Nueva codificación algebraica de la estructura molecular y su aplicación en el ajuste de modelos predictivos
134
0
0
Texto completo
(2) If the wheel doesn't work, reinvent it. Christoph Steinbeck1. 1 http://www.steinbeck-molecular.de/steinblog, 2009. I.
(3) Dedicatoria. A la Revolución.. II.
(4) Agradecimientos A mis padres. A mi novia Annalie que soportó todas las noches con la luz encendida. A mis colegas Cesar Raúl y Stephen J. Barigye, que me aclararon la ruta a seguir. A mis tutores Carlos Morell y Yovani Marrero. …y a todos aquellos que de alguna forma contribuyeron con este final.. III.
(5) Resumen El presente trabajo describe un nuevo método de construcción de atributos orientado a codificar la información estructural de las moléculas. Basado en las formas lineal, bilineal y cuadrática, utiliza las matrices de densidad-electrónica grafo-teórica y las propiedades atómicas como ponderaciones, que actúan como matriz operadora y vectores, respectivamente, en el producto cartesiano definido por las formas algebraicas. Se introduce, por vez primera, los formalismos de balanceo y normalización matricial basados en los esquemas doble estocástico y de probabilidad mutua, las restricciones de conectividad aplicadas sobre la matriz operadora, la codificación localizada para distintos grupos o fragmentos químicos predeterminados y la generalización de la combinación lineal para índices atómicos aplicando una serie de operadores globales de agregación. Se presenta un análisis de variabilidad basado en entropía de Shannon, donde la codificación propuesta tiene mejor comportamiento que otras utilizadas en estudios quimioinformáticos y un análisis de componentes principales donde los descriptores moleculares basados en las formas algebraicas, codifican información estructural ortogonal a la capturada por los descriptores del software DRAGON. Para obtener una medida de la utilidad de los nuevos índices se realiza, finalmente, un estudio QSAR/QSPR para la afinidad de acoplamiento a la corticosteroide-binding globulin a partir del conjunto de datos propuesto por Cramer, considerando todos los casos como conjunto de entrenamiento y también dividiéndolos en conjunto de entrenamiento y en conjunto de prueba, obteniéndose parámetros estadísticos de comparables a superiores respecto a establecidas metodologías reportadas en la literatura.. IV.
(6) Tabla de Contenidos Introducción..................................................................................................................................... 1 1 Fundamento teórico ................................................................................................................ 7. 1.1 Química Grafo-Teórica y Topología Molecular .................................................. 7 1.1.1 Representación topológica de las moléculas................................................ 8 1.1.2 Invariante grafo-teórica ................................................................................. 8 1.1.3 Esquemas de normalización .......................................................................... 9 1.2 Formas algebraicas ............................................................................................. 10 1.3 Índices bidimensionales basados en la topología molecular .............................. 13 1.3.1 Propiedades que debe poseer un nuevo índice topológico.......................... 15 1.3.2 Los índices topológicos y sus aplicaciones ................................................. 17 1.4 Métodos estadísticos en la Química Computacional.......................................... 17 1.4.1 Análisis de Variabilidad .............................................................................. 18 1.4.2 Análisis de Componentes Principales ......................................................... 18 1.4.3 Regresión lineal múltiple ............................................................................ 19 1.4.4 Validación estadística de los modelos ........................................................ 22 1.4.5 Validación interna de los modelos .............................................................. 23 1.5 Conclusiones parciales ....................................................................................... 26 2. Índices QuBiLS-MAS: Teoría, extensión y nueva codificación .......................................... 27. 2.1 Representación matricial del grafo molecular .................................................... 27 2.2 Formalización de la matriz no-estocástica ......................................................... 29 2.3 Mecanismos de normalización matricial ............................................................ 29 2.3.1 Simple estocástica ....................................................................................... 30 2.3.2 Doble estocástica ........................................................................................ 30 2.3.3 Probabilidad mutua ..................................................................................... 31 2.4 Restricciones aplicadas a la matriz operadora.................................................... 31 2.5 Vector de propiedades ........................................................................................ 32 2.5.1 Representación basada en vértices .............................................................. 33 2.5.2 Representación basada en aristas ................................................................ 34 2.6 Descriptores algebraicos QuBiLS-MAS ............................................................ 35 2.7 Grupos de átomos para descriptores locales ...................................................... 38 2.8 Operadores de agregación .................................................................................. 40 2.9 Conclusiones parciales ....................................................................................... 41 3. Automatización del proceso de cálculo ToMoCoMD-CARDD QuBiLS-MAS ................... 43. 3.1 Detalles Técnicos ............................................................................................... 43 3.2 Diseño del software ............................................................................................ 43 3.3 Complejidad temporal ........................................................................................ 48 3.3.1 Transformaciones algebraicas de matrices no estocásticas a probabilidad mutua 48 3.3.2 Transformaciones algebraicas de matrices no estocásticas a simples estocásticas ................................................................................................................ 49 3.3.3 Cálculo de las contribuciones atómicas ...................................................... 50 3.4 Análisis de Desempeño ...................................................................................... 51 V.
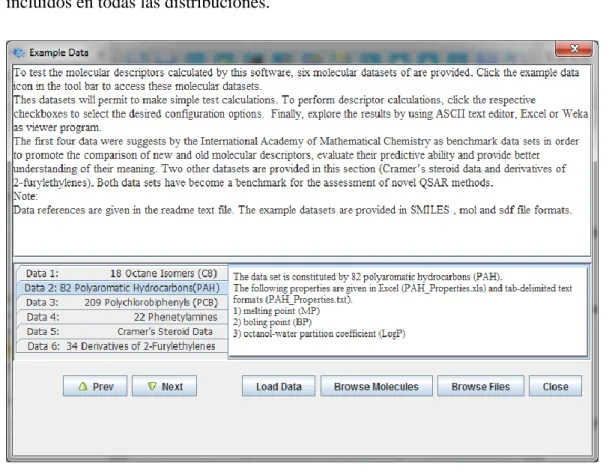
(7) 3.5 Funcionalidades Complementarias .................................................................... 60 3.5.1 Mecanismo automático de control de excepciones ..................................... 60 3.5.2 Decodificador de etiquetas (Descriptor Search)......................................... 60 3.5.3 Datos de ejemplo para principiantes (Example Data) ................................ 62 3.5.4 Modo de procesamiento por lotes (Batch Mode) ........................................ 62 3.6 Conclusiones parciales ....................................................................................... 63 4. Validación de los nuevos índices moleculares...................................................................... 65. 4.1 Análisis de variabilidad y comparación con otros enfoques .............................. 65 4.1.1 Análisis comparativo para átomos de hidrógeno y electrones Lone-Pair .. 66 4.1.2 Análisis comparativo entre las formas algebraicas ..................................... 67 4.1.3 Análisis comparativo respecto a la representación basada en átomos y enlaces 69 4.1.4 Análisis comparativo para los mecanismos de normalización matricial .... 70 4.1.5 Análisis comparativo entre índices totales y locales................................... 72 4.1.6 Análisis comparativo para las propiedades atómicas.................................. 73 4.1.7 Análisis comparativo respecto a los operadores de agregación .................. 76 4.1.8 Análisis comparativo entre restricciones aplicadas a la matriz operadora (cut-off) 79 4.1.9 Análisis comparativo con el software DRAGON ....................................... 81 4.1.10 Análisis comparativo respecto a los descriptores calculados por otras aplicaciones ............................................................................................................... 86 4.2 Análisis factorial por componentes principales ................................................. 87 4.2.1 Independencia lineal según la forma algebraica ......................................... 88 4.2.2 Independencia lineal según el formalismo Matricial .................................. 89 4.2.3 Independencia lineal según la restricción aplicada a la matriz operadora .. 91 4.2.4 Independencia lineal según el operador de agregación............................... 92 4.2.5 Independencia lineal entre los índices QuBiLS-MAS y DRAGON ........ 95 4.3 Modelación de la relación cuantitativa estructura-actividad ............................ 100 4.3.1 Modelación interna según la forma algebraica ......................................... 101 4.3.2 Modelación respecto a otros enfoques reportados en la literatura ............ 102 4.4 Conclusiones parciales ..................................................................................... 105 Conclusiones ............................................................................................................................... 106 Recomendaciones ........................................................................................................................ 107 Referencias bibliográficas ........................................................................................................... 108 Anexos ......................................................................................................................................... 116 A1: Definición matemática de los operadores de agregación ..................................................... 116 A2: Índices cargados en factores exclusivos para DRAGON 3D ............................................... 119 A3: Codificación de las etiquetas para los descriptores QuBiLS-MAS ...................................... 120 A4: Nombre y actividad biológica de las estructuras en la base de datos de Esteroides utilizada en la modelación QSAR ................................................................................................................... 121 A5: Comportamiento de la memoria virtual en las pruebas de rendimiento ............................... 122 A6: Tiempo consumido en segundos por cada experimento ....................................................... 123 A7: Configuración detallada para los proyectos calculados en el análisis QSAR....................... 124 A8: Información suplementaria para la modelación de la relación cuantitativa estructura-actividad ..................................................................................................................................................... 125. VI.
(8) Introducción La Química matemática es el área de la química dedicada a las nuevas y no triviales aplicaciones de las matemáticas a la química, y se ocupa principalmente de los modelos matemáticos para los fenómenos químicos [1], haciendo uso intensivo de las técnicas informáticas más avanzadas para solucionar problemas químicos[2], pero no debe confundirse con la química computacional, la cual es otra rama de la química que utiliza ordenadores para ayudar a resolver problemas bio-químicos, usando los resultados de la química teórica, incorporados en algún software para calcular las estructuras y las propiedades de moléculas [3]. Mientras sus resultados normalmente complementan la información obtenida en experimentos químicos, pueden, en algunos casos, predecir fenómenos. químicos no observados previamente. La química computacional es. ampliamente utilizada en el diseño de nuevos medicamentos y materiales[4]. La investigación que nos ocupa se encuentra sobre la unión de ambas disciplinas, con un énfasis particular en la manipulación de la información en la estructura química, utilizando diferentes parámetros numéricos, que a lo largo de este trabajo son denominados de forma equivalente índices o descriptores moleculares (DMs). Formalmente se define un descriptor molecular como: “el resultado final de un procedimiento lógico y matemático que transforma información química codificada dentro de una representación simbólica de una molécula en un número de utilidad o el resultado de algún experimento estandarizado” [5]. El término utilidad empleado en la definición anterior tiene un doble sentido: este significa que el número puede brindar una visión más profunda en la interpretación de propiedades moleculares y/o puede formar parte de un modelo que posibilite la predicción de alguna propiedad de interés en nuevas moléculas [5]. Los descriptores moleculares pueden ser obtenidos a partir de diferentes teorías, tales como: química-cuántica, teoría de información, química orgánica, teoría de grafos, entre otras; y han sido ampliamente utilizados en diversos estudios y campos científicos, entre los que se pueden mencionar los estudios de la relación cuantitativa estructura actividad/propiedad QSAR/QSPR (del inglés, Quantitative structureactivity/property relationship), estudios de similitud/disimilitud, el diseño de fármacos y 1.
(9) la toxicología. Atendiendo a la naturaleza de su definición, los descriptores moleculares pueden ser clasificados atendiendo a su dimensión (denotada en todos los casos por la letra D), en 0D o de conteo (que tienen en consideración la fórmula química), 1D o basados en fragmentos químicos de las moléculas, 2D o basados en rasgos topológicos de la estructura molecular, y en 3D o geométricos. La definición local o por fragmentos (grupos) de átomos, como también se reporta en la literatura, es una de las características deseables para todo descriptor molecular [5], debido a que muchas de las propiedades o incluso actividades biológicas dependen más de los rasgos estructurales para determinadas zonas moleculares que de la molécula como un todo. Es importante resaltar, que los índices moleculares de definición local reportados hasta el momento para obtener índices globales o por tipos de átomos, solamente utilizan un enfoque aditivo (la suma de las partes es igual al total) [6-9]. Una de las principales aplicaciones de los descriptores moleculares se encuentra en el descubrimiento de nuevos fármacos. Esta actividad por lo general involucra métodos para estudios QSAR/QSPR, los cuales se enfocan en determinar correlaciones entre los índices moleculares y la actividad farmacológica de las estructuras químicas. Estos métodos pueden ser categorizados en QSAR bidimensionales (2D-QSAR) y tridimensionales (3DQSAR). Los procedimientos 2D-QSAR comúnmente utilizan información química derivada de la constitución e información topológica de las moléculas (descriptores 0D2D) [10]. Por su parte los métodos 3D-QSAR, consideran propiedades físico-químicas de los compuestos químicos y su conformación bio-activa [11-13], empleando para ello los llamados índices geométricos (3D), denominados así, por proporcionar información estructural relacionada con la representación tridimensional de la estructura molecular. En los reportes [6, 7, 14-18], Marrero-Ponce y colaboradores, introdujeron nuevos conjuntos de descriptores moleculares de relevancia para estudios QSAR/QSPR y diseño racional de fármacos, mediante los cuales describen características concernientes a la topología de las moléculas. Estos métodos codifican información de la estructura molecular por medio de las formas algebraicas lineal, bilineal y cuadrática utilizando matrices de densidad-electrónica grafo-teórica, que actúan como matriz operadora en el 2.
(10) producto cartesiano para las formas algebraicas, codificadas hasta el momento solamente mediante el formalismo no estocástico y simple estocástico [7, 8]. Estos índices han sido extendidos a considerar un mayor número de características para las moléculas de pequeño y mediano tamaño, basados en los distintos enfoques que pueden presentar las estructuras químicas [17, 19]. Sin embargo, y al igual que otros índices reportados, únicamente tienen en cuenta relaciones entre pares de átomos y sólo aplican conceptos relacionados con las transformaciones directas sobre las aplicaciones lineales (formas lineal, bilineal y cuadrática). En la actualidad, la cantidad de índices moleculares reportados superan los 3000 [5]. A pesar de ello, la búsqueda de nuevos descriptores sigue siendo de interés científico, en aras de mejorar la diversidad de estos y posiblemente recoger información estructural no codificada adecuadamente por los índices definidos hasta la fecha. Entre las limitaciones existentes de los índices moleculares actuales, se pueden mencionar las siguientes: 1) no existe un descriptor (variable) capaz de capturar, y por lo tanto codificar toda la información química extrínseca e intrínseca de la estructura molecular, 2) aún existen propiedades moleculares descritas inadecuadamente por los índices definidos, 3) varios descriptores necesitan redefinirse a formas más simples o generalizadas en aras de disminuir el costo computacional, sin comprometer su calidad y al mismo tiempo aumentar su versatilidad, 4) una gran cantidad de índices moleculares solamente tienen una definición global, es decir, no pueden ser utilizados para estudios donde sean considerados fragmentos o tipos de átomos de la estructura química, 5) la definición previa de los índices algebraicos únicamente involucra un mecanismo para la normalización de matrices, no se aplican restricciones topológicas sobre estas. y. solamente se propone el enfoque aditivo como método de generalización, además 6) la mayoría de las herramientas informáticas que asumen el cálculo automático de los índices publicados hasta la fecha pertenecen a empresas privadas con carácter comercial y costosas licencias, que restringen la extensión y/o corrección de las limitaciones que presentan. Por todo lo anteriormente planteado se tiene como problema científico el siguiente: se necesita una aplicación informática, que codifique descriptores moleculares con 3.
(11) información relevante de alta capacidad predictiva, que permitan estimar actividad y propiedades moleculares de interés a través de relaciones funcionales sencillas y fáciles de calcular, de forma que resulten útiles en la identificación y el diseño racional de nuevos fármacos, así como para los disímiles estudios de la química computacional. Este problema se desglosa en las siguientes preguntas de investigación: 1. ¿La definición de nuevos mecanismos de normalización matricial con restricciones topológicas, de algoritmos localizados mediante fragmentos de átomos y de nuevas propiedades sobre las ponderaciones atómicas, mejoran la calidad de los índices propuestos respecto a los reportados? 2. ¿Cómo extender el enfoque aditivo utilizado como generalización del cálculo de los índices moleculares, para que distinga otras características relevantes respecto las definidas en la literatura? 3. ¿Qué efecto provoca aplicar las propuestas presentadas en la modelación de la relación cuantitativa estructura-actividad? Para darle solución al problema científico se planteó el siguiente objetivo general de investigación que consiste en: definir nuevos índices moleculares de dos dimensiones en una herramienta de cálculo automatizada, que dote a los investigadores de otra alternativa para enfrentar problemas complejos, especialmente en el área de la predicción de actividad biológica con la modelación asistida por computadora. Este objetivo general fue desglosado en los siguientes objetivos específicos: 1. Extender la codificación de los descriptores algebraicos bidimensionales, incorporando mecanismos de normalización matricial más sensibles, nuevas restricciones sobre la matriz operadora y diferentes propiedades atómicas. 2. Aplicar nuevos operadores de agregación como algoritmos de generalización sobre el cálculo de los índices propuestos.. 4.
(12) 3. Diseñar e implementar una herramienta informática para automatizar, de forma eficiente, el proceso de cálculo. 4. Evaluar los índices propuestos respecto a los reportados en la literatura acorde a la calidad de la información que estos capturan y a la utilidad en la predicción de la actividad biológica en compuestos orgánicos. Después de haber evaluado el marco teórico se formularon las siguientes hipótesis de investigación como respuestas a las preguntas de investigación: H1: Los algoritmos de normalización con probabilidad mutua y doble estocástico para el balanceo de las matrices operadoras, aumentan la variabilidad de los índices propuestos respecto a los reportados en la literatura y mejoran el poder predictivo de los modelos obtenidos para la predicción de la actividad biológica. H2: El uso de los operadores de agregación como generalización del enfoque aditivo, propuesto originalmente, mejora la codificación de las estructuras químicas, obteniendo índices colineales y ortogonales respecto a los reportados en la literatura. Esta investigación muestra resultados cuya novedad científica radica en la obtención de nuevos índices topológicos bidimensionales, totales y locales a partir de los conceptos de formas lineales, bilineales y cuadráticas, que codifican la información estructural con más sensibilidad. Además, se proponen varios operadores de agregación sobre los índices atómicos (denominados también como LOVIs: siglas en inglés de LOcal Vertex InvariantS) que generalizan la obtención de descriptores moleculares como una combinación lineal de los mismos. Por otra parte, se introducen por primera vez descriptores algebraicos calculados con algoritmos de balanceo y normalización matricial, con restricciones de conectividad aplicadas sobre la matriz operadora y que son perceptivos para distintos grupos o fragmentos químicos predeterminados. Este trabajo posee como valor práctico el desarrollo de una herramienta informática, donde se encuentra implementada toda la teoría correspondiente al cómputo de los nuevos índices moleculares. Además, tiene incorporado varios módulos complementarios. 5.
(13) que contribuyen 1) al curado y limpieza de las bases de datos de las estructuras químicas que pueden ser analizadas, 2) a la decodificación para las etiquetas univocas asociadas a los índices propuestos, y 3) a procesar por lotes distintas configuraciones de los índices algebraicos. Como valor metodológico, este trabajo aporta la aplicación de una serie de procedimientos para evaluar la calidad de nuevos descriptores moleculares, tales como, análisis de variabilidad basado en entropía de Shannon, análisis de componentes principales y estudios QSAR/QSPR. Además, el uso de operadores de agregación como generalización de la combinación lineal de las contribuciones atómicas puede utilizarse en descriptores moleculares cuyo cálculo puede ser descompuesto en índices atómicos. La tesis está estructurada en cuatro capítulos. En el Capítulo 1 se tratan de manera general, los fundamentos de las formas lineales, bilineales y cuadráticas, así como la definición de los índices bidimensionales que usan la química grafo-teórica y topología molecular. También se presenta el marco experimental estadístico para validar los resultados obtenidos. Seguidamente, en el Capítulo 2, son definidos los procedimientos para el cálculo de los nuevos índices moleculares, detallando de esta forma el concepto de vector molecular y los enfoques matriciales que son utilizados. En el Capítulo 3 se aborda el diseño del software implementado y las pruebas de rendimiento realizadas a este. Finalmente en el Capítulo 4, se muestran los resultados de las diferentes validaciones y pruebas estadísticas realizadas a los índices propuestos. Este documento culmina con las Conclusiones, Recomendaciones, Referencias Bibliográficas y Anexos.. 6.
(14) 1 Fundamento teórico Este capítulo presenta la teoría de grafos aplicada a la modelación topológica de las estructuras moleculares, los conceptos y definiciones relacionadas con los índices topológicos bidimensionales reportados en la literatura, así como su clasificación, requisitos principales y las características más relevantes. Son expuestas además, las diferentes variantes para representar matemáticamente la topología molecular y su modelación mediante una aplicación algebraica. Finalmente, se abordan las técnicas estadísticas utilizadas para validar los resultados de los índices algebraicos que se proponen en esta investigación.. 1.1 Química Grafo-Teórica y Topología Molecular La teoría de grafos ha sido ampliamente aplicada a diversos campos de la ciencia [20]. Un grafo se expresa usualmente como vértices interconectados por aristas [20, 21], donde cada vértice del grafo se representa un objeto y la arista que conecta dos vértices representa la relación entre estos dos objetos. En la química grafo-teórica los objetos del grafo pueden representar orbitales, átomos (o sus núcleos), enlaces, grupos de átomos, moléculas, o colecciones de moléculas. Las aristas de un grafo químico simbolizan las interacciones entre objetos químicos y se usan para definir enlaces químicos, reacciones, mecanismos de reacciones, modelos cinéticos, u otra relación o transformación de los objetos químicos. Sobre la química grafo-teórica y sus aplicaciones podemos encontrar abundante literatura [22-26], donde afloran las principales aplicaciones de los grafos químicos: 1) los índices topológicos (ITs) y otros índices estructurales para los estudios QSAR [9, 27-34], 2) el enfoque de orbitales moleculares de Hückel [35, 36], 3) la enumeración de isómeros, 4) la percepción de simetría estructural y codificación de compuestos químicos [37-39], 5) los grafos cinéticos y de reacción [40], y por último 6) el diseño de síntesis asistida por computadora [41]. De las aplicaciones antes mencionadas, centramos el propósito del presente trabajo en la relacionada con la obtención de descriptores estructurales para el diseño molecular. Por tanto, se presentan las definiciones más relevantes reportados en la literatura en el campo 7.
(15) de la química grafo-teórica y en especial del uso de descriptores moleculares basados en la teoría de grafos en los estudios QSAR, y se enuncian una serie de conceptos y términos matemáticos que serán utilizados en el desarrollo de esta tesis y son imprescindibles para la comprensión de los resultados tanto de la literatura como los alcanzados en la presente investigación.. 1.1.1 Representación topológica de las moléculas La representación topológica de un objeto es aquella que brinda información sobre el número de elementos que lo componen y sus conectividades[42, 43]. En ese sentido, se define a la topología como aquella parte del álgebra que estudia las posiciones e interconexiones de los elementos dentro de un conjunto[44]. Si la topología es aplicada a las moléculas, da lugar a la topología molecular. Por tanto, consideremos en este trabajo que una representación topológica de una molécula puede ser obtenida utilizando un grafo molecular.. 1.1.2 Invariante grafo-teórica Una invariante grafo-teórica es aquella propiedad del grafo que no depende del orden de numeración de los elementos del mismo, las cuales pueden obtenerse por manipulación algebraica del grafo. Como se ha señalado, los grafos moleculares no son una representación numérica de la estructura química y aunque las matrices sí constituyen una representación algebraica, tienen como desventaja que no constituyen invariantes grafoteóricas, ya que su construcción depende de la numeración dada a los vértices del grafo. Sin embargo, una simple invariante como el número de vértices, puede obtenerse a partir de la matriz de adyacencia [45]. Resulta evidente, que para los estudios QSAR, el diseño de fármacos, el tamizaje virtual, etc.; se necesitarían obtener índices numéricos que caractericen estructuralmente los grafos moleculares y que estos índices sean inevitablemente invariantes. Estos descriptores invariantes son los llamados ITs los cuales serán el objeto principal de esta tesis.. 8.
(16) 1.1.3 Esquemas de normalización Como se puede apreciar, las matrices constituyen la herramienta matemática más común para codificar la información estructural de las moléculas [5], teniendo especial interés las relacionadas con la topología molecular. Sin embargo, es inusual utilizar transformaciones probabilísticas sobre estas matrices. A pesar de ello, matrices estocásticas son definidas en el marco de trabajo MARCH-INSIDE [46, 47] y en los descriptores ToMoCoMD-CARDD 2D [19, 48]. En el caso de los descriptores moleculares MARCH-INSIDE, que codifican información relativa a la distribución de los electrones en la molécula, basado en un enfoque simple estocástico acerca de la idea de ecualización de la electronegatividad (principio de Sanderson) [49]. Por su parte, los índices ToMoCoMD-CARDD 2D definen la matriz estocástica de densidad electrónica grafo-teórica, la cual describe cambios en la distribución de los electrones en el tiempo a través de un backbone molecular. Para esta matriz es considerado un caso hipotético, en el cual un conjunto de átomos están inicialmente libres en el espacio y distribuidos alrededor de los núcleos atómicos. En este sentido, los electrones en un átomo arbitrario pueden moverse a otros átomos diferentes mediante una red de enlace químico. Por otra parte, Carbó-Dorca [50] también empleó un escalamiento estocástico aplicado a Matrices de Similitud Cuántica (QSM), bajo el principio de que cualquier fila o columna de una matriz QSM puede fácilmente convertirse en una distribución de probabilidad discreta, realizando así un escalamiento simple estocástico, donde la suma de los elementos de cada fila (o columna) son usados como un factor de escala. Formalmente, pueden definirse las matrices estocásticas como matrices cuadradas para las cuales la suma de cada fila (matrices estocásticas derechas), o la suma de cada columna (matrices estocásticas izquierdas), es igual a 1, significando esto que los elementos de las filas o columnas consisten en número reales no negativos que pueden ser interpretados como probabilidades [51].. 9.
(17) 1.2 Formas algebraicas En matemática, una aplicación vectoriales. y. se denomina isomorfismo entre los espacios. sobre un mismo campo escalar. , si se cumple que. es biyectiva y. además los siguientes axiomas:. ( ̅ (. para todo ̅ ̅. ̅). ( ̅). ( ̅). 1.1. ( ̅). . Esto significa que si se toman dos elementos ̅ ̅ en. y. determina su suma ̅. ̅). y se. ̅ esto corresponde en F a buscar las imágenes de los elementos y. sumarlas. Lo mismo sucede con el producto por un escalar, debido a que si la imagen ( ̅ ) de un vector ̅ se desea multiplicar por un escalar , basta multiplicar ̅ por buscar la imagen Esta aplicación. y. ̅. sólo realiza transformaciones si el espacio de llegada es una copia del. espacio de partida. Sin embargo, en ocasiones surgen correspondencias entre espacios que no son idénticos estructuralmente, pero estas correspondencias trasladan en cierta medida, la estructura de uno de los espacios a otro (por ejemplo,. ). A estas. funciones se les denominan aplicaciones lineales, y las mismas cumplen también con los axiomas de linealidad especificados en la Ecuación 1.1. En particular, si el espacio de llegada de una aplicación lineal es un número real. entonces se le conoce como. forma lineal. En un espacio vectorial. de dimensión finita n, la forma lineal queda completamente. determinada por los valores que se asignan a los elementos de una base. De esta manera, *̅ ̅. si ̅. ∑. ̅̅̅+ es una base de , y cualquier vector ̅ ̅ entonces:. 10. está determinado por.
(18) 1.2. donde, (. ). son las coordenadas del vector ̅ en la base E, y ( ̅) puede. expresarse respecto a la base. *̅ ̅. ̅ + del espacio vectorial F de dimensión m,. como sigue:. 1.3. y sustituyendo 1.3 en 1.2 se obtiene:. 1.4. De manera análoga a la forma algebraica lineal, es definida la forma bilineal, como una aplicación. la cual es lineal en todos sus argumentos tomados. separadamente. Es decir, que esta función satisface las condiciones de linealidad mostradas en la Ecuación 1.5, para cualquier escalar ̅ ̅ ̅̅̅ ̅̅̅ ̅̅̅̅. y cualquier vector. ̅̅̅̅ que pertenecen al espacio vectorial .. 1.5. 11.
(19) Por lo tanto, si *̅ ̅. es un espacio vectorial real en ̅̅̅+ es un sistema base de. cualquier vector (. ). ̅ y ̅ de. (. ) y el conjunto de vectores. , se pueden definir formas no ambiguas para. , siendo sus coordenadas (. ). y. respectivamente. Esto significa que los vectores ̅ y ̅ son expresados. como combinaciones lineales respecto a la base E del espacio vectorial V, como es mostrado a continuación:. 1.6. Por tanto,. 1.7. y si son tomados los coeficientes. como n x n escalares de. ( ̅. ̅ ), es decir,. 1.8. entonces,. 1.9. 12.
(20) De esta manera, la ecuación especificada para la forma bilineal, puede ser escrita como una ecuación matricial simple (ver Ecuación 1.9), donde , - es un vector columna de las coordenadas de ̅ sobre un sistema base canónico de. y , - es la transpuesta del. vector columna , - de las coordenadas de ̅ en la misma base de forma bilineal. es simétrica si ( ̅ ̅). ( ̅ ̅). ̅ ̅. . Finalmente, una. .. Por otro lado y a partir de la definición simétrica de una forma bilineal, es determinada la forma cuadrática, como una función. ( ̅). donde, (. ̅). ( ̅). dada por:. ̅. ( ̅ ̅). 1.10. .. 1.3 Índices bidimensionales basados en la topología molecular Los índices bidimensionales (2D) basados en la topología molecular son el resultado numérico de alguna invariante „extraída‟ del grafo molecular. Es decir, los índices topológicos (ITs) son descriptores moleculares que se obtienen de una invariante grafoteórica [52-56]. Por tanto, los ITs son números calculados a partir de la representación de una molécula como un grafo, siendo independientes de la numeración de los vértices y aristas en el grafo molecular. Estos índices codifican información estructural contenida en la representación bidimensional (2D) de la molécula. Esta descripción „topológica‟ de la molécula contiene información de la conectividad entre átomos (o enlaces) en la molécula y codifica la talla, forma, ramificación, heteroátomos y la presencia de enlaces múltiples [27-29, 31-34, 57]. La información de la molécula contenida en los ITs puede usarse en la descripción de propiedades fisicoquímicas y biológicas [26, 58-60]. 13.
(21) Un resumen completo sobre los ITs es realmente imposible, debido a la gran cantidad de estos índices que han sido publicados en la literatura y al número de ellos que cada año son introducidos. La mayoría de los ITs propuestos están relacionados con la matriz de adyacencia de vértices, de distancias o de combinaciones de estas. No obstante, en la actualidad los ITs han sido clasificados acorde a su naturaleza en, primera, segunda y tercera generación [61], lo cual facilita su estudio. Un tratamiento abarcador de todos los descriptores moleculares disponibles fue recientemente publicado por Todeschini y Consonni [62]. Los ITs de primera generación son números enteros basados en propiedades del grafo como un todo, tales como las distancias topológicas. Los índices más representativos de esta clase son el índice W de Wiener,[63] el índice Z de Hosoya,[64] y los índices B y C de Balaban.[65] De todos estos ITs solo W ha sido usado extensamente en estudios QSAR y en el descubrimiento de nuevos fármacos. El índice de Wiener fue definido en 1947 y desde entonces ha constituido una importante fuente de inspiración para el desarrollo de nuevos ITs. Los resultados obtenidos con el índice W en la modelación molecular propició la definición de varios índices relacionados con este. Algunos de estos son el cuasi-Wiener índice,[66] el índice de Kirchhoff,[67] el índice RDSUM,[68] y el hiper índice de Wiener [69, 70], entre otros. La suma de los grados de cada enlace fue introducido, al mismo tiempo que el índice de Wiener, por Platt; por lo cual este es conocido como el índice F de Platt [71]. Otro de los ITs de primera generación que también ha sido ampliamente utilizado en estudios QSAR, es el índice de Zagreb desarrollado en 1975 utilizando las valencias de los átomos [72]. Los índices topológicos de segunda generación son números reales basados en las propiedades del grafo íntegro. La mayoría de los ITs utilizados en los estudios QSAR y en el diseño/descubrimiento de fármacos en estos momentos, pertenecen a esta clase. Los conjuntos de descriptores moleculares más útiles de esta clase, son los llamados índices de conectividad molecular [28, 29, 73] Estos índices están basados en una invariante grafo-teórica introducida por Randic años atrás, para computar un índice de „ramificación‟ para los alcanos.[74] Estos índices fueron extendidos por Kier y Hall para tener en cuenta la diferenciación entre heteroátomos y los diferentes subgrafos en la 14.
(22) molécula. Los índices de conectividad molecular de „valencia‟ son expresados de la siguiente forma:[28, 29, 73] La mayoría de los ITs analizados hasta ahora describen la estructura de la molécula como un todo por lo tanto pueden considerarse como descriptores moleculares globales o totales. Kier y Hall, a comienzos de la década del 90, introdujeron un nuevo IT denominado índice del estado electrotopológico (E-estado); basado en una invariante grafo-teórica para un átomo en la molécula, representando la accesibilidad de electrones de este átomo. Este índice puede ser considerado como un descriptor molecular local; el mismo codifica información acerca del ambiente topológico y de las interacciones electrónicas debidas a todos los demás átomos en la molécula [9, 30, 57, 75, 76]. También Carrasco y colaboradores introdujeron recientemente un nuevo índice denominado. índice. del. estado. refractotopológico. (i)[77].. Marrero-Ponce. y. colaboradores propone un nuevo enfoque basado en una representación vectorial y otra matricial de la estructura molecular. Estos a su vez están relacionadas con la representación grafo-teórica por medio de un pseudografo de la estructura química[15, 78-84]. Los ITs de tercera generación son números reales basados en propiedades locales del grafo molecular. Estos índices son de reciente publicación [85-87]. Otros de los ITs de esta clase están basados en la aplicación de la teoría de la información a términos de sumas de distancias o sobre nuevas matrices no simétricas introducidas en la literatura [88-90].. 1.3.1 Propiedades que debe poseer un nuevo índice topológico Varios autores han propuesto propiedades que deben poseer los nuevos ITs, consideradas actualmente de un nivel de sofisticación muy alto, el cual deben alcanzar los nuevos descriptores moleculares diseñados. Estas propiedades son presentadas por Randic [91] así: 1) interpretación estructural directa, 2) buena correlación con al menos una propiedad, 3) buena discriminación entre isómeros, 4) localmente definidos, 5) generalizables a análogos superiores, 6) linealmente independientes, 7) simplicidad, 8) no. 15.
(23) basados en propiedades físico-químicas, 9) no trivialmente relacionados con otros índices, 10) eficiencia de construcción, 11) basados en conceptos estructurales familiares, 12) mostrar una dependencia correcta con el tamaño y 13) tener cambios graduales con cambios graduales en la estructura. En realidad, muchos de estos atributos tienen cierto grado de interrelación. Al emplear los ITs para la caracterización de una molécula intrínsecamente tenemos cierta pérdida de información, ya que se está tratando de representar un objeto bidimensional por un número simple. Esta cuestión es crítica, cuando se trata de una actividad biológica que depende de la interacción estéreo-específica del receptor con un sitio de la molécula. Sin embargo, los ITs pueden contener una sorprendente información estructural sobre las moléculas, lo que los hace de gran utilidad en los fines prácticos de sus aplicaciones. Existen dos posibilidades de resolver la pérdida de información de los descriptores grafo-teóricos. La primera, consiste en la generalización de un descriptor simple a análogos „superiores‟ y segunda, la generalización de la invariante grafoteórica como secuencias de números [91]. La generalización de los índices es necesaria para salvar situaciones en las que un descriptor simple no es suficiente y la investigación de una familia de descriptores estructuralmente relacionados puede resolver el problema. Sucede que en la obtención de una familia de descriptores de diferentes órdenes (familia de descriptores relacionados), como generalización de un simple descriptor, se ha observado que muchos de estos son colineales (varios índices pueden ser expresados como combinación lineal de los restantes, por lo que puede existir „redundancia de la información‟). La independencia lineal u ortogonalidad de los índices es uno de los atributos deseables, pues los descriptores colineales pueden afectar la „estabilidad del coeficiente de correlación‟ y dificulta la interpretación de los modelos obtenidos. En este sentido, es importante también que los descriptores sean ortogonales en relación a los restantes ITs. Esta independencia lineal de los índices significa que los mismos conduzcan a una correlación con una propiedad que no es satisfactoriamente explicada por los otros descriptores existentes en la comunidad científica actual.. 16.
(24) La definición local de los descriptores se refiere al hecho de que estos no sean obtenidos de forma global o total para una estructura molecular, sino que puedan ser definidos sobre determinados fragmentos de la propia estructura. La interpretación en términos estructurales, es otro de los atributos deseables para un nuevo IT, ya que desde el punto de vista de su aplicación en estudios QSAR, solo aquellos índices que estén basados en conceptos estructurales simples ayudarán a interpretar propiedades complejas en términos estructurales.. 1.3.2 Los índices topológicos y sus aplicaciones Las aplicaciones de los ITs han estado dirigidas fundamentalmente hacia la predicción cuantitativa de propiedades físico-químicas y biológicas de compuestos orgánicos, en estudios que se han denominado QSPR y QSAR, respectivamente. Esta división, no es solo formal, porque aunque el método en ambos tipos de estudio es similar, por lo general, la actividad biológica es una propiedad mucho más compleja que las propiedades físico-químicas, debido a la gran cantidad de factores que influyen en la bio-actividad de un compuesto químico. La aplicación de los ITs al diseño y selección de nuevas entidades químicas es probablemente una de las áreas más activas de investigación en la aplicación de tales descriptores a problemas biológicos. Uno de los primeros ejemplos del diseño de nuevos compuestos en el uso de estos índices, fue descrito por la Upjohn & Pharmacy en 1993, y fueron capaces de diseñar una nueva clase de compuestos de la familia de las heteropiperazinas. con. actividad. contra. la. HIV-retrotransferasa.[92-97]. Más. recientemente, Graasy y colaboradores fueron capaces de diseñar y sintetizar un péptido que mostró una actividad inmunosupresora aproximadamente 100 veces mayor que los compuestos líderes ensayados [98].. 1.4 Métodos estadísticos en la Química Computacional La informática química es una disciplina que recopila herramientas matemáticas y estadísticas para tratar con datas químicas complejas[99-102]. Estas técnicas son 17.
(25) utilizadas para la recopilación, la elaboración, el análisis y la caracterización de conjuntos de datos, de forma tal que se pueda obtener información útil de ellas. Las mismas, hoy en día, se interceptan no solo con varios campos de la Matemática y la Estadística clásica, sino también de la Inteligencia Artificial (IA) y otras ramas de la ciencia de la computación[103, 104]. En las próximas secciones serán presentadas solo aquellas técnicas que son de interés en la presente investigación.. 1.4.1 Análisis de Variabilidad El método de Análisis de Variabilidad, propuesto por Godden y colaboradores[105, 106], cuantifica el contenido de información y, por lo tanto, la variabilidad de los descriptores moleculares (DMs). Este método no supervisado está basado en el cálculo de la Entropía de Shannon (SE) [107], bajo el principio de que variables deseables para análisis quimiométricos pudieran poseer elevados valores de entropía como un indicador de su tendencia a cambiar gradualmente con la modificación de la estructura molecular; mientras que variables redundantes pudieran tener valores bajos, siendo cero el límite para aquellas variables que contienen el mismo valor para estructuras diferentes. Esta técnica permite evaluar la calidad de los DMs como entidades independientes y ha sido utilizada en la literatura para comparar el desempeño de conjuntos de DMs implementados en diferentes paquetes computacionales, así como en estudios de diversidad molecular[105, 108].. 1.4.2 Análisis de Componentes Principales El Análisis de Componentes Principales (ACP) es un procedimiento matemático que transforma un conjunto de variables correlacionadas de respuesta en un conjunto menor de variables no correlacionadas, llamadas componentes principales [109, 110]. Este procedimiento es útil cuando al trabajar con varias variables (posiblemente con un gran número de variables), se cree que existe cierto grado de redundancia en las mismas. En este caso, redundancia significa que algunas variables están correlacionadas con otras, tal vez porque están midiendo un mismo hecho. Debido a esta redundancia, puede reducirse las variables observadas en componentes principales, de forma tal, que en su totalidad expliquen la mayor varianza posible de las variables originales. 18.
(26) El primer componente extraído generalmente tiene en consideración la mayor varianza de todos los componentes determinados en el estudio. Por su parte el segundo componente, tendrá dos características fundamentales: 1) que explicará la mayor varianza posible que el primero no tuvo en cuenta, y 2) que no está correlacionado con el primero, es decir, la variables cargadas en él son ortogonales a las cargadas en el primer componente. Estas dos características son presentadas por el resto de los componentes determinados en el análisis. Habitualmente, se calculan los componentes sobre variables originales estandarizadas, es decir, variables con media 0 y varianza 1. Esto equivale a tomar los componentes principales, no de la matriz de covarianzas sino de la matriz de correlaciones (en las variables estandarizadas coinciden las covarianzas y las correlaciones). Así, los componentes son autovectores de la matriz de correlaciones y son distintos de los de la matriz de covarianzas. Si se actúa así, se da igual importancia a todas las variables originales. En la matriz de correlaciones todos los elementos de la diagonal son iguales a 1. Si las variables originales están tipificadas, esto implica que su matriz de covarianzas es igual a la de correlaciones, con lo que la variabilidad total (o la traza) es igual al número total de variables que hay en la muestra. La suma total de todos los autovalores es p y la proporción de varianza recogida por el autovector j-ésimo (componente) queda expresada por. .. Por lo tanto, si se tiene en cuenta que en estudios de informática química se trabaja con un gran conjunto de variables (índices moleculares), el uso del ACP resulta ser de gran utilidad, dado que permite explorar a priori la posible existencia de no colinealidad entre las variables, un requisito importante para nuevos descriptores moleculares y también en la modelación de las propiedades físico-químicas y biológicas.. 1.4.3 Regresión lineal múltiple La Regresión lineal múltiple (RLM) estudia las relaciones entre una variable dependiente y un conjunto de variables independientes (o explicativas). Así mismo, la regresión múltiple remite a la correlación múltiple, que analiza la relación entre una serie de 19.
(27) variables independientes o predictores (X1, X2, ..., Xk), considerados conjuntamente, con una variable dependiente o criterio[111]. Sus fundamentos se hallan en la correlación de Pearson.[112] La recta de regresión múltiple tiene la siguiente forma:. Y = a + b1 X1 + b2 X2 +...+ bk Xk. 1.11. siendo „a‟ la intersección o término constante, Y es la variable dependiente o explicada y b1, b2, …, bn son los parámetros que miden la influencia de las variables explicativas tienen sobre el regresando. Por tanto, la RLM puede utilizarse en la predicción de los valores de la variable dependiente, en base a una combinación de variables independientes [113].. 1.4.3.1 Análisis de la varianza El ANOVA (acrónimos del inglés ANalysis Of VAriance) puede comprobar la hipótesis de que. . La variabilidad total de la variable dependiente se divide entre la parte. atribuible a la regresión y la parte residual. La distancia de un punto cualquiera. a la ̅. se sub-divide en dos partes [112]:. 1.12. donde,. es el valor predicho,. dependiente. La diferencia ( (. el valor observado, y ̅ es la media de la variable ) se denomina residuo de la regresión, y la diferencia. ̅ ) corresponde a la distancia explicada por la regresión, y representa el aumento. en la estimación del. mediante la recta de regresión, ver ecuación 1.11.. En el ANOVA, F viene dada por:. 20.
(28) 1.13. donde, F sigue una distribución de Fisher-Snedecor con grados de libertad asociados a los parámetros E y R respectivamente, siendo variables de la ecuación y. , el número de. la cantidad de instancias. La media cuadrática (MC) se. obtiene dividiendo la suma de cuadrados del ANOVA entre los grados de libertad. Por tanto F permite comprobar si el modelo de regresión se ajusta a los datos y evaluar si se rechaza la hipótesis nula, según la cual coeficiente de determinación (. . Si el modelo se ajusta a los datos, el. ) puede calcularse a partir de la suma de cuadrados. (SC) del ANOVA mediante:. 1.14. Es importante señalar que, la mayoría de las investigaciones QSAR/QSPR han sido realizadas usando la técnica de RLM [114, 115], fundamentalmente por su carácter lineal, paramétrico y su “simplicidad”.. 1.4.3.2 Multicolinealidad entre variables El término multicolinealidad se utiliza generalmente para describir la situación en que un gran número de descriptores moleculares (variables o atributos) están altamente intercorrelacionados. Las variables que se aproximan a ser una combinación lineal de las otras, se denominan multicolineales o colineales [112, 116-118]. Una multicolinealidad alta, produce errores estándares altos en los coeficientes de regresión y dificulta estimar la importancia relativa de los descriptores en el modelo, lo cual afecta la interpretación de las actividades modeladas en términos estructurales. La importancia relativa puede determinarse al valorar el incremento en la R, cuando se añade una variable a la ecuación que ya contiene las demás variables. . El método más utilizado para detectar la 21.
(29) existencia de variables colineales es obtener una matriz de correlaciones entre los descriptores moleculares. Uno de los métodos más utilizados para detectar la interdependencia entre variables, es la tolerancia. Problemas con la redundancia de la información y la colinealidad, han sido ilustrados con el uso de ITs, tales como los índices de conectividad molecular [119, 120]. El nivel aceptable de colinealidad es algo subjetivo y en ese sentido se ha reportado que coeficientes de correlación entre las variables aceptables están en el rango de 0.4-0.9 [121].. 1.4.3.3 Identificación y selección de valores atípicos En estadística, existen varios criterios para identificar si un dato experimental, de un conjunto de datos experimentales, es probable que sea un valor atípico[122, 123] (o también conocido en la literatura como outlier). Los outliers son puntos que se desvían significativamente del modelo encontrado (no se ajustan al modelo) o son pobremente predichos por estos, afectando los parámetros estadísticos del mismo [124]. Generalmente, la identificación de outliers busca un mejoramiento cualitativo del modelo. Un buen ejemplo ha sido mostrado por Cronin y colaboradores en la modelación de la toxicidad de compuestos carbonílicos alifáticos para T. pyriformis [121], donde para un total de 140 compuestos solo se obtuvo un moderado ajuste estadístico R2 = 0.753 y al remover una cantidad de cinco outliers el parámetro R2 aumentó hasta 0.853 [121]. Existen varias técnicas para detectar la presencia de subgrupos y outliers, tales como: los análisis de los residuales estandarizados, el método de Leverage, la estadística DFITS, la distancia de Cook y el método de LOOOD (del inglés Leave One Out Outlier Detection)[125, 126].. 1.4.4 Validación estadística de los modelos El enfoque convencional adoptado en los análisis QSAR, basado en la RLM, es considerar el parámetro R2 („varianza explicada‟), R y s. Las variables como R2 varían entre 0 y 1, donde 1 significa un modelo perfecto (explica el 100% de la variable respuesta Y) y 0 identifica un modelo sin ningún poder de explicación. Entonces un alto. 22.
(30) valor de R2 y una baja s, son condiciones necesarias para la validez del modelo RLM, o sea, como en ANOVA la validez viene dada solo por el ensayo F, si varios modelos pasan esta prueba, el de mayor R2 y/o menor s será el mejor modelo encontrado. Según Kier la significación estadística puede evaluarse de tres formas diferentes:[127] 1) comparación del valor de F con el valor tabulado, 2) determinación de casos bien clasificados en la serie de entrenamiento (SE) y 3) validación externa. Además, los métodos de validación cruzada también pueden aplicarse a este tipo de modelos.[128] Ogino y colaboradores han propuesto otro enfoque para seleccionar la mejor, la cual se selecciona teniendo en cuenta el análisis de la combinación de tres criterios:[129] 1) la combinación de variables que minimice el número de compuestos mal clasificados, 2) el empleo de un número menor de variables, y 3) minimizar la colinealidad entre las variables independientes. Las herramientas que pueden ser utilizadas para acceder a la validación de los modelos QSAR obtenidos por RLM, la mayoría pueden extrapolarse de la siguiente forma: [130] 1) Aleatorización de la variable respuesta (Y- Randomización), 2) validaciones cruzadas, 3) división de la data de compuestos en serie de entrenamiento (SE) y en serie de predicción (SP) y 4) confirmación del poder predictivo utilizando SP externas. A continuación desarrollaremos brevemente solo los puntos referidos a los enfoques de validación de los modelos que son de nuestro interés.. 1.4.5 Validación interna de los modelos La validación cruzada (CV, acrónimos en inglés para cross-validation) opera haciendo un número (K) de reducidas modificaciones al conjunto de compuestos de la data original y entonces calcula la precisión de las predicciones de cada uno de los resultados de los modelos [131, 132]. Se crean K conjuntos de datos modificados tomando uno o más grupos de compuestos de los datos, en donde cada observación (compuestos) se toma una vez, sobre el número total de ciclos. El modelo es ajustado a los nuevos datos, dejando la parte omitida fuera, y estos se evalúan en el modelo para computar las predicciones de los compuestos que fueron excluidos. Este procedimiento se repite para cada conjunto de. 23.
(31) datos modificados. El poder predictivo del modelo puede expresarse como q2, el cual ha sido denominado como la „varianza predictiva‟ o la „varianza de la validación cruzada‟, definida por (. ), y puede ser calculada acorde a la siguiente fórmula:. yi yi q2 1 2 yi y. . 2. . 1.15. . donde yi , yi y y son la actividad observada, la actividad estimada y el promedio (media) para el i-ésimo compuesto, respectivamente. Cuando se utiliza un solo compuesto en cada grupo de CV se generan K grupos y el procedimiento se conoce como “dejando uno fuera” y sus siglas en ingles son LOO (acrónimo de Leave One Out). No obstante, Shao ha mostrado que desde el punto teórico y práctico, el procedimiento de dejar „varios‟ fuera (LSO, Leave Several Out) es preferible al LOO.[133] Este resultado se hace más notable cuando el número de instancias se incrementa. La técnica de LSO siempre deja fuera una porción de los datos creando una perturbación constante en la estructura de los datos. Wold y Eriksson recomiendan utilizar un valor de K alrededor de siete, al utilizar el procedimiento de CV [130]. Como un criterio significativo para acceder a la calidad del modelo puede ser usado el promedio de la medias de los errores en valores absolutos, (Mean Absolute Error), para cada uno de los grupos dejados fuera [134]. En la técnica de re-muestreo (bootstrap) [135, 136], el tamaño original de la data (n) es preservado para el conjunto de entrenamiento, mediante la selección de n objetos con repetición, de manera que, el conjunto de entrenamiento contiene algunos objetos repetidos y el conjunto de evaluación está constituido por lo objetos no seleccionados. Este procedimiento de construir conjuntos de entrenamiento y de predicción aleatoriamente es repetido miles de veces, y en cada iteración se calculan los estadísticos relevantes y el promedio de estos constituye el estimado del re-muestreo. 24.
(32) El método del revuelto (Y-scrambling) es empleado para evaluar la correlación al azar [137, 138]. En esta técnica, se calcula un modelo lineal de regresión para la verdadera variable respuesta (Y), junto con un número de regresiones repetidas (200-300 veces) con las mismas variables pero con la variable dependiente aleatoriamente cambiada ( ̃ ) Luego se calcula para cada modelo la varianza explicada. y se evalúa la correlación. entre la respuesta verdadera y la revuelta de la siguiente manera:. 1.16. donde,. es la varianza explicada para el modelo obtenido con los mismos predictores. teniendo el i-ésimo vector revuelto, y. es la correlación entre los vectores para la. respuesta verdadera y la k-ésima revuelta. Un valor del intercepto cercano a cero implica que el modelo no es obtenido al azar, mientras que un intercepto grande indica que los modelos aleatorios poseen el mismo desempeño que el modelo verdadero, razón por la cual no tendría buena calidad y se pudiera considerar que las variables están aleatoriamente correlacionadas. La validación externa permite evaluar si los modelos obtenidos son generalizables a nuevos compuestos químicos, y de esta forma analizar el “verdadero” poder predictivo de los mismos [137]. Para esto se divide la data en 2 conjuntos: la serie de entrenamiento para construir el modelo, y la serie de predicción, que no es utilizada en la selección de variables ni en el desarrollo del modelo, sino usada exclusivamente para evaluar el modelo tras su formación. El estadístico utilizado para comparar la capacidad predictiva de los modelos de regresión es el conocido. (desviación estándar de los errores. en la predicción) [139] el cual es calculado como se muestra a continuación:. 25.
(33) 1.17. donde, ,. es la cantidad de objetos o instancias en el conjunto de prueba o predicción, y. expresan la respuesta estimada y predicha del i-ésimo caso, respectivamente.. 1.5 Conclusiones parciales En este capítulo fueron expuestas teoría de grafos aplicada en la química computacional y las definiciones relacionadas con los índices topológicos bidimensionales reportados en la literatura hasta la fecha y por último, se mencionaron las técnicas estadísticas a utilizar para validar los índices propios QuBiLS-MAS. A manera de conclusión aflora que los índices topológicos que consideran invariantes grafo-teóricas y/o esquemas de normalización, no siempre son adecuados para estudios quimio-informáticos y que el empleo de conceptos relacionados con aplicaciones lineal, bilineal y cuadráticas pudieran contribuir a obtener descriptores moleculares capaces de captar información distinta, con alta variabilidad y con mejor poder discriminatorio que los actuales.. 26.
(34) 2 Índices QuBiLS-MAS: Teoría, extensión y nueva codificación En este capítulo se presenta la teoría elemental sobre la codificación de las estructuras moleculares para obtener los descriptores QuBiLS-MAS (acrónimo del inglés Quadratic, Bilinear and Linear MapS based on Graph–Theoretic Electronic-Density Matrices and Atomic weightingS), así como las nuevas definiciones matemáticas para los índices locales, los formalismos matriciales empleados y los operadores de agregación utilizados como generalización de la combinación lineal de las contribuciones atómicas.. 2.1 Representación matricial del grafo molecular El concepto de grafo ha sido representado algebraicamente mediante una matriz, por varios autores [140-142]. Esta representación de los compuestos químicos es fundamental para manipular matemáticamente las moléculas y obtener una descripción cuantitativa de su estructura. Si se utiliza una matriz de adyacencia para representar un grafo molecular G con N vértices, esta matriz de adyacencia A=A (G), es simétrica y cuadrada, de N x N elementos, y se define como:. a ij = 1 si i j y eij E (G) 2.1. = 0 si i j ó eij E (G). donde E (G) representa el conjunto de las aristas del grafo G. En la matriz de adyacencia A (G) los elementos de la diagonal principal, o sea los a ij tal que i=j, representan los. vi V (G) . En la Figura 2.1 se representan, a manera de ejemplo, el grafo molecular del 2,3-dimetil-butano y las matrices de adyacencia de orden k (k = 0, 1, 2) asociadas a dicho grafo. El elemento k aij de la matriz de adyacencia de orden k, que se denota como. 27.
(35) Ak (G), significa el número de caminos unitarios de longitud k que unen los vértices vi y vj.. 6. 5 2 1. 3 4. 1 0 0 0 A (G ) 0 0 0 . 0 1 0 0 0 0. 0 0 1 0 0 0. 0 0 0 1 0 0. 0 0 0 0 0 1. 0 0 0 0 1 0. 0 1 0 1 A (G ) 0 0 0 . 1 0 1 0 1 0. 0 1 0 1 0 1. 0 0 1 0 0 0. 0 1 0 0 0 0. 1 0 0 0 1 1 2 A (G ) 0 0 1 0 0 0 . 0 3 0 1 0 1. 1 0 3 0 1 0. 0 1 0 1 0 1. 1 0 1 0 1 0. 0 1 0 1 0 1. Figura 2.1 Grafo molecular de la molécula 2,3-dimetil-butano y las matrices de adyacencia asociadas de orden cero (A0(G)), uno (A1(G)) y dos (A2(G)).. A partir de esta representación se define la matriz de distancia, la cual es el esquema base para la formalización algebraica de los índices moleculares QuBiLS-MAS, partiendo de un grafo G con N vértices se denota D (G) y es una matriz cuadrada de N x N elementos, simétrica y sus elementos dij se definen así:. dij = δij si i ≠ j 2.2. = 0 si i = j. donde δij es la longitud del camino más corto entre vi y vj. Por ejemplo la matriz de distancias topológicas asociada al grafo molecular para 2,3-dimetil-butano de la Figura 2.1, se representa en la Figura 2.2. 0 1 2 3 2 3 . 1 0 1 2 1 2. 2 1 0 1 2 1. 3 2 1 0 3 2. 2 1 2 3 0 3. 3 2 1 2 3 0. Figura 2.2 Matriz de distancias topológicas entre los vértices del grafo molecular del 2,3-dimetil-butano. 28.
(36) 2.2 Formalización de la matriz no-estocástica La estructura de las moléculas se codifica en los índices propios QuBiLS-MAS mediante la matriz no estocástica de orden k del pseudografo molecular G , se denota M k , y constituye una matriz de N x N elementos, simétrica, donde N representa el número total de átomos en la molécula. La definición formal de esta matriz de distancia se expresa en el epígrafe anterior y los coeficientes k mij son los elementos de la matriz no estocástica (NS) de orden k asociada al grafo molecular G , y se definen como:. mij Pij si i j y ek E (G). Lii si i j. 2.3. 0 en otros casos. donde E (G) representa el conjunto de aristas de G ; Pij es el número de aristas (enlaces) entre los vértices (átomos) vi y vj, además Lii es el número de pares de electrones no pareados (lone-pairs electrons)[143] o lazos en el átomo vi. Por tanto los valores de la diagonal principal no son siempre ceros, logrando así una mayor discriminación de las estructuras moleculares. De manera general, los elementos mij Pij de tales matrices representan el número de interacciones fuertes y débiles entre un átomo i y otro átomo j. La matriz NSk proporciona el número de caminos de longitud k que unen los vértices vi y vj.. 2.3 Mecanismos de normalización matricial Con el propósito de normalizar la k-ésima matriz NS se definen tres esquemas de probabilidad asociados con las interacciones inter-atómicas en la estructura química, estos son 1) matrices estocásticas, 2) doble estocásticas y 3) de probabilidad mutua, los cuales serán abordados en esta sección. 29.
(37) 2.3.1 Simple estocástica La matriz simple estocástica (SS) de orden k del pseudografo molecular G , se denota SSk, y se obtiene directamente de la matriz no estocástica NSk. La matriz SSk es cuadrada de orden N y los elementos k s ij que la componen son todos no negativos y se definen[19, 144] por la ecuación 2.4.. k k. sij . k. mij. SUM i. k. . mij. k. i. 2.4. k donde k mij son los elementos de NSk y SUM i es la suma de los elementos de la i-ésima. fila de NSk , y representa el grado del vértice de orden k del átomo i, que finalmente se denota como k i . Debe señalarse que la matriz SSk tiene la propiedad que la suma de los elementos en cada fila es igual a la unidad[51].. 2.3.2 Doble estocástica Estas transformaciones simples estocásticas generan matrices no simétricas, y por lo tanto otro enfoque a considerar sería obtener matrices donde la suma de los elementos de todas las componentes sume 1. Con el objetivo de equilibrar las probabilidades en ambos sentidos para estas matrices bidimensionales, se define la matriz doble estocástica como una matriz con entradas reales no negativas cuyas filas y columnas sumen 1[5]. Este tipo de matriz será referida como la DSk y el procedimiento para calcular la matriz doble estocástica asociada a una no estocástica no es trivial. Sinkhorn postuló que una matriz estrictamente positiva A puede ser escalada a una matriz doble estocástica B por [145]:. 2.5. 30.
(38) donde, Dg es una matriz diagonal. Posteriormente, Sinkhorn y Knopp extendieron este teorema para considerar matrices no negativas y propusieron un algoritmo de iteración para el balanceo de matrices[146]. En este sentido, la matriz DSk puede ser calculada a partir de la matriz NSk usando la ecuación 2.5 y el algoritmo Sinkhorn-Knopp.. 2.3.3 Probabilidad mutua Finalmente se introduce la matriz de probabilidad mutua MPk donde sus elementos se calculan también partiendo de la matriz no estocástica NSk definidos por la ecuación 2.6.. ∑. donde,. ∑. 2.6. denota la probabilidad mutua entre los núcleos atómicos i y j, S significa el. espacio muestral, el cual es calculado por la sumatoria de todos los elementos de NSk. Es importante resaltar que el enfoque simple estocástico ha sido previamente usado en otros trabajos [19, 48], pero los esquemas doble estocástico y de probabilidad mutua son presentados por primera vez como una alternativa para la estrategia de normalización.. 2.4 Restricciones aplicadas a la matriz operadora Para tener en consideración solamente algunos tipos de interacciones entre los átomos (por ejemplo interacciones de corto, mediano y largo alcance), tanto en índices locales como globales se aplican tres restricciones diferentes sobre la matriz operadora que expresa la información del pseudografo molecular. Estas restricciones son aplicadas a todas las matrices (NSk, SSk, DSk y MPk) involucradas en el cálculo de las formas algebraicas que se presentan en el epígrafe anterior. 31.
Figure




+7





Documento similar
The 'On-boarding of users to Substance, Product, Organisation and Referentials (SPOR) data services' document must be considered the reference guidance, as this document includes the
In medicinal products containing more than one manufactured item (e.g., contraceptive having different strengths and fixed dose combination as part of the same medicinal
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
Products Management Services (PMS) - Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in
This section provides guidance with examples on encoding medicinal product packaging information, together with the relationship between Pack Size, Package Item (container)
Package Item (Container) Type : Vial (100000073563) Quantity Operator: equal to (100000000049) Package Item (Container) Quantity : 1 Material : Glass type I (200000003204)
Cedulario se inicia a mediados del siglo XVIL, por sus propias cédulas puede advertirse que no estaba totalmente conquistada la Nueva Gali- cia, ya que a fines del siglo xvn y en
El nuevo Decreto reforzaba el poder militar al asumir el Comandante General del Reino Tserclaes de Tilly todos los poderes –militar, político, económico y gubernativo–; ampliaba
