor Trk Estímulo
apoptótico
Receptor muerte
Adaptador
Procaspasa-8
AIF Smac/ Diablo
IAPs
Caspasa-8 tBid
Bid
Caspasa-2
Procaspasa-2
Núcleo ADN
DFF40/CAD
DFF45/ICAD ?
?
?
Procaspasa-9
Caspasa-9
Apaf-1
dATP
Procaspasa-3
Caspasa-3 Caspasa-7
Procaspasa-7
Apoptosis
Figura 3. Receptor de muerte celular y rutas mitocondriales apoptóticas. Ligandos externos activan al receptor de muerte y acti-van a la caspasa-8 la cual activa a su vez a tBid, traslocando a la mitocondria donde induce la liberación del citocromo c (cit c) al citosol. La activación de la caspasa-2 puede inducir también la activación de Bid y la señalización hacia la mitocondria para que libere cit-c, implicando a la mitocondria como un amplificador de la cascada de activación de las caspasas. Dentro del ci-tosol, el cit-c forma apoptosomas (en presencia de Apaf-1, dATP y procaspasa-9) activando la caspasa-9. El efector caspasa -3 y -7, que pueden ser activados por las caspasas -8 ó -9, se requieren para que se active la endonucleasa DFF40/CAD, responsable de la fragmentación genómica del ADN. Además, Smac/Diablo (liberado por la mitocondria) se une a IAPs, potenciando la acti-vación de las caspasas. AIF es también liberado al citosol y trasloca al núcleo, induciendo la fragmentación del ADN y matando a las células de una manera independiente a las caspasas. En presencia de un estímulo apoptótico (e. Ca2+-calmodulin),
edigraphic.com
dropiridina (MPTP) causa parkinsonismo agudo,14,15 realzan la posibilidad de que factores mitocondriales contribuyen en la patogénesis de EP. Estudios de trasplante genómico han mostrado que el defecto en el complejo I es determinado por el ADN mitocon-drial.16-18 Si este defecto es causado por mutaciones en el ADN mitocondrial o si el polimorfismo funcional deja a la célula susceptible al daño por agentes exter-nos, es desconocido.
Por su parte, la acumulación y agregación de la
α sinucleína podría contribuir ampliamente en la muerte de las neuronas dopaminérgicas y oligoden-drocitos debido al daño en el control de proteínas y la destoxificación.19-21 Estas inclusiones pueden cau-sar daño celular al obstruir el tráfico celular normal y alterar la morfología, así como atrapar otros com-ponentes celulares, lo que eventualmente provoca la muerte neuronal.22 Estudios post mortem en huma-nos indican que las especias reactivas de oxígeno son importantes en la patogénesis de la EP esporádica.23,24 El daño en el complejo I mitocondrial libera la activi-dad de estrés por RL y vuelve a las neuronas vulne-rables a la excitotoxicidad por glutamato.
Efecto del medio ambiente y los factores gené-ticos asociados a la enfermedad de Parkinson.
Las alteraciones en los genes α sinucleína, Parkin y UCH-L1 (ubiquitin C-terminal hydrolase-L1), miem-bros del sistema ubiquitin-proteosomal, en pacientes con enfermedad de Parkinson sugieren que el daño de este sistema puede intervenir en la enfermedad.25 El gen mutante de α sinucleína se encuentra involucra-do en la patogénesis del EP autonómico involucra-dominante. Las deleciones en el gen Parkin han sido identifica-das como la causa primaria en raras formas de EP juvenil autosómico recesivo.26 Las proteínas que son ubiquitinizadas por este sistema, son reconocidas por el proteosoma subunidad 26S y son blanco de degra-dación.21,27-28 La alteración de este sistema modifica la capacidad de procesamiento de las proteínas, causan-do la agregación anormal y contribuye a su acumula-ción en los cuerpos de Lewy. La DJ-1 es una proteína de función desconocida que se encuentra alterada en algunos casos de EP autosómico recesivo.29
Estudios epidemiológicos sugieren que agentes tóxicos ambientales que inhiben al complejo I tam-bién se encuentran involucrados en la EP; el MPTP, el paraquat y la rotenona, son un ejemplo de ello.30-33 La exposición al herbicida paraquat ha sido implicada como un factor de riesgo para la EP. El mecanismo exacto aún se desconoce, pero al parecer induce una
fosforilación secuencial de la cinasa c-jun N-terminal (JNK) y de c-jun, la activación de la caspasa-3 y pro-voca la muerte secuencial de neuronas dopaminérgi-cas tanto in vivo como in vitro.34 La rotenona, otro herbicida, es también capaz de provocar EP. Aunque los estudios epidemiológicos sugieren un papel im-portante a los pesticidas en la patogénesis de la en-fermedad, es claro que existe una predisposición ge-nética particular, potencialmente asociada a la forma como el organismo es capaz de metabolizar neuro-toxinas relacionadas a la dopamina.35
Dopamina e inflamación
El metabolismo de la dopamina por sí mismo es fuen-te de producción de óxido, radicales tóxicos por la auto-oxidación y oxidación enzimática, un mecanismo muy relacionado con la toxicidad a la 6-hidroxidopa-mina. Actualmente se ha propuesto la hipótesis de que los productos intermedios de dopamina, quinona/ semiquinona reactivos químicamente, son altamente neurotóxicos y potencialmente genotóxicos. Esta es una consideración muy importante que sugiere que el tratamiento con levodopa acelera la progresión de la EP. Actualmente, se desarrollan terapias experimen-tales con inhibidores de la monoaminooxidasa.36-37
La biosíntesis de catecolaminas es regulada por la tirosinahidroxilasa (TH), la actividad de esta enzima es regulada a su vez por la concentración del cofactor tetrahidrobiopterina (BH4), cuyo nivel es regulado a su vez, por la actividad de la GTP ciclohidrolasa I (GCH), así, la actividad de GCH indirectamente regu-la regu-la actividad de los niveles de catecoregu-laminas. La ac-tividad de la TH en las neuronas dopaminérgicas ni-groestriatales es más sensible a un decremento en la BH4 y la mutación de GCH resulta en una reducción de la actividad de la GCH, de BH4, TH y dopamina, causando una deficiencia de GCH heredada de forma recesiva o una distonía progresiva heredada de forma dominante, que también es llamada distonía respon-siva a dopa; sin embargo, en los pacientes con EP y EP juvenil parece no estar alterada esta vía.38
re-edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
cientes han mostrado que el FNT-α se encuentra in-crementado 366% en el estriado y 432% en el fluido cerebroespinal de pacientes con EP.38,41-43 Algunos es-tudios sugieren que la intensa reacción glial secunda-ria a la lesión por MPTP no se correlaciona directa-mente con la intensidad de la neurodegeneración.44 La ruta inflamatoria de la ciclooxigenasa 2 (COX-2) ha sido implicada en la neurodegeneración. La COX-2 es expresada y regulada en las células gliales por ci-tocinas y lipopolisacáridos catalizando la formación de prostaglandinas, que promueven la reducción de los niveles de hidroperóxido, resultando en la genera-ción de RL,45 sin embargo, cuando se presentan lesio-nes excitotóxicas, excitación sináptica, muerte neuro-nal por apoptosis e isquemia cerebral, la COX-2 es expresada principalmente en neuronas.46-51
Citotoxicidad por glutamato
Existe evidencia indirecta que sugiere la participación de mecanismos glutamatérgicos en la patogénesis de esta enfermedad. El glutamato, el mayor neurotrans-misor excitador en el sistema nervioso central de ver-tebrados, es conocido por tener actividad neurotóxica cuando se encuentra presente en exceso en las sinap-sis. Dos mecanismos generales protegen a las neuro-nas de esta toxicidad: el primero es una rápida remo-ción por parte de proteínas de membrana que lo acarrean, conocidas como transportadoras de ami-noácidos excitatorios (EAAT) y el segundo es por el metabolismo y reciclaje del glutamato por astrocitos sinápticos vía glutamina sintetasa, una reacción que requiere de ATP. Cuando los niveles extracelulares de glutamato son altos (0.5-1.0 mM), el metabolismo del glutamato sufre un cambio y entra al ciclo de de-saminación oxidativa generadora de ATP, en la que participa la glutamatodeshidrogenasa. Esta enzima requiere de ADP para su actividad y comienza a ser funcional cuando los niveles de energía celular se en-cuentran bajos. Se ha observado que uno de los transportadores de glutamato, el EAAT3 específico de neuronas, se encuentra en altas concentraciones en las neuronas dopaminérgicas mesencefálicas. Me-diante estudios por inmunocitoquímica se ha encon-trado que las neuronas dopaminérgicas se tiñen de forma intensa para la enzima glutamatodeshidroge-nasa. Estos datos apuntan a que el glutamato pudie-ra tener algún papel en la fisiopatología de la EP, aún faltan más estudios para determinar el papel exacto que juega en esta patología.52 Actualmente se sabe que la exposición de las neuronas al glutamato provoca una despolarización mitocondrial asociada a
un incremento del influjo de Ca²+ dentro de la mito-condria. También se sabe que la actividad de los re-ceptores a glutamato, los NMDA, inducen un rápido influjo de Ca²+ mitocondrial, disminución del consu-mo de O2 e inhibición de la actividad de los complejos I, II/III y IV de la cadena respiratoria.53
Papel de los metales en la enfermedad de Parkinson
La actividad de la superoxidodismutasa (Mn-SOD y/o Cu/Zn-SOD) controla los niveles de ion superóxido (O2¯•), produciendo peróxido de hidrógeno (H
2O2). Aunque el H2O2 no es muy reactivo, si se encuentra con Fe2+ o Cu+, forma un radical hidróxilo (OH•) al-tamente reactivo y de corta vida por medio de la reac-ción de Fenton-Haber Weiss.54 La peroxidación de los lípidos de la membrana y la modificación oxidativa de varias proteínas de membrana y asociadas a ella, ocu-rren en las enfermedades neurodegenerativas. El es-trés oxidativo asociado a la membrana es promovido por metales con actividad redox, más notablemente el hierro y el cobre. Los niveles de hierro están incre-mentados en poblaciones neuronales vulnerables en la EA y EP. Las manipulaciones dietéticas y farmacológi-cas del hierro y cobre modifican el curso de la enfer-medad en modelos de EA y EP en ratones, por lo que esto sugiere un papel de estos metales en la patogénesis de la enfermedad.55 El incremento del contenido de hie-rro en la sustancia nigra de humanos y neuromelanina en presencia de hierro, juegan un papel importante en la generación de RL.56 El desferol, un quelante de hie-rro, ha demostrado ser un potente agente neuroprotec-tor en modelos de EP con 6-hidroxidopamina.57
Metil-fenil-tetrahidropiridina (MPTP) y su relación con la enfermedad de Parkinson
deri-edigraphic.com
vado, el 1-metil-4-fenilpiridina (MPP+), por una monoaminaoxidasa en las células gliales. El MPP+ es tomado por las células que procesan dopamina hacia los sitios de recaptura y concentrada amplia-mente dentro de las matrices mitocondriales carga-das negativamente. Dentro de la mitocondria, el MPP+ inhibe el complejo enzimático I de la cadena de transporte de electrones (ETC), lo cual causa una degeneración específica de las neuronas cateco-laminérgicas en la sustancia nigra y el locus ceru-leus, reduciendo la producción de ATP en las mito-condrias.60 Esta reacción resulta en toda una gama de alteraciones clínicas e histopatológicas reminis-centes de la EP esporádica.61-63
Se ha encontrado que existe un decremento de al-rededor del 30 al 40% en la actividad del complejo I en la sustancia nigra de pacientes con EP.64-68 Defec-tos localizados en el ADN mitocondrial para el com-plejo I de plaquetas de pacientes con EP, son transfe-ribles a líneas celulares carentes de mitocondrias. Estos defectos son asociados con la producción de RL, incremento a la susceptibilidad al MPP+ y daño al almacén mitocondrial de calcio.
Relativamente tarde en el proceso de apoptosis, la activación de las endonucleasas da como resulta-do la fragmentación del ADN celular, evidente en electroforesis en geles de agarosa como un barrido de ADN de aproximadamente 180 pares de bases.69 Específicamente, los extremos terminales 3’OH re-sultantes de la fragmentación del ADN pueden ser detectados con la técnica de TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-X 3´nick end-labeling). In vivo, el núcleo apoptótico fue detectado en ratones después de 5 días de trata-miento con MPTP (5x30 mg/kg),70 mientras que no pudo ser detectado después de un día de régimen de exposición con MPTP.71
El desarrollo de un inadecuado metabolismo mito-condrial puede alterar una gran variedad de procesos celulares y mecanismos homeostáticos. La falla del transporte de electrones en la ETC puede dar por re-sultado la depleción de ATP, formación de especies reactivas de oxígeno o RL, salida de calcio con despo-larización mitocondrial y formación de poros de tran-sición con canales a través de las dos membranas de la mitocondria. El poro de transición mitocondrial provee un paso citoplásmico a moléculas que usual-mente se concentran dentro de la mitocondria, como el citocromo C. Éste puede activar las cascadas de se-ñalización de la muerte celular programada mediante su contribución en la activación de cisteínas, en la vía enzimática secuestradora de aspartato
(caspa-sas).72 Las caspasas son una familia de proteasas de cisteína con un sustrato específico para el ácido as-pártico. Se ha mostrado que hay activación de la cas-pasa-3 después de la aplicación de MPP+ en neuro-nas del cerebelo73-74 o después de la administración de MPTP in vivo.75 La activación de esta cascada enzi-mática resulta en la autodigestión del contenido celu-lar (Figura 3).14,76
El papel de las neurocininas en la enfermedad de Parkinson
Las neurocininas en los mamíferos son un grupo de neuropéptidos que incluyen a la sustancia P (neuro-cinina-1, NK-1), sustancia K (NK-2) y la neuromedi-na (NK-3). Sus efectos biológicos como neurotrans-misores, neuromoduladores o factores parecidos a moléculas neurotróficas son mediados por tres distin-tos receptores a neurocininas llamados: receptor a SP (NK-1R), NK-2R y NK-3R. Varias líneas de inves-tigación han mostrado que las neurocininas se en-cuentran involucradas en la patogénesis de la EP. El decremento de los niveles de sustancia P y de la in-munorreactividad a la sustancia P que ha sido en-contrada en los tejidos de la sustancia nigra y el es-triado de animales con Parkinson también se encontró en análisis post mortem de pacientes con EP. Las neurocininas ejercen un efecto protector so-bre las neuronas y los receptores a las neurocininas 1 y 3 se encuentran de forma abundante en neuronas colinérgicas y dopaminérgicas de los ganglios basa-les, indicando que estas neuronas se encuentran bajo regulación fisiológica de las neurocininas. La admi-nistración de agonistas de los receptores NKs en mo-delos de Parkinson en ratones tratados con MPTP, crea una modulación en la actividad motora. Éstas constituyen moléculas que se encuentran asociadas al funcionamiento y sobrevida de neuronas de los ganglios basales, en particular de las neuronas dopa-minérgicas.77
Terapias de neuroprotección y restauración
edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
de la enfermedad. Las imágenes de la tomografía por emisión de positrones (PET) y la tomografía por emisión de fotones simple (SPECT) es una herra-mienta útil en el diagnóstico preclínico de la enfer-medad, pero su uso como método de detección en la población, no es posible. Recientemente se han desa-rrollado técnicas de investigación utilizando estos modernos métodos de imagen para seguir el compor-tamiento de células implantadas en modelos de tras-plantes. En este sentido, se están creando grupos de estudio de pacientes con síntomas tempranos asocia-dos al EP, alteraciones olfativas, disfunción neuro-cognitiva sutil, anormalidades del control visuomo-tor y en menor grado, desórdenes de personalidad y de humor, que han sido sugeridos como marcadores que preceden o acompañan las alteraciones iniciales de la enfermedad.80-83
¿El tratamiento con levodopa promueve la lesión?
Estudios de oxidación en pacientes con EP sugieren la influencia del tratamiento con levodopa (LD) como un tratamiento con un efecto pro-oxidativo. Numero-sos estudios in vitro han mostrado un incremento en la oxidación y subsecuente neurotoxicidad secunda-ria al tratamiento con LD. El metabolismo de la do-pamina y LD catalizados por la MAO-B produce RL tóxicos84 que in vitro pueden iniciar la disrupción de la membrana celular por peroxidación lipídica85-86 y promover la muerte de neuronas dopaminérgicas y de otras neuronas en cultivos in vitro.87-90 Debido a es-tos mecanismos, la LD podría ser tóxica para las neuronas dopaminérgicas y acelerar la neurodegene-ración en la EP.88,91,92 Al presentarse altos niveles de estrés oxidativo, se ha observado incremento de la oxidación de lipoproteínas en plasma y en líquido ce-rebroespinal, decremento de los niveles plasmáticos de grupos proteína-sulfhidrilo y bajos niveles de α -to-coferol en líquido cerebroespinal de pacientes con la EP comparados con pacientes con otras enfermedades neurológicas y con los controles. El tratamiento con LD no cambia de forma significativa la oxidación de lipoproteínas en plasma, pero sí incrementa la auto-oxidación y disminuye los niveles de antioxidantes plasmáticos, con mayor significancia para el ubiqui-nol-10, efecto que no se vio cuando se utilizaron ago-nistas de dopamina. Existen otros estudios en los que la LD no parece acelerar la enfermedad.93,94 Aún son necesarios más estudios al respecto para poder deter-minar la influencia real que el tratamiento con levo-dopa ejerce sobre las neuronas levo-dopaminérgicas.
Terapias antioxidantes
El ubiquinol-10 es la forma reducida de la coenzima Q10 y actúa como antioxidante, siendo oxidada a ubi-quinona-10. La coenzima Q10 ha sido reportada sin cambios95 o reducida96 en pacientes con EP. En otro estudio la relación entre ubiquinol-10 y ubiquinol-10/ coenzima Q fue menor en pacientes con EP sin trata-miento con LD. Esto podría implicar un déficit de ubiquinol-10 en pacientes con PD tratados con LD. Estos datos proveen las bases para dar una terapia de suplementación con Q10 en pacientes que reciban tratamiento con LD.93 Estudios recientes muestran que no existe una correlación entre los valores de la coenzima Q10 sérica y el riesgo para la EP.95,97
Se han usado otros antioxidantes como el adenín-dinucleótido de nicotinamida (forma reducida, NADH) que es necesario para la generación de ATP por la mitocondria con resultados parciales. La adminis-tración de glutatión no ha reportado buenos re-sultados debido a la hidrólisis completa que sufre el compuesto en el tracto gastrointestinal; recien-temente se han desarrollado estudios con la mela-tonina, la cual es producida principalmente por la glándula pineal, debido a su potente actividad an-tioxidante endógena.98-100
Terapias no dopaminérgicas
Recientemente se reportó que el dextrometorfán (DM), un agente antitusivo usado ampliamente, atenúa in vi-tro la neurodegeneración dopaminérgica inducida por una endotoxina. En cultivos mesencefálicos sometidos a tratamiento con MPTP, el DM reduce de forma sig-nificativa tanto la producción de radicales superóxido en el medio extracelular como a las especies reactivas de oxígeno intracelulares. El efecto neuroprotector del DM fue observado en animales silvestres, pero no en animales mutantes deficientes en NADPH oxidasa, in-dicando que esta enzima es un mediador crítico para la actividad neuroprotectora del DM.101,102
edigraphic.com
asociado con el inicio de disquinesias después de una terapia de largo plazo con levodopa.104
El modafinil, medicamento usado en alteraciones del sueño ha mostrado tener un efecto protector con-tra los efectos nocivos causados por la adminiscon-tración de MPTP. Inhibe preferentemente la liberación de GABA en el estriado y previene el decremento de dopa-mina, serotonina y noradrenalina en el estriado y los niveles de glutatión y GABA en la sustancia nigra. Parte de este efecto protector se debe a la modulación nigroestriatal de GABA y la modulación de la libera-ción de noradrenalina y serotonina en el estriado.105
El MPTP induce apoptosis a través de muchas vías de señalización, una de ellas es a través de la ci-nasa c-jun N-terminal. Actualmente se está investi-gando un bloqueador llamado SP600125, un inhibi-dor reversible que compite con el ATP para unirse a la cinasa y que tiene gran selectividad por JNK.106
Terapias con antiinflamatorios
Otra terapia en investigación es el uso de agentes an-tiinflamatorios,38 de los cuales el meloxicam, un inhi-bidor de la COX2, ha dado buenos resultados en ani-males40, 107 aunque en humanos los resultados no han sido satisfactorios.108 Esto podría indicar que los mo-delos experimentales no reflejan exactamente los pro-cesos neurodegenerativos o que la muerte neuronal conlleva toda una cascada de procesos que no son re-sueltos por una sola terapia.
Factores neurotróficos y terapias restaurativas
Los factores neurotróficos en virtud de sus propieda-des de neuroprotección, muestran un gran potencial como agentes terapéuticos en enfermedades neurode-generativas. El factor neurotrófico derivado de las cé-lulas gliales (GDNF) es uno de los agentes promoto-res más potentes de sobrevivencia para las neuronas dopaminérgicas mesencefálicas;109,110 también el factor neurotrófico derivado del cerebro (BDNF) y el factor derivado de las células gliales tienen efectos simila-res.111,112 La cascada de señalización intracelular es mediada por un sistema de receptor multicomponente que comprende el GDNFα (componente α del receptor de la familia GDNF) y el receptor tirosina cinasa, los cuales son abundantes en la sustancia nigra.113 La administración directa dentro de la sustancia nigra
produce una restauración funcional y bioquímica parcial después del tratamiento con MPTP.114-116 De-pendiendo del diseño experimental, el GDNF tiene efectos neuroprotectores y restaurativos.117,118
El BDNF promueve la sobrevida de todos los tipos mayores de neuronas afectados en la AD y la EP como las neuronas hipocampales, neocorticales, colinérgicas septales, neuronas basales del cerebro anterior y neu-ronas dopaminérgicas de la sustancia nigra.119
Terapias con vitaminas antioxidantes
Debido a que el estrés oxidativo es un importante me-canismo de muerte celular en la EP, las vitaminas C, E y A, valiosos agentes antioxidantes, se han asociado con los mecanismos fisiopatológicos de la enfermedad. La determinación de estas vitaminas en grupos de pa-cientes con la EP mostró que los niveles son similares a los encontrados en pacientes sanos.120 En estudios de suplementación con vitamina E, se ha observado que se pueden proteger del daño estriatal causado por la 6-hidroxidopamina.121 Estudios in vitro han mostra-do que la vitamina E protege a las neuronas contra los efectos neurotóxicos causados por el glutamato.122 La suplementación con vitamina E y C incrementa los intervalos de dosificación de levodopa.123
Se han sugerido las dietas balanceadas que conten-gan cantidades adecuadas de frutas, vegetales junto con suplementos de S-adenosín metionina, vitaminas C, B6, B12 y folato.36,123 En un estudio realizado por VanItallie y Nufert (2003),124 se sugiere el uso de una dieta hipercetogénica, debido a que las cetonas son usadas en forma eficiente por la mitocondria para la generación de ATP y podría ayudar a proteger a las neuronas vulnerables del daño inducido por RL.
Se sabe que el líquido cefalorraquídeo de pacientes con EP tiene niveles bajos de tiamina libre.125 La co-carboxilasa o pirofosfato de tiamina (PPT) es una coenzima derivada de la tiamina que se encuentra in-volucrada en la mayoría de los procesos respiratorios y reacciones del metabolismo intermedio de las célu-las;126 y se ha comprobado que se encuentra en com-plejos multienzimáticos y participa en la piruvato descarboxilasa, transcetolasa, α-oxoácido descarboxi-lasa, citocromos, acetolacetato sintetasa y transceto-lasa.127,128 Otro aspecto en el cual interviene el PPT, es en la síntesis del ATP, por lo que su deficiencia ocasiona la inhibición del metabolismo energético129-131 así como daño por hipoxia.132,133 Debido a sus efectos en la reducción del metabolismo anaerobio y la dismi-nución de la formación de RL, la administración del PPT podría disminuir los eventos fisiopatológicos que promueven la lesión de la sustancia nigra.
edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
dopaminérgicos del sistema nervioso central se encuen-tran afectados, pero sí lo están partes de otros sistemas que involucran a diferentes neurotransmisores.
La combinación de terapias antiapoptóticas junto con terapias de restauración parecen ser prometedo-ras. De forma interesante en los humanos, la fase subclínica puede durar alrededor de 5 años antes del inicio de los síntomas, según análisis funcionales de imágenes y estudios patológicos. Los cambios en la terapéutica deben de encaminarse a la identificación de estadios subclínicos o de los síntomas iniciales y un inicio temprano de la terapia. Es probable que el mejor tratamiento sea una combinación de los antes expuestos.
REFERENCIAS
1. Gelb D, Oliver E, Gilman S. Diagnostic criteria for Parkinson’s disease. Arch Neurol 1999; 56: 33-39.
2. Dunnett S, Björklund A. Prospects for new restorative and neuroprotective treatments in Parkinson’s disease. Nature 1999; 399 s1: A32-A39.
3. Forno L. Neuropathology of Parkinson’s disease. J Neuro-pathol Exp Neurol 1996; 55: 259-272.
4. Uhl G, Hedreen J, Price D. Parkinson’s disease: loss of neurons from the ventral tegmental area contralateral to therapeutic surgical lesions. Neurology 1985; 35: 1215-1218.
5. Bryant P, Geis C, Moroz A, O´Nelly B, Bogey R. Stroke and neurodegenerative disorders. 4. Neurodegenerative di-sorders. Arch Phys Med Rehabil 2004; 85(S1): S21-S33. 6. Halliwell B. Reactive oxygen species and the central
nervo-us system. J Neurochem 1992; 59: 1609-1623.
7. Turrens J, Alexandre A, Lehninger A. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 1985; 237: 408-414.
8. Beckman J, Crow J. Pathological implications of nitric oxi-de superoxioxi-de and peroxynitrite formation. Biochem Soc Trans 1993; 21: 330-334.
9. Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J, Smith C, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 1992; 298: 431-437.
10. Fitzmaurice P, Ang L, Guttman M, Rajput A, Furukawa Y, Kish S. Nigral glutathione deficiency is not specific for idio-pathic Parkinson’s disease. Mov Disord 2003; 18: 969-976. 11. Reichmann H, Janetzky B. Mitochondrial dysfunction –a
pathogenetic factor in Parkinson’s disease. J Neurol 2000; 247 S2: 1163-1168.
12. Schapira A, Cooper J, Dexter D, Jenner P, Clark J, Mars-den C. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989; 8649: 1269.
13. Arai H, Furuya T, Mizuno Y, Mochizuki H. Inflammation and infection in Parkinson’s disease. Histol Histopathol 2006; 21: 673-678.
14. Langston J, Ballard P, Tetrud G, Irwin J. Chronic Parkin-sonism in humans due to a product of a meperidine-analog synthesis. Science 1983; 4587: 979-980.
15. Dauer W, Przedborski S. Parkinson’s Disease: mechanisms and models. Neuron 2003; 39: 889-909.
16. Gu M, Cooper J, Taanman J, Schapira A. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann Neurol 1998; 44: 177-186. 17. Swerdlow R, Parks J, Miller S, Tuttle J, Trimmer P,
Shee-han J, et al. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol 1996; 40: 663-671.
18. Swerdlow R. Mitochondrial DNA-related mitochondrial dysfunction in neurodegenerative diseases. Arch Pathol Lab Med 2002; 126: 271-280.
19. Goedert M. Alpha-synuclein and neurodegenerative disea-ses. Nat Rev Neurosci 2001; 2: 492-501.
20. Dawson T, Mandir A, Lee M. Animals model of PD: pieces of the same puzzle? Neuron 2002; 35: 219-222.
21. Dawson T, Dawson V. Molecular pathways of neurodegene-ration in Parkinson’s disease. Science 2003; 302: 819-822. 22. Giasson B, Lee V. Are ubiquitination pathways central to
Parkinson’s disease? Cell 2003; 114: 1-8.
23. Jenner P, Olanow C. Understanding cell death in Parkinson’s disease. Ann Neurol 1998; 44 s1: 572-584. 24. Zhang Y, Dawson V, Dawson T. Oxidative stress and
gene-tics in the pathogenesis of Parkinson’s disease. Neurobiol Dis 2000; 7: 240-250.
25. Warner T, Schapira A. Genetic and environmental factors in the cause of Parkinson’s disease. Ann Neurol 2003; 53 s3: S16-S25.
26. Riess O, Jakes R, Kruger R. Genetic dissection of familial Parkinson’s disease. Mol Med Today 1998; 10: 438-444. 27. Fleming S, Fernagut P, Chesselet M. Genetic mouse
mo-dels of parkinsonism: strengths and limitations. NeuroRx 2005; 2: 495-503.
28. Lim K, Dawson V, Dawson T. The cast of molecular cha-racters in Parkinson’s disease. Ann N Y Acad Sci 2003; 991: 80-92.
29. Chung K, Dawson V, Dawson T. New insights into Parkinson’s disease. J Neurol 2003; 250(S3): 15-24. 30. Manning-Bog A, Mc Cormack A, Uversky V, Fink A, Di
Monte D. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and al-pha synuclein. J Biol Chem 2002; 277: 1641-1644.
31. Sherer T, Betarbet J, Greenamyre J. Environment, mito-chondrial and Parkinson’s disease. Neuroscientist 2002; 8: 192-197.
32. Sherer T, Kim J, Betarbet J, Greenamyre J. Subcutaneous rotenone exposure causes highly selective dopaminergic de-generation and alpha-synuclein aggregation. Exp Neurol 2003; 179: 9-16.
33. Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nature Rev Neurosci 2003; 4: 365-275.
34. Peng J, Xo M, Stevenson F, Hsu M, Andersen J. The her-bicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem 2004; 279; 32626-32632.
35. Paolini M, Sapone A, González F. Parkinson’s disease, pes-ticides and individual vulnerability. Trends PharmacolSci 2004; 25: 124-129.
edigraphic.com
37. Henchcliffe C, Schumacher H, Burgut F. Recent advances in Parkinson’s disease therapy: use of monoamine oxidase inhibitors. Expert Rev Neurother 2005; 5(6): 811-821. 38. Nagatsu T, Ichinose H. Molecular biology of
catecholami-ne-related enzymes in relation to Parkinson’s disease. Cell Mol Neurobiol 1999; 19: 5766.
39. Barcia C, Fernández Barreiro F, Poza M, Herrero M. Parkinson’s disease and inflammatory changes. Neurotox Res 2003; 5: 411-418.
40. Hartmann A, Hunot S, Hirsch E. Inflammation and dopa-minergic neuronal loss in Parkinson’s disease: a complex matter. Exp Neurol 2003; 184: 561-564.
41. Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirs-ch E. ImmunocytoHirs-chemical analysis of tumor necrosis fac-tor and its recepfac-tors in Parkinson’s disease. Neurosci Lett 1994; 172: 151-154.
42. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) in-creases both in the brain and in the cerebrospinal fluid from Parkinsonism patients. Neurosci Lett 1994; 165: 208-210. 43. Hunot S, Dugas N, Faucheux B, Hartmann A, Jardieu M,
Debre P, et al. Fc epsilonRII/CD23 is expressed in Parkinson’s disease and induces in vitro, production of ni-tric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci 1999; 19: 3440-3447.
44. Hurley S, O´Banion M, Song D, Arana F, Olschowka J, Haber S. Microglial response is poorly correlated with neu-rodegeneration following chronic, low dose MPTP admi-nistration in monkeys. Exp Neurol 2003; 184: 659-668. 45. Mirjany M, Ho L, Pasinetti G. Role of cyclooxygenase-2 in
neuronal cell cycle activity and glutamate-mediated excito-toxicity. J Pharmacol Exp Ther 2002; 301: 494-500. 46. Adams J, Collaco-Moraes Y, de Belleroche T.
Cyclooxyge-nase-2 induction in cerebral cortex: an intracellular res-ponse to synaptic excitation. J Neurochem 1996; 66: 6-13. 47. Tocco G, Freirer-Moar S, Schreiber T, Sakhi S, Aisen P,
Pasinetti G. Maturational regulation and regional induc-tion of cyclooxygenase-2 in rat brain: implicainduc-tions for Alzheimer’s disease. Exp Neurol 1997; 144: 339-349. 48. Ho L, Osaka H, Aisen P, Pasinetti G. Induction of
cyclooxygenase (COX)-2 but not COX-1 gene expression in apoptotic cell death. J Neuroimmunol 1998; 89: 142-149. 49. Planas A, Soriano M, Justicin C, Rodríguez-Farre E.
In-duction of cyclooxygenase-2 in the rat brain after a mild episode of focal ischemia without tissue inflammation or neural cell damage. Neurosci Lett 1999; 275: 141-144. 50. Wersinger C, Sidhu A. An inflammatory pathomechanism for
Parkinson’s disease? Curr Med Chem 2006; 13: 591-602. 51. Hald A, Lotharius J. Oxidative stress and inflammation in
Parkinson’s disease: is there a causal link? Exp Neurol 2005; 193: 279-90.
52. Plaitakis A, Shashidharan P. Glutamate transport and me-tabolism in dopaminergic neurons of substantia nigra: im-plications for the pathogenesis of Parkinson’s disease. J Neurol 2000; 247(S2): 1125-1135.
53. Rego A, Santos M, Oliveira C. Glutamate-mediated inhibi-tion of oxidative phosphorylainhibi-tion in cultured retinals cells. Neurochem Int 2000; 36: 159-166.
54. Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, et al. Overexpression of cooper/zinc superoxi-de dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pa-thway of caspase activation. J Neurosci 2002; 22: 209-217. 55. Mattson M. Metal-catalyzed disruption of membrane
protein and lipid signaling in the pathogenesis of
neu-rodegenerative disorders. Ann N Y Acad Sci 2004; 1012: 37-50.
56. Grünblatt E, Mandel S, Berkuzki T, Youdim M. Apomor-phine protects against MPTP-induced neurotoxicity in mice. Mov Disord 1999; 14: 612-618.
57. Youdim M, Stephenson G, Shachar D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators. Ann N Y Acad Sci 2004; 1012: 306-325. 58. Beal M. Experimental models of Parkinson’s disease. Nat
Rev Neurosci 2001; 2: 325-334.
59. Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005; 2: 484-494. 60. Singer T, Castagnoli N, Ramsay R, Trevor A. Biochemical
events in the development of parkinsonism induced by MPTP. J Neurochem 1987; 49: 1-8.
61. Zigmond M, Stricker E. Animal models of Parkinsonism using selective neurotoxins: clinical and basic implications. Int Rev Neurobiol 1989; 31: 1-79.
62. Beal M. Mitochondria, oxidative damage and inflammation in Parkinson’s disease. Ann N Y Acad Sci 2003; 991:120-131. 63. Eberhardt O, Schulz J. Apoptotic mechanisms and
antia-poptotic therapy in the MPTP model of Parkinson’s disea-se. Toxicol Lett 2003; 139: 135-151.
64. Bindoff L, Birch-Martin M, Cartlidge N, Parker W, Turn-bull D. Mitochondrial function in Parkinson’s disease. Lan-cet 1989; 1: 49.
65. Schapira A, Cooper J, Dexter D, Clark J, Jenner P, Mars-den C. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 1990; 54: 823-827.
66. Mann V, Cooper J, Krige F, Daniel S, Schapira A, Marsden C. Brain, skeletal muscle and platelet homogenate mito-chondrial function in Parkinson’s disease. Brain 1992; 115: 333-342.
67. Janetzky B, Hauck S, Youdim M, Riederer P, Jellinger K, Pantucek F, et al. Unaltered aconitase activity, but decrea-sed complex I activity in substantia nigrapars compacta of patients with Parkinson’s disease. Neurosci Lett 1994; 169: 126-128.
68. Klivenyi P, Andreassen O, Ferrante R, Lancelot E, Reif D, Beal M. Inhibition of neuronal nitric oxide synthase pro-tects against MPTP toxicity. Neuroreport 2000; 11: 1265-1268.
69. Hartley A, Stone J, Heron C, Cooper J, Schapira A. Com-plex I inhibitors induce dose-dependent apoptosis in PC12 cells: relevance to Parkinson’s disease. J Neurochem 1994; 63: 1987-1990.
70. Tatton N, Nish S. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treated mice using terminal deoxy-nucleotidyl transferase labeling and acridine orange stai-ning. Neuroscience 1997; 77: 1037-1048.
71. Jackson-Lewis V, Jakowec M, Burke R, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 1995; 4: 257-269. 72. Jordan J, Cena V, Prehn J. Mitochondrial control of
neu-ron death and its role in neurodegenerative disorders. J Physiol Biochem 2003; 59: 129-141.
73. Du Y, Dodel R, Bales K, Jemmerson R, Hamilton-Byrd E, Paul S. Involvement of a caspase-3-like cysteine protease in 1-methyl-4-pyridinium-mediated apoptosis of cultured ce-rebellar granule neurons. J Neurochem 1997; 69: 1382-1388.
di-edigraphic.com
SUSTRAÍDODE-M.E.D.I.G.R.A.P.H.I.C
:ROP ODAROBALE FDP
VC ED AS, CIDEMIHPARG
ARAP
ACIDÉMOIB ARUTARETIL :CIHPARGIDEM
fferentially affects striatal glutamate synaptic function. Exp Neurol 2003; 180: 73-86.
75. Eberhardt O, von Coellin R, Kügler S, Lindenau J, Ra-thke-Hartlieb S, Gerhardt E, et al. Protection by synergis-tic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotro-phic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J Neu-rosci 2000; 20: 9126-9134.
76. Rego A, Oliveira C. Mitochondrial dysfunction and reacti-ve oxygen species in excitotoxicity and apoptosis: implicatio-ns for the pathogenesis of neurodegenerative diseases. Neurochem Res 2003; 28: 1563-1574.
77. Chen L, Yung K, Chan Y. Neurokinin peptides and neuro-kinin receptors as potential therapeutic intervention tar-gets of basal ganglia in the prevention and treatment of Parkinson’s disease. Curr Drug Targets 2004; 5: 197-206. 78. Schapira A, Olanow C. Neuroprotection in Parkinson’s
di-sease: mysteries, myths, and misconceptions. JAMA 2004; 291: 358-364.
79. Mandel S, Grunblatt E, Riederer P, Gerlach M, Levites Y, Youdim M. Neuroprotective strategies in Parkinson’s di-sease: an update on progress. CNS Drugs 2003; 17: 729-762.
80. Brooks D. Neuroimaging in Parkinson’s disease. J Am Soc Exp Neuro Ther 2004; 1: 243-254.
81. Tapia-Núñez P, Chaná-Cuevas P. Diagnóstico de la enfer-medad de Parkinson. Rev Neurol 2004; 38: 61-67.
82. Wolters E, Francot C, Bergmans P, Winogrodzka A, Booji J, Berendse H, et al. Preclinical (premotor) Parkinson’s di-sease. J Neurol 2000; 247(S2): 11103-11109.
83. Acton P, Zhou R. Imaging reporter genes for cell tracking with PET and SPECT. Q J Nucl Med Mol Imaging 2005; 49(4): 349-360.
84. Basma A, Morris E, Nicklas W, Seller H. L-dopa cytotoxici-ty to PC12 cells in culture is via its autoxidation. J Neuro-chem 1995; 64: 825-832.
85. Dexter D, Carter C, Wells F, Javoid-Agid F, Agid Y, Lees A, et al. Basal lipid peroxidation in substantia nigra is increa-sed in Parkinson’s disease. J Neurochem 1989; 52: 381-389. 86. Li C, Werner P, Cohen G. Lipid peroxidation in brain:
inte-ractions of L-dopa/dopamine with ascorbate and iron. Neu-rodegeneration 1995; 4: 147-153.
87. Tanaka M, Sotomatsu A, Kanai H, Hirai S. Dopa and do-pamine cause cultured neuronal death in the presence of iron. J Neurol Sci 1991; 101: 198-203.
88. Ziv I, Jancovic J, Rowe D, Xie W, Appel S, Lee W. Neuro-protection by pramipexole against dopa. Life Sci 1999; 64: 1275-1285.
89. Jones D, Gunasekar P, Borowitz J, Isom G. Dopamine-in-duced apoptosis is mediated by oxidative stress and is en-hanced by cyanide in differentiated PC12 cells. J Neuro-chem 2000; 74: 2296-2304.
90. Doggrell S. Recent important trials of pharmacotherapy in Parkinson’s disease. Expert Opin Pharmacother 2005; 6: 1025-1028.
91. Fahn S. Is levodopa toxic? Neurology 1996; 47: S184-S195. 92. Oh J, Chase T. Glutamate-mediated striatal dysregulation and the pathogenesis of motor response complications in Parkinson’s disease. Amino Acids 2002; 23: 133-139. 93. Buhmann C, Arlt S, Kontush A, Möller-Bertram T,
Sper-ber S, Oechsner M, et al. Plasma and CSF markers of oxi-dative stress are increased in Parkinson’s disease and in-fluenced by antiparkinsonian medication. Neurobiol Dis 2004; 15: 160-170.
94. Suchowersky O, Gronseth G, Perlmutter J, Reich S, Ze-siewicz T, Weiner W. Practice parameter: neuroprotective strategies and alternative therapies for Parkinson's disea-se (an evidence-badisea-sed review). Report of the Quality Stan-dards Subcommittee of the American Academy of Neuro-logy. Neurology 2006; 66: 976-982.
95. Jiménez-Jiménez F, Molina J, de Bustos F, García-Redon-do A, Gómez-Escamilla C, Martínez-Salio A, et al. Serum levels of coenzyme Q10 in patients with Parkinson’s disea-se. J Neural Transm 2000; 107: 177-181.
96. Matsubara T, Azuma T, Yoshida S, Tamagami T. Serum coenzyme Q10 level in Parkinson syndrome. In: Folkers, K., eds. Biochemical and clinical aspects of coenzyme Q. Japón: Elsevier Science, 1991: 159-166.
97. Götz M, Gerstner A, Harth R, Dirr A, Janetzky B, Kuhn W, et al. Altered redox of state of platelet coenzyme Q10 in Parkinson’s disease. J Neural Transm 2000; 107: 41-48. 98. Weber C, Ernst M. Antioxidants, supplements, and Parkinson’s
disease. Ann Pharmacother 2006; 40: 935-938.
99. Kedar N. Can we prevent Parkinson’s and Alzheimer’s di-sease? J Postgrad Med 2003; 49: 236-245.
100. Mayo J, Sainz R, Tan D, Antolin I, Rodriguez C, Reiter R. Me-latonin and Parkinson’s disease. Endocrine 2005; 27: 169-78. 101. Liu Y, Qin L, Li G, Zhang W, An L, Liu B, et al.
Dextro-methorphan protects dopaminergic neurons against infla-mmation-mediated degeneration through inhibition of mi-croglial activation. J Pharmacol Exp Ther 2003; 305: 212-218.
102. Zhang W, Wang T, Qin L, Gao H, Wilson B, Ali S, et al. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. FA-SEB J 2004; 18: 589-591.
103. Przuntek H. Non-dopaminergic therapy in Parkinson’s di-sease. J Neurol 2000; 247(S2): 1119-1124.
104. Calon F, Dridi M, Hornykiewicz O, Bedart P, Rajput A, Di Paolo T. Increased adenosine A2A receptors in the brain of Parkinson’s disease patients with dyskinesias. Brain 2004; 127: 1075-1084.
105. Xiao Y, Fu J, Dong Z, Yang J, Zeng F, Zhu L, et al. Neuro-protective mechanism of modafinil on Parkinson disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Acta Pharmacol Sin 2004; 25: 301-305.
106. Wang W, Shi L, Xie Y, Ma C, Li W, Su X, et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res 2004; 48: 195-202.
107. Teismann P, Vila M, Choi D, Tieu K, Wu D, Jackson-Lewis V, et al. COX-2 and neurodegeneration in Parkinson’s di-sease. Ann N Y Acad Sci 2003; 991: 272-277.
108. Grünblatt E, Mandel S, Youdim M. MPTP and 6-hydroxydopamine-induced neurodegeneration as models for Parkinson’s disease: neuroprotective strategies. J Neu-rol 2000; 247(S2): 1195-1202.
109. Kirik D, Georgievska B, Björklund A. Localized striatal de-livery of GDNF as a treatment for Parkinson’s disease. Nat Neurosci 2004; 7: 105-110.
110. Lin L, Doherty D, Lile J, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain do-paminergic neurons. Science 1993; 260: 1130-1132. 111. Kholodilov N, Yarygina O, Frances T, Zhang H, Sulzer D,
edigraphic.com
112. Fumagalli F, Racagni G, Riva MA. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson’s di-sease. Pharmacogenomics J 2006; 6: 95-104.
113. Baloh R, Enomoto H, Jonson E, Milbrandt J. The GDNF family ligands and receptors-implications for neural deve-lopment. Curr Opin Neurobiol 2000; 10: 103-110. 114. Tomac A, Lindquist E, Lin L, Ögren S, Young D, Hoffer B,
et al. Protection and repair of the nigrostriatal dopaminer-gic system by GDNF in vivo. Nature 1995; 373: 335-339. 115. Gash D, Zhang Z, Ovadia A, Cass W, Simmerman L,
Ru-sell D, et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996; 380: 252-255.
116. Gill S, Patel N, Hotton G, O’ Sullivan K, McCarter R, Bunnage M, et al. Direct brain infusion of glial cell line-de-rived neurotrophic factor in Parkinson disease. Nat Med 2003; 9: 589-595.
117. Kearns C, Cass W, Smoot K, Kryscio R, Gash D. GDNF pro-tection against 6-OHDA: time dependence and requirement for protein synthesis. J Neurosci 1997; 17: 7111-7118. 118. Sullivan A, Opacka-Juffry J, Blunt S. Long-term
protec-tion of the rat nigrostriatal dopaminergic system by glial cell line-derived neurotrophic factor against 6-hydroxydo-pamine in vivo. Eur J Neurosci 1998; 10: 57-63.
119. Murer M, Yan Q, Raisman-Vozari R. Brain-derived neu-rotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neuro-biol 2001; 63: 71-124.
120. Paraskevas G, Kapaki E, Petropoulou O, Anagnostouli M, Vagenas V, Papageorgiou C. Plasma levels of antioxi-dants vitamins C and E are decreased in vascular Parkin-sonism. J Neurol Sci 2003; 215: 51-55.
121. Cadet J, Jackson-Lewis V, Fahn S. Vitamin E attenuates the toxic effects of intrastriatal injection of 6-hydroxydo-pamine (6-OHDA) in rats: behavioral and biochemical evi-dence. Brain Res 1989; 476: 10-15.
122. Perumal A, Gopal V, Tordzro W, Cooper T, Cadet J. Vita-min E attenuates the toxic effects of 6-hydroxydopaVita-mine on free radical scavenging systems in rat brain. Brain Res Bull 1992; 29: 699-701.
123. Martin A, Prior R, Shukitt-Hale B, Cao G, Joseph J. Effect of fruits, vegetables, or vitamin E-rich diet on vita-mins E and C distribution in peripheral and brain tissues: implications for brain function. J Gerontol A Biol Sci Med Sci 2000; 55: 144-151.
124. VanItallie T, Nufert T. Ketones: metabolism’s ugly duc-kling. Nutr Rev 2003; 61: 327-341.
125. Jiménez-Jiménez F, Molina J, Hernández A, Fernández-Vivancos E, de Bustos F, Barcenilla B, et al. Cerebrospinal fluid levels of thiamine in patients with Parkinson’s disea-se. Neurosci Lett 1999; 271: 33-36.
126. Frey P. Coenzymes and radicals. Science 2001; 5551: 2489-2490.
127. Hawkins C, Borges R, Perham R. A common structural motif in thiamin pyrophosphate binding enzymes. FEBS Lett 1989; 255: 77-82.
128. Shikata H, Koyama S, Egi Y, Yamada K, Kawasaki T. Cytosolic adenylate kinase catalyzes the synthesis of thia-mine triphosphate from thiathia-mine diphosphate. Biochem Int 1989; 18: 933-941.
129. Meghal S, O´Neal R, Koeppe R. Effect of thiamine defi-ciency, pyrithiamine and oxythiamine on pyruvate meta-bolism in rat and brain in vivo. J Nutr Sci Vitaminol 1977; 23: 385-393.
130. Butterworth R. Effects of thiamine deficiency on brain me-tabolism: implications for the pathogenesis of the Wernicke-Korsakoff syndrome. Alcohol Alcohol 1989; 24: 271-279. 131. Bettendorff L, Mastrogiacomo F, LaMarche J, Dozic S,
Kish S. Brain levels of thiamine and its phosphate esters in Friedreich´s ataxia and spinocerebellar ataxia type 1. Mov Disord 1996; 11: 437-439.
132. Larrieu A, Kao R, Yazdanfar S, Redovan E, Silver J, Ghosh S, et al. Preliminary evaluation of cocarboxylase on myocardial protection of the rat. Heart Ann Thorac Surg 1987; 43: 168-171.