METODOLOGÍA SINTÉTICA
APLICADA A LA SÍNTESIS DE
FÁRMACOS
Tema 5
Enfermedades infecciosas: síntesis de
antibióticos y antifúngicos
Tema 5. Enfermedades infecciosas: síntesis de antibióticos y antifúngicos
5.1. Antibióticos 1
5.2. Bacterias 1
5.2.1. Bacteras Gram-positivas y Gram-negativas 2 5.3. Mecanismos de acción de los antibióticos 5
5.3.1. Acción sobre la pared celular: inhibición de la síntesis del
peptidoglicano 6
5.3.1.a Penicilinas 8
5.3.1.b. Cefalosporinas 12
5.3.1.c. Bacitracina (neomicina) 15
5.3.1.d. Vancomicina 16
5.3.2. Inhibidores de la síntesis de ácidos nucleicos 17 5.3.2.a. Metronidazol: inhibidor de la síntesis de ADN 17 5.3.2.b. Quinolonas y fluoroquinolonas: inhibición de la girasa 18 5.3.2.c. Rifampicina: inhibición de ARN polimerasa 22 5.3.3. Inhibición de la síntesis proteica de la bacteria 23
5.3.3.a. Inhibidores 50S 24
5.3.3.a.1. Eritromicina 24
5.3.3.a.2. Cloranfenicol 25
5.3.3.a.3. Lincomicina y clindamicina 26
5.3.3.a.4. Oxazolidinonas 27
5.3.3.b. Inhibidores 30S 29
5.3.3.b.1. Tetraciclinas 29
5.3.3.b.2. Estreptomicina 30
5.3.3.b.3. Espectinomicina 31
5.3.3.b.4. Kanamicina 31
5.3.4. Interacción con la membrana citoplasmática bacteriana: polimixinas 32 5.3.5. Sulfamidas: antimetabolitos bacteriostáticos 33 5.4. Mecanismos de resistencia a los antibióticos 36 5.4.1. Infecciones graves provocadas por bacterias 37
5.5. Síntesis de quinolonas 39
5.5.1. Síntesis de ofloxacina 39
5.5.1.a. Análisis retrosintético 39
5.5.1.b. Síntesis 40
5.5.1.c. Cuestiones 41
5.5.2. Síntesis de levofloxacina 41
5.5.2.b1. Síntesis de levofloxacina mediante separación de
diastereoisómeros 41
5.5.2.b2. Síntesis de levofloxacina mediante el empleo del pool quiral 42
5.5.2.c2. Cuestiones 42
5.5.2.b3. Síntesis alternativa de levofloxacina mediante el empleo
del pool quiral 43
5.5.2.c3. Cuestiones 43
5.5.3. Síntesis de mesilato de pazufloxazina 43 5.5.3.b. Síntesis de mesilato de pazufloxacina 44
5.5.3.c. Cuestiones 45
5.5.4. Síntesis de moxifloxacina 46
5.5.4.a. Análisis retrosintético 46
5.5.4.b. Síntesis 46
5.5.4.b.1. Síntesis de la octahidro-pirrolopirrolidina 5.35 47 5.5.4.b.2. Síntesis de la dihidroquinolona 5.36 48
5.5.4.b.3. Pasos finales 48
5.5.4.b.4. Pasos finales: alternativa sintética 48
5.5.4.c. Cuestiones 49
5.5.5. Síntesis de gemifloxacina 49
5.5.5.a. Análisis retrosintético 49
5.5.5.b. Síntesis 50
5.5.5.b.1. Síntesis de la pirrolidina 5.50 50 5.5.5.b.2. Síntesis de la quinolinona 5.49 y pasos finales 51
5.5.6. Síntesis de prulifloxacina 52
5.5.6.a. Análisis retrosintético 52
5.5.6.b. Síntesis 53
5.5.6.c. Cuestiones 54
5.6. Síntesis de oxazolidinonas 54
5.6.1. Sïntesis de linezolida 54
5.6.1.a. Análisis retrosintético 55
5.6.1.b. Síntesis 56
5.6.1.c. Cuestiones 57
5.6.2.b. Síntesis alternativa de linezolida 57
5.6.2.c. Cuestiones 58
5.7. Síntesis de cloranfenicol 59
5.7.a.1. Análisis retrosintético 59
5.7.b.1. Síntesis 59
5.7.a.2. Análisis retrosintético alternativo 62
5.7.b.2. Síntesis 62
5.7.c.2. Cuestiones 63
5.7.d. Reacción de epoxidación asimétrica de Sharpless 63 5.7.e.Resolución cinética enantioselectiva de Sharpless 65
6. Antifúngicos 68
6.1. Infecciones fúngicas 68
6.2. Micosis superficiales 68
6.3. Micosis profundas o sistémicas 69
6.4. Fármacos antimicóticos 69
7. Síntesis de antifúngicos 74
7.1. Síntesis de miconazol y econazol 74
7.1.a. Análisis retrosintético 74
7.1.b. Síntesis 75
7.2. Síntesis de ketoconazol 76
7.2.a. Análisis retrosintético 76
7.2.b. Síntesis 77
7.2.c. Cuestiones 78
7.3. Síntesis de fluconazol 79
7.3.a. Análisis retrosintético 80
7.3.b. Síntesis 80
7.3.c. Cuestiones 81
7.4. Síntesis de itraconazol 81
7.4.a. Análisis retrosintético 82
7.4.b. Síntesis 83
7.4.c. Cuestiones 85
7.5. Síntesis de terbinafina 85
7.5.a. Análisis retrosintético 86
7.5.b. Síntesis 86
5.1. Antibióticos
En términos biológicos un antibiótico es una sustancia química producida por un ser vivo que, a bajas concentraciones, mata o impide el crecimiento de ciertas clases de microorganismos sensibles, generalmente bacterias. Ejemplos de antibióticos naturales son los propóleos, sustancias secretadas por las abejas, con las cuales éstas recubren las paredes de sus colmenas a fin de protegerlas de bacterias y hongos.
El término antibiótico abarca en la actualidad a cualquier compuesto, ya sea de origen natural o sintético, capaz de matar a las bacterias (antibiótico bactericida) o detener su crecimiento (antibiótico bacteriostático).
El arranque de la historia moderna de los antibióticos se sitúa en 1897, cuando Ernest Duchesne descubrió en Francia las propiedades antibióticas de la especie Penicillium, aunque su trabajo pasó casi inadvertido entre la comunidad científica. En 1909 Paul Ehrlich desarrolló en Alemania el antibiótico de corto espectro Salvarsan, que se empleó en el tratamiento de la sífilis.
El momento crucial en el descubrimiento de antibióticos corrió a cargo del médico británico Alexander Fleming cuando, el 22 de septiembre de 1928, al inspeccionar sus cultivos descubrió que un hongo había colonizado espontáneamente una de las placas de Petri sembradas con Staphylococcus aureus. Fleming observó que las colonias bacterianas que se encontraban alrededor del hongo, identificado posteriormente como Penicillium notatum, eran transparentes debido a una lisis bacteriana. Aunque no pudo purificar el material obtenido, denominado posteriormente penicilina, publicó su descubrimiento en 1929 en la revista British Journal of Experimental Pathology.
Más de 10 años después, Ernst Chain y Howard Walter Florey produjeron una forma purificada de la penicilina y la utilizaron en seres humanos. Por sus trabajos sobre la penicilina A. Fleming, E. Chain y H. Florey fueron galardonados con el premio Nobel de Medicina en el año 1945.
5.2. Bacterias
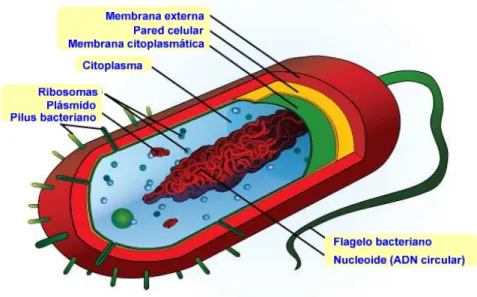
Las bacterias son microorganismos unicelulares que presentan un tamaño de algunos micrómetros de largo (entre 0,5 y 5 μm, por lo general) y diversas formas, incluyendo esferas, barras y hélices. Las bacterias son procariotas y, por lo tanto, a diferencia de las células eucariotas (las constituyentes de los animales y las plantas) carecen de un núcleo delimitado por una membrana, aunque presentan un nucleoide, una estructura elemental que contiene una gran molécula circular de ADN.
El citoplasma de las bacterias carece de orgánulos delimitados por membranas y en él se pueden encontrar:
a) Ribosomas: utilizados en la síntesis de proteínas.
b) Plásmidos: pequeñas moléculas circulares de ADN que coexisten con el nucleoide.
c) Vacuolas: gránulos que contienen sustancias de reserva.
caso está compuesta por peptidoglicano, denominado también mureína. La pared celular está rodeada por la segunda membrana lipídica, denominada membrana externa.
El espacio comprendido entre la membrana citoplasmática y la pared celular, se denomina espacio periplásmico.
Figura 5.1. Representación esquemática de una bacteria de tipo bacilo Gram-negativa
La membrana citoplasmática de las bacterias es similar a la de las plantas y los animales, y está formada por una bicapa lipídica, compuesta fundamentalmente de fosfolípidos en la que se insertan moléculas de proteínas. La membrana citoplasmática de las bacterias realiza numerosas funciones entre las que se incluyen las de barrera osmótica, transporte, biosíntesis, transducción de energía, centro de replicación de ADN y punto de anclaje para los flagelos.
Las bacterias disponen de una pared celular que rodea a su membrana citoplasmática. Las paredes celulares bacterianas están hechas de peptidoglicano, sustancia compuesta por cadenas de polisacárido enlazadas por péptidos que contienen aminoácidos de la serie D. Estos aminoácidos no se encuentran en las proteínas, por lo que protegen a la pared bacteriana de la mayoría de las peptidasas.
5.2.1. Bacteras Gram-positivas y Gram-negativas
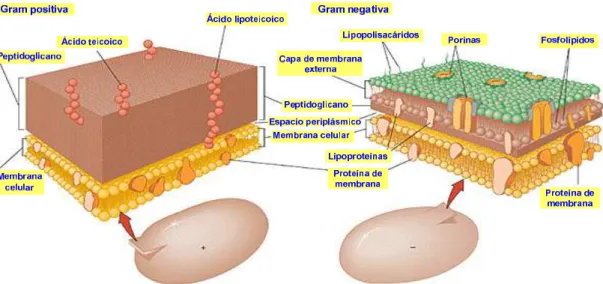
Las bacterias se clasifican en Gram-positivas y Gram-negativas en función de la reacción de la pared celular a la tinción de Gram, un tipo de tinción diferencial empleado en microbiología para la visualización de bacterias. El método debe su nombre al bacteriólogo danés Christian Gram, que desarrolló la técnica en 1884. Así, las bacterias Gram-positivas son la que se visualizan de color morado en la tinción, mientras que las bacterias Gram-negativas se visualizan de color rosa, rojo o grosella.
parte derecha de la figura 5.2 se visualizan las bacterias Escherichia coli (Gram negativas) tras ser teñidas con la tinción de Gram.
Figura 5.2. Visión al microscopio de bacterias Gram-positivas y Gram-negativas después de la tinción de Gram
Las bacterias Gram-positivas contienen una membrana citoplasmática rodeada de una gruesa pared celular compuesta de numerosas capas de peptidoglicano (véase la parte superior de la figura 5.3).
Por el contrario, las bacterias Gram-negativas presentan una membrana citoplasmática rodeada de una fina pared celular de peptidoglicano rodeada por una segunda membrana lipídica (la membrana externa) que contiene lipopolisacáridos y lipoproteínas (véase la parte inferior de la figura 5.3).
Figura 5.3. Representación de las paredes celulares de bacterias
Figura 5.4. Comparación de las paredes bacterianas
Los ácidos teicoicos son glicopolímeros aniónicos que se encuentran unidos covalentemente a la pared de peptidoglicano mediante un enlace fosfodiéster con el grupo hidroxilo C6 del ácido N-acetilmurámico del peptidoglicano. Las estructuras más comunes de los ácidos teicóicos están compuestas de un disacárido ManNAc(β1→4)GlcNAc que se une por el hidroxilo C4 del residuo de ManNAc a fosfato de glicerol, que a su vez se une a una cadena mucho más larga que consta de repeticiones de fosfato de glicerol o ribitol (véase la figura 5.5).1
Figura 5.5. Representación estructural de una pared de ácido teicoico
Los ácidos teicoicos están cargados negativamente y por lo tanto contribuyen a la carga negativa de la pared celular de las bacterias Gram-positivas.
Otros componentes característicos de las paredes de las bacterias Gram-positivas son los ácidos lipoteicoicos cuyas estructuras se indican en la figura 5.6. Los ácidos lipoteicoicos atraviesan totalmente la capa de peptidoglicano y se anclan a ésta mediante interacciones hidrofóbicas, al contrario que los acidos teicoicos que se unen covalentemente al peptidoglicano.
HO O P O O P O OX O O OH O O n X=D-alanina O O HO H HO HO O O O HO H HO HO O O O
C15H31
O
C15H31
Figura 5.6. Representación estructural de un ácido lipoteicoico
En la figura 5.7 se representa un fragmento de la pared celular de una bacteria Gram-positiva, en la que se puede observar la inserción de los ácidos teicoicos en la red de peptidoglicano y la penetración de los ácidos lipoteicoicos a lo largo de la malla de peptidoglicano y su anclaje en la membrana plasmática.
Figura 5.7
Las diferencias en la estructura de la pared celular dan lugar a diferencias en la susceptibilidad antibiótica. Por ejemplo, la vancomicina puede matar solamente a bacterias Gram-positivas y es ineficaz contra patógenos Gram-negativos, tales como Haemophilus influenzae o Pseudomonas aeruginosa.
5.3. Mecanismos de acción de los antibióticos
Los mecanismos de acción de los fármacos antibióticos son muy variados e incluyen la:
1) Inhibición de la síntesis del peptidoglicano constituyente de la pared celular de las bacterias.
2) Inhibición de la síntesis de ácidos nucleicos bacterianos mediante bloqueo del modo de acción de girasas y polimerasas.
3) Inhibición de la síntesis de proteínas bacterianas mediante interacción con el ribosoma bacteriano.
4) Interacción con la membrana citoplasmática bacteriana.
5) Inhibición de la síntesis del ácido fólico.
Figura 5.8. Modos de acción antibacteriana
5.3.1. Acción sobre la pared celular: inhibición de la síntesis del peptidoglicano
Muchos antibióticos actúan sobre el peptidoglicano, el principal componente de la pared celular, ya sea inhibiendo su formación o su organización. La deficiente formación de la pared celular provoca la debilitación de ésta, lo que conlleva su ruptura con la consiguiente muerte de la bacteria.
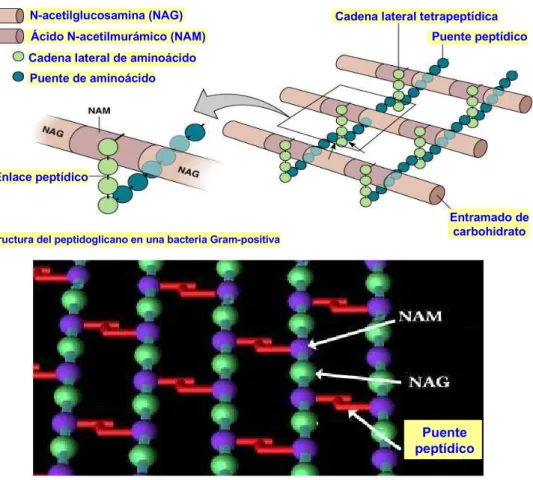
El peptidoglicano, o mureína, es un copolímero formado por una secuencia alternante de N-acetil-glucosamina y de ácido N-acetilmurámico, que se unen mediante enlaces β-1,4.
Enlazado al ácido N-acetilmurámico se encuentra una cadena peptídica de entre tres y cinco aminoácidos de longitud (véase la figura 5.9).
En bacterias Gram-positivas la pared celular es relativamente gruesa (entre 15-80 nanometros) y está formada por una serie de capas de peptidoglicano. El grupo ácido del ácido N-acetilmurámico se encuentra conectado con una cadena peptídica que contiene L-alanina, D-glutamato, L-lisina (usualmente) y D-alanina.
Figura 5.9. Estructura del peptidoglicano de una bacteria Gram-negativa
Las cadenas peptídicas esterificadas con el ácido N-acetilmurámico se entrecruzan formando una malla tridimensional mediante unión covalente del residuo de D-alanina de una cadena con el residuo de ácido meso-diaminopimélico de otra (véase la figura 5.10).
Figura 5.10. Entrecruzamiento de las cadenas de peptidoglicano
N-acetilglucosamina (NAG)
Ácido N-acetilmurámico (NAM)
Cadena lateral de aminoácido Puente de aminoácido
Enlace peptídico
Estructura del peptidoglicano en una bacteria Gram-positiva
Cadena lateral tetrapeptídica Puente peptídico
Entramado de carbohidrato
Puente peptídico
Figura 5.11. Representaciones de la malla de peptidoglicano
5.3.1.a Penicilinas
Las penicilinas pertenecen a la familia de antibióticos β-lactámicos. Estos compuestos ejercen su acción antibiótica interfiriendo la construcción de la pared celular de la bacteria.
Figura 5.12. Estructura de la penicilina G
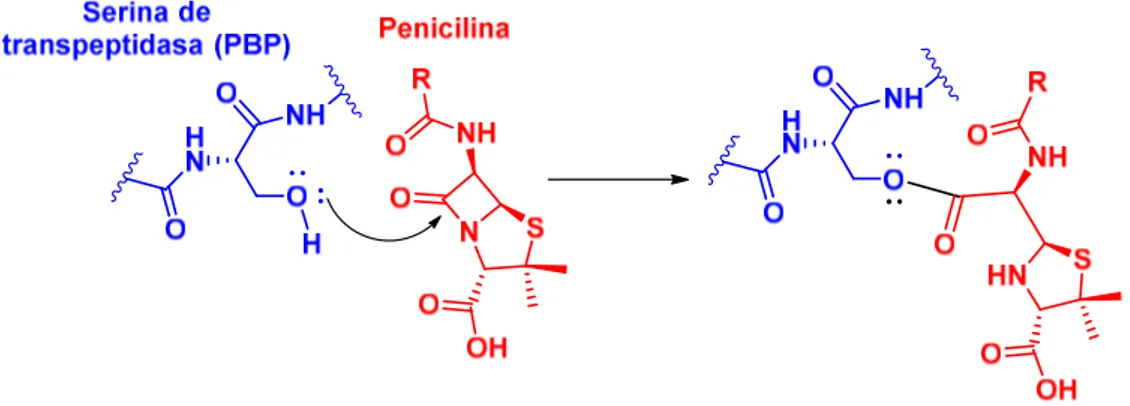
Las penicilinas inhiben la síntesis de la pared celular al interrumpir la reacción de transpeptidación necesaria para la biosíntesis del peptidoglicano. Las penicilinas inactivan las denominadas proteínas fijadoras de penicilina (PBPs del inglés Penicillin Binding Proteins), situadas en la superficie interna de la membrana bacteriana.
Las PBPs son enzimas con acción transpeptidasa, carboxipeptidasa y endopeptidasa y están implicadas en los estadios terminales de la síntesis de la pared bacteriana y de su organización durante el crecimiento y división.
acción de la transpeptidasa mediante la unión covalente de la penicilina en el centro activo de la enzima (figura 5.13).
Figura 5.13.Interacción covalente de las penicilinas con las transpeptidasas
En la figura 5.14 se indican los pasos de inactivación de la PBP por la penicilina. En el paso 1 la PBP se aproxima a la capa de peptidoglicano en crecimiento. En el paso 2 la enzima se une a la cadena peptídica y provoca el entrecruzamiento. En el paso 3 se observa cómo la PBP de disocia de la pared de peptidoglicano una vez ha generado el entrecruzamiento.
En el paso 4 se indica la entrada de la penicilina en el centro activo del PBP. En el paso 5 el anillo β-lactámico de la penicilina, representado como la parte superior de la letra P, se une covalentemente al centro activo de la PBP bloqueando la acción enzimática.
En la figura 5.15 se puede apreciar la unión de una penicilina a una transpeptidasa bacteriana.
Figura 5.15. Penicilina (en blanco) unida a una transpeptidada bacteriana
El aumento de la presión intracelular, como consecuencia de una inadecuada formación de la pared celular, provoca la muerte de la bacteria (figura 5.16).
Figura 5.16. Representación de la destrucción de una bacteria mediante lisis de la pared celular
Algunas especies de hongos del género Penicillium sintetizan de forma natural penicilinas, como la penicilina G (véase la figura 5.12).
hongo Penicillium. En el medio de cultivo se añade un exceso de un precursor de la cadena lateral y el microorganismo la introduce en el ácido 6-aminopenicilánico, el núcleo central de todas las penicilinas (figura 5.17).
Figura 5.17. Ácido 6-aminopenicilánico
Las penicilinas semisintéticas se obtienen mediante modificación química de derivados de la penicilina G. Así, la cadena lateral de la penicilina G se elimina mediante reacción con la enzima penicilina acilasa. Esta reacción enzimática proporciona el ácido 6-aminopenicilánico, que se modifica químicamente para dar lugar a un antibiótico con características mejoradas. Las penicilinas semisintéticas presentan, entre otras propiedades, mayor estabilidad al pH ácido, y mayor resistencia a betalactamasas. Entre este tipo de penicilinas está la penicilina V (véase la figura 5.18), que se puede administrar por vía oral, porque, al contrario que la penicilina G que debe administrarse de forma parenteral, resiste la hidrólisis ácida del estómago.
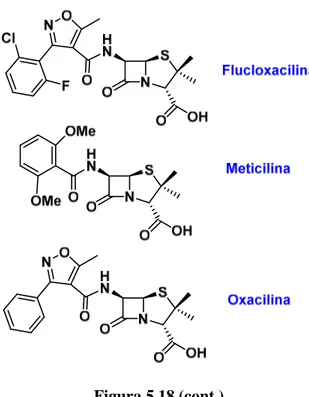
En la figura 5.18 se indican las estructuras de algunas penicilinas semisintéticas.2
Figura 5.18
Figura 5.18 (cont.)
5.3.1.b. Cefalosporinas
Las cefalosporinas son una clase de antibióticos betalactámicos con una estructura química bastante similar a la de las penicilinas. El núcleo central de las cefalosporinas es el ácido 7-aminocefalosporánico (figura 5.19). Las cefalosporinas inhiben la transpeptidación por unión a las proteinas PBPs implicadas en la última fase de la formación de la pared celular.
Figura 5.19. Ácido 7-aminocefalosporánico
La primera cefalosporina fue aislada en 1948 por el científico italiano Giuseppe Brotzu de cepas del hongo Cephalosporium acremonium que crecía en una alcantarilla de la isla de Cerdeña. G. Brotzu advirtió que las cepas del hongo producían una sustancia eficaz contra la salmonela (Salmonella typhi) causante de la fiebre tifoidea. Además, el filtrado sin procesar del hongo curaba infecciones por estafilococo. Del líquido de cultivo del hongo se obtuvieron tres antibióticos diferentes, denominados P, que eran eficaces contra bacterias Gram-positivas y contra bacterias Gram-negativas.
La cefalosporina C fue aislada en la Escuela de Patología Sir William Dunn de la Universidad de Oxford. Una molécula más eficaz derivada de la cefalosporina C fue comercializada por la compañía Eli Lilly en la década de los sesenta del siglo pasado.3
Las cefalosporinas se agrupan en generaciones según sus características antimicrobianas. Las primeras cefalosporinas se han clasificado en el grupo denominado de primera generación (figura 5.20).
Figura 5.20. Cefalosporinas de 1ª generación
Las cefalosporinas de espectro extendido se han clasificado como cefalosporinas de segunda generación. Cada nueva generación de cefalosporinas tiene más potencia frente a bacterias Gram-negativas.
Figura 5.21. Cefalosporinas de 2ª generación O H N N S COOH O N O N S HOOC
H2N Cefixima
O H N N S COOH O OAc N MeO N S
H2N Cefotaxima
O H N N S COOH O S N MeO N S H2N
N N H N O O Ceftriaxona O H N N S COO O N N O N S H2N
HOOC
Ceftazidina
Figura 5.22. Cefalosporinas de 3ª generación
5.3.1.c. Bacitracina (neomicina)
La bacitracina (neomicina) es un antibiótico de naturaleza peptídica producido por cepas de la variedad Tracy de la bacteria Bacillus subtilis. Fue aislada en 1943 de tejido dañado y polvo callejero desbridado de una fractura abierta de tibia que presentaba la niña Margaret Tracy como consecuencia de una accidente de tráfico. En la herida se halló una cepa de Staphylococcus aureus. Poco tiempo después, en un segundo cultivo había desaparecido el estafilococo pero en su lugar se halló una bacteria previamente desconocida, denomina Bacillus subtilis variedad Tracy, que producía un poderoso antibiótico.
La bacitracina se prescribe contra bacterias Gram-positivas, especialmente en heridas y mucosas, porque inhibe la formación de la pared celular de estos microorganismos.
Figura 5.24. Estructura de la bacitracina
La bacitracina actúa sobre el fosfato de bactoprenol, el compuesto encargado de transportar los monómeros de NAM-difosfato de uridina (UDP) y el NAG-difosfato de uridina a lo largo de la membrana bacteriana. La estructura del bactoprenol se indica en la figura 5.25.
Figura 5.25. Estructura del bactoprenol
La bacitracina se une al fosfato de bactoprenol e impide la desfosforilación de éste cuando regresa a la membrana citoplasmática (figura 5.26). Así, las moléculas de bactoprenol que no han perdido el segundo grupo fosfato no pueden unir nuevos monómeros y no pueden insertarlos en el peptidoglicano. Como consecuencia, la pared bacteriana en formación es débil y la bacteria muere por lisis osmótica.4
Citoplasma
Membrana citoplasmática Bacitracina
Bactoprenol
Periplasma
Figura 5.26. Modo de acción de la bacitracina
5.3.1.d. Vancomicina
La vancomicina es un glicopéptido biosintetizado por Nocardia orientalis (véase la estructura en la figura 5.27).
Figura 5.27. Estructura de la vancomicina
La vancomicina ejerce su acción antibiótica impidiendo la síntesis de la pared celular bacteriana mediante inhibición de la acción de las transpeptidasas. Así, durante el crecimiento de la pared bacteriana, las enzimas denominadas autolisinas se encargan de introducir en el peptidoglicano los bloques de construcción NAG-NAM-péptido. Estos bloques se unen a la parte final de la pared bacteriana en formación mediante la intervención de transglicosidasas. Finalmente, las transpeptidasas unen el péptido de un monómero NAG-NAM-péptido con el de otro monómero para dotar de fortaleza a la pared bacteriana mediante la formación de entrecruzamientos.
La vancomicina se une a los péptidos de los monómeros de peptidoglicano bloqueando la formación de entrecruzamientos en el peptidoglicano, lo que conduce a la formación de una pared bacteriana débil que provoca la muerte de la bacteria por lisis osmótica (figura 5.28).
Citoplasma
Membrana citoplasmática
Transpeptidasa Periplasma
Bloqueo del entrecruzamiento
Figura 5.28. Modo de acción de la vancomicina 5.3.2. Inhibidores de la síntesis de ácidos nucleicos
5.3.2.a. Metronidazol: inhibidor de la síntesis de ADN
El metronidazol es un antiparasitario del grupo de los nitroimidazoles que se emplea en el tratamiento de las infecciones provocadas por protozoarios y bacterias anaeróbicas. El metronidazol se prescribe también para el tratamiento de enfermedades dermatológicas como el acné rosácea.
contra bacterias anaeróbicas y protozoos, debido a la facilidad que exhiben estos microorganismos en la conversión intracelular del metronidazol en su forma activa.
Figura 5.29. Estructura del metronidazol
5.3.2.b. Quinolonas y fluoroquinolonas: inhibición de la girasa
Las quinolonas y las fluoroquinolonas actúan sobre la replicación del ADN bacteriano bloqueando o inhibiendo las enzimas topoisomerasa II (girasa) y topoisomerasa IV. Las topoisomerasas son las enzimas que actúan sobre la topología del ADN, ya sea enrollándolo, para permitir que se almacene de manera más compacta, o desenrollándolo en el proceso de replicación o lectura del mismo. La ADN girasa reduce la tensión molecular producida por el enrollamiento del ADN y lleva a cabo cortes de doble cadena que después son unidos por las ligasas.
Figura 5.30. Estructura general de quinolonas
La ADN-topoisomerasa II (ADN-girasa) es la diana de las quinolonas en el caso de bacterias Gram-negativas, mientras que la ADN-topoisomerasa IV, que fue descubierta tiempo después de que fuera descubierta la ADN-girasa, es el objetivo de las quinolonas en el caso de bacterias Gram-positivas (véase la figura 5.31).
También se han descubierto especies bacterianas en las cuales el mecanismo de acción de las quinolonas involucra la inhibición de ambos enzimas.
El primer fármaco antibacteriano de tipo quinolona fue el ácido nalidíxico (figura 5.32), que se introdujo en el mercado en el año 1962 para el tratamiento de las infecciones urinarias.5 Después de este fármaco se comercializaron otras quinolonas, como la cinoxacina
o el ácido pipemídico (figura 5.32).
Figura 5.32
En los años 1980 se introdujeron las fluoroquinolonas, antibacterianos con mejores actividades y propiedades farmacocinéticas que las quinolonas, ya que presentaban mayores tiempos de vida media y mejores eficacias orales. De entre esta segunda generación de antibacterianos quinolónicos cabe destacar la norfloxacina, la perfloxacina y la ciprofloxacina (figura 5.33).
Figura 5.33
El rápido desarrollo de resistencia a los antibióticos está obligando a investigar nuevas estructuras quinolónicas. En la figura 5.34 se representan las estructuras de algunas fluoroquinolonas de tercera generación que están siendo estudiadas desde el punto de vista clínico.
Figura 5.34
Desde el punto de vista químico el mecanismo de acción de las fluoroquinolonas involucra la interacción de las funciones carbonilo, carboxilo y flúor con residuos de ácido aspártico, serina y lisina de las girasas y topoisomerasas y con los residuos de purina, guanina y magnesio(II) presentes en el ADN. Estas interacciones crean un "cerrojo" para las topoisomerasas, las cuales no pueden efectuar el movimiento (impulsado por ATP) de apertura de las hebras de ADN, bloqueándose de esta forma los procesos biológicos necesarios para la vida de la bacteria.
A continuación se describirán con un poco más de detalle las relaciones entre la estructura y la actividad de las quinolonas.
a) Efecto quelante
La presencia de un grupo carboxilo en la posición C3, contigua al carbonilo cetónico, permite a las quinolonas la formación de quelatos metálicos (figura 5.35).
Figura 5.35. Efecto quelante de las quinolonas
Los quelatos metálicos de las quinolonas con los iones de metales de valencia superior como aluminio (III), magnesio (II), calcio (II), hierro (II y III) y cobre (II) conlleva la formación de complejos metálicos insolubles en agua que pueden interferir con los niveles óptimos de concentración en sangre del antibiótico. Esto supone un inconveniente desde el punto de vista de formulación de la droga y desde el punto de vista de la interacción con alimentos (especialmente derivados lácteos), interacción con otros medicamentos (como los antiácidos a base de aluminio y magnesio) e interacción con suplementos alimenticios que contengan sales de hierro como fuente de hierro adicional.
El problema de la quelación se puede evitar administrando conjuntamente un medio ácido, para prevenir así la formación del carboxilato al favorecer la forma de ácido carboxílico. Si tal co-administración no es posible, entonces el paciente no debe comer nada 1 hora antes o 1 hora después de la administración de la quinolona.
b) Carácter ácido-base
N O
COO N
F N O
Levofloxacina
N O
COO N
F N O N
O
COOH N
F N O
Base Ácido
H H
Figura 5.36. Carácter anfótero de las quinolonas
En medio alcalino la quinolona tiene carga negativa, lo que favorece su solubilidad en agua. A medida que el pH baja hacia valores más ácidos se alcanza el punto isoeléctrico de la molécula. La forma zwitteriónica, que se alcanza a valores de pH casi neutros, es la forma que más se logra absorber a los pH neutros requeridos para la absorción del medicamento.
En la tabla 5.1 se resumen las relaciones entre la estructura y la actividad antibiótica de las quinolonas.
Tabla 5.1
Posición Efecto
1 Controla la potencia antibiótica: el grupo ciclopropilo es óptimo
2 El átomo ideal para esta posición es el átomo de hidrógeno
3-4 El grupo carboxilo en C-3 y el grupo carbonilo en C-4 son ideales para la unión de la quinolona a la girasa
5 Controla la potencia antibiótica es este orden NH2>OH>CH3>H
6 Controla la potencia antibiótica: lo ideal es el átomo de F
7 El grupo piperidinilo aumenta la potencia contra Gram(-)
El grupo aminopirrolidinilo aumenta la potencia contra Gram(+)
8 La sustitución en esta posición influencia la actividad anaeróbica
Figura 5.37 5.3.2.c. Rifampicina: inhibición de ARN polimerasa
La ARN polimerasa es una enzima cuyas cadenas polipeptídicas se unen a un factor que confiere especificidad para el reconocimiento de los sitios promotores requeridos para iniciar la transcripción del ADN.
La rifampicina es una antibiótico bactericida del grupo de las rifamicinas. Es un componente semisintético derivado de Amycolatopsis rifamycinica, previamente conocido como Amycolatopsis mediterranei y Streptomyces mediterranei.
La rifampicina está normalmente indicada en el tratamiento de las infecciones por Mycobacterium, incluyendo la tuberculosis y la lepra. En combinación con el ácido fucsídico se emplea en el tratamiento de Staphylococcus aureus resitente a meticilina (MRSA). Se usa en la profilaxis de Neisseria meningitidis (meningitis) y en el tratamiento de las infecciones por Listeria, Neisseria gonorrhoeae, Haemophilus influenzae y Legionella pneumophila. Junto con la doxiciclina, la rifampicina es la terapia de elección para el tratamiento de la brucelosis.
Figura 5.38. Estructura de la rifampicina
En la figura 5.39 se describe la molécula de ARN polimerasa unida a la rifampicina. El recuadro muestra con más detalle la unión de la rifampicina a la enzima.
Figura 5.39. Rifampicina unida a ARN polimerasa
5.3.3. Inhibición de la síntesis proteica de la bacteria
La síntesis proteica es uno de los procesos más frecuentemente afectados por la acción de los antimicrobianos, y su inhibición selectiva es posible gracias a las diferencias estructurales entre los ribosomas bacterianos y los eucariotas.
Los ribosomas son corpúsculos celulares que utilizan las instrucciones genéticas contenidas en el ácido ribonucleico (ARN) para enlazar secuencias específicas de aminoácidos y formar así proteínas. Los ribosomas se encuentran en todas las células y también dentro de dos estructuras celulares llamadas mitocondrias y cloroplastos. Casi todos los ribosomas flotan libremente en el citoplasma (el contenido celular situado fuera del núcleo), aunque muchos están enlazados a redes de túbulos envueltos en membranas que constituyen el llamado retículo endoplasmático.
Los ribosomas bacterianos están formados por dos subunidades (30S y 50S) que contienen ARN ribosómico (ARNr 16S en la subunidad 30S, y ARNr 5S y ARNr 23S en la subunidad 50S) y diversas proteínas llamadas S (small o pequeña, en la subunidad 30S) o L (large o grande, en la subunidad 50S).
Figura 5.40. Subunidades ribosómicas 30S y 50S
subunidad 50S, y constituyen el complejo de iniciación de la síntesis de proteínas. En este complejo hay dos sitios activos, el locus A (aminoacil), en el que se fijan los aminoacil-ARNt, y el locus P (peptidil), donde se engarza la cadena polipeptídica en formación y donde se ubicará el formilmetionil-ARNt que inicia la cadena peptídica, y el sitio E donde se coloca el ARNt antes de salir del ribosoma (figura 5.40).
5.3.3.a. Inhibidores 50S 5.3.3.a.1. Eritromicina
El antibótico macrólido eritromicina fue descubierto en 1952 como producto del metabolismo de una cepa de Streptomyces erythreus (designación cambiada luego a Saccharopolyspora erythraea), obtenido originalmente de una muestra de tierra recolectada en el archipiélago de las Filipinas. El antibiótico fue comercializado en 1952 bajo el nombre de Ilosone®, por el nombre de la región de Iloilo, en las Filipinas, de donde provenía el microorganismo del que fue aislado.
La eritromicina se ha usado para el tratamiento de infecciones del tracto respiratorio superior, la piel y tejidos blandos causadas por organismos susceptibles, principalmente bacterias Gram-positivas, y especialmente en pacientes alérgicos a las penicilinas. La eritromicina es el medicamento de elección para el tratamiento de infecciones por Mycoplasma pneumoniae, Legionella pneumophila, difteria, coqueluche, conjuntivitis o neumonía por Chlamydia trachomatis y angiomatosis bacilar y también es una alternativa segura a las tetraciclinas en el tratamiento de infección pélvica por clamidia durante el embarazo.
Figura 5.41. Estructura de la eritromicina
La eritromicina entra en el citoplasma bacteriano e inhibe la síntesis proteica al unirse reversiblemente a la subunidad 50S del ribosoma bacteriano impidiendo la translocación de péptidos desde el sitio receptor en el ribosoma hasta el sitio donador. La eritromicina solo es activa contra los microorganismos que están dividiéndose activamente.
Figura 5.42. Unión de la eritromicina (en rojo) a la subunidad 50S del ribosoma de la bacteria Deinococcus radiodurans
5.3.3.a.2. Cloranfenicol
El cloranfenicol se aisló en 1947 de una cepa de Streptomyces venezuelae, aunque en la actualidad se obtiene por síntesis química.
N CHCl2
OH HO
H O O2N
Figura 5.43. Estructura del cloranfenicol
Figura 5.44. Modo de acción del cloranfenicol
El cloranfenicol podría inhibir también la síntesis proteica en células eucariotas, lo que explicaría en gran medida algunos aspectos de su toxicidad. De hecho, los graves efectos secundarios del cloranfenicol, como el daño a la médula ósea (incluyendo anemia aplásica) limitan su uso en infecciones muy graves, como la meningitis, el tifus y la fiebre tifoidea
La resistencia al cloranfenicol se debe al gen cat. Este gen codifica una enzima llamada "cloranfenicol acetiltransferasa" que desactiva el cloranfenicol enlazando uno o dos grupos acetilo derivados de la acetil-S-coenzima A a los grupos hidroxilo del cloranfenicol. Esta acetilación impide la unión del cloranfenicol al ribosoma bacteriano.
5.3.3.a.3. Lincomicina y clindamicina
La lincomicina es un antibiótico natural que se obtiene de la bacteria Streptomyces lincolnensis. La lincomicina es, generalmente, activa frente a bacterias Gram-positivas como Streptococcus pyogenes, estreptococos del grupo vitridans, Corynebacterium diphtheriae y frente a bacterias Gram-positivas anaerobias como Propionibacterium acnes, Clostridium tetan y Clostridium perfringens
La lincomicina no es activa frente a la mayoría de las cepas de Enterococcus faecalis, ni frente a la Neisseria gonorrhoeae, Neisseria meningitidis, Haemophilus influenzae ni frente a microorganismos Gram-negativos.
La clindamicina es un antibiótico semisintético obtenido por sustitución del hidroxilo en la posición 7(R) de la lincomicina por un cloro. La clindamicina se emplea en el tratamiento de infecciones provocadas por bacterias Gram-positivas aeróbicas e infecciones como septicemias y peritonitis provocadas por bacterias Gram-negativas anaeróbicas, entre las que se incluyen las de los géneros Bacteroides y Fusobacterium.
La lincomicina y la clindamicina son inhibidores de la síntesis de las proteínas bacterianas al antagonizar la peptidiltransferasa, enzima que añade un resto peptídico unido al ARNt al siguiente aminoácido. También se cree que estos dos antibióticos inhiben la translocación de los ribosomas y evitan la disociación del peptidil-ARNt del ribosoma bacteriano, al unirse reversiblemente al punto P de la subunidad 50S del ribosoma. Por este motivo, estos compuestos son fundamentalmente bacteriostáticos, si bien puede ser bactericidas cuando se encuentran en concentraciones elevadas.
5.3.3.a.4. Oxazolidinonas
Las oxazolidinonas representan una de las últimas familias de antimicrobianos incorporadas a la práctica clínica. Así, las oxazolidinonas inhiben la síntesis proteica e impiden la formación del complejo de iniciación 70S, formado por formilmetionil-ARNt, ARNm, diversas proteínas y las subunidades ribosómicos 30S y 50S.
La primera oxazolidinona empleada como agente antibiótico fue la cicloserina (4-amino-1,2-oxazolidin-3-ona), una droga de segunda línea empleada contra la tuberculosis desde 1956.
Figura 5.46. Estructura de la cicloserina
La linezolida es una oxazolidinona bacteriostática frente a bacterias Gram-positivas, incluidas cepas multirresistentes de S. aureus y Enterococcus spp., pero carece de actividad frente a la práctica totalidad de las bacterias Gram-negativas. Fue el primer antibiótico comercializado del grupo de las oxazolidinonas y suele reservarse para el tratamiento de infecciones bacterianas graves donde fracasan otros antibióticos por haber generado resistencia. La linezolida está indicada en infecciones de la piel y tejidos blandos, neumonía nosocomial e infecciones por Enterococcus faecium resistente a vancomicina, siempre que se evidencie o se sospeche resistencia a meticilina.
La linezolida se comercializa en España con el nombre de Zyvoxid®, en forma de comprimidos, polvo para suspensión oral o solución para perfusión intravenosa.
La linezolida se fija a la subunidad ribosómica 50S, en el centro peptidiltransferasa dentro del ARN ribosómico 23S (dominio V), distorsionando así el punto de unión del formilmetionil-ARNt e impidiendo la formación del complejo de iniciación.
Ribosoma bacteriano
Antibiótico ARNt
Aminoácido enlazado a ARNt
ARNm La linezolida se fija a la unidad 50S ribosómica impidiendo la formación del
complejo de iniciación
Figura 5.48
En la figura 5.49 se describe la interacción de la linezolida con el centro activo del ribosoma bacteriano. Los nucleótidos que interaccionan con el fármaco se muestran en gris y los átomos de carbono de la linezolida se muestran en color salmón, los de oxígeno en color rojo, los de nitrógeno en color azul oscuro y el de flúor en color azul claro.
Figura 5.50. Colocación de la linezolida en el ribosoma bacteriano
En el año 2002, la compañía AstraZeneca introdujo la posizolida (AZD2563, figura 5.51) que presenta excelente actividad contra todas las bacterias Gram-positivas, sin importar su resistencia a otras clases de antibióticos.
Figura 5.51. Estructura de la posizolida
5.3.3.b. Inhibidores 30S 5.3.3.b.1. Tetraciclinas
Las tetraciclinas constituyen un grupo de antibióticos, unos naturales y otros obtenidos por semisíntesis, que abarcan un amplio espectro de actividad antimicrobiana. Químicamente son derivados de la tetracenocarboxamida, núcleo tetracíclico, de donde deriva el nombre del grupo.
Las tetraciclinas naturales se extraen de las bacterias del género Streptomyces. De Streptomyces aurofaciens se extraen la clortetraciclina y la demetilclortetraciclina. La oxitetraciclina se extrae de S. rimosus. La tetraciclina, representante genérico del grupo (véase la figura 5.52), se extrae del S. viridifaciens, aunque también se puede obtener de forma semisintética. Una característica común a las tetraciclinas es su carácter anfotérico, lo que les permite formar sales tanto con ácidos como con bases, utilizándose usualmente los clorhidratos solubles.
Las tetraciciclinas se emplean en el tratamiento de infecciones de la piel, como acné y rosácea, en infecciones urogenitales, como gonococia y sífilis, en infecciones gastrointestinales, como disentería, cólera, amebiasis, úlcera gástrica, infecciones periodontales, en infecciones respiratorias, como faringoamigdalitis, bronquitis y algunas formas de neumonía atípica. También se emplean en otras infecciones, como la fiebre recurrente, fiebre Q, tifus exantemático, tifus endémico, actinomicosis y brucelosis.
Las tetraciclinas bloquean la traslación bacteriana mediante unión reversible a la subunidad ribosómica 30S. Esta unión distorsiona los anticodones del ARNt que no pueden alinearse correctamente con los codones del ARNm (figura 5.53).
Figura 5.53. Modo de acción de las tetraciclinas
Las tetraciclinas también inhiben a las metaloproteasas de matriz (MMPs) enzimas involucrados en la angiogénesis de tumores. Aunque todavía no se conocen muy bien los mecanismos por los cuales las tetraciclinas actúan en la angiogénesis sus efectos están demostrados, por lo que se podrían utilizar como sustancias en el tratamiento del cáncer.
5.3.3.b.2. Estreptomicina
La estreptomicina, antibiótico de la familia de los aminoglucósidos, fue aislada por primera vez en 1943 de la actinobacteria Streptomyces griseus.
La estreptomicina está indicada en el tratamiento de diversas formas de tuberculosis producidas por la bacteria Mycobacterium tuberculosis. Por lo general, esta sustancia se administra con otros antituberculosos. La estreptomicina también se emplea en el tratamiento profiláctico de la endocarditis bacteriana, de la brucelosis y en el granuloma inguinal causado por Donovania granulomatis.
Los antibióticos aminoglucósidos, como la estreptomicina, se unen a la subunidad 30S del ribosoma bacteriano. Esta unión interfiere en la elongación de la cadena peptídica y provoca lecturas incorrectas del código genético. El resultado es la inhibición de la síntesis proteica y/o la formación de proteínas anómalas. Algunas de éstas son proteínas de membrana no funcionales, que forman canales que permiten el ingreso de más moléculas del fármaco antibiótico en la bacteria, provocando finalmente la muerte celular (figura 5.55).
Figura 5.55. Modo de acción de los antibióticos aminoglucósidos
5.3.3.b.3. Espectinomicina
La espectinomicina es un antibiótico bacteriostático que fue descubierto en 1962 de una cepa de Streptomyces spectabilis.
O O
O
OH
H H N HO
N
HO H
O H3C
H
H3C H
Espectinomicina
Figura 5.56. Estructura de la espectinomicina
Su actividad principal es frente a bacterias Gram-negativas, estando indicado de forma casi exclusiva en uretritis gonocócicas como alternativa a los antibióticos de primera elección como los betalactámicos y las quinolonas.
5.3.3.b.4. Kanamicina
Figura 5.57. Estructura de la kanamicina
La kanamicina se prescribe para el tratamiento de infecciones provocadas Staphylococcus aureus, Proteus, Escherichia coli, Shigella, Mycobacterium tuberculosis, etc. Se muestra eficaz en el tratamiento de mastitis, septicemias, nefritis, neumonías, enteritis, actinobacilosis, tuberculosis especialmente causada por especies resistentes a la eritromicina, etc. La kanamicina es más efectiva en el tratamiento de las infecciones urinarias cuando el pH de la orina es alcalino. La kanamicina también se administra, generalmente en combinación con neomicina oral, para la preparación preoperatoria del colon.
5.3.4. Interacción con la membrana citoplasmática bacteriana: polimixinas
Las polimixinas son decapéptidos catiónicos, ramificados y cíclicos. Las polimixinas A, B, C, D y E se aislaron en 1947 a partir de diferentes cepas de Bacillus polymyxa, aunque la mayoría de ellas (A, C y D) eran demasiado tóxicas para su uso terapéutico utilizándose, al principio, sólo la polimixina B.
En 1959 se comercializó la polimixina E (colistina®, figura 5.58) para el tratamiento de las infecciones bacterianas.
H N O H N O NH O NH O NH O N H O HN CH2 CH2
H2N
NH2
H2N O
HN OH O H2N
NH OH HN
O H2N
N O
Figura 5.58. Estructura de la colistina® (polimixina E)
aeruginosa. Normalmente las especies de Proteus y Serratia marcescens son resistentes, mientras que la sensibilidad de Bacteroides fragilis es variable.
Las polimixinas destruyen las membranas celulares de las bacterias al reaccionar con los fosfolípidos de éstas e incrementar su permeabilidad. Las resistencias son más bien raras, pero pueden aparecer cuando el antibiótico, debido a cambios en la membrana externa, no llega a la membrana citoplasmática.
5.3.5. Sulfamidas: antimetabolitos bacteriostáticos
Un antimetabolito es una sustancia que bloquea el metabolismo impidiendo que tengan lugar ciertas reacciones químicas intracelulares. En las células, los antimetabolitos son tomados por los metabolitos a los cuales se parecen, por lo que son procesados en las células de una manera similar a la de los compuestos normales. Sin embargo, su incorporación en determinadas biomoléculas impide a las células realizar sus funciones vitales, por lo que éstas son incapaces de crecer y sobrevivir.
Las sulfonamidas son antimetabolitos bacteriostáticos inhibidores de la síntesis del ácido fólico. Estos compuestos fueron los primeros fármacos eficaces en el tratamiento sistémico de las infecciones bacterianas, siendo el primero de todos ellos el protonsil cuya estructura se indica en la figura 5.59. En realidad el prontosil no es una sulfamida sino un colorante azoico, que fue sintetizado en los laboratorios de la empresa Bayer a inicios de los años 1930.
En 1932 el pátologo alemán Gerhard Domagk demostró que el prontosil protegía el organismo de los ratones de laboratorio contra el ataque de estreptococos de la especie Streptoccocus pyogenes. El doctor Domagk usó el prontosil en su propia hija, quien estaba a punto de sufrir la amputación de un brazo por una infección. Más tarde se vio que también actuaba contra las bacterias causantes de la escarlatina, la meningitis y ciertas enfermedades venéreas. En 1939 G. Domagk fue galardonado con el premio Nobel de Medicina y Ciencias Fisiológicas, que no pudo recibir por impedírselo el gobierno nacionalsocialista. Terminada la Segunda Guerra Mundial aceptó el premio, el cual le fue entregado en 1947.
La actividad antibiótica del prontosil era menor en las infecciones causadas por otros cocos y, curiosamente, no tenía ningún efecto in vitro, ejerciendo su acción antibacteriana sólo en animales vivos. En 1935 se descubrió que el prontosil era metabolizado in vivo, liberando la 4-aminobencenosulfonamida, que era el compuesto biológicamente activo (véase la figura 5.59). Este descubrimiento permitió abaratar los costos de preparación del antibiótico, puesto que no era necesario sintetizar el colorante azoico sino la propia sulfonamida, y permitió una más rápida biodisponibilidad del fármaco en los pacientes.
La 4-aminobencenosulfanilamida era eficaz contra bacterias Gram-negativas, meningococos y gonocos y su descubrimiento inició la era de los antibióticos sulfa. Al ser los únicos antibióticos efectivos en los años previos a la penicilina, las sulfonamidas fueron utilizadas extensivamente en la Segunda Guerra Mundial como desinfectantes de las heridas de guerra. El polvo de sulfa era parte del botiquín de primeros auxilios de los soldados estadounidenses y se les instruía para que esparcieran el polvo de sulfamida sobre cualquier herida abierta.
El ácido tetrahidrofólico, también conocido como coenzima F, folato H4 o FH4, es una coenzima derivada del ácido fólico (vitamina B9) de particular importancia en el metabolismo de los aminoácidos y de la síntesiss de purinas. El ácido tetrahidrofólico participa en la transferencia de grupos metilo, formilo, metileno y formimino. Su escasez en el organismo puede causar anemia.
En humanos el ácido tetrahidrofólico se produce a partir de ácido dihidrofólico mediante la intervención de la enzima dihidrofolato reductasa. Sin embargo, en bacterias el ácido tetrahidrofólico se biosintetiza mediante una ruta metabólica que empieza con la generación de ácido dihidropteróico por reacción, catalizada por la enzima dihidropteroato sintetasa, entre pteridina y ácido p-aminobenozico (véase la figura 5.60). El ácido dihidropeteróico reacciona con ácido glutámico y se convierte en ácido dihidrofólico, que se convierte en ácido tetrahidrofólico por acción de la enzima dihidrofolato reductasa.
Las sulfonamidas son estructuralmente semejantes al ácido p-aminobenzoico (PABA) y se comportan como antimetabolitos de esta sustancia, bloqueando la formación de ácido dihidropteróico por unión competitiva a la pteridina. Las bacterias no son capaces de extraer tetrahidrofolato del medio, por lo que dependen de su propia síntesis para conseguirlo. Si la síntesis esta bloqueada se produce un déficit metabólico en la bacteria, facilitando su muerte e impidiendo su reproducción
El número de sulfamidas sintetizadas es muy numeroso y suelen clasificarse de acuerdo con su farmacocinética. En la figura 5.61 se indican las estructuras de algunas sulfamidas empleadas como agentes antibacterianos.
El sulfametoxazol se administra en combinación con un fármaco denominado trimetroprima, que bloquea la formación del ácido tetrahidrofólico por inhibición de la enzima dihidrofolato reductasa.
N N
OMe OMe OMe NH2
H2N
Trimetroprima
Figura 5.62. Estructuras de la trimetroprima
Las sulfonamidas tienen un amplio espectro de acción contra bacterias Gram-positivas y Gram-negativas. Sin embargo, las cepas resistentes a las sulfonamidas se han vuelto comunes y la utilidad de estos agentes ha disminuido considerablemente.
5.4. Mecanismos de resistencia a los antibióticos
El contacto repetitivo de las bacterias con los antimicrobianos provoca una respuesta defensiva de éstas, creando, por distintas vías, mecanismos de resistencia.
Uno de los mecanismos más conocidos y más frecuentes en el fenómeno de la resistencia bacteriana es la producción, por parte de las bacterias, de enzimas que son capaces de inactivar a los antibióticos. Ya en 1940, A. P. Abraham y E. Chain, científicos que participaron en el aislamiento de la penicilina, habían identificado las penicilinasas, y cuatro años después, Kirby comprobó que eran la causa principal de la resistencia del Staphylococcus aureus a la penicilina. Esta bacteria se convirtió, a mediados de la década de 1950, en una verdadera pesadilla por su alta incidencia en la sepsis intrahospitalaria.
Otro mecanismo, antaño menos relevante pero que en los últimos años ha venido adquiriendo una mayor importancia, es el fenómeno de las mutaciones. El caso específico del Streptococcus pneumoniae es uno de los más preocupantes, ya que ésta ha dejado de ser una bacteria con alta sensibilidad a la penicilina, para convertirse en un germen multirresistente, a través, específicamente, de una mutación que provoca cambios en las proteínas fijadoras de penicilinas (PBP).
Las bombas de eflujo, que expulsan el agente antimicrobiano fuera de la bacteria, también han ido creando un creciente número de bacterias resistentes y multirresistentes.
A pesar de que en el fenómeno de resistencia están involucradas todas las bacterias, existen algunas especies que están mostrándose particularmente resistentes a los antibióticos, como es el caso de Streptococcus pneumoniae, Staphylococcus aureus y epidermiditis, enterococos, pseudomonas, klebsiellas y enterobacter, hasta el punto de que las posibilidades para combatir las cepas de neumococo-resistente a penicilina, estafilococo-resistente a meticilina y enteroco-resistente a vancomicina se han reducido de forma alarmante.
Los principales mecanismos moleculares y bioquímicos que conducen a la resistencia a los antibióticos se resumen a continuación:
2) Modificación de la diana del antibiótico, y modificación de la accesibilidad a la diana, incluyendo la disminución de la permeabilidad de la membrana externa de las bacterias Gram-negativas.
3) Acción de las bombas de eflujo, que expulsan el agente antimicrobiano fuera de la bacteria.
En la figura 5.63 se representan esquemáticamente los mecanismos de resistencia bacteriana a los antibióticos.
Figura 5.63. Principales mecanismos de resistencia a los antibióticos
5.4.1. Infecciones graves provocadas por bacterias
Actualmente se considera que las bacterias Gram-positivas aerobias son las principales causantes de las infecciones graves hospitalarias, constituyendo cerca del 60% de los casos. Esta preponderancia, con relación a otros grupos bacterianos, ha ido acrecentándose durante los últimos 25 años como consecuencia de la convergencia de diversos factores, entre ellos la mayor utilización de procedimientos invasivos y la mayor prevalencia de microorganismos resistentes a los antibióticos convencionales.6
Entre los patógenos predominantes en las infecciones graves hospitalarias se pueden citar los estafilococos coagulasa negativo, Staphyloccocus aureus y Enterococcus spp. Específicamente, Staphyloccocus aureus es el principal agente causal de cuadros de osteomielitis e infecciones de piel y de tejidos blandos, así como uno de los más comunes en las neumonías hospitalarias. En este sentido, prácticamente el 50% de las bacteriemias asociadas con cateterismos, de las infecciones de piel y tejidos blandos e infecciones bacterianas de vías bajas son atribuibles a este microorganismo tan ubicuo en el medio hospitalario.
Igualmente, los enterococos han ido incrementando su protagonismo en infecciones graves, fundamentalmente debido a su gran capacidad para desarrollar resistencia frente a diversos grupos de antibióticos, así como la de transferirla a otros microorganismos. También el neumococo Streptococcus pneumoniae es una ubicua causa de enfermedad, especialmente en niños pequeños, ancianos y pacientes afectados por cuadros de inmunodepresión patológica o inducida farmacológicamente.
También hay que señalar que los estreptococos del grupo Viridans, responsables principales de la endocarditis, son cada vez más frecuentes como agentes etiológicos de otras infecciones complicadas, especialmente en pacientes inmunodeprimidos.
Otras infecciones graves son las provocadas por Staphylococcus aureus resistentes a meticilina (MRSA), que suponen el 24% de las infecciones hospitalarias registradas en España en el año 2001. De ellas, el 80% era además resistente a otros antibacterianos como la ciprofloxacina, la clindamicina, el sulfametoxazol-trimetoprima, etc. Otro tanto puede decirse de los estafilococos coagulasa-negativo con sensibilidad disminuida a la teicoplanina, cuya incidencia ha sido estimada en más del 22%.
El Sistema Europeo de Vigilancia de Resistencias Antimicrobianas (EARSS) reportó en 2002 datos similares a los descritos para España. En este sentido, se ha indicado que el 22% de las cepas aisladas de Staphylococcus aureus eran resistentes a meticilina, alcanzando las tasas más elevadas en Grecia (44%), Irlanda (42%), Malta (43%) y Gran Bretaña (44%). Por su parte, las tasas más altas de cepas de Enterococcus faecium resistente a vancomicina se observaron en Grecia (19%), Italia (21%) y Croacia (22%).
Desde el aislamiento en 1996 de la primera cepa clínica de Staphylococcus aureus con sensibilidad disminuida a la vancomicina (VISA, vancomycin-intermediate S. aureus o GISA, glycopeptide-intermediate S. aureus ), se ha ido asociando con frecuencia creciente su mayor prevalencia con la aparición de fracasos terapéuticos asociados al uso de antibióticos glucopeptídicos (vancomicina, teicoplanina), especialmente en unidades de cuidados intensivos, donde este tipo de antibióticos es considerado como de primera línea.
5.5. Síntesis de quinolonas 5.5.1. Síntesis de ofloxacina
La ofloxacina es un antibiótico efectivo contra un gran número de bacterias Gram-positivas y Gram-negativas, por lo que se considera un antibiótico de amplio espectro. La ofloxacina viene en preparación ótica y se emplea en el tratamiento de infecciones en el oído, tanto en adultos como en niños.
5.5.1.a. Análisis retrosintético
En el esquema 5.1 se indica un análisis retrosintético para la ofloxacina. El proceso comienza con la desconexión del anillo de metilpiperazina, que se instalará mediante reacción de SNAr de la N-metilpiperacina 5.1 sobre el intermedio 5.2. La desconexión de la
función acilo en el intermedio 5.2 se basa en una reacción de SEAr y conduce al diester 5.3.
Este compuesto, por desconexión de la parte de metilenmalonato basada en un proceso de adición Michael-retroMichael, que se ha indicado como MRM en el esquema 5.1, origina el fragmento 5.4 y la benzoxacina 5.5. La benzoxazina se obtendrá de la imina 5.6 por hidrogenación, la cual derivará de la aminocetona 5.7. Una operación de interconversión de grupo funcional sobre 5.7 conduce a la nitrocetona 5.8, que se obtendrá por reacción de O-alquilación del nitrofenol 5.9. Este compuesto se sintetizará a partir del trifluoronitrobenceno 5.11. N O COOH N F N O Ofloxacina N O COOR NH F N O F + 5.1 5.2
SEAr
N O F O F RO O OR NH O F O F RO O OR X 5.3 5.4 5.5 + N F O F 5.6 imina NH2 F O F O 5.7 NO2 F O F O O-alquilación NO 2 F OH F O X + 5.8 5.9 5.10 NO2 F F F 5.11 MRM SNAr
IGF
IGF IGF
5.5.1.b. Síntesis
La síntesis de la ofloxacina se indica en el esquema 5.2. El proceso comienza con la obtención 2,3-difluoro-8-nitrofenol 5.9 mediante reacción SNAr del
1,2,3-trifluoro-4-nitrobenceno 5.11 con KOH en dimetilsulfóxido.7 La reacción de O-alquilación del hidroxilo
fenólico de 5.9 con cloroacetona proporciona la nitrocetona 5.8. Cuando este compuesto se hidrogena en presencia de Ni Raney como catalizador se obtiene directamente la benzoxacina 5.5. El calentamiento de 5.5 en presencia del etoximetilenmalonato 5.4 conduce al compuesto 5.3, que se somete a la reacción SEAr intramolecular mediante calentamiento
en presencia de éster polifosfórico (PPA). Esta reacción proporciona el éster tricíclico 5.2, que se convierte en el ácido 5.12 por saponificación. La ofloxacina se obtiene mediante reacción SNAr del ácido 5.12 con la metilpiperazina 5.1.
Ofloxacina N O COOEt NH F N O F 5.1 5.2 N O F O F EtO O OEt NH O F O F EtO O EtO EtO 5.3 5.4 5.5 NO2 F O F O NO2 F F F O Cl 5.8 5.11 5.10 KOH 10%. DMSO
rt (29%) NO
2
F OH F
, KI K2CO3, acetona
(65%)
Ni Raney, H2 EtOH, rt (90%) 5.9 140-145ºC PPA 140-150ºC (64% 2 etapas)
HCl 12 M AcOH, reflujo (94%) N O COOH F O F
DMSO, 100ºC (62%) 5.12
Esquema 5.2
5.5.1.c. Cuestiones
1) ¿Cuál es el mecanismo de formación del 2,3-difluoro-8-nitrofenol 5.9? ¿Cómo se puede explicar la regioselectividad de esta reacción?
2) Explique mecanísticamente la formación de la benzoxacina 5.5 a partir de la nitrocetona 5.8.
3) Proponga un mecanismo que explique la formación del compuesto 5.3.
4) Explique mecanísticamente la formación de la ofloxacina por reacción entre la metilpiperazina 5.1 y el compuesto 5.12. ¿Cómo se puede explicar la regioselectividad de esta reacción?
5.5.2. Síntesis de levofloxacina
El enantiómero S de la ofloxacina se denomina levofloxacina, y es entre 8 y 128 veces más potente que la mezcla racémica.
N O
COOH N
F N O
Ofloxacina
N O
COOH N
F N O
Levofloxacina
La levofloxacina se comercializó en 1996 como antibiótico contra la neumonía, la bronquitis crónica y contra las infecciones de la piel y del tracto urinario. Unos años después de su comercialización la lista de indicaciones de la levofloxacina se incrementó para incluir el tratamiento de la neumonía causada por Streptococcus pneumoniae resistente a penicilina y la neumonía causada por Staphylococcus aureus, Pseudomonas aeruginosa, Serratia marcescens, Haemophilus influenzae, Kliebsella pneumoniae y Escherichia coli.
5.5.2.b. Síntesis de levofloxacina
5.5.2.b1. Síntesis de levofloxacina mediante separación de diastereoisómeros
Para la síntesis de la levofloxacina se prepara el compuesto 5.5 en forma ópticamente activa mediante la secuencia de reacciones indicada en el esquema 5.3. Así, la reacción de amidación con el cloruro de la (S)-N-tosilprolina 5.13, seguida de cristalización, proporciona la amida 5.14 ópticamente activa. La hidrólisis básica de 5.14 permite la obtención del compuesto 5.5 en forma ópticamente activa. Este compuesto se emplea como precursor quiral en la síntesis de la levofloxacina.
Esquema 5.3
La aplicación sobre el compuesto (S)-5 de la secuencia sintética indicada en el esquema 5.2 proporciona la levofloxacina.
5.5.2.b2. Síntesis de levofloxacina mediante el empleo del pool quiral
Muchas síntesis de compuestos ópticamente activos se llevan a cabo empleando productos naturales, o derivados de ellos (pool quiral), como materiales de partida o como reactivos en alguno de los pasos sintéticos. Este es el caso de la síntesis de la levofloxacina que se indica en el esquema 5.5, en la que se instala el centro estereogénico con el empleo del (S)-alaninol.8 En esta síntesis se utiliza el cetoéster 5.15 como material de partida.9 Este
compuesto se convierte en el etoximetilenceteoester 5.16 por calentamiento con formiato de trietilo en anhidrido acético. La reacción de 5.16 con (S)-alaninol 5.17 proporciona el enaminocetoéster 5.18, que se transforma en la dihidroquinolinona 5.19 por reacción con hidruro sódico en dimetilsulfóxido. Cuando 5.19 se calienta en presencia de KOH se obtiene el compuesto (S)-5.2 por saponificación de la función éster y reacción SNuAr intramolecular.
Finalmente, la reacción de (S)-5.2 con la metilpiperacina conduce a la levofloxacina.
Esquema 5.5
5.5.2.c2. Cuestiones
1) Explique mecanísticamente la formación del etoximetilenocetoéster 5.16.
2) Explique mecanísticamente la formación de la dihidroquinolinona 5.19.
8 Mitscher, L. A.; Sharma, P. N.; Chu, D. T. W.; Shen, L. L.; Pernet. A. G. J. Med. Chem. 1987, 30, 2283-2286.