Derivada del Grafo Molecular como una Novedosa Vía para la Generación de Descriptores Moleculares 2D/3D: Teoría y Aplicación QSPR
65
0
0
Texto completo
(2) Âj{xÇ çÉâ ÜxtÄÄç ãtÇà áÉÅxà{|Çz àÉ {tÑÑxÇ? à{x. ã{ÉÄx âÇ|äxÜáx vÉÇáÑ|Üxá áÉ à{tà çÉâÜ ã|á{ vÉÅxá àÜâxÊ Paulo Coelho, “The Alchemist”.
(3) En la vida de cualquier ser humano existen personas sin las cuales, ese tránsito extasiado y efímero por el mundo no tiene sentido. Yo no soy la excepción y por eso quiero hacer un merecido y sencillo homenaje a las principales personas que conspiran (o alguna vez lo hicieron) para que yo tenga sueños realizables. A todas ellas quiero dedicar el fruto del árbol, que algunos de ellos sembraron en mi y otros cultivaron, para que yo pudiera alimentarme de su sabia maravillosa que no es más que amor, ciencia y poesía. Dedico esta tesis a: Mi Madre Tania E. Santiago Carlos Gracias por existir y ser fuente de inspiración para todo lo que hago en la vida. Gracias por los sacrificios y desvelos que has tenido para no preocuparme y facilitarme la vida. Espero nunca defraudarte y que siempre estés orgullosa de mi. Gracias por todo… A la memoria de tres mujeres excepcionales: Mi tía Isabel Mis abuelas Ángela y Angelina Que hicieron del niño un joven de bien. Me brindaron todo el cariño y la seguridad a la que un ser humano puede aspirar y aun viven conmigo guiando cada uno de mis pasos en la vida, pues sus enseñanzas amanecen conmigo en todas las auroras de mi vida. A mis Hermanos: Carlos E. Ventura Santiago Rayner Martínez Borroso Quisiera ser para ellos un ejemplo a seguir. Ojalá y siempre perciban lo importante que son para mí. Al mejor de mis mejores amigos: Carlos A. Ventura Torres Gracias por escuchar mis problemas y la mayor parte de las veces solucionarlos. Gracias por quererme como a un hijo. Espero no fallarte nunca. A mi novia: Gretel León Platero Por su paciencia, su amor y sobre todo su apoyo, sin el cual no habría superado muchos momentos difíciles de mi vida académica y personal. A mi papá: Oscar Martínez Borges Para que se sienta siempre orgulloso de mí. A mi abuelo: Cesar Santiago Suástegui Por su apoyo siempre que lo he necesitado y sus sabios consejos. A mis tías y tío: Nancy Santiago Carlos, Yuliet Martínez Mena y Vladimir Santiago Carlos Por su cariño incondicional, su presencia y apoyo en todos los momentos de mi vida. A otras personas importantes: Mis primas y primos Mantengamos siempre la misma unión y que el cariño y la confianza siempre medien entre nosotros como hasta ahora. A mis amigos: Yoan Hidalgo Rosa, Alexey Díaz Gómez y Ariel García Tamayo A ellos también gracias por ser como hermanos para mí y siempre comportarse como tal. Por estar a mi lado siempre que los he necesitado, en los buenos y malos momentos, a los tres muchas gracias..
(4) Muchas personas han sido cómplices del desarrollo de este trabajo y de mi vida científica, estudiantil y académica. A todos “MUCHAS GRACIAS”. Muchas Gracias a: Mis Tutores: Dr. Yovani Marrero Ponce Por brindarme su amistad y conocimientos, para guiarme en el apasionante mundo de la investigación científica. Por hacer de mí, un joven con gran amor por las ciencias y una incalculable curiosidad y deseos de vencer los distintos retos que pone el conocimiento científico en las mentes humanas. Dr. Enrique R. Molina Pérez Por su amistad y apoyo incondicional. Además de abrirme la puertas al hermoso mundo de las investigaciones en la Química Teórica y el Diseño de Fármacos. Mis Amigos y Compañeros de Estudios: Lisdelys Rodríguez, Yoslainy Echevarría, Manuel A. Treto, Luis M. Peralta, Yoan Hidalgo, Reinier Tumbarell. Por acompañarme en los buenos y malos momentos de mi vida universitaria. Por servir de sostén y contraparte a mis ideas, como yo lo he sido con ellos. Porque todos somos un pequeño pero muy unido grupo de hermanos(as) sin lazos sanguíneos. Mis profesores: Por el ejemplo y la dedicación con que depositaron en mí los más actuales y refinados conocimientos de Química y otras ciencias vitales para desempeñarme como un buen profesional. Mis compañeros del CAMD-BIR Unit: Por su ayuda y colaboración siempre que la he necesitado. Aun quedan muchas personas que han aportado su grano de arena para que este trabajo haya llegado a feliz término y este día sea realmente memorable para mí. No menciono nombres para no caer en la obligación de dar una prioridad o peor aun, olvidar a alguien. Todos ellos saben quienes son, a ustedes “mucha gracias”.. La gratitud es el más legítimo pago al esfuerzo ajeno, es reconocer que todo lo que somos, es la suma del sudor de los demás. Que un hombre solo no vale nada, y que la dependencia humana, además de necesaria, es hermosa. José Martí.
(5) SINTESIS En el presente trabajo se ha propuesto una novedosa metodología matemática para describir la estructura molecular obteniéndose así toda una nueva familia de descriptores moleculares topológicos. Este enfoque se basa en una representación matricial del grafo molecular y en el cálculo de las correspondientes derivadas del grafo para n-uplas de elementos, así como derivadas de orden superior y mixtas. En esta primera aplicación se utilizan las derivadas para duplas basadas en átomos (pares de átomos) a partir de las cuales se obtienen las correspondientes derivadas totales-locales para cada átomo que sirven de base para el cálculo de Normas de Minkowski y Geométricas de cada molécula. Estas normas permiten establecer correlaciones entre la estructura de los compuestos, con diferentes propiedades de los mismos (químicas, físicas, químico-físicas y biológicas, etc). Estos nuevos índices han sido definidos también teniendo en cuenta el orden y tipo de subgrafos utilizados para generar la matriz de Incidencia Generalizada “Q” (matriz utilizada en este enfoque para representar la topología molecular y aplicada por primera vez en la definición de un descriptor molecular), además de la posibilidad que brindan de ser aplicados tanto de forma total como local para átomos o agrupaciones atómicas específicas. Esta gama de posibilidades mencionadas nos abren las puertas a la creación de nuevas familias de descriptores moleculares, utilizando la derivada del grafo molecular, y nos permite contar con una nueva herramienta de utilidad práctica para la realización de estudios QSAR/QSPR/QSTR. Los descriptores a los cuales está dedicado este trabajo, fueron implementados en un programa desarrollado en MATLAB, que aunque esta en una fase inicial, permite al investigador realizar cálculos teóricos en un tiempo relativamente corto y con un bajo costo computacional. Esta primera aplicación ha demostrado, hasta el momento, que estos descriptores moleculares (DMs) son útiles para el diseño molecular y permiten obtener modelos matemáticos más sencillos, interpretables y robustos que muchos de los que han sido descritos en la literatura. En el futuro se desarrollarán otras aplicaciones de los nuevos índices aplicados a otros problemas más complejos de la química-médica actual. En este sentido, los nuevos índices de derivada se han utilizado para modelar varias propiedades químico-físicas de una serie de octanos, obteniéndose resultados satisfactorios para todas las propiedades modeladas. También se obtuvieron buenos modelos (R2 = 99.5 % y s = 2.24) en la descripción de la temperatura de ebullición de 28 alcoholes alifáticos, que han sido utilizados previamente por otros autores. Se realizó un estudio para conocer como se comportan determinados parámetros durante la descripción matemática de la estructura molecular y que nos ayudan a interpretar mejor los resultados obtenidos. Estos estudios se basaron en conocer: 1) mejor ponderación 2) mejor orden y 3) mejor norma, para la descripción de propiedades químico-físicas como la temperatura de ebullición de alcoholes alifáticos. Resultando la electronegatividad según la escala de Mulliken la mejor ponderación utilizada y el orden 1 el mejor orden para describir esta propiedad. Las Normas ofrecieron resultados muy semejantes todas. Además se desarrolló un proceso de interpretación con el objetivo de conocer el tipo de información químico-física que recoge esta novedosa metodología matemática durante la descripción estructural de las moléculas, comprobándose que existe una gran linealidad entre la información estérica y electrónica de las moléculas con esta descripción. El resultado de la comparación con otros métodos resultó muy satisfactorio. Esta y otras aplicaciones (todavía en proceso) validan hasta el momento la aplicación de este novedoso método teórico, para ser usado en el diseño “racional” automatizado de fármacos y otras aplicaciones de la química-física..
(6) GLOSARIO 2D 3D ANOVA CoMFA Fexp IDGM ITs LGO LNO LOO MAE MON NEM q2 QSAR QSTR QSPR R R2 s scv Te VC P. Δ Y (X ) P. XY. Bidimensional Tridimensional Análisis de Varianza Comparative Molecular Field Analysis Razón de Fisher experimental Índices de Derivada del Grafo Molecular Índices Topológicos Procedimiento de validación interna cruzada ‘leave-group-out’ Procedimiento de validación interna cruzada ‘leave-n-out’ Procedimiento de validación interna cruzada ‘leave-one-out’ Error medio absoluto Número motor de octanos Nueva Entidad Molecular Coeficiente de correlación al cuadrado del procedimiento de validación cruzada LOO Quantitative Structure Activity Relationships Quantitative Structure Toxicity Relationships Quantitative Structure Property Relationships Coeficiente de correlación Coeficiente de determinación o coeficiente de correlación al cuadrado Desviación estándar Desviación estándar del procedimiento de validación cruzada LOO Temperatura de ebullición Validación cruzada Derivada LT de orden “Y” para el átomo “X”, usando ponderación “P” Norma “p” de Minkoswki (p = 1,2,3 … n). Z P. ξY. Norma geométrica de orden “Y”, usando ponderación “P”.
(7) “No hay que pedirle precisión filosófica en el lenguaje que es lo que la educación añade al genio; pidámosle inspiración que es lo que da el genio” José Martí.
(8) 1. INTRODUCCIÓN Entre los aspectos más interesantes y útiles de la Química está su capacidad de crear nuevas moléculas desconocidas por el hombre, con enormes aplicaciones a distintas esferas de la vida humana. Actualmente existen mas de 26 millones de compuestos conocidos, y aunque un gran número de estos (>1, 000, 000) está disponible en diferentes bases de datos químicas, una gran cantidad de estos compuestos no ha encontrado aun aplicaciones farmacológicas, agroquímicas, industriales o de algún otro tipo. Los elevadísimos costos (más de 800 millones de dólares)1 que presentan los métodos tradicionales de ‘prueba y error’ para la obtención de Nuevas Entidades Moleculares (NEM) y su baja efectividad [se necesita ensayar sobre 10 mil compuestos, de los cuales sólo 10 pasaran los ensayos y solo uno llega a convertirse en medicamento útil en terapéutica],2, 3 han dirigido la atención de los investigadores hacia soluciones mucho más asequibles, rápidas, económicas y efectivas. Debe señalarse, además; que el tiempo transcurrido, desde que se sintetiza el principio activo hasta que se introduce en el mercado, es de 12 a 15 años y que 7 de cada 10 medicamentos no recupera su costo.3 Notemos que este tipo de ensayos experimentales no solo tienen altos costos en términos de recursos materiales, humanos y de tiempo, sino que debemos tener en cuenta también el aspecto de tipo ético que conlleva la investigación con animales y su posterior sacrificio. En todo caso, nuevos paradigmas para el descubrimiento molecular han sido introducidos recientemente, por las principales industrias farmacéuticas, basados en el uso de grandes bibliotecas de compuestos químicos y de sistemas robóticos para realizar ensayos biológicos.4, 5 De tal modo los sistemas HTS (acrónimo de High-Throughput Screening), permiten la síntesis y ensayo de miles de compuestos cada día pero representa un costo inalcanzable para la mayoría de los países.4, 6 Por todo lo antes mencionado, la industria Farmacéutica ha reorientado las estrategias de búsqueda hacia métodos que permitan la identificación y la optimización de nuevos compuestos líderes de un modo efectivo (en el menor tiempo posible y a un costo razonable). En este sentido, el enfoque de diseño/descubrimiento de fármacos asistido por computadoras ofrece una alternativa al mundo real de síntesis y evaluación.7 Este procedimiento engloba todas las técnicas asistidas por ordenadores usadas en el diseño, descubrimiento y optimización de compuestos con propiedades deseadas8 y ha jugado un rol fundamental en el desarrollo de un número de fármacos que están ahora en el mercado.9 Este tipo de estudio se basa en el uso de un mundo “virtual” de hipótesis, generadas por computadoras y probadas en la práctica. Este tipo de procedimiento ‘in sílico’ evita los procesos actuales de síntesis y bioensayos, los cuales se hacen solamente después de la exploración de los conceptos iniciales con modelos computacionales.7 Por tanto, podemos plantear que los estudios QSAR/QSPR/QSTR (siglas en inglés acrónimos de Quantitative Structure Activity/Property/Toxicity Relationships) se han convertido en una importante área de investigación en la química computacional y teórica.10, 11 Este tipo de estudios se encuentra en la intersección entre la biología, la química, la matemática y la computación, tienen dos objetivos fundamentales. El primero: brindar una vía para estimar, con un aceptable grado de precisión, la actividad/propiedad/toxicidad estudiada a nuevos compuestos. El segundo: es obtener una interpretación en términos estructurales de la actividad/propiedad/toxicidad estudiada. El paradigma enarbolado en los estudios QSAR/QSPR/QSTR (en lo adelante se utilizará solo el término QSAR) está relacionado con el hecho de que las propiedades físicas, físico-químicas, químicas, biológicas y toxicológicas de los compuestos orgánicos dependen en último termino de la estructura molecular.12, 13 Existen muchos parámetros químico-físicos de las moléculas que son una expresión cuantitativa de la estructura molecular, también dicha estructura puede ser descrita usando diferentes métodos y estrategias matemáticas. Estas descripciones de las moléculas son conocidas como índices o descriptores moleculares (DMs) y los resultados numéricos por ellos expresados pueden ser relacionados estadísticamente con determinadas propiedades (actividades) moleculares y en estudios de similitud/disimilitud molecular. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR.
(9) Capítulo 1. Introducción. En la actualidad, existe un gran número de descriptores moleculares que pueden ser usados en estudios QSAR.14 Los descriptores moleculares son ‘términos que caracterizan un aspecto específico de una molécula’ y contienen información estructural derivada de la representación estructural de las moléculas bajo estudio.8 La naturaleza de los descriptores, depende de cual haya sido el proceder utilizado para la definición de los mismos, pudiendo tener en cuenta rasgos topológicos (2D), geométricos (3D), electrónicos de las moléculas, etc. Algunos de estos descriptores sin embargo, tienen ‘más información’ de propiedades físico-químicas que de los rasgos estructurales de la molécula. Estos incluyen los basados en la determinación experimental de propiedades físico-químicas, tales como la mayoría de las constantes de los sustituyentes, hidrofóbicas, electrónicas y estéricas.15 En contraste, los llamados índices topológicos (ITs) tienen la información estructural contenida en una representación bidimensional de las moléculas (generalmente el grafo molecular con los átomos de hidrógenos suprimidos) sin considerar ningún rasgo físico-químico de las moléculas.16,. 17. La. mayoría de estos índices pueden considerarse como descriptores estructurales explícitos. Otro grupo de descriptores, llamados químico-cuánticos describen rasgos electrónicos de las moléculas basados en el uso de la función de onda molecular. Los descriptores geométricos tienen información de los rasgos estructurales 3D de las moléculas en una vía explícita,18 tales como distancia y ángulos de enlaces o en una vía implícita, en forma de descriptores topográficos.19 Los ITs han comenzado a ocupar un lugar importante dentro del conjunto de descriptores moleculares utilizados en los estudios QSAR, siendo probablemente el diseño/descubrimiento de nuevos compuestos bioactivos, una de las más activas áreas de investigación donde se aplican estos descriptores a problemas biológicos.13, 17, 20-39 Con el objetivo de desarrollar procesos mas ‘racionales’ de investigación en el diseño de fármacos o en la modelación de otras propiedades de interés, la definición de nuevos descriptores moleculares40 es un campo prometedor en la química médica actual. En nuestra universidad existen diferentes grupos de investigación que han desempeñado un papel relevante en ese sentido, los cuales dirigen sus investigaciones al desarrollo de nuevos principios activos para la industria farmacéutica en las ramas de la medicina veterinaria, humana y la sanidad vegetal. Uno de estos grupos ha sido el Grupo de Modelación Molecular y Diseño de Fármacos (MSDD, por sus siglas en inglés) del Centro de Bioactivos Químicos (CBQ); en este grupo existe una tradición en el uso de métodos basados en la teoría de grafos, en la que se utilizan aproximaciones muy eficientes de representación de la estructura molecular, implementadas en varios paquetes computacionales, entre los que se destacan el MODEST (MOlecular DESing Tool) y el TOSS-MODE (TOpological Sub-Structural MOlecular DEsing). Otro grupo, perteneciente a la Facultad de Química y Farmacia de la propia universidad, que presenta resultados relevantes en este campo es el Grupo de Descubrimiento Molecular Asistido por Computadora e Investigaciones Bioinformáticas (CAMD-BIR Unit, por sus siglas en inglés). Estos grupos han orientado sus investigaciones hacia la utilización de diferentes métodos para los estudios QSAR, priorizando el desarrollo y aplicación de nuevos descriptores grafo-teóricos para el diseño molecular de compuestos orgánicos potencialmente bioactivos.23-39 Teniendo en cuenta lo planteado anteriormente, fueron definidas recientemente tres nuevas familias de ITs a partir de la aplicación de conceptos de la matemática discreta y el algebra lineal a la química.31-34 Estos descriptores están basados en el cálculo de formas cuadráticas, lineales y bilineales; los que por su analogía con las mismas, han sido denominados como índices cuadráticos, lineales y bilineales moleculares, respectivamente. Estos ITs han sido aplicados en diversos estudios QSAR/QSPR con resultados satisfactorios.31-35, 37, 41 Estos nuevos DMs fueron generalizados para ser utilizados en estudios que involucren. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 12.
(10) Capítulo 1. Introducción. bio-macromoléculas, tales como ácidos nucleicos y proteínas; obteniendo resultados superiores a los obtenidos por otros enfoques.42, 43 Sin embargo, no siempre estos y otros índices utilizados, muestran un desempeño totalmente satisfactorio para la predicción de ciertas propiedades. De hecho no se puede esperar que un conjunto específico de índices sea superior absolutamente a otros conjuntos posibles y/o pueda producir buenos resultados en todos los problemas. Por ello, consideramos adecuado proponer un nuevo enfoque para la obtención de nuevas familias de ITs 2D, útiles para el diseño molecular y para los estudios QSPR/QSAR, que posibiliten realizar una descripción adecuada de las moléculas y de esta forma contar con una nueva herramienta para el estudio químico-físico-matemático de las sustancias, su estructura, sus propiedades y sus posibles transformaciones, aspectos que resultan de primordial interés para los químicos teóricos, sintéticos y médicos. Además pueden ser aplicados en la solución de diferentes problemas de corte tecnológico: 1) en la industria química de nuevos materiales, 2) industria alimentaría, 3) industria farmacéutica, por solo citar algunos. En esta última (industria farmacéutica) han sido aplicados satisfactoriamente muchos ITs, los cuales posibilitan obtener, con un gasto mínimo de recursos y en el menor tiempo posible candidatos a fármacos con grandes posibilidades de llegar a convertirse en un medicamento de prescripción clínica. La mayor parte de los ITs utilizados actualmente han sido definidos de forma global (ver capítulo 2), lo cual es una gran limitación pues muchas propiedades/actividades de las moléculas dependen de una combinación de aspectos locales y totales de la estructura molecular, más que con solo rasgos globales de la molécula. La definición local de los ITs (para un determinado fragmento o núcleo base, átomo o grupo de átomos en una molécula) es una de las 13 propiedades deseadas para un nuevo índice propuesta por Randic.67 Es válido destacar, además, que la mayor parte de los ITs han sido definidos a partir de la matriz de adyacencia y de distancia topológica (ver capítulo 2), mientras que la matriz de incidencia (ver capítulo 2) nunca ha sido utilizada para la generación de ITs. Las dos primeras matrices han sido utilizadas con el propósito de generar nuevos índices y tienen propiedades interesantes que han favorecido su uso con este propósito. Así por ejemplo, los principales ITs definidos hasta el momento hace uso de ellas (momentos espectrales, índices de Randic y de valencia, el índice del estado electrotopológico, el descriptor de Wiener, etc) o de una combinación de las mismas, como es el caso de los índices de carga (para más información ver capitulo 2). En contraste, la matriz de incidencia posee características no deseadas (ej., no cuadrada y asimétrica) para los químicos matemáticos y no ha sido fuente de ninguno de los ITs definidos hasta el momento. Esta matriz será usada en este trabajo como fuente de nuevos ITs, como será mostrado a lo largo del presente reporte.. Por todo lo anterior se plantea el siguiente problema científico: Los descriptores moleculares existentes en la actualidad no posibilitan siempre una descripción totalmente adecuada de la estructura molecular y es necesario definir nuevos DMs bidimensionales (2D), que recojan nuevos contenidos de información y permitan describir y/o estimar diferentes propiedades químicofísicas y biológicas de compuestos orgánicos y así posibiliten el estudio de sistemas químicos con un gasto mínimo de recursos materiales y tiempo, además de abrir una puerta a nuevas posibles interpretaciones químico-físicas de los fenómenos naturales y de laboratorio.. Como vía para solucionar el problema científico se formula la siguiente hipótesis: Es posible aplicar conceptos de la Matemática Discreta y el álgebra lineal a la química para definir nuevas familias de descriptores moleculares con contenidos de información diferentes a los existentes y que. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 13.
(11) Capítulo 1. Introducción. puedan ser utilizados en el diseño ‘racional’ de fármacos y otras aplicaciones destinadas a otros sectores (puramente químico, alimentario, industrial, etc).. Para demostrar la anterior hipótesis y dar respuesta al problema científico planteado, se proponen los siguientes objetivos: Objetivo general Definir, implementar computacionalmente, interpretar en términos estructurales y químico-físicos así como aplicar en estudios QSPR una nueva familia de descriptores moleculares 2D basados en la aplicación de conceptos de la matemática discreta y el álgebra lineal a la química y que ayuden a resolver – o al menos resolver mejor – problemas de predicción de propiedades físicas, química, químico-físicas y biológicas de nuevos compuestos. Objetivos específicos ¾ Definir un suceso generado por un modelo que devenga en la confección de una matriz de incidencia que lo describa. ¾ Definir total y localmente nuevos DMs basados en el concepto de Derivada de un Grafo Molecular (usando la matriz de incidencia generalizada, para cada orden y/o tipo de sub-grafo). ¾ Implementar computacionalmente los nuevos índices. ¾ Realizar una interpretación químico-física y en términos estructurales de los nuevos índices propuestos. ¾ Aplicar una de las nuevas familias de ITs al diseño molecular mediante la realización de estudios QSPR comparativos, que permitan validar la calidad de los nuevos ITs.. La novedad científica de este trabajo está fundamentada en la definición de una nueva familia de ITs, basados en el cálculo de derivadas del grafo molecular (para átomos y enlaces) respecto a un suceso prefijado. En el mismo se utilizan, por primera vez, las matrices de incidencia de subgrafos moleculares (generalizada para átomos y enlaces) y de frecuencia de relaciones en la definición de un ITs. Esta invariante grafo-teórica ha sido extendida para derivadas de n-uplas, de orden superior y mixtas, conformando así una nueva familia de ITs. Finalmente, los DMs totales y locales (para grupos de átomos o enlaces) se definen usando las Normas 1, 2 y 3 de Minkowski y la Norma Geométrica, a partir de vectores conformados por ITs atómicos.. En esta tesis se realizan los siguientes aportes: Aporte Teórico: Se definen nuevos índice usando una nueva invariante grafo-teórica, basada en el cálculo de las Derivada del Grafo, para lo cual se definen nuevas representaciones matriciales nunca antes utilizadas en la generación de un ITs, tales como las matrices de incidencia, de incidencia generalizada y de frecuencia de relaciones. Este DMs ha sido definido de forma local (para átomos o agrupaciones atómicas) y total (considerando toda la molécula) para lo cual se introduce el uso de las Normas 1, 2 y 3 de Minkowski y Geométrica [estas normas pueden recoger información de todos los átomos de la molécula (descriptor global) o de agrupaciones atómicas determinadas (descriptores locales)]. Aporte Práctico: Se desarrolló un programa en MATLAB, capaz de calcular de forma rápida y automatizada los valores de las derivadas de las moléculas representadas como grafos moleculares. Se realizaron aplicaciones en la modelación de propiedades químico-físicas de. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 14.
(12) Capítulo 1. Introducción. compuestos orgánicos demostrándose las potencialidades de estos índices para estudios QSPR/QSAR y el diseño de fármacos. Aporte Metodológico: Se plantea una metodología matemática novedosa para la descripción estructural de las moléculas orgánicas, basada en el cálculo de derivadas del grafo usando un suceso prefijado. Por tanto, nuevos sucesos generaran nuevas ITs al generar nuevas matrices de incidencia y sus respectivas matrices de frecuencia. Además el uso de normas para obtener índices globales (y también locales) a partir de contribuciones atómicas también puede ser usado por otros índices definidos localmente constituyendo nuevas invariantes moleculares totales (y locales, para átomos del mismo tipo, etc).. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 15.
(13) "Podrán morir las personas, pero jamás sus ideas." Ernesto Che Guevara.
(14) 2. QUÍMICA GRAFO-TEÓRICA y MÉTODOS ESTADÍSTICOS (QUIMIOMÉTRICOS) EN EL DISEÑO MOLECULAR 2.1. Química Grafo-Teórica y Topología Molecular en el Diseño Molecular 2.1.1. Introducción a la Química Grafo-Teórica y a la Topología Molecular La teoría de grafos ha sido ampliamente aplicada a diversos campos de la ciencia.1 Un grafo se expresa usualmente como vértices interconectados por aristas.1, 2 Donde cada vértice del grafo se representa un objeto y la arista que conecta dos vértices representa la relación entre estos dos objetos. En la química grafo-teórica los objetos del grafo pueden representar orbitales, átomos (o sus núcleos), enlaces, grupos de átomos, moléculas, o colecciones de moléculas. Las aristas de un grafo químico simbolizan las interacciones entre objetos químicos y se usan para definir enlaces químicos, reacciones, mecanismos de reacciones, modelos cinéticos, u otra relación o transformación de los objetos químicos. En la literatura existente sobre la química grafo-teórica y sus aplicaciones;3-7 podemos encontrar la mayoría de las principales aplicaciones de los grafos químicos, como son: 1) los índices topológicos (ITs) y otros índices estructurales para los estudios QSAR;8-16 2) el enfoque de 18. 3) la enumeración de isómeros, percepción de simetría estructural y. codificación de compuestos químicos;. 4) grafos cinéticos y de reacción;22 y 4) el diseño de síntesis asistida. orbitales moleculares de Hückel;17,. 19-21. por computadora.23 De todas estas aplicaciones mencionadas, la de interés para el presente trabajo es aquella relacionada con la obtención de descriptores estructurales para el diseño molecular. Por tanto, antes de realizar un análisis de los resultados más importantes reportados en la literatura en el campo de la química grafo-teórica y en especial del uso de descriptores moleculares basados en la teoría de grafos en los estudios QSAR, daremos una breve reseña de la teoría de grafos en donde enunciaremos una serie de conceptos y términos matemáticos que serán utilizados en el desarrollo de esta tesis y que son imprescindibles para la comprensión de los resultados tanto de la literatura como los alcanzados en el presente trabajo. 2.1.2. Representación topológica de moléculas. La representación topológica de un objeto es aquella que brinda información sobre el número de elementos que lo componen y sus conectividades. En ese sentido, se define a la topología como aquella parte del álgebra que estudia las posiciones e interconexiones de los elementos dentro de un conjunto.24 Si la topología es aplicada a las moléculas, da lugar a la topología molecular. Por tanto, consideremos en este trabajo que una representación topológica de una molécula puede ser obtenida utilizando un grafo molecular. A continuación se darán algunas de las principales definiciones en teoría de grafos. 2.1.3. Los grafos, conceptos generales sobre los mismos. Ante todo, comenzaremos por introducir matemáticamente el concepto de grafo. Sea V un conjunto finito de vértices y E el conjunto de aristas que unen pares no ordenados de los elementos de V. En términos matemáticos un grafo es definido como G = (V, E).1, 4 El número de vértices en un grafo es designado como n y el número de aristas por m. Otra de las formas de definir un grafo es la siguiente: consideremos un conjunto no vacío V = {vi / i =1, 2,…,n}, un conjunto no vacío E = {ei / i =1, 2,…,m} y una aplicación θ, la cual asocia a cada elemento de E con un par no ordenado de elementos de V. Esta aplicación es denominada la aplicación de incidencia asociada con un grafo y le da singularidad al grafo para un conjunto V dado. Por tanto, para cada ei existe un par no ordenado [vi, vj] tal que θ(e) = [vi, vj]. En este caso los dos conjuntos E y V junto con la aplicación θ forman un grafo. Sin embargo, con el propósito de obtener una representación más real de la topología de la molécula, nosotros necesitamos identificar los diferentes átomos en la misma, lo cual es posible ‘etiquetándolos’ con sus Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR.
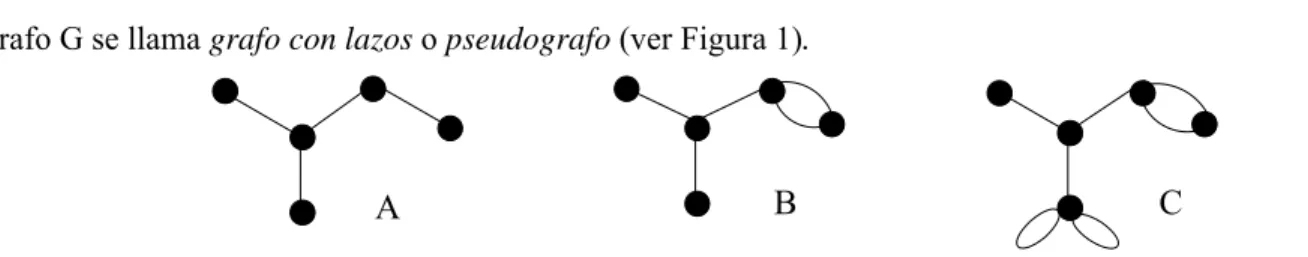
(15) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. símbolos químicos o con una significación característica, la cual denominaremos “peso” del átomo. Entonces, el grafo se transformaría en un grafo ‘etiqueteado’ o ponderado. En este sentido, a cada vértice vi del grafo G = (V, E) le podemos adicionar en correspondencia el peso wi de un conjunto de pesos W = {wi / i =1, 2, …}. Como resultado obtenemos un conjunto de los vértices ponderados {(vi,wi) / i =1, 2, …, n}, con ello, no hace falta que todos los pesos sean distintos. De la misma forma, podemos poner el peso pi del conjunto de pesos P = {pi / i =1, 2, …} en correspondencia a cada elemento del conjunto E. Como resultado obtenemos un conjunto de las aristas ponderadas {(ei,pi) / i =1, 2,…,m}; además, no hace falta que sean distintos todos los pesos.25 Si en G hay pares repetidos (aristas múltiples, o sea vértices que están unidos por más de una arista), entonces el grafo G se llama grafo con aristas múltiples o multigrafo. Las aristas de la forma {Balaban, 1998 #43}, se denominan lazos o bucles. Si en G hay lazos (pueden también existir aristas múltiples), entonces el grafo G se llama grafo con lazos o pseudografo (ver Figura 1).. A. B. C. Figura 1. A) Grafo simple; B) Multigrafo; y C) Pseudografo. En este trabajo usaremos pseudografos, por ser la condición más general de los grafos, los cuales pueden ser orientados o no orientados. Los pseudografos serían orientados si los pares de vértices fueran ordenados. En el trabajo actual solo usaremos pseudografos no orientados, por lo que las definiciones que a continuación daremos, están relacionadas con la teoría de pseudografos (se sobrentiende pseudografos no orientados). Estas definiciones de los términos más utilizados en los grafos moleculares son muy útiles para describir varias características estructurales de estos.1, 4, 26, 27 Si ak = {vi, vj} es una arista, entonces los vértices vi, vj, se llaman extremos de la arista ak. Si los vértices vi y vj son los puntos finales de ei se denota como ei∼[vi, vj], lo que se lee como ‘ei es incidente con vi y vj’ (enlace que los une). Los vértices vi, vj se llaman adyacentes si existe una arista ak tal que ak = {vi, vj}∈ V, (o sea, si existe una arista que los une). Dos aristas se denominan adyacentes si ellas tienen un vértice en común. En un multigrafo, el grado del vértice vi [δ(vi)] es el número de aristas del multigrafo que son incidentes al vértice vi. En un pseudografo, el grado del vértice vi es igual al número total de aristas (que no sean lazos) incidentes a este vértice, más el número de lazos incidentes a él. Un camino (P) es una sucesión de aristas con vértices comunes. La longitud (l) de un camino es el número de aristas del mismo. Así por ejemplo, camino de longitud cero (P0) es una sucesión de vértices que P. contiene solo un vértice. Un subgrafos es una sección del grafo, subestructura o parte de este. Kier y Hall45 clasifican los subgrafos según su orden o tipo. Así los subgrafos pueden clasificarse como path (camino, senda) si todos los vértices tienen grado menor o igual a dos, si todos los vértices poseen grado superior a dos, entonces se clasifica como cluster (grupo) y si existen vértices que cumplan con una gama variada de grados (menores y mayores que dos) en el mismo subgrafo, entonces estamos en presencia de un subgrafo de tipo path-cluster. Si el subgrafo es cíclico se dice que es de tipo chain. El orden de un subgrafo viene expresado por el número de aristas que este posea.. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 18.
(16) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. 2.1.3.1. Empleo de matrices para representar grafos moleculares. Los grafos moleculares son ampliamente usados para representar la estructura química de compuestos orgánicos. Sin embargo, los grafos moleculares son una representación no numérica de la estructura química; y la obtención de los descriptores moleculares para nuestros propósitos requieren una descripción numérica de los grafos moleculares. Los grafos pueden representarse matricialmente en forma algebraica.8,. 28, 29. La. descripción numérica de la estructura de los compuestos químicos, es esencial para la manipulación computacional de las moléculas y para los cálculos de los índices moleculares que de ella se derivan. Así por ejemplo, la matriz de adyacencia A = A(G) del grafo G con n vértices, es la matriz cuadrada simétrica nxn y los elementos [A]ij se definen de la siguiente forma: [A(G)]ij = 1 si i≠j y eij ∈ E = 0 si i=j o eij ∉ E donde E representa el conjunto de las aristas de G. En la matriz de adyacencia A(G) la fila i y columna i corresponden al vértice vi de G. Como un ejemplo sencillo, en la Figura 2 se muestra el grafo molecular y la matriz de adyacencia del 1-etil-2-metil-ciclopropano. Una propiedad interesante de A(G) viene dada por el hecho de que la matriz Ak(G) da el número de caminos unitarios de longitud k que unen los vértices vi y vj. Esto permite interpretar los elementos aij como el número de caminos unitarios, de orden (longitud) k, entre los vértices vi y vj (ver Figura 2). v1 v2 v3 v4 v5 v6. 1. 5 4. v2. v3. v4. v5. v6. v1 v2. v3. v4. v5. v6. v1. 1. 0. 0. 0. 0. 0. v1. 0. 1. 0. 0. 0. 0. v1. 1. 0. 1. 1. 0. 0. v2 0 6 A = v3 v4 v5. 0 0. 1 0. 0 1. 0 0. 0 0. 0 v2 1 0 A = v3. 1 0. 0 1. 1 0. 1 1. 0 0. 0 0. v2 2 A = v3. 0 1. 3 1. 1 2. 1 1. 1 1. 0 0. 0 0. 0 0. 0 0. 1 0. 0 1. 0 0. v4 v5. 0 0. 1 0. 1 0. 0 1. 1 0. 0 1. v4 v5. 1 0. 1 1. 1 1. 3 0. 0 2. 1 0. v6. 0. 0. 0. 0. 0. 1. v6. 0. 0. 0. 0. 1. 0. v6. 0. 0. 0. 1. 0. 1. 3 2. v1. Figura 2. Grafo molecular y matrices de adyacencia de la molécula de 1-etil-2-metil-ciclopropano.. La matriz de adyacencia A(Gw) del grafo molecular G con vértices y aristas ponderadas (con n vértices) es la matriz simétrica nxn (cuadrada) y los elementos [A (w)]ij se definen de la siguiente forma:29 [A(Gw)]ij = V(w)wi si i=j = E(w)wij si eij ∈ E = 0 si eij ∉ E donde V(w)wi es el peso del vértice vi, E(w)wij es el peso de la arista eij, y w es un determinado peso o etiqueta que se utilice para computar Vw y Ew. La matriz de distancia D = D(G) de un grafo G con n vértices, es la matriz simétrica nxn (cuadrada) y los elementos [D]ij se definen de la siguiente forma:8, 28 [D]ij = dij si i≠j = 0 si i=j donde dij es la longitud del camino más corto entre los vértices vi y vj de G. Por ejemplo, la matriz de distancias topológicas entre vértices del grafo molecular de la molécula de 1-etil-2-metil-ciclopropano (ver Figura 2) se representa en la Figura 3.. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 19.
(17) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. v1. v2. v3. v4. v5. v6. v1. 0. 1. 2. 2. 3. 4. v2 D = v3. 1 2. 0 1. 1 0. 1 1. 2 2. 3 3. v4 v5. 2 3. 1 2. 1 2. 0 1. 1 0. 2 1. v6. 4. 3. 3. 2. 1. 0. Figura 3. Matriz de distancias topológicas entre vértices de la molécula de 1-etil-2-metil-ciclopropano.. Para un grafo G, se define la matriz diagonal, DEG = DEG(G), donde la entrada i-ésima es igual al grado del vértice vi, δi y todos los demás elementos son iguales a cero. Matriz de incidencia: para un grafo G, con una ordenación de vértices (v1, v2, … vn) y aristas (e1, e2, … em),. ( ). entonces la matriz de incidencia del grafo para esa ordenación es la matriz de n filas y m columnas B = bij Definida por la condición de que: ⎧1 b ij = ⎨ ⎩0. si. vi ∈ e j. si. vi ∉ e j. Como ejemplo podemos citar que siendo G=({A,B,C,D},{A,B},{B,C},{C,D},{A,D},{B,D}) (Ver Figura 4) la matriz de incidencia de G, respecto a la ordenación de sus vértices A,B,C,D y sus aristas {A,B},{B,C},{C,D},{A,D},{B,D}, es la matriz:. Figura 4. Grafo y matriz de incidencia que lo representa.. Existe un gran número de matrices que caracterizan estructuralmente a un grafo molecular y han servido para computar varios ITs. Algunos ejemplos son la matriz Laplaciana L(w, G), la matriz de Detour, [Δ]ij,30 la matriz de Detour-distancia, [Δ-D]ij,30 la matriz de distancia-valencia [Dval(p, q, w, G)]ij,31 la matriz de resistencia-distancia [Ω(w)]ij,32 la matriz de conductancia eléctrica [EC(w)]ij,32, la matriz de Wiener inversa [RW(w, G)]ij,35 la matriz de Szeged [Szu]ij,36 entre otras. 2.1.4. Invariante grafo-teórica. Una invariante grafo-teórica es aquella propiedad del grafo que no depende del orden de numeración de los elementos del mismo, las cuales pueden obtenerse por manipulación algebraica del grafo. Como se ha señalado, los grafos moleculares no son una representación numérica de la estructura química y aunque las matrices sí constituyen una representación algebraica, tienen como desventaja que no constituyen invariantes grafo-teóricas, ya que su construcción depende de la numeración dada a los vértices del grafo. Sin embargo, una simple invariante como el número de vértices, puede obtenerse a partir de la matriz de adyacencia.37 Resulta evidente, que para los estudios QSAR, el diseño de fármacos, el tamizaje virtual, etc; se necesitarían obtener índices numéricos que caractericen estructuralmente los grafos moleculares y que estos índices sean inevitablemente invariantes. Estos descriptores invariantes son los llamados ITs los cuales serán el objeto principal de esta tesis. 2.1.5. Índices bidimensionales (2D) basados en la topología molecular. De lo anterior se deduce claramente que debe ser un resultado numérico de alguna invariante ‘extraída’ del grafo molecular. Es decir, los índices topológicos (ITs) son descriptores moleculares que se obtienen de Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 20.
(18) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. una invariante grafo-teórica.. 38-42. Por tanto, los ITs son números calculados a partir de la representación de una. molécula como un grafo, siendo independientes de la numeración de los vértices y aristas en el grafo molecular. Estos índices codifican información estructural contenida en la representación bidimensional (2D) de la molécula. Esta descripción ‘topológica’ de la molécula contiene información de la conectividad entre átomos (o enlaces) en la molécula y codifica la talla, forma, ramificación, heteroátomos y la presencia de enlaces múltiples.8-10, 13-16, 43 La importante información de la molécula contenida en los ITs puede usarse en la descripción de propiedades físico-químicas y biológicas.7, 44-46 Un resumen completo sobre los ITs es realmente imposible, debido a la gran cantidad de estos índices que han sido publicados en la literatura y al número de ellos que cada año son introducidos. Por tanto, nosotros nos concentraremos solamente en los ITs que más ampliamente han sido aplicados a diferentes problemas relacionados con la estructura y aquellos que sean más importantes para los objetivos del presente trabajo. La mayoría de los ITs propuestos están relacionados con la matriz de adyacencia de vértices, de distancias o de combinaciones de estas. No obstante, en la actualidad los ITs han sido clasificados acorde a su naturaleza en, primera, segunda y tercera generación,47 lo cual facilita su estudio. Un tratamiento abarcador de todos los descriptores moleculares disponibles fue recientemente publicado por Todeschini y Consonni.37 2.1.5.1. Índices topológicos de primera generación. Los ITs de primera generación son números enteros basados en propiedades del grafo como un todo, tales como las distancias topológicas. Los índices más representativos de esta clase son el índice W de Wiener,47 el índice Z de Hosoya,48 y los índices B y C de Balaban.49 De todos estos ITs solo W ha sido usado extensamente en estudios QSAR y en el descubrimiento de nuevos fármacos. El índice de Wiener fue definido en 1947 y desde entonces ha constituido una importante fuente de inspiración para el desarrollo de nuevos ITs.. 1 N W (G ) = ∑ 2 i =1. N. ∑ [D(G)] j =1 j ≠i. (2.1). ij. Los resultados obtenidos con el índice W en la modelación molecular propició la definición de varios índices relacionados con este. Algunos de estos son el cuasi-Wiener índice,50 el índice de Kirchhoff,51 el índice RDSUM,52 y el hiper índice de Wiener,53, 54 entre otros. La suma de los grados de cada enlace fue introducido, al mismo tiempo que el índice de Wiener, por Platt; por lo cual este es conocido como el índice F de Platt.55 Otro de los ITs de primera generación que también ha sido ampliamente utilizado en estudios QSAR, es el índice de Zagreb desarrollado en 1975 utilizando las valencias de los átomos (Gutman y col, 1975).56. M1 =. n. ∑ [δ (vi )]2. i =1. (2.2). ( ). M 2 = δ (vi )δ v j. (2.3). 2.1.5.2 Índices topológicos de segunda generación. Estos índices son números reales basados en las propiedades del grafo íntegro. La mayoría de los ITs utilizados en los estudios QSAR y en el diseño/descubrimiento de fármacos en estos momentos, pertenecen a esta clase. El conjunto de descriptores moleculares más útiles de esta clase, son los llamados índices de conectividad molecular9,. 10, 57. Estos índices están basados en una invariante grafo-teórica introducida por. Randic 25 años atrás, para computar un índice de ‘ramificación’ para los alcanos.58 Estos índices fueron extendidos por Kier y Hall para tener en cuenta la diferenciación entre heteroátomos y los diferentes subgrafos Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 21.
(19) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. en la molécula. Los índices de conectividad molecular de ‘valencia’ son expresados de la siguiente forma:9, 10, 57. (2.4) donde, s es el número de subgrafo de tipo t (camino, cluster, camino-cluster y cadena) con m aristas, n es el número de vértices del subgrafo y δj ves la valencia atómica computada utilizando la siguiente fórmula (Ec. 1.8):. (2.5) donde Zi es el número de electrones de valencia del átomo i, Zi son todos los electrones del átomo i y Hi es el v. número de átomos de hidrógeno unidos a este átomo. Balaban introdujo en 1982 como modificación de los índices de conectividad el índice J de Balaban, el cual es un índice de conectividad basado en las distancias promedios y fue definido de la siguiente forma:59. J=. [. ]. −1 / 2 m D(vi )D(v j ) ∑ μ + 1 bonds. (2.6). donde D(vi) es la suma de todas las distancias topológicas relacionadas con el átomo i. Es decir, la suma de todas las entradas de las filas o columnas de la matriz de distancia (D) correspondiente para un átomo i; μ es el número ciclomático. Gálvez y colaboradores han introducido una serie de descriptores grafo-teóricos con el objetivo de completar la información estructural contenida en los índices de conectividad molecular. Entre estos descriptores, de probada utilidad en el diseño de fármacos, el índice de carga aparece entre los más interesantes.60 Los ITs de carga Gk y Jk son definidos de la siguiente forma: n −1. Gk = ∑. n. ∑ CT. ij. i =1 j =i +1. δ (k , d ij ). J k = Gk (n − 1). (2.7) (2.8). donde CTij = mij – mji, donde m son los elementos de la matriz auxiliar M definida como: M = AxD*. La matriz D* es la matriz de los inversos de las distancias cuadradas, en la cual sus entradas en la diagonal se toman como 0 y δ es la delta de Kronecker. La mayoría de los ITs analizados hasta ahora describen la estructura de la molécula como un todo por lo tanto pueden considerarse como descriptores moleculares globales. Kier y Hall, a comienzos de la década del 90, introdujeron un nuevo IT denominado índice del estado electrotopológico (E-estado); basado en una invariante grafo-teórica para un átomo en la molécula, representando la accesibilidad de electrones de este átomo. Este índice puede ser considerado como un descriptor molecular local; el mismo codifica información acerca del ambiente topológico y de las interacciones electrónicas debidas a todos los demás átomos en la molécula. El índice del E-estado para un átomo i en una molécula se define como:11, 12, 41, 61, 62 S i = I i + ∑ ΔI ij. (2.9). j. La suma es el total de los otros átomos j dentro del grafo molecular. El término de perturbación de un átomo i por un átomo j se define de la siguiente forma (Ec. 1.13):. (. ). ΔI ij = I i − I j / d ij2 Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. (2.10) 22.
(20) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. donde, d ij es la distancia topológica que separa los átomos i y j en el grafo desprovisto de hidrógenos. Los valores intrínsecos son definidos como (Ec. 1.14):. [. ]. I i = (2 / N i )2 δ i v + 1 / δ i. (2.11). donde N es el número cuántico principal para los electrones de valencia del átomo i. El valor de Si para un átomo i puede ser visto, como una suma de efectos de los átomos del esqueleto molecular a varias distancias de i sobre este último:. S i = I i + ∑ (I i − I j ) / 4 + ∑ (I i − I j ) / d ij2 j. (2.12). j. El primer término es un valor intrínseco definido con anterioridad, el segundo son las contribuciones de los átomos enlazados y el último representa las contribuciones de los átomos no enlazados. Adicionalmente, investigadores de nuestro país han realizado apreciables aportes en la introducción de nuevos ITs. Desde mediados de los 90 Estrada y col. han definido varios ITs, entre los que destaca como de mayor interés en estudios quimio-bioinformáticos los momentos espectrales de la matriz de adyacencia entre aristas [E(G)] de un grafo molecular.63-73 De esta forma el momento espectral de orden k se define como la traza de la k-ésima potencia de la matriz E y su símbolo es μk.68 A su vez, la traza se define como la suma de los valores de la diagonal principal. Lo anterior se puede expresar matemáticamente de la siguiente manera:. m. μ k = Tr ( E k ) = ∑ k eii. (2.13). i =1. donde, keii son los elementos de la diagonal principal de la matriz Ek. De la definición dada de E y μk, se observa que los momentos espectrales son números enteros positivos. Para lograr diferenciar las moléculas con heteroátomos a través de los momentos espectrales, las aristas del grafo molecular son ponderadas con determinados pesos (dij), lo que aumenta notablemente las posibilidades y versatilidad del método. Mediante los dij se pueden introducir de manera natural, en la diagonal principal de la matriz, propiedades medias de los enlaces como son: la distancia, la polarizabilidad, el dipolo medio de enlace, etc. Además, se han definido los momentos espectrales locales de la matriz de adyacencia entre enlaces del grafo molecular desprovisto de hidrógeno,67 pudiendo estar la matriz ponderada o no, en la diagonal principal. Carrasco y col. introdujeron recientemente un nuevo índice denominado índice del estado refractotopológico (ℜi).74 El mismo es un índice hibrido y se define como el valor de refractividad intrínseca de un átomo i, mas un termino de perturbación para un grafo sin hidrógenos suprimidos. Matemáticamente se define de la siguiente manera:. ℜ i = ARi + ΔARi. (2.14). donde ARi el valor de refractividad intrínseca de un átomo i y ΔARi es un termino de perturbación definido por:. Δ AR. i. =. n. ∑. j =1. ( AR. i. − AR j ) / rij. 2. (2.15). Marrero-Ponce y col. desarrollaron un nuevo enfoque basado en una representación vectorial y otra matricial de la estructura molecular. Estas están a su vez están relacionadas con la representación grafo-. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 23.
(21) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. teórica por medio de un pseudografo de la estructura química. Estos nuevos índices se conocen como índices cuadráticos por analogía a las formas cuadráticas y se definen matemáticamente de la siguiente manera:75-82. qk (x ) =. n. n. ∑ ∑ i =1. k. j =1. m ij x i x j = [X]tMk[X]. (2.16). donde, mij = mji (matriz cuadrada simétrica), n es el número de átomos de la molécula y x1,…,xn son las coordenadas del vector molecular ( x ) en la base canónica {e1,…,en} de Rn. La definición local de estos descriptores se basa en invariantes grafo-teóricas para un fragmento FR dado, dentro de un seudografo específico y es la siguiente:75-82. q kL ( x ) =. n. n. ∑ ∑ i =1. j =1. k. m ijL x i x j = [X]t MkL [X]. (2.17). donde n es el número de átomos del fragmento de interés y kmijL es el elemento de la fila “i” y columna “j” de la matriz MkL ≡ Mk(G, FR) [ qkL(x) ≡ qk(x, FR)]. Esta matriz se extrae de la matriz k-ésima potencia de M y contiene la información referida a los vértices del fragmento FR de interés y también de su entorno molecular. De igual forma, estos autores han definido otros grupos de nuevos índices moleculares algebraicos basados en aplicaciones lineales y formas bilineales, los cuales están siendo utilizados de forma satisfactoria en estudios QSAR/QSPR y el descubrimiento de nuevos compuestos bioactivos.31, 33,34 2.1.5.3 Índices topológicos de tercera generación. Los ITs de tercera generación son números reales basados en propiedades locales del grafo molecular. Estos índices son de reciente publicación.83-85 Otros de los ITs de esta clase están basados en la aplicación de la teoría de la información a términos de sumas de distancias o sobre nuevas matrices no simétricas introducidas en la literatura.86-88 Sin embargo, nosotros no discutiremos este tipo de ITs porque estos no han tenido una gran aplicación en estudios QSAR y en el diseño/descubrimiento de nuevos fármacos. 2.1.6. Propiedades que debe poseer un nuevo índice topológico. Randic propuso una lista de propiedades que deben poseer los nuevos ITs,89 considerada actualmente de un nivel de sofisticación muy alto, el cual deben alcanzar los nuevos descriptores moleculares diseñados. Estas propiedades son las siguientes: 1) interpretación estructural directa, 2) buena correlación con al menos una propiedad, 3) buena discriminación entre isómeros, 4) localmente definidos, 5) generalizables a análogos superiores, 6) linealmente independientes, 7) simplicidad, 8) no basados en propiedades físico-químicas, 9) no trivialmente relacionados con otros índices, 10) eficiencia de construcción, 11) basados en conceptos estructurales familiares, 12) mostrar una dependencia correcta con el tamaño y 13) tener cambios graduales con cambios graduales en la estructura. En realidad, muchos de estos atributos tienen cierto grado de interrelación. A continuación, comentaremos algunas de estas propiedades ‘deseables’ a través, también, de algunos problemas o desventajas que presentan algunos ITs. Al emplear los ITs para la caracterización de una molécula intrínsecamente tenemos cierta pérdida de información, ya que se está tratando de representar un objeto tridimensional por un número simple. Esta cuestión es crítica, cuando se trata de una actividad biológica que depende de la interacción estereoespecífica del receptor con un sitio de la molécula. Sin embargo, los ITs pueden contener una sorprendente información estructural sobre las moléculas, lo que los hace de gran utilidad en los fines prácticos de sus aplicaciones. Existen dos posibilidades de resolver la pérdida de información de los descriptores grafo-teóricos. La primera, consiste en la generalización de un descriptor simple a análogos ‘superiores’ y segunda, la generalización de la invariante grafo-teórica como secuencias de números.89 La generalización de los índices es necesaria para Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 24.
(22) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. salvar situaciones en las que un descriptor simple no es suficiente y la investigación de una familia de descriptores estructuralmente relacionados puede resolver el problema. Sucede que en la obtención de una familia de descriptores de diferentes órdenes (familia de descriptores relacionados), como generalización de un simple descriptor, se ha observado que muchos de estos son colineales (unos indices pueden ser expresados como combinación lineal de los restantes, por lo que puede existir ‘redundancia de la información’). La independencia lineal u ortogonalidad de los índices es uno de los atributos deseables, pues los descriptores colineales pueden afectar la ‘estabilidad del coeficiente de correlación’ y dificulta la interpretación de los modelos obtenidos. En este sentido, es importante también que los descriptores sean ortogonales en relación a los restantes ITs. Esta independencia lineal de los índices significa que los mismos conduzcan a una correlación con una propiedad que no es satisfactoriamente explicada por los otros descriptores existentes. Una de las principales deficiencias que aún presentan la mayoría de los ITs está relacionada con su selectividad estructural; o sea, la capacidad de los mismos para diferenciar entre estructuras que no sean isómeras. La sensibilidad de la discriminación de isómeros, surge debido a que la diferenciación de isómeros garantiza que estos descriptores puedan ser utilizados en estudios donde las propiedades moleculares varían para diferentes isómeros y en general, en los estudios de aquellos aspectos de las propiedades moleculares que dependen del tamaño de la molécula. De forma general, la selectividad de los ITs disminuye a medida que aumenta el número de vértices en el grafo que representa el esqueleto carbonado molecular, por ejemplo el andamiaje carbonado de un alcano. Razinger y colaboradores mostraron además que no existe una relación directa entre la discriminación de isómeros y la utilidad de un índice para correlacionar con propiedades físicas de interés.90 Este resultado, demuestra que una cuestión importante en el diseño de un nuevo IT, es que este contenga información estructural valiosa y no sólo que sea altamente discriminante para isómeros. La definición local de los descriptores se refiere al hecho de que estos no sean obtenidos de forma global para una estructura molecular, sino que puedan ser definidos sobre determinados fragmentos de la propia estructura. La interpretación en términos estructurales, es otro de los atributos deseables para un nuevo IT, ya que desde el punto de vista de su aplicación en estudios QSAR, solo aquellos índices que estén basados en conceptos estructurales simples ayudarán a interpretar propiedades complejas en términos estructurales. 2.1.7. Los índices topológicos y sus aplicaciones. Las aplicaciones de los ITs han estado dirigidas fundamentalmente hacia la predicción cuantitativa de propiedades físico-químicas y biológicas de compuestos orgánicos, en estudios que se han denominado QSPR y QSAR, respectivamente. Esta división, no es solo formal, porque aunque el método en ambos tipos de estudio es similar, por lo general, la actividad biológica es una propiedad mucho más compleja que las propiedades físico-químicas, debido a la gran cantidad de factores que influyen en la bioactividad de un compuesto químico.. La aplicación de los ITs al diseño y selección de nuevas entidades químicas es probablemente una de las áreas más activas de investigación en la aplicación de tales descriptores a problemas biológicos. Uno de los primeros ejemplos del diseño de nuevos compuestos en el uso de estos índices, fue descrito por la Upjohn & Pharmacy en 1993. Ellos fueron capaces de diseñar una nueva clase de compuestos de la familia de las heteropiperazinas con actividad contra la HIVretrotransferasa.91-96 Más recientemente, Graasy y col. fueron capaces de diseñar y sintetizar un. Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 25.
(23) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. péptido que mostró una actividad inmunosupresora aproximadamente 100 veces mayor que los compuestos líderes ensayados.97 2.2. Métodos Estadísticos (Quimiométricos) en el Diseño Molecular 2.2.1. Introducción a los Métodos Quimiométricos en el Diseño Molecular Los estudios QSAR constituyen un enfoque que permite entender como la variación estructural afecta la propiedad/actividad biológica de un conjunto de compuestos. En estos estudios, los descriptores moleculares (X) se correlacionan con una variable respuesta (Y). Es decir, este análisis puede definirse como una aplicación de métodos matemáticos y estadísticos al problema de encontrar una ecuación empírica de la forma Yi = fi(X1, X2, ...Xn), donde Yi son las propiedades y/o actividades biológicas de la molécula, y X1, X2, ...Xn son propiedades estructurales experimentales o calculadas (descriptores moleculares) de los compuestos. En este sentido, cada compuesto puede representarse como un punto en un espacio multidimensional, en los cuales los descriptores X1, X2, ...Xn son coordenadas independientes del compuesto. El objetivo más usual de este análisis es incrementar el entendimiento del sistema biológico bajo investigación o predecir la propiedad estudiada a un objeto (compuesto) no utilizado en la obtención del modelo. 2.2.2. Quimiometría.. El término quimiometría, surgió en la década del 70 y se define como la disciplina química que utiliza métodos estadísticos y matemáticos para seleccionar y optimizar los métodos analíticos y preparativos, así como procedimientos para el análisis e interpretación de los datos.100 2.2.3. Metodología general empleada en los estudios QSAR. Los principios de la metodología QSAR pueden describirse mediante los siguientes pasos comunes:100 1) Formulación del problema, se determina el objeto de análisis y el nivel de información requerido, 2) Parametrización cuantitativa de la estructura molecular de los compuestos químicos orgánicos/secuencia de biopolímeros, 3) Medición de la propiedad de interés (‘efectos biológicos’), 4) Escoger el tipo de modelo QSAR que se va a desarrollar, 5) Selección de los compuestos (diseño estadístico de la serie), 6) Análisis matemático de los datos y Validación interna y externa de los modelos obtenidos, 7) Interpretación de los resultados y Aplicación de los modelos desarrollados al diseño/descubrimiento de un nuevo compuesto líder, desarrollando procedimientos de tamizaje virtuales. Sin embargo, el desarrollo de cualquier QSAR es un ciclo interactivo. 2.2.4. Regresión lineal múltiple (RLM). La RLM estudia las relaciones entre una variable dependiente y un conjunto de variables independientes. Así mismo, la regresión múltiple remite a la correlación múltiple, que se representa por R. Es decir, la correlación múltiple analiza la relación entre una serie de variables independientes o predictores (X1, X2, ..., Xk), considerados conjuntamente, con una variable dependiente o criterio. Sus fundamentos se hallan en la correlación de Pearson.101 La recta de regresión múltiple tiene la siguiente forma: Y = a + b1 X1 + b2 X2 +...+ bk Xk. (2.18). siendo ‘a’ un valor constante. Como puede observarse, la RLM puede utilizarse en la predicción de los valores de la variable dependiente, en base a una combinación de variables independientes. 2.2.4.1. Principio de la parsimonia para seleccionar el número optimo de variables. La R2 aumenta en la medida en que se añaden variables a la ecuación; pero a partir de cierto punto el incremento de R2 para cada nueva variable que se añade, es insignificante. Un buen modelo no debe presentar ni demasiadas variables, ni debe olvidar las que sean verdaderamente relevantes. Es decir, debe cumplir el Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 26.
(24) Capitulo 2. Química Grafo-Teórica y Métodos Quimiométricos en el Diseño Molecular. principio de la parsimonia, según el cual un fenómeno debe ser descrito con el número mínimo de elementos posibles. Diversos procedimientos se han propuesto para seleccionar el número óptimo de variables a incluir en la ecuación, como por ejemplo la ‘forward selection’, ‘backward elimination; y ‘stepwise selection’.102 Este último método es el más utilizado (es una combinación de los dos anteriores) y sigue un proceso de selección de variables paso a paso. 2.2.4.2. Incremento de R2 y correlación parcial. Se llama incremento de R2 a una estimación de la importancia relativa que tiene la variable que acaba de entrar en este paso para predecir el criterio. El incremento de R2 viene dado por:. Rc2 = R 2 − Ri2. (2.19). donde Ri2 es el coeficiente de correlación múltiple al cuadrado cuando todas las variables, excepto la i (la que acaba de entrar en este paso), están incluidas en la ecuación. Por lo tanto, la Ri2 en un paso determinado coincide con la R2 del paso anterior. Un coeficiente Ri2 alto significa que esta variable proporciona información importante que no está contenida en las otras variables. 2.2.4.3. Análisis de la varianza. El ANOVA (ANalysis Of VAriance) sirve para comprobar la hipótesis de que R2 = 0. La variabilidad total de la variable dependiente se divide entre la parte atribuible a la regresión y la parte residual. La distancia de un punto cualquiera Yi a la Y se sub-divide en dos partes:101. (. )(. Yi − Y = Yi − Yi + Yi − Y. ). (2.20). siendo Yi el valor predicho por la ecuación de predicción. El valor Yi − Yi , denominado residual de la regresión sería cero si la recta pasase exactamente por encima del punto Yi. El otro valor, Yi − Y , corresponde a la distancia explicada por la regresión y representa el aumento en la estimación de Yi mediante la recta de regresión. En el ANOVA, F viene dada por:. F=. MCregresion MCresidual. (2.21). Esta F sigue una distribución F de Snedecor con grados de libertad v1 = υ, v2 = n- υ -1; siendo υ el número de variables de la ecuación. La media cuadrática (MC) se obtiene dividiendo la suma de cuadrados por los grados de libertad. La F sirve para comprobar si el modelo de regresión se ajusta a los datos y permite evaluar si se rechaza la hipótesis nula, según la cual, R2 = 0. Es interesante observar, que si el modelo se ajusta a los datos, el coeficiente de determinación (R2) se puede calcular a partir de las suma de cuadrados (SC) del ANOVA mediante:. R2 = 1−. SC residual SCtotal. (2.22). 2.2.4.4. Importancia de la tolerancia en la RLM. La tolerancia es una medida del grado de asociación lineal entre las variables independientes.103 Para la variable i, la tolerancia es igual a 1- Ri2 , donde Ri2 es la correlación múltiple al cuadrado entre la variable i considerada como variable dependiente y las demás variables independientes. Valores bajos en la tolerancia, indican que la variable i puede ser considerada como una combinación lineal de las otras variables Oscar Martínez Santiago – Derivada del Grafo: Novedosa vía para generar DMs 2D/3D: Teoría y Aplicaciones QSPR. 27.
Figure



+7





Documento similar