Estudio de la Reconstitución de Poblaciones de Linfocitos T Mediante Marcadores Epigenéticos
189
0
0
Texto completo
(2) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . Diseño y formato de tesis Paula Andrea Correa Vanegas.. Diseño y edición de portada René Jaramillo Restrepo.. 1.
(3) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . Los Doctores Ricardo Pujol-Borrell y Roger Colobran Oriol, Investigadores del Instituto de Investigación del Hospital Universitari Vall d’Hebron (VHIR), Unidad de Inmunología, del Banc de Sang i Teixits - Laboratorio de Investigación Biomédica en Inmunología, LIRAD. Además, son Profesores Asociados del Departamento de Biología Celular, Fisiología e Inmunología de la Universidad Autónoma de Barcelona UAB,. Certifican que:. La tesis titulada: “Estudio de la Reconstitución de Poblaciones de Linfocitos T mediante marcadores epigenéticos” ha sido realizada por Paula Andrea Correa Vanegas, bajo su dirección y guía, es aprobada para ser presentada y defendida para aplicar por el grado de Doctor en Inmunología de la Universidad Autónoma de Barcelona. Barcelona, 1 de Febrero de 2013. Dr. Ricardo Pujol-Borrell. Dr. Roger Colobran Oriol. 3.
(4) AGRADECIMIENTOS. Quiero agradecer la formación, el apoyo y la confianza que me han brindado mis dos directores, el Dr. Ricardo Pujol-Borrell y el Dr. Roger Colobran, quienes me acompañaron en cada fase de mi trabajo de tesis y me ayudaron a sortear las dificultades que pudieron surgir en estos 4 años de investigación. Doy las gracias con un especial afecto a las Dras. Marta Vives y Eva Martínez por su calidez, acompañamiento científico y apoyo personal. Gracias también a Marco Fernández, Pilar Armengol, Gerar Requena, Eugeni Aragall y Amanda Rus, por su soporte técnico, por la paciencia y experiencia que compartieron conmigo en el desarrollo de las técnicas moleculares y de citometría. Gracias a Irma, Cristina, Patricia, Carla, Aina y Jorge por su complicidad y amistad en estos años de trabajo juntos y en general a todo el grupo de trabajo del Servicio de Inmunología y de Endocrinología del Hospital Universitario German Trias i Pujol. Y finalmente, pero no menos sentido, gracias a Silvia Ventura y Albert Tomas, Julia Tomas, Fosca y Bruna, por su soporte, confianza, amistad y por convertirse en nuestra familia en Barcelona. A todos les estaré eternamente agradecida.. 4.
(5) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . Tengo que decir que muchas veces me preguntaron y me pregunte: vale la pena?....se justifica el esfuerzo?...y, nunca tuve la respuesta definitiva para estas preguntas, pero siempre me motivo la convicción de que sólo el saber y el conocimiento te hacen libres y que esa libertad no la ponen en tus manos sino que hay que salir a buscarla. Entonces, te das cuenta de que en el camino de esa búsqueda, vas encontrando la felicidad, el amor, los amigos, la experiencia y la vida misma. Por eso al final de esta etapa de formación puedo responder que, si vale la pena y que es el mejor momento para agradecerle a todas las personas que me ayudaron y me apoyaron para cumplir este sueño, porque de lo único que estoy segura es que sola jamás hubiera podido.. A mi Familia y muy especialmente, a mí esposo y amigo: René. . . 5.
(6) Table de Contenido . ABREVIATURAS 1. INTRODUCCIÓN ...................................................................................................................... 12 1.1. . El Timo ................................................................................................................................ 12 . 1.2 . Estructura y Función Tímica ............................................................................................ 12 . 1.3. . Desarrollo y diferenciación de Células T ....................................................................... 14 . 1.4. . Reordenamiento y expresión del TCR ........................................................................... 16 . 1.5. . Selección positiva ............................................................................................................. 18 . 1.6. . Selección negativa ............................................................................................................ 18 . 1.7. . Células T SP4 o SP8 ........................................................................................................ 19 . 1.8. . Regulación de los correceptores CD4 y CD8 ............................................................... 22 . 1.9. . Marcadores celulares de timocitos ................................................................................. 25 . 1.10. . Salida de células T del timo a periferia (Thymic Output) ........................................ 28 . 1.11 Monitoreo de la función tímica ........................................................................................ 30 1.13 Cuantificación de TREC ................................................................................................... 32 1.14 Spectrotyping ..................................................................................................................... 33 1.15 Reconstitución linfocitaria ................................................................................................ 35 1.16 Maduración postímica ...................................................................................................... 37 1.17 Epigenética y linfocitos T ................................................................................................. 40 1.18 Modificación de las histonas ........................................................................................ 41 1.18 MicroRNAs: ........................................................................................................................ 42 1.19 Metilación del DNA ........................................................................................................... 43 1.19.1 . Características y funciones de las CGIs ................................................................ 44 . 1.19.2 . Regulación de la metilación del DNA ..................................................................... 48 . 1.20 Métodos de estudio de patrones de metilación del DNA ............................................ 55 2 . HIPÓTESIS ................................................................................................................................ 60 . 3 . OBJETIVOS ............................................................................................................................... 62 . 4 . MÉTODOS ................................................................................................................................. 64 4.1 . Identificación in sílico de islas CpG ................................................................................ 64 . 4.2 . Muestras y tejidos ............................................................................................................. 65 . 4.3 . Tratamiento de las muestras y obtención de células ................................................... 66 . 4.4 . Tinción con anticuerpos y separación de soblaciones selulares por FACS ............. 69 . 4.5 . Extracción de DNA genómico y RNA total .................................................................... 71 . 6.
(7) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . 4.6 . Tratamiento del gDNA con bisulfito ................................................................................ 71 . 4.7 . Diseño de oligonucleótidos y PCR nested .................................................................... 73 . 4.8 . Clonación ........................................................................................................................... 77 . 4.9 . Secuenciación ................................................................................................................... 81 . 4.10 Análisis de metilación ....................................................................................................... 82 4.11 Síntesis de cDNA y análisis de expresión génica por PCR cuantitativa en tiempo real ............................................................................................................................................. 83 4.12 Monitoreo y cuantificación de la metilación del DNA con PCR y sondas FRET ..... 85 4.13 Cuantificación del contenido de TRECs en los diferentes estadios de desarrollo de linfocitos T ............................................................................................................................. 86 4.14 Análisis estadístico ........................................................................................................... 87 RESULTADOS .......................................................................................................................... 89 . 5 . Resultados I: Analisis de metilacion en poblaciones de linfocitos T ............................. 89 . 5.1 . 5.1.1 . Identificación y ubicación de las CGIs en los genes CD4, CD8A y CD8B ....... 90 . 5.1.2 . Marcaje y separación de poblaciones de células T ............................................. 92 . 5.1.3 . Análisis cuantitativo de la metilación en CGIs de los genes CD4 y CD8A por secuenciación del DNA tratado con Bisulfito. ....................................................... 97 . 5.2 . 5.2.1 . Perfiles de metilación del extremo 5’ de la CGI-1 en el gen CD8A analizados por qPCR-FRET en el desarrollo de los linfocitos T. ......................................... 131 . 5.2.2 . Gradiente de metilación del gen CD8A en diferentes poblaciones de células T . ................................................................................................................................... 140 . 5.3 . 6 . Resultados II: Diseño de una prueba molecular basada en qPCR-FRET .............. 127 . Resultados III: Analisis de expresión de los correceptores CD4 y CD8 ................. 143 . 5.3.1 . Expresión de CD4 y CD8 en timo ......................................................................... 144 . 5.3.2 . Expresion de CD4 y CD8 en linfocitos de sangre de cordón y sangre periférica (adultos) .................................................................................................................... 145 . DISCUSION ............................................................................................................................. 151 6.1 Selección de los marcadores celulares para la separación de poblaciones de células T humanas. ......................................................................................................................... 153 6.2 . Perfiles de metilación de los genes CD4 y CD8A y características de sus CGIs . 154 . 6.3 . Nuevas metodologías y la PCR-FRET en el análisis de metilación del DNA ........ 159 . 6.4 . La metilación del gen CD8A como biomarcador de células RTEs. ......................... 162 . 6.5 La expresión de los correceptores CD4 y CD8 en las poblaciones de células T analizadas ........................................................................................................................... 163 6.6 Una modificación química diferente en el DNA de mamíferos puede ser confundida con la metilación. ............................................................................................................... 164 7 . CONSLUSIONES ................................................................................................................... 166 . 7.
(8) Referencias ...................................................................................................................................... 168 Anexos .............................................................................................................................................. 189 . 8.
(9) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . ABREVIATURAS APC:. Antigen presenting cell.. CB:. Cord blood.. cDNA:. Copy DNA.. CGIs:. CpG islands, regiones del DNA (Islas) ricas en citosina-guanina.. CGIsS:. CpG islands shores.. CLP:. Common lymphoid progenitor.. CpG:. Citosina-fosfato-guanina.. CpGPAP:. CpG island predictor analysis platform.. cTEC:. Células epiteliales tímicas corticales.. DMR:. Differentially methylated regions.. DN:. Double negative thymocytes.. DNAss:. DNA single strand.. DNMTS:. DNA metil transferasas.. DP:. Double positive thymocytes.. ETP:. Early thymic progenitor.. FACS:. Fluorescence Activated Cell Sorting.. FRET:. Fluorescence resonance energy transfer.. FSC:. Fetal serum cow.. gDNA:. Genomic DNA.. HAT:. Histona acetil transferasa.. HCPGs:. High content of CpG.. HDACs:. Histona deacetilasas.. HRM:. High resolution melting.. ICPGs:. Intermediate content of CpG.. ITAM:. Immune receptor tyrosine-based activation motif.. LCPGs:. Low content of CpG.. LT:. Linfocito T.. 9.
(10) MBD:. Methylated binding domain.. MHC:. Major histocompatibility complex.. miRNA:. micro RNAs. MS-qFRET:. Metilation specific quantitative FRET. MS-HRM:. Metilation sensitive High resolution melting. MSP:. Methylation specific primers.. mTEC:. Células epiteliales tímicas medulares.. NKT:. Natural killer T cells.. Pb:. Pares de bases en el DNA.. PBMC:. Peripheral blood mononuclear cells.. PBS:. Phosphate buffer saline.. PBSF:. PBS fetal. PCR:. Polymerase chain reaction.. PTK7:. Protein tyrosine kinase 7.. qRT-PCR:. Quantitative real time – polymerase chain time.. RTE:. Recent thymic emigrant.. SNP:. Single nucleotide polymorphism.. SP:. Single positive.. SMRT:. Single-molecule real time sequencing. TCR:. T cell receptor. Receptor de célula T. TRECs:. T cell receptor excision circles.. Treg:. Células T reguladoras.. TSP:. Thymus seeding progenitor.. TSS:. Transcription star site.. UCSC:. University of California-Santa Cruz.. UTR:. Untranslated region.. Wt:. Wild type.. 10.
(11) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . INTRODUCCIÓN. 11.
(12) 1. INTRODUCCIÓN. 1.1.. El Timo. El timo es un órgano linfoide primario que brinda un microambiente especializado a los progenitores de células T provenientes de médula ósea. Su función se basa en dar educación y maduración a los timocitos antes de que estos sean exportados a la periferia para establecerse como células T maduras y funcionales. El timo junto con la médula ósea son los encargados del mantenimiento homeostático del sistema inmune periférico.. 1.2.. Estructura y Función Tímica. El timo es un órgano lobulado dividido por septos y fibras y compuesto principalmente de estroma. El estroma es un elemento no hematopoyético de tejido conectivo, que provee el microambiente adecuado para el desarrollo de las células T y está dividido en dos componentes histológicos, uno externo cortical y una interno medular (Figura 1.1). . Figura 1.1. M: tejido medular, C: tejido cortical, L: lóbulo, BV: vaso sanguíneo, C: corpúsculos de Hasall. Pathology Department. University of Massachusetts Amherst.. 12.
(13) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . El timo contiene las células T en todas las fases de maduración y son denominadas timocitos. La célula T más inmadura entra al timo desde la médula ósea a través de los vasos sanguíneos ubicados en la región cortico medular y desde allí comienzan su proceso de maduración, pasando luego por la porción cortical. Esta zona se encuentra densamente poblada de timocitos inmaduros y células epiteliales tímicas corticales (cTECs). Una vez los timocitos comienzan la maduración, migran hacia la médula tímica que está conformada por fibroblastos, células endoteliales, macrófagos derivados de médula ósea, células dendríticas y células epiteliales tímicas medulares (mTECs). Las mTECs tienen un papel muy importante en la función tímica, pues son ellas las encargadas de expresar un ámplio abanico de antígenos propios (muchos de ellos no ubiquos sino restringidos a tejidos específicos) que se presentarán a las células T para educarlas en la respuesta antígeno-específica del sistema inmunológico (1). En esta región los timocitos aprenden la tolerancia a lo propio, la restricción dependiente de antígenos unidos a MHC y completan el reordenamiento del TCR. Además del componente celular, la médula tímica posee tejido conectivo, matriz extracelular y los corpúsculos de Hassall, que son capas concéntricas de células reticulares planas y granulares con alto contenido de queratina, encargadas de albergar remanentes de células tímicas en proceso apoptótico. Es en la médula tímica donde se encuentran los timocitos en estadios más maduros, incluyendo aquellos que ya son aptos para salir a la periferia; esta salida de células T se hace a través de una rica red de vasos linfáticos eferentes, que drenan las células hacia los ganglios linfáticos periféricos. El timo sufre una marcada involución asociada con la edad, hasta convertirse en un órgano atrófico. En la edad adulta se pierde gran parte del tejido epitelial tímico, lo que genera un desarrollo menos eficiente de las células T o timopoyesis, una reducción en las células T recién emigrantes del timo (RTEs) y una reducción en la eficacia de la respuesta inmunológica. En consecuencia, puede haber un detrimento en la salud del individuo, por patologías tales como infecciones oportunistas, autoinmunidad o aumento en la incidencia de cáncer (2,3). La función tímica además de la edad, puede verse. 13.
(14) modificada por otros procesos no fisiológicos como son los tratamientos con esteroides, la irradiación y la malnutrición (caquexia) (4,5).. 1.3.. Desarrollo y diferenciación de Células T. Las células T humanas se originan a partir de un precursor pluripotente en la médula ósea o el hígado fetal. El precursor migra al timo, donde adquiere la identidad de célula T e inicia el proceso de diferenciación. Los progenitores que migran cada día son relativamente pocos, pero están adaptados para proliferar rápidamente una vez llegan al microambiente tímico (6). La proliferación celular en esta primera fase es inicada por acción de la IL-7, producida por las células epiteliales tímicas (7).. Luego de la primera proliferación, estas células pasan por una serie de fenotipos celulares muy discretos para alcanzar las características y funciones propias del linaje T. Este proceso es complejo e involucra cambios en la expresión de muchos genes, factores de transcripción y factores de diferenciación, como se puede observar en la figura 1.2 (8-12).. El primer estadio de diferenciación dentro del timo humano es la célula denominada progenitor linfoide común (CLP). El CLP llega a través de un vaso sanguíneo ubicado en la unión cortico-medular del timo. Esta célula aún tiene la capacidad de diferenciarse hacia células B, T, NK o célula dendrítica (13,14). El CLP se diferencia hacia célula proT. Este estadio se caracteriza por la expresión en superficie del receptor de IL-7 (IL-7R) y el CD34. Se ha descrito que esta fase es altamente proliferativa. Posteriormente, aparecen las fases conocidas como pre-T (pre-T1 a pre-T4), donde se generan los cambios intratímicos más significativos. Durante estas fases comienza a evidenciarse la regulación negativa de la molecula CD34 y se sugiere que se inicia el commitment del linaje T. También se inicia el reordenamiento del TCR y aparece la expresión de los correceptores CD4 y CD8. Los timocitos continúan su maduración en la que completan. 14.
(15) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . el reordenamiento de los loci del TCR. Además, inician la expresión en superficie de la molecula CD3. A partir de aquí, pasan a la fase de DP donde ya pueden expresar un TCRab maduro, lo cual es interpretado por la célula como una señal de sobrevida y expansión celular (14-17). A partir de la fase de DP, las células T inician los procesos de selección positiva y selección negativa que dan lugar a la célula T CD4 o CD8 (SP4 o SP8), que saldrá a periferia. De acuerdo con lo anterior, en la diferenciación de los timocitos tienen lugar tres fases claves que son: i) el reordenamiento de los loci del TCR; ii) la selección positiva y iii) la selección negativa. En ellas intervienen grupos de genes, factores de transcripción y citoquinas.. Figura 1.2. Estadios del desarrollo temprano en timocitos. Los precursores emigran de medula ósea, ingresan al timo como precursor linfoide común (CLP) y comienzan su diferenciación pasando por los estadios de DN, DP y SP.Adaptado de Graux C, et al. Leukemia, 2006.. 15.
(16) 1.4.. Reordenamiento y expresión del TCR. Las células T maduras se caracterizan por la expresión en su membrana del complejo TCR/CD3. El TCR es un heterodímero transmembrana compuestos de dos cadenas ab o gd. Todas las proteínas a, b, g o d están compuestas por un dominio variable y uno constante. Los dominios variables son muy polimórficos y tienen la función del reconocimiento específico de los antígenos que son presentados por las moléculas MHC. En el humano los genes que codifican para las cadenas a y d (TCRA y TCRD), están en el cromosoma 14q11 y los que codifican para la cadena b y g (TCRB y TCRG), están localizados en el cromosoma 7q34 y 7q15 respectivamente. Los genes que codifican para varias de las cadenas del TCR, están conformados por diferentes segmentos, entre ellos el V (variable), el D (diversidad), el J (joining) y el C (constant). Durante el desarrollo de los timocitos, los genes y loci del TCR sufren un estricto reordenamiento que tiene una secuencia lineal donde se codifican unidades completas y se alcanza la exclusión de secuencias y alelos, a través de elecciones al azar entre varios de los segmentos (V, D, J) y por la delección de nucleótidos extras en la porción J de los genes. Estos reordenamientos son los que garantizan la alta variabilidad del TCR y generan el repertorio específico de células T que tendrá cada individuo. Las células T pueden expresar en su superficie diferentes tipos de receptores, el TCR ab o TCR gd. La expresión del TCR gd en la superficie de células T humanas comienza a partir de la semana 9 de gestación, mientras que la expresión del TCR ab se da a la semana 10. Mientras tanto los timocitos contienen los genes del TCR en estado inactivo y no se expresan. La recombinación solo se inicia después de que se activan los genes RAG1 y RAG2 que son esenciales para el reordenamiento de las regiones variables. Las señales del TCR a su vez son esenciales para la sobrevida de la célula T. El TCR se asocia con la molécula de superficie CD3 y la cadena z formando un complejo que activa una cascada de señales luego del primer contacto con el antígeno especifico.. 16.
(17) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . Dentro de esta cascada las proteínas quinasas p56Lck y p59Fyn median la interacción entre el TCR y los correceptores CD4 y CD8. Además, se fosforila el residuo ITAM presente en el CD3 y la cadena z, lo que lleva al reclutamiento de otras quinasas tales como la ZAP-70 y la PI3K. Esta cascada de señales activa otros genes, entre ellos los de citoquinas y factores de crecimiento y diferenciación para que se de la expansión clonal (18,19). Cuando se generan todos estos eventos, el timocito expresa en su superficie el heterodímero TCR ab, el CD4 y el CD8. También se da la expresión del receptor de quimiocinas CCR7 que guía la migración del timocito DP hacia la medula tímica (8) y apaga la expresión de los genes RAG1 y RAG2. Existe también la variación de la célula T que reordena su TCR hacia gd. Este hecho puede darse si, durante el reordenamiento del gen que codifica para la cadena a que tiene incluido el locus para la cadena g, se incluye este locus y no el locus a, sólo así podrá expresarse un TCRgd (20) (Figura 1.3). . Figura 1.3. Reordenamiento del TCR en humanos. Adaptado de Abbas AK, 2007.. 17.
(18) 1.5.. Selección positiva. La selección positiva de la célula T es dependiente del reconocimiento de antígenos presentados por el complejo MHC (Major Histocompatibility Complex). Con este proceso se garantiza la eliminación de células potencialmente nocivas para el sistema inmune. La célula T después de expresar el TCR en su superficie ya está preparada para reconocer cualquier péptido antigénico (propio o extraño), que sea presentado en el contexto de una molécula MHC. Las moléculas MHC son reconocidas como propias por las células T desde que están el timo y son expresadas principalmente, en la superficie de las células presentadoras de antígeno (APCs), que en el contexto del timo serian las TEC y algunas células dendríticas (DCs). Por tanto la selección positiva es el proceso en el que la célula T DP, cuyo TCR interactúa de forma débil o con baja afinidad frente al péptido propio presentado por una moléculas MHC, es estimulada para sobrevivir y dividirse, mientras que los timocitos cuyo TCR no reconoce ninguno de los péptidos MHC de clase I o clase II presentados por las APC, son inducidos a morir por apoptosis. Esto asegura que las células T maduras estarán restringidas por el MHC que expresa cada individuo. Durante la selección positiva también se fija la restricción del linfocito por las moléculas MHC de clase I o de clase II, asegurando que las células T CD8 solo reconozcan péptidos que son presentados por el MHC clase I, mientras que las células T CD4 solo estarán capacitadas para reconocer péptidos presentados por el MHC clase II y expresaran altos niveles de TCR en la superficie celular (TCRhigh) (7,21).. 1.6.. Selección negativa. La selección negativa es el proceso en el cual los timocitos cuyo TCR interactúa con los péptidos presentados por las moléculas MHC de una manera fuerte y con gran avidez (largo tiempo y difícil disociación) sean eliminados. Así, se asegura que las células T. 18.
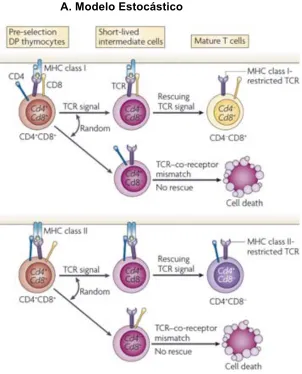
(19) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . que son reactivas contra los antígenos propios presentes en altas concentraciones en el timo, sean eliminadas antes de salir a periferia. En esta fase, las células T estarán restringidas por un MHC propio y además toleran los antígenos propios que encontraran en periferia. Este proceso se denomina tolerancia central. En este punto los timocitos ya expresarán el CD4 o el CD8 en su superficie y no ambos, por lo que pasan a la siguiente fase de maduración como células T single positive (SP) 4 o SP8.. 1.7.. Células T SP4 o SP8. Como se mencionó antes, durante la selección positiva se da la diferenciación del timocito a SP4 o SP8. Esta selección lleva un intrincado cambio en la expresión de correceptores celulares. Requiere de una estricta regulación negativa sobre la expresión bien sea de CD4 o de CD8, según la identidad que haya adquirido la célula T, ya que los timocitos DP expresaban ambos correceptores.. La regulación negativa se inicia sobre el CD8, antes que sobre el CD4. Se inicia con un fenotipo intermedio CD4+CD8lowTCRhigh. Posteriormente, este estadio derivará hacia SP4, mientras que los timocitos que se comprometen hacia SP8 pasan por dos estadios intermedios mas que son CD4lowCD8lowTCRhigh y CD4lowCD8+TCRhigh, antes de ser SP8 (22, 23). En este punto las células T ya están maduras para salir del timo y presentan una correlación perfecta entre el MHC clase I o clase II y la expresión de CD8 o CD4. Sin embargo, se han planteado dos hipótesis sobre la decisión del compromiso de la célula T hacia CD4 o CD8. Una teoría propone que es un fenómeno completamente estocástico mientras la otra sugiere que la decisión es instructiva y se toma a partir de la intensidad de la señal con la cual responda el TCR al interactuar con el complejo MHCpéptido (23). Ambas teorías han sido muy discutidas y hasta el momento no se ha probado lo suficiente ninguna de ellas en humanos. . 19.
(20) El modelo de selección estocástica sugiere que la finalización de la expresión del gen de cada correceptor durante la fase de DP sucede completamente al azar. Luego, solo sobreviven las células T en las cuales coinciden la especificidad del TCR y el correceptor expresado y son estas células las que se diferencian al estadio SP. Sin embargo, esta hipótesis implicaría que la elección del linaje CD4/CD8 fuese un proceso altamente ineficiente, lo que se contradice con algunos estudios experimentales que sugieren que la eficiencia de tal proceso sería del 90% (23). Otro factor que va en contra de esta hipótesis es que los factores de regulación de la transcripción de los genes CD4 y CD8 son completamente diferentes y por tanto la opción de apagar o encender la expresión de uno u otro no sería la misma (24,25). El modelo instructivo de la selección del linaje CD4/CD8 postula que durante la selección positiva, la señal directa que da el TCR a las células T DP es la que suspende la expresión del correceptor que no corresponde con la señal emitida. Por tanto este modelo plantea que la señal del TCR está restringida por la molécula MHC con la que interactúa y que su intensidad es regulada por la interacción del complejo TCR/CD4 o TCR/CD8. Esta señal es regulada por el residuo intracitoplasmático de cada uno de los correceptores. El residuo intracitoplasmático del CD4 se une con mayor afinidad a la quinasa Lck, que el residuo del CD8 (26,27). Entonces, la unión TCR-CD4 es fuerte, mientras que la unión TCR-CD8 es más débil, pero son estas uniones las que dan la instrucción del gen que debe expresarse para la decisión del linaje y el paso a célula T SP. Sin embrago, algunos estudios experimentales concluyen que la instrucción de la interacción del TCR con el correceptor es válida pero no suficiente para que defina la célula T como CD4 o CD8 (28). Muchos estudios concluyen que la instrucción por señales tiene más que ver con el número de células T CD4 o CD8, pero que no interfiere con la decisión de linaje (29,30) (Figura 1.4). En conclusión, más estudios deben ser realizados en humanos, para definir claramente los procesos por los cuales las células se comprometen hacia el linaje CD4 o CD8 y conservan su fenotipo en periferia y durante el resto de su vida.. 20.
(21) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . A. Modelo Estocástico. B.. Modelo Instructivo basado en la fuerza de la señal. Figura 1.4. A. Modelo estocástico de selección del linaje CD4 o CD8. B. Modelo instructivo de intensidad de la señal. Adaptado de Singer A et al, Nat Rev Immunol, 2008.. 21.
(22) 1.8.. Regulación de los correceptores CD4 y CD8. Las células T ab en timo tienen estadios claramente definidos a partir de la expresión de los correceptores CD4 y CD8 en la superficie celular. Estos correceptores son glicoproteínas que se unen a la membrana e interactúan con los dominios proximales de las moléculas MHC clase I y clase II, mientras que sus dominios citoplasmáticos reclutan miembros de la familia de Lck. Durante el desarrollo de las células T, estas hacen la elección del linaje entre CD4 y CD8, y este hecho implica como se menciono antes, la terminación de la expresión del gen contrario al de la identidad que asumirá la célula T. Además, induce la expresión de una cascada de genes propios de cada programa funcional, bien sea de células T helper CD4 o citotóxicas CD8. Muchos esfuerzos se han hecho para resolver la regulación de la expresión de los genes CD4 y CD8. Experimentos realizados a nivel nuclear muestran una compleja red de elementos reguladores positivos para ambos genes, al igual que otra red de elemento reguladores negativos. Pero actualmente se acepta que la expresión del CD4 está regulada principalmente de forma negativa por la función de un silenciador, mientras que la regulación del CD8 es primordialmente regulada por la interacción de múltiples enhancers. El gen CD4 se sabe hasta el momento que posee elementos reguladores upstream del promotor que son dos enhancers, uno proximal y otro distal, conocidos como E4pro y E4dis, respectivamente. Además, posee un silenciador denominado S4. La regulación positiva del CD4 está controlada principalmente por el E4pro, mientras que el elemento principal durante la regulación negativa es el S4. Algunos estudios identificaron dos elementos reguladores mas, uno de ellos un enhancer ubicado en el primer intrón (Ein) del gen y un enhancer que solo está presente en las células T DP (EDP) (31). Funcionalmente se observó que el E4pro posee sitios de unión para proteínas Heb y E2a, requeridas para la activación de los LT, mientras que el E4dis está. 22.
(23) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . involucrado en la activación del gen LAG3 (Gen de activación de linfocitos 3), el cual se encuentra cercano al gen CD4 tanto en humanos como en ratón. El E4in por su parte parece intervenir en la función del S4. Hasta el momento los estudios sugieren que los enhancer pueden tener diferente función e importancia en la regulación de la expresión de CD4 en diferentes estadios del desarrollo a partir de células T DP. Algunos experimentos sugieren que el S4 se requiere estrictamente en el silenciamiento del gen CD4 en los estadios de DP a SP, pero que en periferia ya no es necesario para que el LT mantenga su identidad (33,34). Al parecer no es necesario reprimir el locus CD4 a partir de que la célula T SP8 sale del timo, lo que hace concluir que se produce una herencia del silenciamiento cuando la clona se expande. In vivo se encontró que el S4 del locus CD4 tiene motivos funcionales críticos que incluyen sitios de unión para RUNX3, MYB y HES1, los cuales son indispensables para que el silenciador actúe (3234) (Figura 1.5 A y C). En contraste con el mecanismo de regulación del CD4, la expresión del CD8 es regulada por los enhancers E8I-E8IV, localizados en el extremo 5’ del promotor del gen CD8a. El E8I controla la expresión inmediatamente después de que los timocitos pasen la selección positiva y este control es activo en LT CD8ab y LT CD8aa intraepiteliales (35). Al parecer la acción del E8I es dependiente de la conformación de la cromatina del gen, sin embargo la acción de este solo factor no es suficiente para inducir la expresión de los genes CD8a y CD8b, lo que sugiere que debe trabajar en conjunto con otros elementos reguladores. Los demás enhancers parecen actuar de modo especifico en cada estadio del desarrollo: el E8II funciona en DP y SP8, el E8III, solo actúa sobre los DP y el E8IV potencia la acción del E8II para regular los timocitos DP, SP8 y SP4 (36) (Figura 1.5 B y C).. 23.
(24) A. Regulación de la expresión del gen para el correceptor CD4. B. Regulación de la expresión del gen para el correceptor CD8. C. Interacción de los elementos reguladores para CD4 y CD8 durante la diferenciación de células T. Figura 1.5. Mecanismos de regulación de la expresión de los genes CD4 y CD8. A. Enhancers (E4dis, E4prox, E4DP), silenciador (S4) y factores nucleares que se unen a cada segmento. B. Enhancers involucrados en el control de la expresión del CD8 en los diferentes estadios del desarrollo y los factores nucleares que participan. C. Red de interacciones y estadios de los elementos de regulación de los genes CD4 y CD8 durante la elección del linaje. Adaptado de Singer A. et al Nat rev immunol. 2008.. 24.
(25) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . Además, de los enhancer se sugiere la posible existencia de un silenciador que ayude a controlar la actividad del E8II y IV, pero hasta el momento no se ha identificado. El compendio de estudios existentes hasta ahora se resume en que, el primer paso para el control de la expresión del CD4 o el CD8 se inicia por la fuerza y duración de la señal que recibe el TCR al contacto con el antígeno. Luego se completan los procesos por la activación de vías intracelulares en las que participan diferentes genes y factores de transcripción como se resume en la Figura 1.5 C. Dos de los principales factores de transcripción identificados hasta ahora son el ThPOK y el RUNX3, específicamente expresados en timocitos CD4 y CD8 durante la diferenciación. El gen ThPOK codifica para un miembro de la familia de factores de transcripción Pok, que corresponden a una serie de dedos de zinc encargados de atraer e interactuar con otros factores de transcripción (37,38). Así, el ThPOK, se conoce como el POK de transcripción T helper. Estudios en modelos murinos muestran que una mutación de este gen desencadena una ausencia de LT CD4 mientras que los niveles de CD8 permanecen normales o aumentados. Algunos autores han sugerido también que este factor de transcripción puede. actuar. sobre. el. estadio. intermedio. de. células. CD4+CD8lowTCRhigh,. direccionándolas aberrantemente hacia SP8. En condiciones normales en cambio, se observó que el ThPOK es necesario y suficiente para que se genere el linaje SP4 y se reprima el SP8, según el programa de señales del complejo TCR-MHC-péptido (39). La expresión de ThPOK es inducida por acción del GATA3, aunque no está claro si es por una acción directa o si está mediada a su vez por el RUNX3, que es un gen importante en el silenciamiento de las proteínas TOX, durante la diferenciación hacia LT CD4 (21). 1.9.. Marcadores celulares de timocitos. El desarrollo y maduración del linaje T desde que empieza en la médula ósea o el hígado fetal hasta que se diferencian los timocitos en linfocitos T periféricos, se. 25.
(26) acompaña de un fenotipo, dado por las moléculas de superficie que expresan en cada uno de sus estadios. El linaje linfoide proviene del mismo precursor que las demás células hematopoyéticas y es identificado por la molécula CD34. Algunos autores especulan que si el CD34 aparece acompañado del marcador CD10, estas células son precursoras directas de células T y B (40,41). Algunos investigadores encontraron que el 14% de las células en médula ósea tenían el fenotipo CD34+CD10+CD45RA+CD43+ e IL7RA+. Estas células expresan transcritos para PAX5 e IGB. Además poseen transcriptos para GATA3 y pTa específicos de la diferenciación hacia célula T. Este fenotipo se acepta actualmente para identificar el CLP (42). Posteriormente, se identificaron los marcadores CD7+, CD38low y CXCR4+ como marcadores de precursores linfoides en sangre de cordón neonatal, con potencial para diferenciarse hacia célula T (43). Adicionalmente al CLP, se ha tratado de identificar el progenitor de establecimiento tímico (Thymus Seeding Progenitor, TSP) y el progenitor temprano en timo (Early Thymic Progenitor, ETP). Algunos autores sugieren que las células CD34+CD45RA+CD7+, corresponden al TSP, mientras que las células CD34+CD38low con reordenamiento del TCR corresponden al ETP (44,45). Otros estudios muestran que no hay una sola célula que pueda migrar al timo, sino varias, con fenotipo diferente. Esto se respalda en el hecho de que se encontraron células CD34+CD19+ en timo (46). En resumen se podría pensar que un grupo de precursores tienen la capacidad de dirigirse e instaurarse en timo y que son multipotenciales, pero que aun no están restringidos al linaje de célula T por lo que esta cualidad la adquieren en una fase posterior del desarrollo tímico. Los estadios transicionales de la célula T en el timo han sido caracterizados fenotípicamente, por medio de la evaluación de los reordenamientos del gen TCR (45, 47). Se ha encontrado que la célula CD34+CD1a+ es la primera población que se. 26.
(27) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . compromete al desarrollo del linaje de célula T, perdiendo toda capacidad de transformarse en otra célula (48-51).. Luego de que se establece el linaje de célula T, después del estadio pre-T, como se mencionó en apartados anteriores. El fenotipo de los timocitos se ha establecido de acuerdo a lo que se observa en la figura 1.6. De acuerdo con Blom et al. Hay un estadio de célula CD4 intermedio que denominan small CD4 ISP (immature single positive) en el que las células son CD34+CD1a+CD3e+CD2+CD5+CD7+IL-7Ra+. Es en este estadio donde se expresa por primera vez el correceptor CD4 y continúa madurando hacia estadios intermedios, antes de su paso a timocito DP (52).. Los estadios más tempranos del desarrollo de la célula T también se han analizado a nivel molecular y la característica mas clara es el avance del reordenamiento de los loci TCR, donde se identificó que la secuencia era TCRd>g>b>a (47, 45).. 27.
(28) Figura 1.6. Marcadores celulares en el desarrollo temprano de células T humanas. Adaptada de Blom B, et al. Annu Rev Immunol 2006. 1.10.. Salida de células T del timo a periferia (Thymic Output). . El sistema de vigilancia inmunológica genera las señales necesarias para que haya migración de células T desde el timo a la circulación periférica para así mantener el repertorio de LT y la homeostasis periférica. El mecanismo por el cual los timocitos salen del timo, es poco conocido. Hasta ahora la evidencia muestra que las quimocinas. 28.
(29) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . tienen un papel muy importante en este proceso, pero también es sabido que esta acción por sí sola no sería suficiente para mantener la homeostasis. Algunos estudios sugieren que las células T salen del timo siendo aún inmaduras y que completan su desarrollo en órganos linfoides periféricos (53-55). Estos estudios se han enfocado en identificar y cuantificar las células T que recién salen del timo (Recent Thymic Emigrants, RTEs), las cuales son claves en la valoración de la función tímica y la reconstitución periférica (53,55). Hasta el momento se sabe que los RTEs salen con un fenotipo maduro en cuanto al perfil de TCR, CD4 y CD8. Los RTEs que son funcionalmente competentes 16 horas después de estar en periferia (56-60) y entre el 10-20% sufren divisiones celulares extratímicas. Los estudios también sugieren que un 50% de las células que están listas para salir del timo, sufren una división justo antes de ser RTEs. Estas células que se han dividido recientemente van a periferia pocas horas después de completar la síntesis del DNA. Esta fase de expansión intratímica tardía demuestra que los requerimientos diarios de LT en periferia son altos (61). Otros estudios realizados por Boursalian et al sugieren que la célula T continua su diferenciación post tímica con maduración progresiva en fenotipo y función. Por tanto, se acepta que la maduración final de los RTEs se da en órganos linfoides periféricos (62). La mayoría de estudios de output tímico muestran que no es un fenómeno al azar y que requiere de una serie de eventos de maduración antes de que la célula RTE pueda salir a periferia. Sin embargo, en humanos la diferenciación fenotípica y funcional entre los RTEs y las células T naive de larga vida ha tenido poco éxito. En general, los estudios sobre el output tímico y los RTEs en humanos están metodológicamente muy limitados. Ademas, las señales que regulan la migración de timocitos maduros a la periferia aún no están completamente claras. Se han propuesto varios procesos para la salida de células T hacia periferia: quimiorepulsión del estroma tímico, señales de quimioatracción desde la periferia o pérdida de respuesta a las señales de retención tímica (63). En estos procesos estarían involucradas las siguientes quimocinas y sus receptores:. 29.
(30) CXCL12 y su receptor CXCR4 (64), CCL19 y CCR7 (65), CCL25 y CCR9 y CCL22 y CCR4. El output en un timo adulto puede reducirse hasta un 95% comparado con lo que un timo joven puede emitir y la tasa de exportación celular es insuficiente para reemplazar la pérdida diaria de células T naive en periferia. Por tanto se hace necesario activar la proliferación homeostática periférica de células T maduras y células T memoria para tratar de compensar la pérdida de función tímica (66).. 1.11. Monitoreo de la función tímica. Relativamente poco se conoce acerca del número y composición normal de células en el timo humano y cómo varían sus parámetros de acuerdo con la edad, la respuesta inmunológica y los estados de salud y enfermedad. Hasta hace poco la medición de la función tímica se veía limitada a la determinación de los niveles de LT en el pool de células periféricas, pero esto no reflejaba de forma cuantitativa la contribución de RTEs a sangre periférica ni permitia determinar la diversidad del repertorio de células T periféricas (67,68). Luego se diseñaron una serie de métodos de imagen con emisión de fluorescencia o de radioactividad y algunos con marcaje celular específico, para evaluar el tamaño y celularidad del timo (69,70). Aún continúan siendo usados en protocolos clínicos. La inmunofenotipificación y la citometría multicolor se han convertido en una herramienta para medir el pool de células T (71,72). Muchos antígenos de superficie celular han sido explorados como marcadores de RTEs en diferentes especies. Por ejemplo, Kong et al describió el chT1 como un marcador de superficie de RTEs en aves de corral, e identificó que era expresado por las células que habían emigrado del timo en un tiempo máximo de dos semanas (73). Sin embargo, hasta el momento no se ha. 30.
(31) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . identificado ningún homólogo de esta molécula de superficie en murinos ni en humanos. En humanos en cambio se encontró que el CD45RA (la isoforma de peso molecular alto de tirosin fosfatasa) y el CD62L (L-Selectina), son los marcadores de mayor utilidad para identificar los células T naive tempranas (74). La desventaja de estos marcadores es que no son exclusivos de RTEs o LT naive de novo, puesto que los LT de memoria que son CD45RA- CD45RO+, pueden revertir a CD45RA+ si han pasado un largo tiempo y no han iniciado su proceso de apoptosis. Estas células continúan siendo células de memoria efectoras funcionales, pero fenotípicamente podrían confundirse con una célula naive (75). Por tanto las poblaciones de células T con el marcador CD45RA+ no garantizan una correspondencia indiscutible con RTEs. Otros estudios sugieren que deben combinarse los marcadores CD45RA+/CD45RO-, CD62L+, CD27+, CD95low para la identificación de los RTEs CD4+ (76) o el CD103 y CD11a en el caso de los RTEs CD8+ (72). Por otra parte se reportó que la combinación de los marcadores CD31 y PTK7 podían identificar claramente los RTEs CD4+, pero no los RTE CD8+ (7780). Actualmente, sigue habiendo controversia en cuanto a la metodología que permite diferenciar entre RTEs y células T naive; pero el fenotipo más aceptado para las células T näive es el CD3+CD4+/CD8+CD45RA+CD62L+ (81), mientras que para RTEs CD4 es el CD3+CD4+/CD8+CD45RA+CD62L+CD31+ y para RTEs CD8 aun no se define claramente. Otros métodos menos comunes para la identificación de RTEs en humanos han sido probados en protocolos investigativos o en pacientes infectados por VIH. Estos métodos consisten en el marcaje de la purina desoxirribosa, con un isotipo radioactivo, el deuterio glucosa (D-Glucosa), el cual se combina con una separación de células por citometría y un análisis de espectrometría de masas para evaluar la síntesis de DNA en las células T. Esta tecnología está restringida a protocolos clínicos y requiere de expertos en espectrometría de masas, lo que lo convierte en una metodología impráctica para el estudio e identificación rutinaria de los RTEs (82). Análisis de la función tímica a nivel molecular. 31.
(32) 1.13 Cuantificación de TREC Los estudios de imagen y de inmunofenotipo, así como los marcajes in vivo sirven para monitorear la timopoyesis pero, sin embargo, han sido insuficientes para medir la maduración y la cantidad y calidad de los RTEs que salen del timo para repoblar el pool de LT en periferia. Douek et al encontraron que como resultado del reordenamiento de segmentos de los genes que codifican para el TCR, ciertas secuencias eran expulsadas produciendo un bioproducto de DNA episomal llamado T-cell receptor rearrangement excision circle (TREC) (83,84). Estos TRECs son moléculas estables, no se duplican durante la mitosis y se diluyen con cada división celular (85-87). Los TRECs son producidos por un segmento génico que codifica para la cadena d del TCR (TCRD), ubicada dentro del locus TCRA, tanto en humanos como en ratones (85-87). Como funcionalmente el reordenamiento del TCRA requiere primero de la delección del locus TCRD, es esto lo que genera la formación del DNA circular sj-TREC (88). Los sjTREC se miden por técnicas moleculares como la PCR en tiempo real, en la que se cuantifica el numero de moléculas de DNA episomal (89). Después de la selección positiva, cada célula T de novo que sale del timo conducirá el mayor numero de sjTRECs posibles, por tanto se puede correlacionar la proporción de sjTRECs de una célula con la cantidad de progenie clonal que tiene. Sin embargo, los sjTRECs presentan desventajas como marcador de función tímica, ya que pueden haber moléculas de TRECs que se conservan en algunas LT naive de larga vida. Estas moléculas constituirían un resultado falso positivo a la hora de correlacionarlas con el número de RTEs. Por el contrario, puede haber divisiones celulares de los RTEs inmediatamente después de salir a periferia, lo que subestimaría el número de moléculas de TRECs, con respecto al porcentaje real de RTEs (89-93). Estos factores de confusión llevaron a estudiar y desarrollar otras técnicas para detectar la producción de células T de timo. Por tanto se analizó el bioproducto del reordenamiento del gen TCRB que es otro círculo de DNA de exclusión denominado el bTREC. Este es. 32.
(33) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . inversamente proporcional a la división de la célula, por lo que la proporción entre el número de copias de sjTRECs/bTRECs, puede dar una medida más directa del output tímico (88) (Figura 1.7A y 1.7B). Sin embargo, como los TRECs no se replican, la mitad de las células que sufren división después de cada reordenamiento del TCR, no poseerán el mismo número de TRECs (ni b, ni sj), por lo que no serán identificadas como RTEs y esto dificultará la evaluación cuantitativa del número de RTEs que se encuentran en periferia, dando como resultado un valor subestimado del número real de células que han salido del timo. Por tanto los TRECs, continúan siendo una estrategia imperfecta de identificación y cuantificación de los RTEs.. 1.14 Spectrotyping El proceso de reordenamiento de las regiones VDJ durante la timopoyesis resulta en la expresión de un único TCR en cada célula T para generar las nuevas poblaciones de RTEs y células T naive que conforman el repertorio de LT en periferia (94). Basado en este reordenamiento se generó otro método molecular de análisis de la timopoyesis, conocido como Spectrotyping. Esta técnica estudia la tercera región hipervariable que se encuentra entre la región V y J de la cadena B del TCR, que se conoce con el nombre de tercera región determinante de complementariedad (CDR3). Los resultados del Spectrotyping permiten evaluar la diversidad del repertorio del TCR en periferia, el cual será alto si la reconstitución es de carácter tímico y bajo si es producto de homeostasis periférica.. El método consiste en determinar por técnicas de PCR y RT-PCR, a partir del cDNA de LT, el tamaño y variabilidad de la región CDR3 de la cadena b del TCR. Este sitio es el que determina el contacto del TCR con el complejo péptido/MHC y tiene un papel clave en el reconocimiento antigénico. La gran diversidad del CDR3 se genera por la inserción al azar de nucleótidos durante el reordenamiento de la cadena b del TCR (95-97). Por lo. 33.
(34) tanto, a mayor variabilidad de tamaño del cDNA, mayor policlonalidad del pool de células T.. 1. Conformación de los bTRECs y de los sjTRECs. 2. Análisis de proporciones de bTRECS/sjTRECS para evaluar el output tímico. Figura 1.7. A. Conformación de los sjTRECs y los bTRECs. B. Generacion y seguimiento del DNA episomal, producto del reordenamiento del TCR en timocitos. Adaptado de Ferrando-Martinez R, J immunol Methods, 2010.. 34.
(35) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . 1.15 Reconstitución linfocitaria. La reconstitución inmunológica depende de dos vías: la primera es la generación de nuevas células T a partir de progenitores hematopoyéticos y timopoyesis; la segunda corresponde a la expansión en periferia de LT maduros residuales que responden a reestimulación antigénica y a citoquinas homeostáticas (98). El mantenimiento homeostático y la diversidad del repertorio T en la periferia dependen de un balance dinámico entre estas dos vías y de la generación de linfocitos funcionales durante la edad adulta de los individuos. La vía tímica es más lenta pero garantiza un repertorio policlonal más alto. En ella se conserva la diversidad del TCR y hay generación de células T naive de novo. Esto garantiza una repoblación de LT CD4, CD8 y células T reguladoras. La vía periférica en cambio, puede activarse rápidamente y asegurar una reconstitución inmediata pero inestable, menos sostenida (3-6 meses) y con características oligoclonales, que puede constituir un desbalance importante de la respuesta inmune frente a algunos antígenos (99). Las dos vías de reconstitución pueden ser evaluadas por la combinación de marcadores celulares como el CD45RA, CCR7, CD62L con marcadores moleculares como el contenido. de. sjTRECs. en. las. células.. Aquellas. células. con. fenotipo. CD45RA+CCR7+CD62L+ y un alto contenido de sjTRECs se considera que provienen de células T naive de novo que acaban de pasar por el proceso de timopoyesis. En cambio las células con bajo contenido de sjTRECs y además con características oligoclonales de TCR, se acepta que provienen principalmente de reconstitución periférica por expansión de clonas de LT residuales maduros (100). La calidad y el tiempo que tarda en darse la reconstitución por la vía de la timopoyesis,. depende de la edad del individuo y del estímulo generado por el desbalance periférico. Por ejemplo, en niños que se han sometido a quimioterapia, la renovación del pool de células T por timopoyesis puede tardar tan solo 6 meses. En adolescentes, este. 35.
(36) proceso puede tomar entre 12 y 24 meses (101,102) y en adultos la renovación de células T naive de novo se limita significativamente, tardando entre 3 y 5 años (103105). Otros factores pueden alterar el proceso de reconstitución: es el caso de la radiación, la cual puede causar daños a nivel de la médula ósea que pueden verse reflejados en la duración y calidad de la timopoyesis, de forma independiente de la edad (106,107). Las citoquinas, principalmente la IL-7 y su receptor (IL-7R) pueden también afectar la reconstitución inmunológica por vía tímica (108-110). Algunos autores también han reportado que disminuciones en la hormona del crecimiento y en el IGF-1 (insulinelike grow factor-1), también pueden afectar la función tímica (111-113). La. reconstitución. inmunológica. periférica,. también. llamada. expansión. homeostática periférica (114), ocurre principalmente en procesos de depleción aguda, con una caída del pool de células T, en muy corto tiempo, con inducción de la conversión de células T naive a células T activadas, induciendo proliferación de las clonas de periferia (114). Algunos han propuesto que esta vía puede activarse de manera independiente de citoquinas y antígenos pero otros autores observan que la IL7 y la IL-15 son capaces de actuar sobre algunas células T naive en periferia y sobre células T memoria obligandolas a dividirse (115, 116), lo cual podría explicar los altos niveles de IL-7R en las células T naive en periferia (117). Estudios en humanos muestran que hay una correlación inversa entre los niveles de IL-7 en suero y la reconstitución de células T después de una linfodepleción, mientras que altos niveles de IL-7 en periferia, se asocian con severa linfopenia (118). La IL-15 también es determinante en el mantenimiento homeostático de LT, principalmente CD8 de memoria. La reconstitución periférica es diferente para los LT CD4 que para los LT CD8. La reconstitución por timopoyesis parece ser mas importante en la reconstitución de poblaciones de LT CD4 que en las de LT CD8 y se conoce también que solo las células T naive de novo tienen la capacidad de reconstituir el pool de LT CD4 memoria central. 36.
(37) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . (119). Los LT CD8, en cambio, se recuperan principalmente por la vía de reconstitución periférica y tienen una tendencia oligoclonal. También se ha encontrado que hay reconstitución tímica del linaje CD8 pero, al parecer, con una dinámica diferente de la observada con las subpoblaciones de LT CD4. Además, se encontró que la reconstitución por vía tímica, de la línea CD8 se da con un repertorio de TCR restringido a largo tiempo.. 1.16 Maduración postímica El primer estadio periférico de las células T es el de célula RTE. Por esto, el número y la identificación de RTEs en son importantes pero hasta el momento muy complejos, ya que no hay biomarcadores claramente definidos para diferenciar entre RTEs y LT naive, lo cual sería de gran valor tanto en condiciones fisiológicas como en algunas condiciones clínicas como la inmunosupresión (bien sea de origen infeccioso, por linfoabrasión, trasplante o quimioterapia), la colitis ulcerativa (123), la leucemia mieloide crónica (124) o la tiroiditis autoinmune (125), entre otras.. La mayoría de estudios sobre RTEs se desarrollan en modelos murinos, por las dificultades técnicas que se encuentran para su estudio en humanos. Estudios en ratones muestran que el pico máximo de RTEs se alcanza a las 3 semanas de edad y que la proporción con respecto al total de células T en periferia es del 100% entre el nacimiento y las 3 semanas y decrece a un 20% a las 6 semanas después de salir del timo, a partir de ese momento comienza el proceso de involución tímica. En ratones los RTEs se acumulan en el bazo y nódulos linfoides periféricos y allí maduran hacia LT naive (126,127).. Se observó que los RTEs compiten por nichos vacios en los órganos linfoides periféricos con los LT naive de larga vida, que aún no han sido activados por ningún antígeno (126). Tambien sabemos que se ubican preferencialmente en espacios. 37.
(38) linfopénicos, lo que parece favorecer su sobrevida y desarrollo hacia LT naive. Una vez los RTEs encuentran su espacio se mantienen allí y comienzan su maduración hacia células T naive; el primer paso es aumentar la expresión del CD24 (127), una molécula esencial para la proliferación homeostática de los RTEs. Además, incrementan altos niveles de expresión de TCR y CD2, en cambio expresan bajos niveles de MHCII, CD45RO, IL-7R y CD28 (127-129). Los RTEs CD4 y CD8 también varían en su fenotipo y comportamiento. Los CD8 proliferan al llegar al nicho periférico, tienen altos niveles de CCR9 y producen menos niveles de citoquinas. Los RTEs CD4 tienen una menor proporción de proliferación, una menor producción de IL-2, IL-4 e IFNg y expresan bajos niveles de CD25 (127,130, 131) y tienen alterada la capacidad para diferenciarse en los linajes de células T helper (Th1, Th2, Th17, Treg), especialmente hacia el linaje Th2. (131). Además, algunos estudios encontraron que la regulación epigenética de los loci de la citoquinas IL-2 e IL-4 pueden direccionar la diferenciación de los RTEs CD4 a LT naive (127, 131,132). Estudios en RTEs humanos, identificados mediante el contenido de TRECs, sugieren que estas células proliferan en periferia, como respuesta a IL-2, IL-4 e IL-7, y su expansión es completamente independiente del TCR. Los RTEs humanos tienen altos niveles de CCR7 en su superficie, a través del cual reciben las señales quimiotácticas para dirigirse y permanecer en los órganos linfoides secundarios. Además, observaron que tienen la apoptosis disminuida hasta en un 33% para garantizar el mantenimiento del pool de células en periferia (54). Los RTEs CD4 y CD8 permanecen en el nódulo linfático hasta que completan su maduración y se convierten en células T naive. Los LT naive permanecerán en ese estado hasta que reconozcan el antígeno específico, presentado por células dendríticas (DC) que llegan al nódulo linfático. Cuando el LT naive reconoce el péptido, se forma un complejo APC-MHC I/II con el LT CD4/CD8 que genera la primera señal inmunológica y. 38.
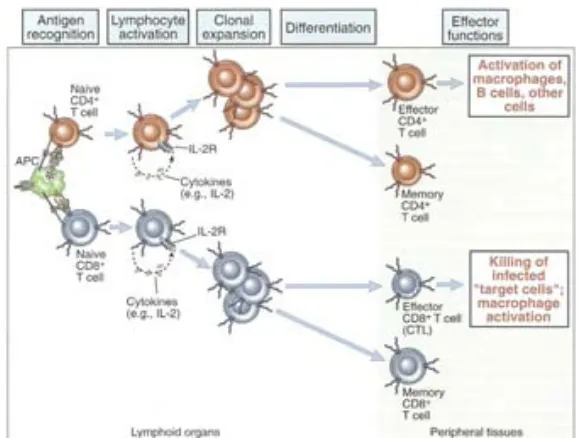
(39) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . luego recibe la segunda señal de activación a través de la molécula B7. Esto hace que se active la producción de IL-2 por parte del LT, que ejerce una acción autocrina para activar la expansión clonal de la célula T activada. Se acepta que a partir de esta expansión el LT deja de ser naive y continúa hacia el proceso de diferenciación (Figura 1.8). En este proceso de diferenciacion participan citoquinas claves como son la IL-4, IL10 e IFN-g. Algunos de los LT activados dejan el órgano linfoide donde ocurrió la activación y entran en circulación como células efectoras. Otras células, particularmente las CD4+, permanecen en el órgano linfoide para inducir diferenciación del linfocito B hacia célula plasmática secretora de anticuerpos específicos contra el antígeno que indujo la activación de la célula T. Las células T efectoras pueden migrar hacia cualquier sito del organismo donde haya una infección o una inflamación. Alli, se encuentran con el antígeno específico que las activó. Muchas de estas células actúan bien como secretoras de citoquinas y activadoras de otras células inmunológicas (como es el caso de los LT CD4), o bien destruyendo las células afectadas (como en el caso de los LT CD8). En esta fase los receptores de superficie cambian y pasan a ser LT (CD4 o CD8), CD45RA-, CD45RO+, CD31- y la expresión de quimocinas y sus receptores también se modifica, especialmente la del CCR7. . Figura 1.8. Diferenciación y activación de las células T periféricas. Adaptada de Abbas, AK. 2007.. 39.
(40) Después de que los LT CD4 y CD8 cumplan su función, muchas de estas células entran en apoptosis y son eliminadas para recobrar los niveles basales de LT en circulación, mientras que otro grupo se diferencia hacia células T de memoria que serán las encargadas de responder rápidamente frente a un segundo reto contra el mismo antígeno para el cual fueron educadas. La función de las células T de memoria ya solo dependerá de la primera señal del TCR, ya que en este caso las señales coestimuladoras no son requeridas.. 1.17 Epigenética y linfocitos T En los últimos 50 años se avanzó en la comprensión de los mecanismos moleculares que regulan la expresión génica en eucariotas y se identificaron nuevos procesos que intervenían en dicha regulación. Estos nuevos proceso parecían relacionados con la diversidad morfológica y funcional de las células y tejidos, y se les denomino epigenética, que es “el estudio de los cambios heredables meióticos y/o mitóticos en la función génica que no pueden ser explicados por cambios en la secuencia del DNA”. Los diferentes procesos que esta definición involucra, están basados en la modificación de la cromatina y la accesibilidad al DNA, por medio de varios aspectos: i) modificaciones de las histonas, que pueden regular el inicio de la transcripción. ii) la acción de los microRNAs (miRNAs) que son pequeñas moléculas de RNA con aproximadamente 22 nucleótidos que participan en la regulacion negativa de su gen blanco, al cual son complementarios. Los miRNA pueden unirse creando una interferencia postranscripcional, y iii) la metilación del DNA, que es el proceso epigenético de células eucariotas más avanzadas y que se describe como el más común en mamíferos. La metilación está involucrada en identidad celular, control de la expresión génica específica de tejido, modificaciones celulares dependientes de la edad, la alimentación y el ambiente. Tambien, comienza a tomar fuerza como biomarcador en algunas patologías.. 40.
(41) Estudio de reconstitución de poblaciones de linfocitos T, mediante marcadores epigenéticos . 1.18. Modificación de las histonas. Este mecanismo es conocido por el profundo efecto que tiene en el control de la expresión génica y se basa en la observación de que el DNA no está “desnudo” sino que, por el contrario, se encuentra formando un complejo íntimamente ligado a las histonas que se encargan de mantenerlo plegado y comprimido. Esta estructura conforma la cromatina que da forma y estabilidad a los cromosomas.. La cromatina es una estructura conformada por unidades que se repiten y que reciben el nombre de nucleosoma. El nucleosoma, a su vez, se conforma por un octámero de proteínas que contiene dos moléculas de cada histona (H2A, H2B, H3 y H4). Alrededor del octamero de histonas se plega una cadena de DNA de 147 pares de bases (pb) de longitud. Los nucleosomas son estructuras dinámicas que pueden tener muchas configuraciones, lo que hace que la cromatina adquiera diferentes características a lo largo del genoma y pueda presentarse de dos maneras: accesible o abierta, que en tal caso se conoce como eucromatina y se asocia con el favorecimiento del inicio de la transcripción génica; o cerrada y condensada, en cuyo caso se denomina heterocromatina y está asociada con la regulación negativa de algunos genes. Se conoce que los cambios en la cromatina se regulan por modificación de las histonas, y tales modificaciones pueden ser acetilación, metilación, fosforilación, ubiquitinización, deaminación, e isomerización de las prolinas presentes en cada proteina (133). En mamíferos. las. modificaciones. que. sufren. las. histonas. son. principalmente. postransduccionales y se encuentran más comúnmente en sus extremos C-terminal o N-terminal. La combinación de modificaciones en las histonas, son propias de cada tejido y de cada tipo de célula, siendo interpretados como el código epigenético de la maquinaria celular o “código de histonas”. Las modificaciones más comunes en mamíferos son la acetilación y la metilación. La acetilación es el proceso mejor entendido hasta el momento; se sabe que las lisinas (K) en las posiciones 9 y 14 de todas las histonas pueden ser acetiladas por medio de las enzimas histona-. 41.
Figure




+7




Outline
Maduración postímica 37
Características y funciones de las CGIs 44
Regulación de la metilación del DNA 48
Métodos de estudio de patrones de metilación del DNA 55
Perfiles de metilación del extremo 5’ de la CGI-1 en el gen CD8A analizados
Perfiles de metilación de los genes CD4 y CD8A y características de sus CGIs 154
Documento similar
In medicinal products containing more than one manufactured item (e.g., contraceptive having different strengths and fixed dose combination as part of the same medicinal
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
Package Item (Container) Type : Vial (100000073563) Quantity Operator: equal to (100000000049) Package Item (Container) Quantity : 1 Material : Glass type I (200000003204)
Cedulario se inicia a mediados del siglo XVIL, por sus propias cédulas puede advertirse que no estaba totalmente conquistada la Nueva Gali- cia, ya que a fines del siglo xvn y en
No había pasado un día desde mi solemne entrada cuando, para que el recuerdo me sirviera de advertencia, alguien se encargó de decirme que sobre aquellas losas habían rodado
Abstract: This paper reviews the dialogue and controversies between the paratexts of a corpus of collections of short novels –and romances– publi- shed from 1624 to 1637:
