Predicción de toxicidad acuática de compuestos orgánicos sobre Poecilia reticulata
79
0
0
Texto completo
(2) Aguas profundas son las palabras de la boca del hombre, y arroyo que rebosa, la fuente de la sabiduría. Prov: 18.4.
(3) A mis padres por traerme al mundo y luego enseñarme a vivir, por mostrarme el camino con su infinito amor, y al resto de mi familia, especialmente a mis abuelos. A mi novia por no faltar a su promesa de amarme en las buenas y en las malas y por brindarme su apoyo incondicional cada día de mi vida. Y a todas aquellas personas que de una forma u otra me han ayudado en la vida..
(4) “La gratitud es el mas legitimo pago al esfuerzo ajeno y es reconocer que todo lo que somos es el resultado del sudor de los demás….”. Mis más sinceros agradecimientos para todas aquellas personas que de una manera u otra me han ayudado a culminar exitosamente mis estudios y este trabajo. Quisiera agradecer especialmente a mi familia y a mi novia por confiar en mí y por todo el apoyo y el amor que me han brindado durante todo el transcurso de mi carrera. A mi tutor J. Alberto por su apoyo, ánimo y dirección durante el desarrollo de este trabajo. Al Grupo de Diseño de Fármacos, por su atención y toda la ayuda que me ha brindado para el desarrollo de esta tesis. A mis compañeros de aula especialmente a Leyanis y Oremia por estar conmigo en los momentos buenos y malos durante estos cinco años de mi vida estudiantil. A todos muchas gracias..
(5) ABSTRACT. The main aim of the study was to develop quantitative structure-toxicity relationship (QSTR) models for the prediction of aquatic toxicity using atom-based non-stochastic and stochastic quadratic indices. The used dataset consist of 300 organic compounds, separated into training and test sets, for which toxicity data to the fresh water fish Poecilia Reticulata (guppy) were available. Using multiple linear regression, two statistically significant QSTR models were obtained with non-stochastic (R2 = 0.807 and s = 0.649) and stochastic (R2 = 0.808 and s = 0.636) quadratic indices. A leave-group-out (LGO) cross-validation procedure was carried out achieving values of q2 = 0.789 (scv = 0.667) and q2 = 0.791 (scv = 0.652) for each model, respectively. In addition, an external validation test set was performed, which yields significant values of R2pred of 0.836 and 0.801, correspondingly. The non-stochastic and stochastic quadratic indices appear to provide an interesting alternative to costly and time-consuming experiments for determining toxicity. Finally, the QSTR models developed in this work were used to predict the ecotoxicological risk of several organics compound of interest..
(6) RESUMEN. El objetivo fundamental de este estudio fue desarrollar relaciones cuantitativas estructuratoxicidad (QSTR) para la predicción de la toxicidad acuática utilizando los índices cuadráticos estocásticos y no estocásticos basados en relaciones de átomos. La bases de datos recopilada de la bibliografía esta formada por 300 compuestos orgánicos, separada en serie de entrenamiento y serie de predicción, para los cuales había sido reportado el valor de toxicidad (Log LC50) acuática sobre el pez Poecilia Reticulata (guppy). Empleando un análisis de regresión lineal múltiple, dos modelos estadísticamente significativos, fueron obtenidos con los índices cuadráticos no-estocásticos (R2 = 0.807 y s = 0.649) y estocásticos (R2 = 0.808 y s = 0.636). Para verificar la robustez y predictibilidad de los modelos empleamos la técnica de validación cruzada, dejandogrupo-fuera mostrando valores de q2 = 0.789 (scv = 0.667) y q2 = 0.791 (scv = 0.652) para cada modelo, respectivamente. Adicionalmente, el poder predictivo del modelo fue analizado empleando una serie de predicción externa donde se obtuvieron valores significativos de R2pred de 0.836 y 0.801 para el modelo no-estocastico y estocastico, respectivamente. Estos resultados nos permiten plantear que índices cuadráticos pueden ser empleados como alternativa para los ensayos experimentales los cuales son altos consumidores de tiempo y dinero además de la necesidad de emplear animales de laboratorio. Finalmente, los modelos desarrollados fueron utilizados para predecir el potencial ecotoxicológico de un grupo de reactivos de la base de datos de ocioso y caducos de la Universidad Central de Las Villas..
(7) INDICE DE CONTENIDOS Pág. RESUMEN INDICE GLOSARIO INTRODUCCION .................................................................................................................. 1 Objetivo General ................................................................................................................. 4 Objetivos Específicos ...................................................................................................... 4 1. REVISION BIBLIOGRAFICA ......................................................................................... 5 1.1.1 Características generales, biológicas y reproductivas del Poecilia Reticulata ...... 5 1.1.2 Bioensayos regulatorios e investigativos. Papel de Poecilia Reticulata en la investigación ecotoxicológica.............................................................................................. 6 1.1.3 Estudios QSAR empleando al Poecilia Reticulata como biomarcador. ................. 8 1.2. Índices cuadráticos moleculares no-estocásticos y estocásticos basados en relaciones de átomos. ........................................................................................................ 10 1.3. Métodos Estadísticos (Quimiométricos) en el Diseño Molecular............................ 14 1.3.1. Introducción a los Métodos Quimiométricos en el Diseño Molecular............. 14 1.3.2. Quimiometría. ..................................................................................................... 14 1.3.3 Metodología general empleada en el los estudios QSAR. ................................. 14 1.3.4. Regresión lineal múltiple (RLM). ...................................................................... 15 1.3.5. Multicolinealidad entre variables con el uso de RLM. .................................... 17 1.3.6. Compuestos ‘outliers’ y técnicas para la selección de los mismos. ................. 17 1.3.7. Validación estadística de los modelos QSAR. .................................................. 18 1.3.8. Análisis de conglomerados (análisis de clusters). ............................................. 20 1.4. Regulaciones de los Métodos QSAR. ....................................................................... 20 MATERIALES Y MÉTODO .............................................................................................. 23 2.1 Obtención de la base de datos de toxicidad acuática sobre (Poecilia. Reticulata) . 23 2.2 Método Computacional. TOMOCOMD-CARDD software ...................................... 23 2.3. Análisis Estadístico de los Datos. Análisis de RLM. ............................................... 25 RESULTADOS Y DISCUSIÓN .......................................................................................... 27 3.1 Obtención de los Modelos en la Predicción de la Toxicidad Acuática. .................. 27 3.2. Cumplimiento de los principios de la OECD. ......................................................... 35 3.3. Empleo de los modelos desarrollados para la predicción del potencial ecotoxicológico de compuestos Ocioso y Caducos de la UCLV. ................................... 39 CONCLUSIONES................................................................................................................. 43 RECOMENDACIONES ...................................................................................................... 44 REFERENCIAS BIBLIOGRÁFICAS ................................................................................ 45. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(8) GLOSARIO AC: Análisis de conglomerado (del inglés cluster) ADL: Análisis Discriminante Lineal CAS: Servicio de Registro de Compuestos Químicos (por sus siglas en Ingles) LC50: Concentración letal media DA: Dominio de aplicación EHS: División de Medioambiente, Salud y Seguridad (por sus siglas en ingles) EPA: Agencia de Protección del Medioambiente (por sus siglas en Ingles) ETA: Índices topo-químicos extendidos de átomos IT: Indice topológico LGO: validación cruzada dejando-grupo-fuera (leave-group-out) LOO: validación cruzada dejando-uno-fuera (leave-one-out) LSO: validación cruzada dejando-varios-fuera (leave-several-out) MAE: (Mean Absolute Error) MC: Media cuadrática N: Número de compuestos empleados en el modelo OECD: Siglas en ingles de Organización para la Cooperación y el Desarrollo (Organization for Economic Cooperation and Development) QSAR: Quantitative Structure Activity Relationships QSPR: Quantitative Structure Property Relationships QSTR: Quantitative Structure Toxicity Relationships R2: Coeficiente de correlación RLM: Regresión Lineal Múltiple s: Desviación estándar SC: Suma de cuadrados SE: Serie de entrenamiento SP: Serie de predicción TETRATOX: TOMOCOMD: (TOpological MOlecular COMputer Design). VC: Validación cruzada.
(9) INTRODUCCION Cada año, como el resultado de nuevos procesos biológicos industriales o naturales, nuevos productos químicos son producidos y/o identificados. Pero no todos estos compuestos son seguros (1). Por lo que nuestro ambiente acuático está bajo un desafío constante de un gran numero de contaminantes provenientes de múltiples fuentes, desde aguas residuales de los hogares, derrames accidentales durante la transportación, las descargas ilegales, hasta accidentes industriales. Estos desafíos combinados requieren una vigilancia constante de aquellas entidades responsables de la calidad ambiental actual y la conveniencia del agua para consumo humano, así como un esfuerzo profundo en la evaluación del riesgo de los actuales y posibles contaminantes (2). Estos problemas son objeto de estudio de la ecotoxicología, que es la ciencia que estudia el destino y los efectos de los contaminantes en los ecosistemas tratando de explicar las causas y prever los riesgos probables. La ecotoxicología prospectiva evalúa la toxicidad de las sustancias antes de su producción y uso. La ecotoxicología retrospectiva se ocupa de confirmar y cuantificar los daños de la sustancia en el ecosistema. El efecto causado por un tóxico dependerá de su toxicidad inherente (capacidad de causar algún efecto nocivo sobre un organismo vivo), del grado de exposición, que a su vez dependerá de la cantidad que ingrese, de cuánto pase a los distintos compartimentos del ecosistema y de su persistencia (3). Los efectos adversos causados por un agente tóxico pueden presentarse repentinamente, causando la muerte de algunos organismos, o provocar cambios sutiles que se manifiestan luego de meses o años. Se debe recordar que un agente químico, dependiendo del nivel de exposición puede provocar diferentes respuestas en un organismo receptor, o sea determinados niveles de un agente pueden tener un efecto benéfico o curativo. Sin embargo niveles superiores de esta misma sustancia pueden provocar efectos adversos e incluso la muerte del receptor (4). Existen nociones de toxicidad desde mediados del siglo XVI. El médico suizo Paracelso (1493-1541) escribió “Todas las sustancias son venenosas. No hay nada, que no sea venenoso. La dosis diferencia un veneno de un remedio” (5). Debido a esta larga historia podría pensarse que existe disponibilidad de una gran cantidad de datos de toxicidad para su uso hoy en día, sin embargo nada más lejos de la verdad (6). Cada año se agregan millares de compuestos al Servicio de Registro de Compuestos Químicos (CAS por sus siglas en Ingles); por lo que muchas agencias medioambientales están envueltas en esta tarea. De hecho, a Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(10) Introducción. 2. finales del 2007 mas de 33 millones de compuestos habían sido registrados en el CAS (muchos de los cuales son compuestos orgánicos sintéticos) y se estima que su incremento anual esta entre 500 y 1000 nuevos compuestos cada año (7). Los inventarios de sustancias industriales realizadas en diferentes países arrojaron como resultado un contenido de aproximadamente 100.000 sustancias en Europa, 75.000 en Estados Unidos y 23.000 en Canadá. (8) Estos numerosos contaminantes orgánicos son el resultado directo del uso creciente de compuestos químicos, como herbicidas, fungicidas, productos del hogar, pesticidas, solventes industriales entre otros. Muchos de estos productos han sido denominados como peligro potencial por la Agencia de Protección del Medioambiente (EPA por sus siglas en Ingles) (9). Como se planteó anteriormente, el gran reto de la ecotoxicología es determinar o predecir los efectos adversos de los agentes químicos sobre los organismos y el medio ambiente. Las regulaciones de seguridad medioambiental exigen que cada nuevo producto que se obtenga, sea probado cuidadosamente antes de entrar al mercado para verificar cualquier efecto negativo en el medioambiente (10). Las pruebas experimentales para determinar el efecto de las sustancias representa la fuente más fiable para obtener dichos datos. Desgraciadamente la realización de tales análisis requiere de un elevado consumo de recursos materiales y de tiempo; estos ensayos no permiten evaluar una gran cantidad de compuestos y son poco prácticos debido al gran número de nuevos compuestos fabricados anualmente.(10) Siendo necesario conocer el impacto ecotoxicológico de los compuestos hay que buscar nuevas alternativas a la determinación experimental de propiedades toxicológicas.(11) En años recientes, los estudios cuantitativos estructura-toxicidad (QSTR por sus siglas en ingles) han emergido como una herramienta útil en este tipo de estudios; dada las aplicaciones exitosas de los estudios cuantitativos estructura-actividad (QSAR por sus siglas en ingles) en otros campos tales como farmacología, química y el diseño racional de fármacos. La necesidad de procesar muchos datos, donde la mayor parte del tiempo proviene de diferentes fuentes y no poseen el mismo significado biológico, ha llevado al desarrollo de muchos modelos sofisticados. Debido a que el ambiente acuático es muy vulnerable a los contaminantes, se ha dedicado un interés especial a la valoración de la toxicidad acuática.(10) Los estudios QSAR/QSTR ofrecen las ventajas de una mayor velocidad y de un costo más bajo, especialmente cuando se compara con los estudios experimentales. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(11) Introducción. 3. Para que un modelo QSAR sea aceptado con fines regulatorios debe cumplir con cinco principios conocidos como “principios de la OECD (Organización para la Cooperación Económica y el Desarrollo, por sus siglas en ingles) para la validación de los modelos cuantitativos de relación estructura-actividad para ser usados con propósitos regulatorios” (12). Entre los muchos estudios que utilizan vertebrados como biomarcadores están, el estudio de mortalidad en peces para Poecilia Reticulata (guppy). En este sentido, un buen número de estudios se ha desarrollado utilizando como biomarcador al Poecilia Reticulata sin embargo los mismos han sido desarrollados empleando una amplia gama de compuestos orgánicos que tienen en común un esqueleto base o determinados fragmentos .por tanto el principal problema de dichos estudios esta en su limitado alcance ya que solo es valido para algunas clases de compuestos. El mayor reto sigue estando en poder obtener modelos QSTR que permitan describir una mayor diversidad estructural simultáneamente. Recientemente, nuestro grupo ha desarrollado un novedoso método químico-computacional conocido por sus siglas acrónimas en ingles: TOMOCOMD (TOpological MOlecular COMputer Design). Este programa permite el cálculo de varias familias de nuevos descriptores moleculares.(13-17) Las cuales han sido empleadas en varios estudios QSAR/QSPR obteniendo satisfactorios resultados.(18-27) Este método es muy flexible y permite el estudio de pequeñas y grandes moléculas tales como proteínas y ácidos nucleicos.(28-31) Los resultados obtenidos hasta ahora con los descriptores TOMOCOMDCARDD nos permiten suponer que pueden ser una herramienta útil para la predicción del potencial ecotoxicológico de sustancias de interés. Por todo lo anterior se plantea el siguiente problema científico: Los modelos QSTR utilizados hasta el momento y que emplean al Poecilia Reticulata como biomarcador han sido obtenidos a partir de bases de datos limitadas y por tanto no son útiles para la predicción de la toxicidad de otros compuestos químicos. Para dar respuesta a la problemática científica nos planteamos la siguiente hipótesis Si se aplican otros enfoques grafo-teóricos podremos obtener modelos matemáticos a partir de bases de datos más extensas y que a su vez sean efectivos, sencillos, interpretables y robustos, de modo que puedan ser utilizados en la predicción del potencial ecotoxicológico de diferentes sustancias de interés. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(12) Introducción. 4. Para demostrar la anterior hipótesis y dar respuesta a la problemática científica planteado, se proponen los siguientes objetivos: Objetivo General 9 Obtener, utilizando los descriptores TOMOCOMD-CARDD, modelos o sistemas de cribado computacional capaces de estimar el potencial ecotoxicológico de diferentes sustancias de interés, tanto su influencia negativa para los seres humanos como para el medio ambiente Objetivos Específicos 9 Coleccionar datos de toxicidad acuática de diferentes compuestos químicos sobre Poecilia Reticulata, para construir una base de datos propia. 9 Obtener modelos QSTR utilizando el ensayo de toxicidad aguda al pez Poecilia Reticulata como biomarcador de la toxicidad acuática de compuestos orgánicos, empleando un nuevo enfoque computacional. 9 Validar la calidad estadística y predictiva de los modelos obtenidos, a través de procesos de validación tanto interna como externa. 9 Determinar el Dominio de Aplicación de los Modelos 9 Tamizar los reactivos de la base de datos de productos ociosos y caducos de la UCLV para ordenarlos según su toxicidad.. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(13) Revisión Bibliográfica. 5. 1. REVISION BIBLIOGRAFICA 1.1.1 Características generales, biológicas y reproductivas del Poecilia Reticulata Los guppys (Poecilia reticulata) son pequeños peces tropicales pertenecientes a la familia Poecilidae, se caracterizan por su afinidad a climas tropicales (32-35), ya que son originarios de las corrientes costeras del nordeste de Sudamérica. Deben su nombre a Robert John Lechmere Guppy que los introdujo en el comercio de acuario, es un pez muy popular en el ámbito acuarístico. Puesto que hoy en día se ha logrado desarrollar una gran cantidad de variedades que van desde cambios en su coloración hasta su tipo y forma de la cola. Entre las variedades más comerciales se encuentran: king cobra, flamingo, mitad negro, cabeza de jade, metálico y multicolor (36). Aunque es originario del Caribe, puede ser encontrado en forma nativa desde Venezuela y el norte de Brasil hasta México, abarcando Guyana Británica y Surinam, Trinidad y Tobago y Barbados (37). Los guppys son sumamente variables tanto fenotípica como genéticamente. Sexualmente los machos exponen una serie asombrosa de puntos y rayas diferentemente coloreadas, tal que cada macho casi parece único (Ver Figura 1), haciendo al guppy uno de los vertebrados más polimorfos conocidos. Aunque las hembras no muestren tal coloración, ellos varían en términos de sus compañeros preferidos tanto dentro de poblaciones como entre poblaciones, haciendo al guppy un sistema poderoso para estudiar la selección sexual (38). En esta especie la reproducción es vivípara, el tiempo de desarrollo usual del embrión dentro de las hembras va de 25 a 30 días aproximadamente. La duración de este periodo depende de la temperatura del agua, la nutrición y la edad del pez. El hecho de que las crías se desarrollen dentro de la madre, proporciona una excelente protección contra peces depredadores y condiciones adversas del entorno, sin embargo, suele ocurrir canibalismo, ya que los padres frecuentemente se comen a sus crías. Por esto, debe prepararse el acuario para separar a los padres de sus crías inmediatamente después de que estas nacen (37). Los P. reticulata son los peces vivíparos que toleran un ámbito más extremo de temperatura,ya que viven en aguas que van desde los 16 ºC hasta los 30ºC,siendo la más adecuada entre 25 y 28ºC. El macho posee una cola muy desarrollada y bien coloreada en forma triangular la mayoría de las veces, pero su cuerpo es de menor tamaño que el de la hembra. Por el contrario, la cola de la hembra no es tan grande como la del macho y tiene en comparación con éste una coloración pobre. Cuando estos peces se encuentran en su etapa Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(14) Revisión Bibliográfica. 6. reproductiva la aleta anal del macho sufre una metamorfosis, convirtiéndose en un gonopodium con el cual fertiliza a la hembra. Por su parte la hembra desarrolla un punto obscuro arriba de su aleta anal, que indica su madurez (37).. Figure 1. Diversidad fenotípica del Guppy. 1.1.2 Bioensayos regulatorios e investigativos. Papel de Poecilia Reticulata en la investigación ecotoxicológica. Los bioensayos de toxicidad aguda o crónica permiten evaluar el efecto de una sustancia química en organismos vivos (39). Las pruebas ecotoxicológicas agudas cuantifican las concentraciones letales de un xenobiótico sobre una especie en particular de la biota (40). Los bioensayos de toxicidad, con agentes contaminantes bajo condiciones de laboratorio, se han incrementado en estos últimos años debido a la brevedad con que se obtiene la información sobre la concentración letal media (LC50) (en mg o mg L-1) y los efectos subletales que afectan negativamente a la biota en los ambientes marinos, estuarinos y dulceacuícolas (39).. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(15) Revisión Bibliográfica. 7. Las pruebas ecotoxicológicas con peces son tradicionalmente empleadas en muchas partes del mundo, ya que éstos juegan un papel importante dentro de la cadena alimenticia (41). Recientemente, hay una tendencia a usar peces de pequeño tamaño como especies centinela para investigaciones ecotoxicológicas e investigaciones biomédicas. Los pequeños peces tienen varias ventajas en estudios de ecotoxicología, ya que ellos son generalmente fáciles para mantener y de reproducirse en condiciones de laboratorio. El tiempo de generación es relativamente corto, y el pez puede producir huevos con regularidad, de ahí proporcionando una variedad de bioensayos (42-45). Los peces consumen y controlan las poblaciones de insectos, microcrustáceos y algas, y permiten de esta forma la recirculación, remoción y resuspensión del material orgánico dentro del ecosistema. Debido a su gran importancia, se han desarrollado una gran variedad de bioensayos que han empleado especies de peces, que son sensibles a la presencia de determinados agentes tóxicos. Los peces son organismos acuáticos extremadamente sensibles a la perturbación ambiental, siendo afectados en su crecimiento y en sus funciones reproductivas (46). Numerosas especies de peces han sido propuestos como bioindicadores para evaluar la ecotoxicidad de sustancias químicas contaminantes como: peces cebra (Danio nuevo Río), pececillo de cabeza gorda (Pimephales promelas), pez del mosquito (Gambusia affinis), guppy (Poecilia reticulata) y medaka japonés (Oryzias latipes), entre otros son comúnmente usados como modelos de peces de agua dulce en estudios ecotoxicológicos (4752). Debido a su alta tasa de reproducción y facilidad de mantenimiento, los guppys son un recurso valioso para la investigación biomédica. Por ejemplo, David Reznick y los colegas explotan diferencias demográficas en la historia de vida como un modelo para entender las fuerzas que forman la variación en envejecido, y en nuestro laboratorio estudiamos al mutante de guppy ‘curveback’ como un modelo hasta ahora único para la escoliosis idiopática familiar (38). Pero los guppys son también uno de los primeros sistemas modelos para el estudio de selección sexual, evolución genética, y ecología. Se han realizado diversos estudios en P. reticulata en el ámbito del aprendizaje y del comportamiento reproductivo (53-55), así como modificaciones en el comportamiento por acción de pesticidas organofosforados (56).. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(16) Revisión Bibliográfica. 8. 1.1.3 Estudios QSAR empleando al Poecilia Reticulata como biomarcador. Varios estudios QSAR se han desarrollado utilizando el ensayo de letalidad de 96h del Poecilia Reticulata como biomarcador (57-60). Sin embargo estos estudios en su gran mayoría tienen un alcance limitado pues parten de bases de datos de series congenéricas o que tienen un sustituyente común para todos los compuestos. Aquí se trataran de manera abreviada los estudios más relevantes publicados hasta el momento, comentando los resultados más sobresalientes, el método empleado así como los descriptores utilizados. En la segunda mitad de la década de los 90 del pasado siglo Verhaar, Urrestarazu y Hermens desarrollaron un estudio QSAR para predecir la toxicidad aguda 172 compuestos orgánicos sobre el pez P. Reticulata empleando la Regresión Lineal Múltiple (RLM) como técnica estadística. Los modelos obtenidos con cuatro variables significativas arrojaron valores de R2=0.928 y un q2=0.920 para la validación cruzada. Este estudio inicio en aquel momento el empleo un nuevo enfoque para entender el mecanismo de toxicidad y encontrar la relación entre los mecanismos de toxicidad y los parámetros fisicoquímicos de los compuestos (57). Posteriormente, Katritzky y Tatham en el año 2001 propusieron aplicar el método CODESSA para la predicción de toxicidad acuática sobre P. reticulata (59). Emplearon una base datos mas extensa la cual fue dividida por clases, basándose en el mecanismo de acción toxica, de la siguiente manera: 90 compuestos en la clase 1 definidos como narcóticos no polares, de la cual se obtuvo un modelo con los siguientes parámetros estadísticos R2=0.955 y s=0.3105, para la clase 2 clasificados como narcóticos polares con una totalidad de 121 compuestos se obtuvo R2=0.918 y s=0.2924 , para la clase 3 (productos químicos reactivos) conformada por una base de datos de 41 compuestos se obtuvo R2=0.848 y s=0.5596, y para la 4ta clase definidos como pesticidas con una data de 31 compuestos resulto una R2=0.755 y una s=0.6569 (59). Adicionalmente, en el año 2002 Seward, Hamblen y Schultz realizaron un estudio de comparación de datos de toxicidad entre P. reticulata y Tetrahymena Pyriformis para un grupo de productos químicos (58). Empleando la base de datos TETRATOX para modelar la capacidad de T. pyriformis de predecir la toxicidad de una especie diferente, en este caso el guppy P. reticulata. Para llevar a cabo dicho estudio se empleó una base de datos de 124 compuestos, se desarrollo un análisis de RLM empleando como variable dependiente logLC50 y el resto como variables independientes. Se identificaron 5 compuestos outliers, Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(17) Revisión Bibliográfica. 9. que una vez retirados de la base de datos mejoraron la relación obteniéndose estadísticamente modelos con un R2 = 0,85 y una s= 0,42 (58). Mas recientemente, en el año 2004, Roy y Ghosh realizaron un estudio de relación cuantitativa estructura toxicidad (QSTR) en el campo de la toxicología acuática con el objetivo de evaluar la seguridad ecológica del pez Poecilia reticulata frente a derivados del benceno (60). Para ello emplearon una base de datos de 92 compuestos basándose y como técnica estadística en una RLM; los descriptores empleados en este trabajo fueron los índices topo-químicos extendidos de átomos (ETA por sus siglas en ingles) aunque también desarrollaron modelos con otros descriptores topológicos y fisicoquímicos para comparar los resultados. Los datos fueron pre-procesados utilizando un análisis de componentes principales para reducir la dimensionalidad e identificar las variables más importantes. El mejor resultado obtenidos con índices topológicos y fisicoquímicos fueron R2 =0.738, q2= 0.718, s=0.340 mientras que al emplear los descriptores ETA el mejor modelo mostró un comportamiento superior con R2 =0.885, q2= 0.865, s=0.23; de la comparación se obtuvieron algunas consideraciones interesantes respecto a la toxicidad de los compuestos en relación a su estructura química (60). Por ultimo un estudio realizado en el año 2005 por Hoover, Acree y Abraham en el cual se desarrollaron modelos para predecir la toxicidad química sobre varias especies de peces incluía también al P. reticulata (guppy) (1). La base de datos de toxicidad reportada para este pez es la segunda más grande de las seis especies de peces consideradas en el dicho estudio, registrándose los valores de LC50 para 148 compuestos. En ese estudio se alcanzaron buenos resultados para los parámetros estadísticos del mejor modelo QSAR desarrollado en el mismo, como son una R2=0.946 con una pequeña desviación estándar de aproximadamente 0.28 unidades. Adicionalmente desarrollaron otros modelos empleando fracciones de esta base de datos alcanzando también buenos resultados (1). De manera general podemos plantear que a pesar de las limitaciones antes mencionadas, este tipo de estudios posibilita la predicción de la toxicidad acuática de productos químicos orgánicos y permite también ayudar en la identificación de compuestos con determinado modo de acción tóxica como son la reactividad química específica, la narcosis no polar y polar entre otros. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(18) Revisión Bibliográfica. 10. 1.2. Índices cuadráticos moleculares no-estocásticos y estocásticos basados en relaciones de átomos. Los índices cuadráticos totales, qk(x) han sido previamente definidos en trabajos de nuestro grupo (14, 17, 20, 61), por lo cual aquí solo se brindara una pequeña reseña de los mismos. Teniendo en cuanta lo anterior podemos plantear que loa índice cuadráticos basados en relaciones de átomos se calculan entonces a partir de la ecuación que se muestra a continuación: qk (x) =. n. n. ∑ ∑ i =1. k. j =1. a ij X i X. j. (1.1). donde aij = aji (matriz cuadrada simétrica), n es el número de átomos de la molécula y X1,…,Xn son las coordenadas del vector molecular (X) en la base canónica qua de Rn. Por tanto, las coordenadas de X son los valores numéricos de una propiedad atómica que caracteriza a cada tipo de átomo en la molécula, pues en la base canónica las coordenadas de cualquier vector coinciden con los componentes del vector (62-68). Los coeficientes kaij son los elementos aij de la k-ésima potencia de la matriz M del seudografo molecular, la cual es utilizada como matriz de la forma cuadrática con respecto a la base canónica. La expresión de qk(x) puede ser escrita como una simple ecuación matricial (14, 17, 20, 61):. q k ( x ) = [X 1. L. X. n. ]. ⎡ a 11 ⎢ M ⎢ ⎢⎣ a n 1. L L. a1n M a nn. ⎤ ⎥ ⎥ ⎥⎦. k. ⎡X1⎤ ⎢ M ⎥ ⎥ ⎢ ⎢⎣ X n ⎥⎦. (1.2). o en una forma matricial más compacta, qk(x) = [X]t Mk [X]. (1.3). donde [X] es un vector columna (una matriz de nx1) de las coordenadas de X en la base canonical de ℜ n, [X]t es la transpuesta de [X] (una matriz de 1xn) y Mk es la k-ésima potencia de M. Como puede apreciarse, los índices cuadráticos totales para una molécula de n átomos son aplicaciones entre espacios, que transforman al vector molecular en k números, en correspondencia con las k-ésimas M matrices utilizadas como matrices de la transformación. Matemáticamente, podemos considerar a los índices cuadráticos como formas cuadráticas q en x1, x2,…,xn variables (q: Rn → R) que utilizan las k-ésimas matrices de los pseudografos moleculares (Mk) como matrices de las formas. En la Tabla 1.1 Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(19) Revisión Bibliográfica. 11. (columna izquierda) se ejemplifica esta representación (M0-M2) para la molécula del 2formil-6-metilbenzonitrilo. Uno de los criterios importantes de la lista de propiedades deseables para un nuevo índice topológico (IT) es la posibilidad de definir localmente los descriptores (69). Es por ello que se ha propuesto una definición local de los índices cuadráticos moleculares. La definición de estos descriptores, invariantes grafo-teóricas para un fragmento FR dado, dentro de un seudografo específico es la siguiente (14, 17, 20, 61): q kL ( x ) =. m. m. ∑ ∑ i =1. k. j =1. a ijL X i X. j. (1.4). donde m es el número de átomos del fragmento de interés y kaijL es el elemento de la fila “i” y columna “j” de la matriz MkL ≡ Mk(G, FR) [ qkL(x) ≡ qk(x, FR)]. Esta matriz se extrae de la matriz k-ésima potencia de M y contiene la información referida a los vértices del fragmento FR de interés y también de su entorno molecular. La matriz MkL = [kaijL] y los elementos kaijL se definen a continuación: k. aijL = kaij si ambos vi y vj son átomos contenidos dentro del fragmente de interés. (1.5). = 1/2 kaij si vi o vj están contenidos en el fragmento de interés pero no ambos = 0 de otra forma Nótese que si una molécula se divide en Z fragmentos moleculares, la matriz Mk puede ser dividida en Z matrices locales MkL, L = 1,...Z y la matriz k-ésima potencia de M es exactamente la suma de las k-ésima potencia de las Z matrices locales. Utilizando este enfoque, los índices cuadráticos totales son la suma de los índices cuadráticos locales de los Z fragmentos: Z. q k ( x) = ∑ q kL ( x). (1.6). L =1. Cada orden de las formas cuadráticas locales tiene un significado particular. Especialmente para los primeros valores de k, contienen información sobre la estructura del fragmento FR en sí. Para valores mayores, contiene información sobre el entorno del fragmento FR considerado dentro del pseudografo molecular (14, 17, 20, 61).. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(20) Revisión Bibliográfica. 12. Tabla 1.1. Cálculo de Mk(G) y Sk(G) para la Molécula del 2-formil-6-metilbenzonitrilo Cuando k Varía entre 0 y 2. 2 1 CHO 3 4. O1. 9 10 CN. 8. C9. C2. C3. N10. C4. 7. 5 N 6. C5. CH3 11. C7 C11. N6. Estructura Molecular. Pseudografo Molecular (G). C9 N10 C11. δi. O1. C2. C3. C4. C5. 0 0 0 0 0 0 0 0 1 0 0. 0 0 0 0 0 0 0 0 0 1 0. 0 0 0 0 0 0 0 0 0 0 1. 1 1 1 1 1 1 1 1 1 1 1. 1 0 0 0 0 0 0 0 0 0 0. 0 1 0 0 0 0 0 0 0 0 0. 0 0 1 0 0 0 0 0 0 0 0. 0 0 0 1 0 0 0 0 0 0 0. 0 0 0 0 1 0 0 0 0 0 0. 0 0 0 0 0 0 0 1 0 3 0. 0 0 0 0 0 0 0 0 3 0 0. 0 0 0 0 0 0 1 0 0 0 0. 2 3 4 3 3 3 4 4 4 3 1. 0 1 0 0 0 0.66 0 0.33 0 0 0 0.25 0.25 0.25 0 0 0 0.33 0.33 0.33 0 0 0 0.33 0.33 0 0 0 0 0.33 0 0 0 0 0 0 0 0.25 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. 0 0 1 0 0 0 1 1 10 0 0. 0 0 0 0 0 0 0 3 0 9 0. 0 0 0 0 0 1 1 1 0 0 1. 6 8 14 10 9 10 12 16 13 12 4. 0.66 0 0.143 0 0 0 0 0 0 0 0. k. aij O1 C2C3 C4 C5 N6 C7C8 M0(G) O1 1 0 0 0 0 0 0 0 C2 0 1 0 0 0 0 0 0 C3 0 0 1 0 0 0 0 0 C4 0 0 0 1 0 0 0 0 C5 0 0 0 0 1 0 0 0 N6 0 0 0 0 0 1 0 0 C7 0 0 0 0 0 0 1 0 C8 0 0 0 0 0 0 0 1 C9 0 0 0 0 0 0 0 0 N10 0 0 0 0 0 0 0 0 C11 0 0 0 0 0 0 0 0 M1(G) O1 0 2 0 0 0 0 0 0 C2 2 0 1 0 0 0 0 0 C3 0 1 1 1 0 0 0 1 C4 0 0 1 1 1 0 0 0 C5 0 0 0 1 1 1 0 0 N6 0 0 0 0 1 1 1 0 C7 0 0 0 0 0 1 1 1 C8 0 0 1 0 0 0 1 1 C9 0 0 0 0 0 0 0 1 N10 0 0 0 0 0 0 0 0 C11 0 0 0 0 0 0 1 0 M2(G) O1 4 0 2 0 0 0 0 0 C2 0 5 1 1 0 0 0 1 C3 2 1 4 2 1 0 1 2 C4 0 1 2 3 2 1 0 1 C5 0 0 1 2 3 2 1 0 N6 0 0 0 1 2 3 2 1 C7 0 0 1 0 1 2 4 2 C8 0 1 2 1 0 1 2 4 C9 0 0 1 0 0 0 1 1 N10 0 0 0 0 0 0 0 3 C11 0 0 0 0 0 1 1 1. C8. 0 0.625 0.071 0.1 0 0 0 0.063 0 0 0. 0.33 0.125 0.287 0.2 0.111 0 0.083 0.125 0.077 0 0. 0 0.125 0.143 0.3 0.222 0.1 0 0.063 0 0 0. 0 0 0.071 0.2 0.333 0.2 0.083 0 0 0 0. N6 S0(G) 0 0 0 0 0 1 0 0 0 0 0 1 S (G) 0 0 0 0 0.33 0.33 0.25 0 0 0 0 S2(G) 0 0 0 0.1 0.222 0.3 0.166 0.063 0 0 0.25. C7. C8. C9. N10. C11. 0 0 0 0 0 0 1 0 0 0 0. 0 0 0 0 0 0 0 1 0 0 0. 0 0 0 0 0 0 0 0 1 0 0. 0 0 0 0 0 0 0 0 0 1 0. 0 0 0 0 0 0 0 0 0 0 1. 0 0 0 0 0 0.33 0.25 0.25 0 0 1. 0 0 0 0 0 0 0 0 0.25 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.25 0 0 0.25 0.25 0.25 0 0 0.25 0 0.75 0 0 1 0 0 0 0 0 0. 0 0 0.071 0 0.111 0.2 0.333 0.125 0.077 0 0.25. 0 0.125 0.143 0.1 0 0.1 0.166 0.25 0.077 0.25 0.25. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata. 0 0 0 0 0 0 0.071 0 0 0 0 0 0 0 0 0 0 0.1 0.083 0 0.083 0.063 0.188 0.063 0.769 0 0 0 0.75 0 0 0 0.25.
(21) Revisión Bibliográfica. 13. Los índices cuadráticos atómicos y de átomo-tipo son dos casos específicos de índices cuadráticos moleculares locales (para FR = átomo y para FR = conjunto de átomos del mismo tipo, respectivamente). O sea, que los índices cuadráticos átomo-tipo se calculan sumando los índices cuadráticos de todos los átomos del mismo tipo en la molécula. En este formalismo, cada átomo en la molécula es clasificado según su tipo, tales como heteroátomos, H-unidos a heteroátomos, halógenos, átomos de carbonos en cadenas alifáticas, átomos aromáticos (anillos aromáticos), entre otros. Adicionalmente los índices cuadráticos estocásticos, sqk(x) presentan las mismas propiedades descritas para sus homólogos no estocásticos y se derivan de estos. Es decir, los k-ésimos índices cuadráticos estocásticos totales y locales se calculan según la misma invariante definida en la ecuación 1.1, pero usando la matriz estocástica de adyacencia entre átomos del pseudografo molecular, Sk(G), como matriz de la forma cuadrática. Sk(G) puede ser obtenida directamente de Mk(G). Los elementos ksij se definen como se muestra en la ecuación 5.30: k k. s ij =. k. aij. SUM i. =. k. aij. k. δi. (1.7). donde kaij son los elementos de la k-ésima potencia de M, y kSUMi es la suma de la fila iésima de Mk o grado del vértice de orden k del átomo i, kδ i . Esta transformación normaliza cada fila de la matriz original y por tanto, sus k-ésimos elementos constituyen las probabilidades de transición con las cuales un electrón se mueve de un átomo i a otro j en un período de tiempo discreto tk.. En la columna derecha de la Tabla 1.1 se muestra, a modo de ejemplo, las matrices estocásticas de orden 0-2 para la molécula del 2-formil-6metilbenzonitrilo. Nótese que los k-ésimos elementos sij toman en consideración la información de la topología molecular en k pasos a través de todo el esqueleto covalente. Así por ejemplo, los valores de 2sij pueden distinguir entre las diferentes formas híbridas de cada átomo. En este sentido, en la Tabla 1.1 (columna derecha) puede observarse que los electrones tienen una mayor probabilidad de regresar a un átomo de nitrógeno sp [p(N10) = 0.75] que a un átomo de nitrógeno sp2 [p(N6) = 0.33] en t2 (k = 2). Un comportamiento similar puede observarse entre los diferentes “estados híbridos” de los átomos de carbono en la molécula 2-formil-6-metilbenzonitrilo (ver Tabla 1.1): Csp3 [p(C11) = 0.25]; Csp2 [p(C2) = 0.625]; Csp2arom [p(C3) = 0.285, p(C4) = 0.3, p(C5) = 0.33, p(C7) = 0.33, p(C8) = 0.25]; y Csp [p(C9) = 0.769]. Esto es un resultado lógico si tomamos en cuenta las propiedades Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(22) Revisión Bibliográfica. 14. electrónicas (por ejemplo su escala de electronegatividad) de cada una de las diferentes hibridaciones de estos átomos.. 1.3. Métodos Estadísticos (Quimiométricos) en el Diseño Molecular 1.3.1. Introducción a los Métodos Quimiométricos en el Diseño Molecular. Los estudios QSAR constituyen un enfoque que permite entender como la variación estructural afecta la propiedad/actividad biológica de un conjunto de compuestos. En estos estudios, los descriptores moleculares (X) se correlacionan con una variable respuesta (Y). Es decir, este análisis puede definirse como una aplicación de métodos matemáticos y estadísticos al problema de encontrar una ecuación empírica de la forma Yi = fi(X1, X2, ...Xn), donde Yi son las propiedades y/o actividades biológicas de la molécula, y X1, X2, ...Xn son propiedades estructurales experimentales o calculadas (descriptores moleculares) de los compuestos. En este sentido, cada compuesto puede representarse como un punto en un espacio multidimensional, en los cuales los descriptores X1, X2, ...Xn son coordenadas independientes del compuesto. El objetivo más usual de este análisis es incrementar el entendimiento del sistema biológico bajo investigación o predecir la propiedad estudiada a un objeto (compuesto) no utilizado en la obtención del modelo.. 1.3.2. Quimiometría.. El término quimiometría, surgió en la década del 70 y se define como la disciplina química que utiliza métodos estadísticos y matemáticos para seleccionar y optimizar los métodos analíticos y preparativos, así como procedimientos para el análisis e interpretación de los datos (70).. 1.3.3 Metodología general empleada en el los estudios QSAR.. Los principios de la metodología QSAR pueden describirse mediante los siguientes pasos comunes (70): 1) Formulación del problema, se determina el objeto de análisis y el nivel de información requerido, 2) Parametrización cuantitativa de la estructura molecular de los compuestos químicos orgánicos/secuencia de biopolímeros, 3) Medición de la propiedad de interés (‘efectos biológicos’), 4) Escoger el tipo de modelo QSAR que se va a desarrollar, 5) Selección de los compuestos (diseño estadístico de la serie), 6) Análisis matemático de los Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(23) Revisión Bibliográfica. 15. datos y Validación interna y externa de los modelos obtenidos, 7) Interpretación de los resultados y Aplicación de los modelos desarrollados al diseño/descubrimiento de un nuevo compuesto líder, desarrollando procedimientos de tamizaje virtuales. Sin embargo, el desarrollo de cualquier QSAR es un ciclo interactivo.. 1.3.4. Regresión lineal múltiple (RLM).. La RLM estudia las relaciones entre una variable dependiente y un conjunto de variables independientes. Así mismo, la regresión múltiple remite a la correlación múltiple, que se representa por R. Es decir, la correlación múltiple analiza la relación entre una serie de variables independientes o predictores (X1, X2, ..., Xk), considerados conjuntamente, con una variable dependiente o criterio. Sus fundamentos se hallan en la correlación de Pearson.(71) La recta de regresión múltiple tiene la siguiente forma: Y = a + b1 X1 + b2 X2 +...+ bk Xk. (1.8). siendo ‘a’ un valor constante. Como puede observarse, la RLM puede utilizarse en la predicción de los valores de la variable dependiente, en base a una combinación de variables independientes.. 1.3.4.1. Principio de la parsimonia para seleccionar el número optimo de variables.. La R2 aumenta en la medida en que se añaden variables a la ecuación; pero a partir de cierto punto el incremento de R2 para cada nueva variable que se añade, es insignificante. Un buen modelo no debe presentar ni demasiadas variables, ni debe olvidar las que sean verdaderamente relevantes. Es decir, debe cumplir el principio de la parsimonia, según el cual un fenómeno debe ser descrito con el número mínimo de elementos posibles. Diversos procedimientos se han propuesto para seleccionar el número óptimo de variables a incluir en la ecuación, como por ejemplo la ‘forward selection’, ‘backward elimination; y ‘stepwise selection’ (72). Este último método es el más utilizado (en combinación con los dos anteriores) y sigue un proceso de selección de variables paso a paso. 1.3.4.2. Incremento de R2 y correlación parcial.. Se llama incremento de R2 a una estimación de la importancia relativa que tiene la variable que acaba de entrar en este paso para predecir el criterio. El incremento de R2 viene dado por: Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(24) Revisión Bibliográfica. 16. Rc2 = R 2 − Ri2. (1.9). donde Ri2 es el coeficiente de correlación múltiple al cuadrado cuando todas las variables, excepto la i (la que acaba de entrar en este paso), están incluidas en la ecuación. Por lo tanto, la Ri2 en un paso determinado coincide con la R2 del paso anterior. Un coeficiente Ri2 alto significa que esta variable proporciona información importante que no está contenida en las otras variables.. 1.3.4.3. Análisis de la varianza.. El ANOVA (ANalysis Of VAriance) sirve para comprobar la hipótesis de que R2 = 0. La variabilidad total de la variable dependiente se divide entre la parte atribuible a la regresión y la parte residual. La distancia de un punto cualquiera Yi a la Y se sub-divide en dos partes:(71). (. )(. Yi − Y = Yi − Yi + Yi − Y. ). (1.10). siendo Yi el valor predicho por la ecuación de predicción. El valor Yi − Yi , denominado residual de la regresión sería cero si la recta pasase exactamente por encima del punto Yi. El otro valor, Yi − Y , corresponde a la distancia explicada por la regresión y representa el aumento en la estimación de Yi mediante la recta de regresión. En el ANOVA, F viene dada por: F=. MC regresion MC residual. (1.11). Esta F sigue una distribución F de Snedecor con grados de libertad v1 = υ, v2 = n- υ -1; siendo υ el número de variables de la ecuación. La media cuadrática (MC) se obtiene dividiendo la suma de cuadrados por los grados de libertad. La F sirve para comprobar si el modelo de regresión se ajusta a los datos y permite evaluar si se rechaza la hipótesis nula, según la cual, R2 = 0. Es interesante observar, que si el modelo se ajusta a los datos, el coeficiente de determinación (R2) se puede calcular a partir de las suma de cuadrados (SC) del ANOVA mediante: R2 = 1−. SC residual SCtotal. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata. (1.12).
(25) Revisión Bibliográfica. 17. 1.3.4.4. Importancia de la tolerancia en la RLM.. La tolerancia es una medida del grado de asociación lineal entre las variables independientes (73). Para la variable i, la tolerancia es igual a 1- Ri2 , donde Ri2 es la correlación múltiple al cuadrado entre la variable i considerada como variable dependiente y las demás variables independientes. Valores bajos en la tolerancia, indican que la variable i puede ser considerada como una combinación lineal de las otras variables independientes. Por tanto, la tolerancia de una variable, en un paso cualquiera del análisis ‘stepwise’, es la proporción de su varianza intra-grupo no explicada por otras variables del análisis.. 1.3.5. Multicolinealidad entre variables con el uso de RLM.. El término ‘multicolinealidad’ se utiliza para describir la situación en que un gran número de descriptores moleculares están altamente intercorrelacionados. Las variables que se aproximan a ser una combinación lineal de las otras, se denominan multicolineales o colineales (71-74). Una ‘multicolinealidad’ alta, produce errores estándares altos en los coeficientes de regresión y dificulta estimar la importancia relativa de los descriptores en el modelo, lo cual afecta la interpretación de las actividades modeladas en términos estructurales. La importancia relativa puede determinarse al valorar el incremento en la R, cuando se añade una variable a la ecuación que ya contiene las demás variables ( Ri2 ). El método más utilizado para detectar la existencia de variables colineales es obtener una matriz de correlaciones entre los descriptores moleculares. Uno de los métodos más utilizados para detectar la interdependencia entre variables, es la tolerancia. Problemas con la redundancia de la información y la colinealidad, han sido ilustrados con el uso de ITs, tales como los índices de conectividad molecular (75, 76). El nivel aceptable de colinealidad es algo subjetivo y en ese sentido se ha reportado que coeficientes de correlación entre las variables aceptables están en el rango de 0.4-0.9 (77).. 1.3.6. Compuestos ‘outliers’ y técnicas para la selección de los mismos.. Los ‘outliers’ son puntos que se desvían significativamente del modelo encontrado (no se ajustan al modelo) o son pobremente predichos por estos, afectando los parámetros estadísticos del mismo (78). Generalmente, la identificación de ‘outliers’ busca un Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(26) Revisión Bibliográfica. 18. mejoramiento cualitativo del modelo. Un buen ejemplo ha sido mostrado por Cronin y col. en la modelación de la toxicidad de compuestos carbonílicos alifáticos para T. Pyriformis (77). En este estudio, para un total de 140 compuestos solo se obtuvo un moderado ajuste estadístico (R2 = 0.753). Sin embargo, al remover cinco outliers R2 aumentó hasta 0.853 (77). Existen varias técnicas para detectar la presencia de ‘outliers’, tales como: los análisis de los residuales estandarizados, los residuales studentizados, el método de Leverage, la estadística DFITS, la distancia de Cook y el método de dejar “varios” fuera (74).. 1.3.7. Validación estadística de los modelos QSAR.. El enfoque convencional adoptado en los análisis QSAR, basado en la RLM, es considerar el parámetro R2 (‘varianza explicada’), R y s. Las variables como R2 varían entre 0 y 1, donde 1 significa un modelo perfecto (explica el 100% de la variable respuesta, Y) y 0 un modelo sin ningún poder de explicación. Entonces un alto valor de R2 y una baja s, son condiciones necesarias para la validez del modelo RLM. O sea, como en ANOVA la validez viene dada solo por el ensayo F, si varios modelos pasan esta prueba, el de mayor R2 y/o menor s será el mejor modelo encontrado. Cuatro herramientas pueden ser utilizadas para acceder a la validación de los modelos QSAR obtenidos por RLM y la mayoría de estas pueden también extrapolarse a la validación de los modelos obtenidos con el Análisis Discriminante Lineal (ADL) (79): 1) Aleatorización de la variable respuesta (Y- Randomización), 2) validaciones cruzadas, 3) división de la data de compuestos en serie de entrenamiento (SE) y en serie de predicción (SP) y 4) confirmación del poder predictivo utilizando SP ‘externas’. A continuación desarrollaremos brevemente solo los puntos referidos a los enfoques de validación de los modelos que son de nuestro interés.. 1.3.7.1. Validación interna de los modelos (Validaciones cruzadas).. La validación cruzada (VC) opera haciendo un número (G) de reducidas modificaciones al conjunto de compuestos de la data original y entonces calcula la precisión de las predicciones de cada uno de los resultados de los modelos(80, 81). Entonces, la VC crea G conjuntos de datos modificados tomando uno o más grupos de compuestos de los datos, en donde cada observación (compuestos) se toma una vez, sobre el número total de ciclos de VC, G. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(27) Revisión Bibliográfica. 19. Entonces el modelo es ajustado a los nuevos datos, dejando la parte omitida fuera, y estos se evalúan en el modelo para computar las predicciones de los compuestos que fueron excluidos. Este procedimiento se repite para cada conjunto de datos modificados. El poder predictivo del modelo puede expresarse como q2, el cual ha sido denominado como la ‘varianza predictiva’ o la ‘varianza de la validación cruzada’, la cual es igual a (1PRESS/SSY), o sea que puede ser calculado acorde a la siguiente fórmula: ∧ ⎛ ⎞ ⎜ yi − yi ⎟ ∑ ⎠ q2 = 1− ⎝ 2 ∑ yi − y. (. 2. ). (1.13). ∧. donde yi , yi y y es la actividad observada, estimada y el promedio (media) para el i-ésimo compuesto, respectivamente. Cuando se utiliza un solo compuesto en cada grupo de VC (lo cual da N grupos), el procedimiento se conoce como “dejando uno fuera” y sus siglas en ingles son LOO (acrónimo de Leave-One-Out). No obstante, Shao ha mostrado que desde el punto teórico y práctico, el procedimiento de dejar ‘varios’ fuera (LSO; Leave-Several-Out) es preferible al LOO.(82) Este resultado puede entenderse al considerar que sucede cuando el número de compuesto, N, se incrementa. La técnica de LSO siempre deja fuera una porción de los datos creando una perturbación constante en la estructura de los datos. Wold y Eriksson recomiendan utilizar un valor de G alrededor de siete, al utilizar el procedimiento de VC.(79) El promedio de la media de los errores en valores absolutos, MAE (Mean Absolute Error), para cada uno de los grupos dejados fuera puede ser usado como un criterio significativo para acceder a la calidad del modelo (83).. 1.3.7.2. Validación de los modelos empleando de una serie de predicción externa.. Usualmente el procedimiento de VC es denominado validación interna, porque todos los compuestos que considera pertenecen a los mismos datos originales. Sin embargo, cuando el número de compuestos es grande, estos pueden dividirse en dos conjuntos separados de entrenamiento o calibración y otro conjunto de validación o predicción (validación externa). Muchos investigadores consideran a los altos valores de q2 (q2 > 0.5) como un indicador del poder predictivo de un modelo QSAR.(84-88) En contraste con estas especulaciones, varios investigadores han demostrado que la “única” condición necesaria y suficiente para poder Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(28) Revisión Bibliográfica. 20. estimar el verdadero poder predictivo de un modelo es comparar los valores predichos y observados de una extensa (suficientemente larga) SP externa.(89-92). 1.3.8. Análisis de conglomerados (análisis de clusters).. El análisis de conglomerados (en inglés cluster) es un técnica multivariante que permite agrupar los caso o variables de un archivo de dataos en función del parecido o similaridad existente entre ellos. Como técnica de agrupación de casos el AC es similar al análisis discriminante. Sin embargo, mientras el análisis discriminante efectúa la clasificación tomando como referencia un criterio o variable dependiente (los grupos de clasificación), el AC permite detectar el numero optimo de grupos y su composición únicamente a partir de la similaridad existente entre los casos; además el AC no asume ninguna distribución especifica para las variables (74). Un método muy utilizado es el conocido como de k medias, que es un método de agrupación de casos que se basa en las distancias existentes entre ellos en un conjunto de variables. El mismo agrupa los casos según su cercanía al centroide (centro multivariado del cluster) del cluster más cercano; así continúa la lectura secuencial del archivo de datos asignando cada caso al centroide más cercano y actualizando el valor de los centroides a medida que se incorporan nuevos casos. El proceso termina cuando todos los casos han sido asignados a uno de los k clusters.. 1.4. Regulaciones de los Métodos QSAR.. La Organización para Cooperación Económica y Desarrollo (OECD) es una organización intergubernamental en la cual los representantes de 30 países industrializados en Norteamérica, Europa y la Asia y región Pacífica, así como la Comisión europea, se encuentran para coordinar y armonizar políticas, hablar de cuestiones de interés mutuo, y trabajar juntos para responder a problemas internacionales. La mayor parte del trabajo de OECD es realizado por más de 200 comités especializados y grupos de trabajo formados por delegados de países miembros. Los comités y los grupos de trabajo son coordinados desde la secretaría de la organización, localizada en París, Francia, que es organizada en diferentes secciones y divisiones. La división de Medioambiente, Salud y Seguridad (EHS por sus siglas en ingles) publica documentos gratuitos en diez series diferentes: Pruebas y Análisis; Buenas Práctica de Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(29) Revisión Bibliográfica. 21. Laboratorio y Conformidad en Monitoreo; Pesticidas y Biocidas; Manejos de Riesgo; Armonización Regulatoria en Biotecnología; Seguridad de Nuevas Comidas y Alimentos; Accidentes Químicos; Liberación de Contaminantes y Registros de Transferencia; Documentos de Guías de Emisión; y la Seguridad en la Fabricación de Nanomateriales. Más información sobre La división de Medioambiente, Salud y Seguridad y publicaciones EHS está disponible en el sitio de Web de la OECD (http://www.oecd.org/ehs/). Los grupos de trabajo de OECD en QSAR y la Reunión Conjunta han concurrido que la validación de estudios QSAR para objetivos reguladores son mejor realizados por las autoridades reguladoras de los países miembros. En el futuro previsible, la aceptación de estudios QSAR como una fuente de alternativa de datos (sin necesidad de realizar pruebas de laboratorio) para la toma de decisiones estará basada en la fiabilidad y la transparencia de un específico QSAR dentro de un contexto regulador específico. Por consiguiente, principios de validación para un modelo QSAR son queridos para dirigir a las agencias reguladoras en la evaluación e interpretación de los mismos durante procesos de toma de decisión específicos en un nivel más alto que los criterios que solo solían juzgar la validez estadística. Sin embargo, la transparencia de la interpretación estadística de un QSAR es la piedra angular para el uso confiable con carácter regulatorio. Como la aceptación modelos QSAR crece para llenar la necesidad de datos, es de esperar que la validez estadística permanezca como un aspecto crucial mientras la interpretación mecanística y explicación de los resultados de los modelos será requerida siempre que sea posible. Los 5 principios de la OECD que debe cumplir un modelo QSAR para ser aceptado con fines regulatorios son: 1. un punto de medición definido; 2. un algoritmo inequívoco; 3. un dominio de aplicación definido; 4. apropiadas medidas de calidad de ajuste, robustez y predictibilidad; 5. una interpretación mecanística de ser posible Un problema crucial de cualquier estudio QSAR es la identificación del dominio de aplicación (DA) de un modelo de clasificación o de regresión. Pues, en efecto solo son validas las predicciones para aquellos compuestos que estén dentro del dominio de aplicación. El DA es aquella región teórica en el espacio químico, definido por los descriptores del modelos y la respuesta modelada, y por todo esto a su vez por la naturaleza de los compuestos de la serie de Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(30) Revisión Bibliográfica. 22. entrenamiento, representado en cada modelo por descriptores moleculares específicos. Se puede decir por lo tanto, que el DA de un modelo QSAR en “el rango dentro del cual es tolerada una nueva molécula (93). El dominio de aplicación de un modelo QSAR es la respuesta y el espacio químico estructural en el cual el modelo realiza predicciones con una adecuada fiabilidad. Por lo que no se puede pretender extrapolar el uso de los modelos QSAR para aquellos tipos de compuestos que están fuera del dominio de aplicación (94). Para la RLM, un enfoque muy empleado es el leverage (h), una medida basada en distancias. A través del enfoque del leverage (95) es posible verificar si un nuevo compuesto esta dentro del dominio de aplicación del modelo, el valor de h (95) de un compuesto mide la influencia de este en el modelo. Los valores de leverage pueden ser calculados para los compuestos de la SE y nuevos compuestos. En el primer caso, son útiles para encontrar aquellos compuestos que influencian los parámetros del modelo, y que lo convierten en un modelo inestable. En el segundo caso, son útiles para chequear el dominio de aplicación del modelo (93, 96). El leverage crítico es el valor de corte realizado para el modelo en una base de datos. Valores por encima de este leverage crítico son considerados no fiables. Solo las estructuras químicas predichas que pertenezcan al dominio de aplicación deben tenerse en cuenta con gran nivel de fiabilidad.. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(31) Materiales y Método. 23. MATERIALES Y MÉTODO 2.1 Obtención de la base de datos de toxicidad acuática sobre (Poecilia. Reticulata). Los compuestos con sus respectivos valores experimentales de concentración letal media (LC50, concentración en mg/L que produce la muerte del 50% de los animales empleados en el ensayo luego de 96 horas de exposición al compuesto) para el pececillo (Poecilia. Reticulata) fueron recopilados de varios artículos publicados (1, 57-59, 97-100). Se empleo como variable dependiente la transformación logarítmica para los valores de LC50 de igual manera que en previos estudios. La recopilación bibliográfica nos permitió construir una base de datos de 300 compuestos de los cuales 229 se emplearon para construir la serie de entrenamiento (SE) y la serie de predicción (SP) 71. Se realizó un análisis de conglomerados a los compuestos que se destinaron para construir la serie de entrenamiento y la serie de predicción con el objetivo de garantizar representatividad estructural en ambas series. La compilación de datos de toxicidad sobre (Poecilia. Reticulata) a partir de reportes bibliográficos recientes (1, 57-59, 97-100), nos permite desarrollar un modelo con un dominio de aplicación mayor que los previamente reportados, lo cual es una característica deseable en este tipo de modelos.. 2.2 Método Computacional. TOMOCOMD-CARDD software. Nuestro grupo de investigación ha introducido recientemente un nuevo programa interactivo para el diseño molecular e investigaciones químio-bioinformáticas. Este programa se denomina TOMOCOMD (acrónimo de TOpological MOlecular COMputer Design) (101) y que ha sido desarrollado en la Universidad Central ‘Marta Abreu’ de Las Villas. En este paquete computacional se ha implementado el cálculo de varias familias de descriptores moleculares, basados en representaciones vectoriales y matriciales de la estructura molecular. El programa está compuesto por cuatro sub-programas, cada uno de ellos con un módulo de visualización y otro de cálculo. Los sub-programas son los siguientes: CARDD (ComputedAided ‘Rational’ Drug Design), CAMPS (Computed-Aided Modeling in Protein Science), CANAR (Computed-Aided Nucleic Acid Research) y CABPD (Computed-Aided BioPolymers Docking), por sus siglas en ingles. En esta tesis se han utilizado los cálculos. obtenidos con el primer sub-programa. Este ‘software’ fue desarrollado basado en una interfase amigable con el usuario, el cual no tiene que dominar a priori ningún conocimiento Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
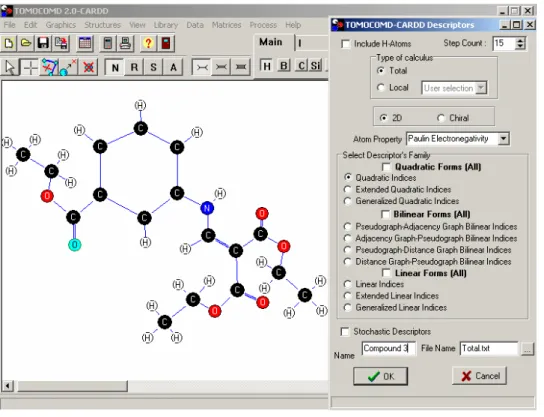
(32) Materiales y Método. 24. de programación computacional. Específicamente, en este trabajo se han utilizado los índices cuadráticos totales y locales de la matriz de adyacencia entre vértices del ‘grafo’ usado para la representación de la estructura química de las moléculas o bio-macromoléculas. Estos han sido empleados en varios estudios QSAR, utilizando el ADL y la RLM como técnicas estadísticas. En la Figura 2.1 se muestra la interfase gráfica del programa de cálculo CARDD.. Los principales pasos para desarrollar un estudio QSAR utilizando el enfoque TOMOCOMD, se resumen brevemente a continuación:. 1. Representar el ‘grafo’ molecular de cada una de las moléculas de la base de datos a analizar, usando el módulo de dibujo del software. Este procedimiento se lleva a cabo seleccionando el átomo deseado perteneciente a diferentes grupos de la tabla periódica en el momento de representar las moléculas. 2. Usar un ‘peso’ (etiqueta) apropiado de átomo, con el propósito de diferenciar cada tipo de átomo en la molécula. 3. Computar los índices cuadráticos totales y locales de la matriz de adyacencia entre vértices del ‘grafo’ que haya sido utilizado para la representación de la estructura química de las moléculas. Este paquete computacional genera una tabla en la cual las filas corresponden a los compuestos (casos) y las columnas a los índices moleculares calculados.. A Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(33) Materiales y Método. 25. B Figura 2.1. TOMOCOMD-CARDD Software: A, Ventana para seleccionar el módulo de trabajo. B, Interfase gráfica del sub-programa de diseño “in silico” de fármacos. 4. Encontrar una o varias ecuaciones QSAR usando técnicas estadísticas adecuadas, tales como RLM, ADL, entre otras. Es decir, se encuentra una relación cuantitativa entre una actividad A y la estructura química codificada con los descriptores calculados. En este caso, la ecuación obtenida debe tomar la siguiente apariencia: A = a0q0(x) + a1q1(x) + a2q2(x) +….+ akqk(x) + c. (2.1). donde A es la medida de la actividad, qk(x) [o qkL(x)] es el k-ésimo índice cuadrático total o local, y los términos ak’s son los coeficientes obtenidos por el análisis estadístico multivariable. 5. Probar la robustez y demostrar el poder predictivo de las ecuaciones QSAR obtenidas usando procedimientos de validación interna y externa.. 2.3. Análisis Estadístico de los Datos. Análisis de RLM. Los modelos QSAR-RLM se obtuvieron con el paquete de programas estadísticos STATISTICA (74). El método de selección de variables utilizado fue el de “pasos hacia delante (‘forward stepwise’). En todos los casos el estadístico F y la tolerancia se usaron Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
(34) Materiales y Método. 26. para el control del proceder de selección. En este sentido, la colinealidad entre variables fue examinada utilizando las matrices de correlaciones entre las variables incluidas en el modelo. Siempre se utilizó, por defecto, como valor mínimo aceptable de tolerancia 0.01. Los estadísticos usados para evaluar la calidad del modelo y el ajuste del mismo a los datos experimentales fueron el coeficiente de correlación múltiple (R) y el cuadrado de su valor (R2, coeficiente de determinación). La desviación estándar (s) y la F de Fischer (y/o el nivel de significación del modelo y de cada variable, p ≤ 0.05) también se tuvieron en cuenta a la hora del ajuste y selección de los modelos desarrollados. La calidad predictiva de las ecuaciones desarrolladas se evaluó utilizando los estadísticos del proceso de validación cruzada (VC, validación interna). En este sentido, fueron aplicados el procedimiento de VC, LOO y LGO. Además, en cada caso se utilizaron series de validación externas, para medir la estabilidad y el poder predictivo de los modelos QSAR obtenidos.. Yunier Perera Sardiñas−Predicción de la toxicidad acuática de compuestos orgánicos en Poecilia reticulata.
Figure


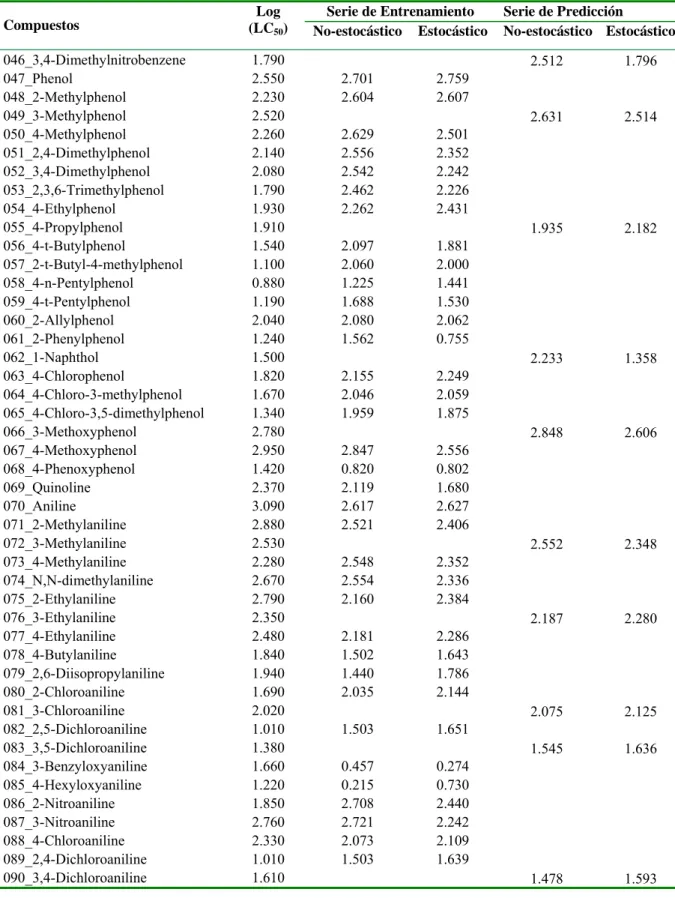
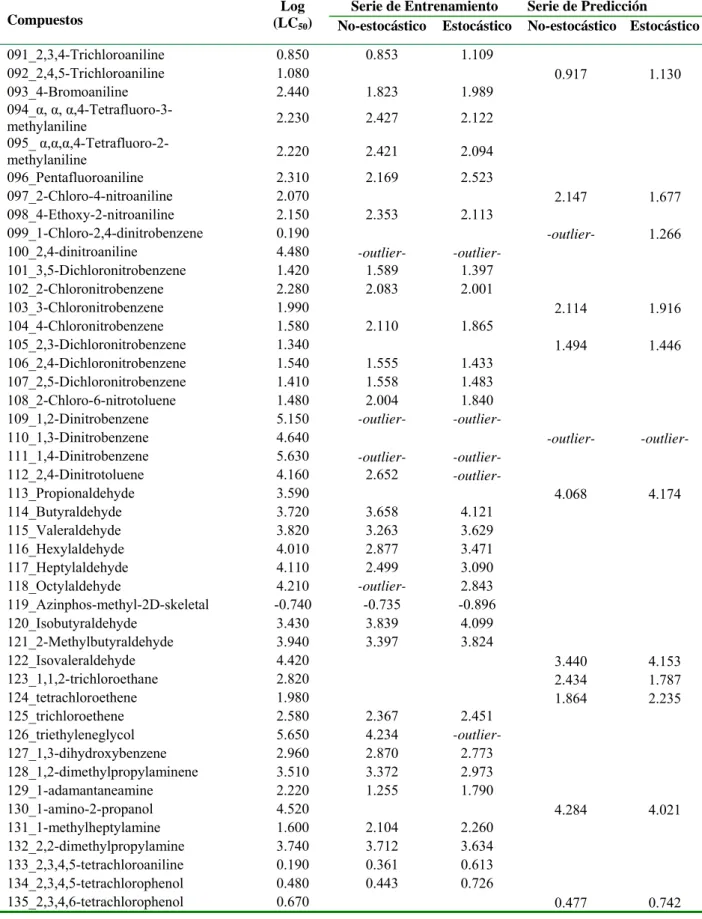
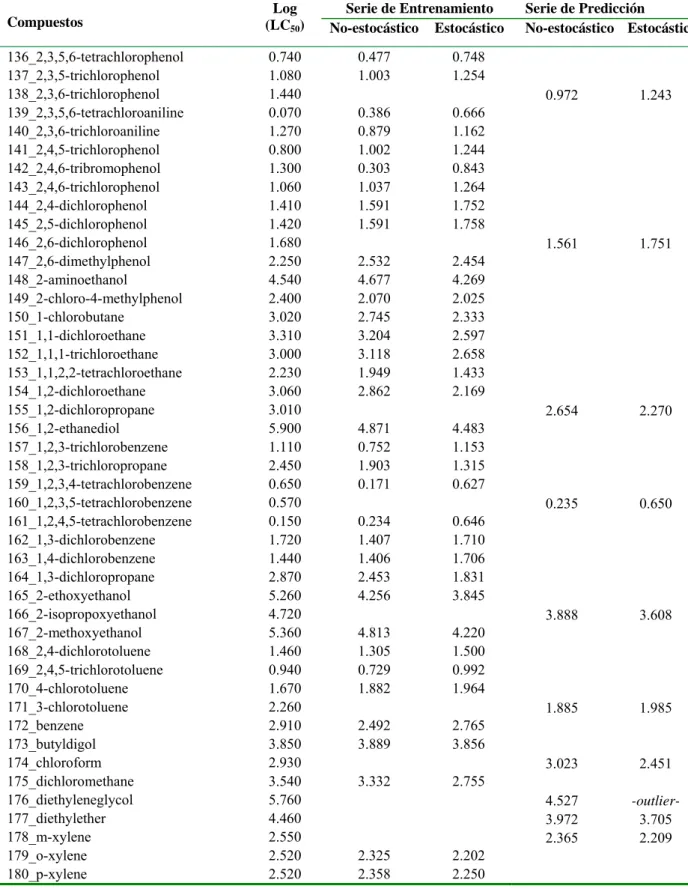
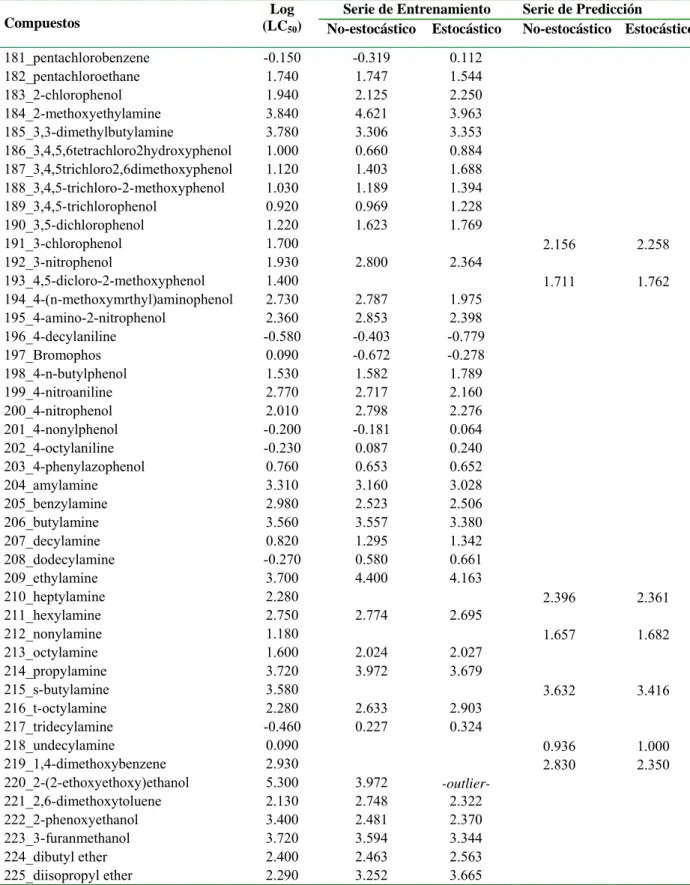
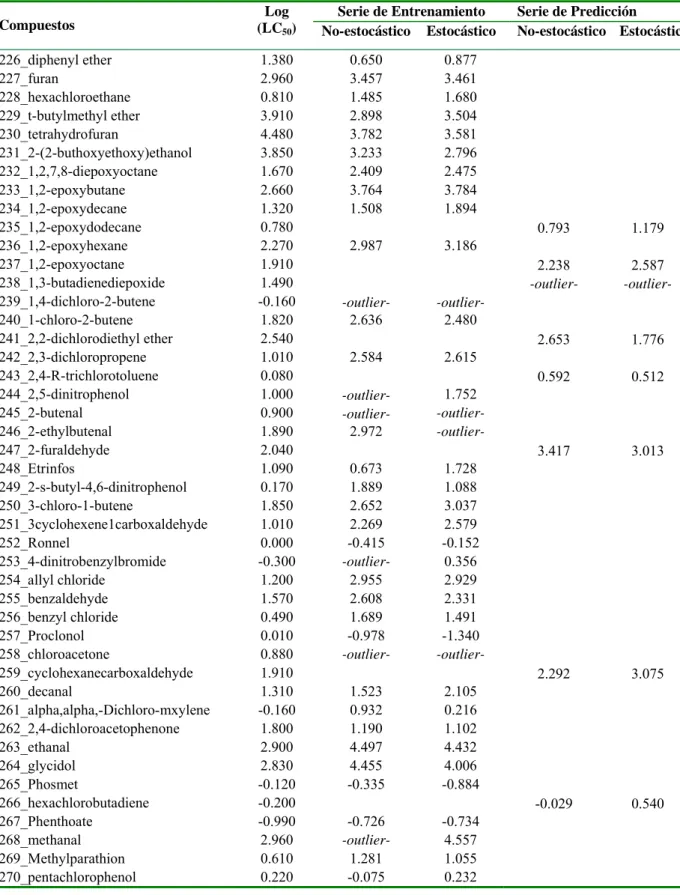
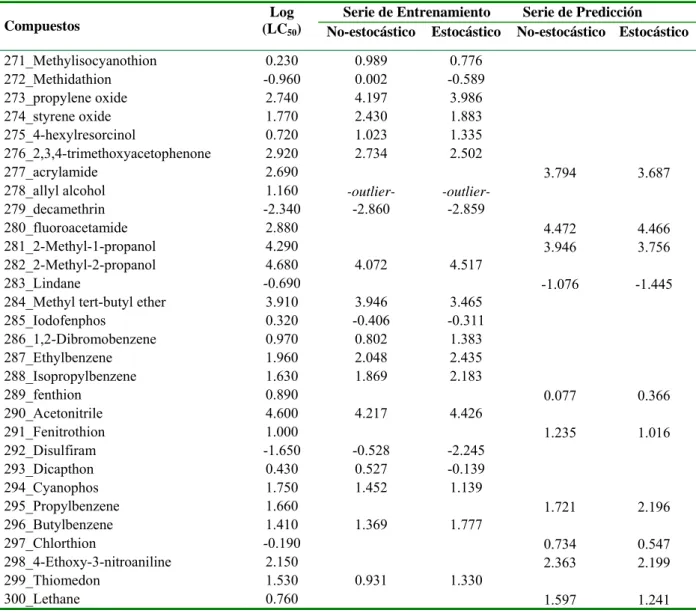
![Table 3.1. Valores experimentales y predichos [Log (LC 50 )] para la serie de entrenamiento y para la serie de predicción](https://thumb-us.123doks.com/thumbv2/123dok_es/7372691.463349/37.892.110.786.198.1110/table-valores-experimentales-predichos-serie-entrenamiento-serie-predicción.webp)
+7
Documento similar
