ANOTACIÓN DEL TRANSCRIPTOMA FOLIAR DE LA UCHUVA (Physalis peruviana L.): FRUTA PROMISORIA DE LA FAMILIA SOLANACEAE
GINA ALESSANDRA GARZÓN MARTÍNEZ
PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS
MAESTRÍA EN CIENCIAS BIOLÓGICAS ÉNFASIS EN BIOTECNOLOGÍA VEGETAL
ANOTACIÓN DEL TRANSCRIPTOMA FOLIAR DE LA UCHUVA (Physalis peruviana L.): FRUTA PROMISORIA DE LA FAMILIA SOLANACEAE
GINA ALESSANDRA GARZÓN MARTÍNEZ
Director:
LEONARDO MARIÑO RAMÍREZ Ph.D. Bioquímica
Codirectora:
LUZ STELLA BARRERO MENESES Ph.D. Genética Molecular Vegetal
PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS
MAESTRÍA EN CIENCIAS BIOLÓGICAS ÉNFASIS EN BIOTECNOLOGÍA VEGETAL
NOTA DE ADVERTENCIA
“La Pontificia Universidad Javeriana no se hace responsable por los conceptos, criterios, opiniones y conclusiones expresados por sus alumnos, solo velará por que el trabajo no tenga ataques personales y únicamente se vea en él, el anhelo de buscar la verdad y justicia”.
_________________________ ___________________________
Ingrid Schuler Ph.D. Manuel Franco Ph.D.
DEDICATORIA
A Dios, por ser mi guía en el camino y permitirme culminar otra etapa de mi vida como profesional.
A mis padres, por su apoyo, paciencia, comprensión, consejos y por enseñarme que es posible alcanzar todo lo que me proponga.
AGRADECIMIENTOS
Al Doctor Leonardo Mariño Ramírez, por su asesoría y dirección de este proyecto, por permitirme hacer parte de este estudio y sumergirme en el campo de la Bioinformática, pero en especial por sus consejos, enseñanzas, paciencia, colaboración, apoyo y confianza.
A la Doctora Luz Stella Barrero, codirectora de este proyecto, por su asesoría y confianza para realizar este estudio, pero en especial por permitirme hacer parte de su equipo de trabajo, valioso apoyo y colaboración.
A Felix Enciso, por su asesoría y correcciones de este proyecto, pero sobre todo por su apoyo incondicional, enseñanzas y amistad.
A la Unidad de Bioinformática y Biología Computacional, del Laboratorio de Genética Molecular Vegetal, de la Corporación Colombiana de Investigación Agropecuaria (CORPOICA), sede Tibaitatá, por las instalaciones y equipos, además del tiempo otorgado para la realización de la Maestría.
Al Ministerio de Agricultura y Desarrollo Rural – MADR, por la financiación de este proyecto y apoyo económico en mis estudios de Maestría como parte del proyecto de investigación No. 072-2008L4787-3281.
Al Doctor Víctor Nuñez por parte del financiamiento de la secuenciación del transcriptoma foliar de uchuva, como parte del proyecto de investigación No. 054/08190-2008L7922-133322, financiado por el MADR.
A Ligia Suescún del Laboratorio de cultivo de tejidos, por suministrar el material vegetal in vitro para la secuenciación del transcriptoma foliar.
A Erika Sánchez, Jaime Simbaqueba y Jaime Osorio, por su amistad, valioso apoyo, buenos consejos y motivación para seguir adelante.
A mis compañeros de universidad y laboratorio, Linda Gómez, Magda Gómez, Yaneth Camargo, Paola Delgadillo, Diana Duarte y Franklin Mayorga, por su constante apoyo.
INDICE GENERAL
1. INTRODUCCIÓN ... 15
2. OBJETIVOS ... 17
2.1 Objetivo general ... 17
2.2 Objetivos específicos ... 17
3. ANTECEDENTES BIBLIOGRÁFICOS ... 18
3.1 Familia Solanaceae ... 18
3.2 Generalidades de la uchuva (Physalis peruviana L.) ... 18
3.2.1. Origen y distribución ... 19
3.2.2 Taxonomía ... 19
3.2.3 Principales problemas del cultivo ... 20
3.2.4 Usos potenciales e importancia ... 20
3.2.5 Producción Nacional ... 21
3.2.6 Estudios morfoagronómicos, moleculares y genómicos ... 22
3.3 Técnicas de secuenciación de siguiente generación ... 23
3.3.1 Pirosecuenciación ó 454 ... 25
3.3.2 Aplicaciones de la pirosecuenciación en transcriptómica de novo en plantas 27 3.4 Análisis computacionales de ESTs ... 29
3.4.1 Ensamblaje de ESTs ... 29
3.4.2 Anotación de secuencias consenso ... 32
4. MATERIALES Y MÉTODOS ... 39
4.1 Localización ... 39
4.2 Material vegetal ... 39
4.3 Extracción de ARN y construcción de la librería normalizada de ADNc ... 39
4.4 Secuenciación y ensamblaje ... 40
4.5 Anotación Funcional ... 41
4.6 Identificación de marcadores moleculares ... 42
4.7 Organización de la información en una base de datos ... 42
5. RESULTADOS Y DISCUSIÓN ... 43
5.1 Síntesis de ADNc y normalización ... 43
5.2 Secuenciación de ESTs y ensamblaje ... 43
5.2.1 Ensamblaje comparativo con papa y tomate ... 46
5.3 Anotación funcional ... 47
5.3.1 Anotación por homología de secuencias BLASTX ... 47
5.3.2 Anotación por términos ontológicos (GO) ... 49
5.3.3 Anotación funcional por KEGG ... 53
5.3.4 Identificación de dominios protéicos... 55
5.3.5 Genes potenciales codificantes de proteínas asociados a diferentes tipos estrés biótico y otros compuestos de interés en uchuva ... 56
5.4 Identificación in silico de marcadores SSRs ... 60
5.5 Base de datos con la información principal de la anotación del transcriptoma foliar de la uchuva ... 63
6. CONCLUSIONES ... 65
7. RECOMENDACIONES ... 66
8. BIBLIOGRAFÍA ... 67
LISTA DE TABLAS
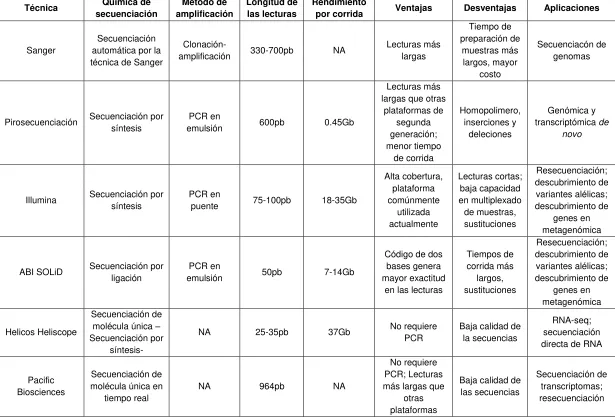
Tabla 1: Técnicas de secuenciación y sus aplicaciones ... 24
Tabla 2: Tipos de BLAST comúnmente usados para anotación. ... 35
Tabla 3: Estadísticas de la secuenciación y ensamblaje ... 44
Tabla 4: Estadísticas del ensamblaje comparativo con papa y tomate ... 46
Tabla 5: Número de isotigs, singletons y unigenes con hits de BLAST contra las bases de datos datos UniProtKB/Swiss-Prot y RefSeq ... 48
Tabla 6: Información general de la anotación funcional del transcriptoma foliar de P. peruviana... 50
Tabla 7: Principales rutas metabólicas específicas asociadas con los unigenes de P. peruviana mediante KEGG... 54
Tabla 8: Dominios protéicos identificados en el transcriptoma de P. peruviana ... 55
Tabla 9: Marcadores microsatélites identificados en los contigs de P. peruviana ... 61
LISTA DE FIGURAS
Figura 1: Procedimiento general de la técnica de secuenciación 454 de Roche. ... 27
Figura 2: Estrategias de ensamblaje de lecturas generadas a partir de técnicas de secuenciación de siguiente generación. ... 30
Figura 3: Ensamblaje basado en el algoritmo OLC en Newbler ... 32
Figura 4: Estructura jerárquica de padres e hijos, de las ontologías génicas. ... 36
Figura 5: Distribución de las lecturas secuenciadas ... 45
Figura 6: Distribución de especies, a partir de los resultados generados por homología de secuencias de los unigenes contra la base de datos UniProtKB/Swiss-Prot y RefSeq. ... 49
Figura 8: Diagrama de Venn de la comparación de los unigenes de P. peruviana con las secuencias de proteína de S. lycopersicum y S. tuberosum. ... 60
LISTA DE ANEXOS
Anexo 1: Cebadores usados para la construcción de la librería normalizada ... 77
Anexo 2: Representación esquemática del flujo de trabajo usado. ... 78
Anexo 3: Estadísticas del ensamblaje realizado con el algoritmo Newbler………..81
Anexo 4: Estadísticas de los términos ontológicos (GO) ... 80
Anexo 5: Enzimas asociadas a rutas metabólicas de la base de datos KEGG. ... 81
Anexo 6: Principales rutas metabólicas asignadas para los unigenes, a partir de la base de datos Kyoto Encyclopedia of Genes and Genomes (KEGG)... 83
Anexo 7: Enzimas asociadas a la ruta biosintética putativa de los witanólidos ... 84
Anexo 8: Ruta biosintética de los fenilpropanoides ... 86
Anexo 9: Estadísticas del BLASTX entre los unigenes de P. peruviana y las proteínas de S. lycopersicum (ITAG2.3) y S. tuberosum (PGSC_DM_v3.4). ... 87
Anexo 10: Marcadores SSRs relacionados con ontologías de “respuesta a estímulos” asociadas a estrés de tipo biótico y abiótico. ... 88
Anexo 11: Sintaxis usada para la creación de la base de datos y su posterior alimentación, mediante lenguaje SQL. ... 92
RESUMEN
ABSTRACT
1. INTRODUCCIÓN
Dentro de la cadena frutícola de Colombia, la uchuva (Physalis peruviana L.), miembro de la familia de las solanáceas, representa una de las especies promisorias del país en los mercados internacionales. Hoy en día es el segundo frutal de exportación, abarcando el 54% de las exportaciones de frutas frescas colombianas, excluyendo el banano (AGRONET, 2012). A nivel comercial, el genotipo colombiano es uno de los más apetecidos por sus mejores características organolépticas (color, sabor), lo que le confiere ventaja en los mercados internacionales (Almanza & Fischer, 1993). Además, esta especie es de gran interés en el campo biomédico, como fuente de vitaminas, compuestos antiinflamatorios y antioxidantes, así como por sus propiedades anticancerígenas (Franco et al., 2007; Ramadan, 2011; Wu et al., 2009; Yen et al., 2010). Sin embargo, problemas como la fusariosis o marchitez vascular, representan una de las mayores limitantes en el cultivo de uchuva (González & Barrero, 2011), que junto a problemas asociados con la calidad del fruto (v.g. rajamiento, (Fischer, 2005a)), ocasionan grandes pérdidas en producción en varias regiones del país.
En los últimos años, la innovación en tecnologías de secuenciación, ha permitido ampliar la investigación genómica en diferentes especies no modelo. El aumento en el volumen de información generado por estás tecnologías, ha estado acompañado por el desarrollo de herramientas computacionales, que permiten el procesamiento y análisis de los datos generados, en busca de información útil para la comunidad científica. Estudios a nivel transcriptómico en plantas, han empezado a ser una herramienta valiosa para la generación de los primeros inventarios de genes en diferentes especies, permitiendo su identificación y descubrimiento, asociación con rutas metabólicas, generación de marcadores moleculares, estudios en variabilidad genética, con miras a estudios de asociación genotipo-fenotipo para atributos de interés, información valiosa para su uso en programas de mejoramiento genético.
1.1 PLANTEAMIENTO DEL PROBLEMA Y JUSTIFICACIÓN
en uchuva, es la implementación de programas de mejoramiento genético apoyados por herramientas moleculares y genómicas. No obstante, pocos estudios se han reportado encaminados a la generación de información genética de este especie (Espinosa et al., 2004; Muñoz et al., 2008), lo que se encuentra a su vez relacionado con la información casi nula a nivel genómico de la misma, que permita la identificación de genes y desarrollo de marcadores moleculares asociados con características de interés, que posibiliten la implementación de programas de selección asistida por marcadores moleculares (MAS) o de selección genómica (Heffner et al., 2010; Varshney et al., 2012).
Sin embargo, gracias a los avances en las tecnologías de secuenciación de siguiente generación (NGS), ha sido posible la secuenciación de genomas completos o porciones de éste, tanto en especies modelo, como organismos no modelo con limitada información genómica. No obstante, características como la poliploidía y el gran tamaño de los genomas de muchas plantas, a causa de la gran cantidad de elementos repetitivos o duplicaciones parciales del genoma, hacen del ensamblaje de novo en especies no modelo uno de los procesos más desafiantes en genómica (Imelfort & Edwards, 2009). Así, en los últimos años, la secuenciación de novo de transcriptomas o porciones codificantes del genoma, se ha convertido en la estrategia más económica y efectiva ampliamente utilizada para la identificación y descubrimiento génico a gran escala en plantas no modelo (Blanca et al., 2011; C. Sun et al., 2010). De esta forma, la generación de información a nivel transcriptómico de organismos huérfanos, pero de gran interés económico, nutricional o ecológico, está permitiendo acelerar y facilitar los programas de mejoramiento, mediante la identificación de herramientas moleculares útiles para estas especies.
2. OBJETIVOS
2.1 Objetivo general
Generar el primer catálogo de genes de uchuva (Physalis peruviana) con información funcional en la especie.
2.2 Objetivos específicos
Secuenciar y ensamblar la primera colección de transcritos de uchuva a partir de una librería de ADNc proveniente del transcriptoma foliar.
Anotar funcionalmente los transcritos de uchuva mediante el uso de herramientas bioinformáticas.
Identificar marcadores microsatélites a partir del transcriptoma foliar de uchuva.
3. ANTECEDENTES BIBLIOGRÁFICOS
3.1 Familia Solanaceae
Las solanáceas son el tercer taxa más importante a nivel comercial en plantas; comprende 96 géneros y alrededor de 9.000-10.000 especies, siendo el género Solanum el más cosmopolita con aproximadamente 2.000 especies. Esta familia se encuentra distribuida a nivel mundial, con una alta diversidad concentrada principalmente en el Neotrópico (Knapp, 2002). En ella, se destacan diversas especies de interés agronómico (Solanum tuberosum, S. lycopersicum, S. melongena), actividad farmacológica (Nicotiana tabacum) y ornamental (Petunia hybrida) (Knapp et al., 2004).
Además de la gran importancia de esta familia como recurso alimenticio, varias de las especies de solanáceas tienen gran interés a nivel científico, como plantas modelo para otras integrantes de la familia; como por ejemplo S. tuberosum y S. lycopersicum, cuyos genomas están siendo ampliamente estudiados, para características de interés agronómico y el esclarecimiento de procesos de evolución, domesticación, desarrollo, biosíntesis de metabolitos secundarios, defensa en plantas, entre otros (Mueller et al., 2009; X. Xu et al., 2011). De esta manera, las herramientas genómicas de especies modelo como las anteriormente mencionadas, permiten una investigación más eficiente en otras especies de solanáceas con limitada información a nivel genómico, partiendo del hecho que los genomas de las solanáceas se caracterizan por altos niveles de conservación en organización y contenido genómico (Mueller et al., 2005).
3.2 Generalidades de la uchuva (Physalis peruviana L.)
3.2.1. Origen y distribución
El origen de la uchuva aún no está esclarecido. Sin embargo, se creé que el centro de origen y diversificación se encuentra en los Andes, posiblemente en países como Colombia, Perú y Ecuador (Bartholomaus et al., 1990; Legge, 1974; Medina, 1991). Esta especie crece como planta silvestre y semisilvestre distribuyéndose en zonas tropicales y subtropicales. Es apta para proteger terrenos en erosión y se considera como planta pionera en suelos recientemente formados, debido a su crecimiento rápido, expansivo y su habilidad de adaptación (Ligarreto et al., 2005).
Actualmente, se produce en varios países, como Sudáfrica, Nueva Zelanda, Australia, Kenia, India, Malasia, China y Colombia, conociéndose más de 80 ecotipos a nivel mundial, con alta variabilidad fenotípica (González et al., 2008; Ligarreto et al., 2005). En Colombia, se encuentra en los departamentos de Antioquía, Boyacá, Cesar, Cundinamarca, Huila, Magdalena, Nariño, Putumayo, Quindío y Tolima, a altitudes de 1.300 a 3.700 msnm. En el país, se siembra en asociación con otros cultivos como frutales, frijol, arveja y maíz (Ligarreto et al., 2005); y se cultivan tres ecotipos: ‘Kenia’, ‘Sudáfrica’ (reintroducidos del África), y ‘Colombia’, que se caracteriza por tener una mejor coloración y mayor contenido de azúcares en el fruto, lo cual le ha conferido mayor demanda en los mercados internacionales (Flores et al., 2000).
3.2.2 Taxonomía
Según la base de datos “Plants Database” del Departamento de Agricultura de los Estados Unidos (USDA, 2012), la clasificación taxonómica de la uchuva es:
Reino: Plantae
Género: Physalis L.
Especie: Physalis peruviana
3.2.3 Principales problemas del cultivo
La uchuva, presenta diferentes problemas de tipo fitosanitario y de calidad que afectan en gran medida la productividad del cultivo. Dentro de los principales inconvenientes de tipo fitosanitario, se encuentra la marchitez vascular o fusariosis, causada por el hongo Fusarium oxysporum. Este hongo, ha ocasionado pérdidas hasta del 60% en el departamento de Cundinamarca y del 40 al 50% en el departamento de Boyacá por posible traslado de semilla infectada por el patógeno (González & Barrero, 2011). Sin embargo, existen además otros tipos de enfermedades que afectan el cultivo, siendo en ciertas ocasiones, una limitante en la producción. Se consideran otras enfermedades causadas por hongos; como la mancha gris (Cercospora sp.), muerte descendente (Phoma sp.), mancha negra de las hojas (Alternaria sp.) y moho gris (Bottytis sp.); causadas por bacterias (Ralstonia sp.), por virus (mosaico de la uchuva), por nemátodos (Meloidogyne sp.) y diferentes clases de insectos plaga (Benavides & Mora, 2005; Zapata et al., 2005).
Por otro lado, los problemas de calidad, rendimiento y almacenamiento son otros de los inconvenientes que enfrentan los productores, ante los estándares de calidad que exige el mercado internacional (Fischer, 2005; Lanchero et al., 2007). El rajado de fruto es uno de los problemas que produce pérdidas importantes hasta del 45% en producción y comercialización; sin embargo el mecanismo que ocasiona este fenómeno no ha sido completamente esclarecido, no obstante, parece estar condicionado a la sensibilidad varietal u otros factores como exceso de agua y efectos nutricionales (Fischer, 2005).
3.2.4 Usos potenciales e importancia
considerados por poseer propiedades farmacológicas y ácidos grasos poliinsaturados, esenciales para la salud humana (Ramadan & Morsel, 2003)
Recientemente, a los extractos de uchuva a partir de partes aéreas de la planta, se le han atribuido propiedades anticancerígenas, antibacterianas y repelente a insectos, gracias a compuestos bioactivos como los witanólidos, fenoles y etanólicos (Fang et al., 2012; Quispe et al., 2009; Wu et al., 2006). Extractos supercríticos de dióxido de carbono (SFE-CO2) mostraron un efecto inhibidor en la proliferación de líneas celulares cancerígenas humanas H661 de pulmón (S. J. Wu et al., 2009) y una actividad antioxidante y antiinflamatoria atribuidas a los altos contenidos de fenoles y flavonoides (Franco et al., 2007; Wu et al., 2006). Así mismo, el compuesto 4-Hidroxi-witanólido E aislado de hojas y tallos de uchuva demostró ser un potencial agente terapéutico contra el cáncer de pulmón (Yen et al., 2010). De esta manera, P. peruviana es una planta promisoria en el campo biomédico así como en el área de alimentos funcionales.
Por otra parte, las bayas de uchuva tienen gran potencial, para consumirse en fresco o como ingrediente en ensaladas, cocteles de frutas, diferentes platos gourmets y licores. Además, puede cocerse para ser usada en pastelería, repostería, y preparación de conservas (Bonilla et al., 2009).
3.2.5 Producción Nacional
La uchuva representa hoy en día para el país, una de las principales especies con mayor proyección en los mercados internacionales. Colombia es el principal productor mundial de esta fruta. Sin embargo, también se cultiva en países como Sudáfrica, Kenia, Brasil, Argentina, Chile, Australia y Nueva Zelanda (Bonilla et al., 2009). En los últimos años la producción de uchuva en el país ha crecido significativamente. Entre los años 1993 y 2010, la producción de este frutal incrementó de 4 toneladas a 12.024 toneladas (AGRONET, 2012). La producción en Colombia, para el año 2010, se concentró en el departamento de Boyacá con un 56,6%, seguido por Antioquia con un 20,1% y Cundinamarca con un 13,5% (AGRONET, 2012).
banano (AGRONET, 2012). Según AGRONET (2012) la Unión Europea es el principal consumidor de esta fruta, concentrando un 93% del mercado, seguido de otros países de Asía y América.
3.2.6 Estudios morfoagronómicos, moleculares y genómicos
En Colombia, las colecciones albergadas en los bancos de germoplasma nacionales, han sido parcialmente evaluadas para atributos morfológicos y agronómicos (González et al., 2008; Lagos et al., 2003; Ligarreto et al., 2005; Palomino, 2010). En la Universidad Nacional de Colombia, se realizó un estudio para la caracterización morfológica de 29 accesiones de P. peruviana de la colección de germoplasma, para caracteres cuantitativos y cualitativos a nivel vegetativo y reproductivo. En este trabajo, se encontró variabilidad fenotípica que permitió establecer material potencialmente importante para siembra y futuros programas de mejoramiento (Palomino, 2010). En CORPOICA, en el centro de Investigaciones C.I. La Selva, se evaluaron morfológicamente 46 accesiones de uchuva, mediante 69 descriptores, que fueron útiles para la diferenciación de las accesiones evaluadas, observando mayor variabilidad en los rasgos cuantitativos (93.1%) que en los cualitativos (72,5%) (González et al., 2008).
En el área de citogenética, Rodríguez y Bueno (2006) caracterizaron cinco ecotipos de P. peruviana, encontrando variabilidad genética entre estos; presentando una dotación cromosómica de 2n = 24 en ecotipos silvestres, 2n = 32 en el ecotipo Colombia y 2n = 48 en el ecotipo Kenia, en los cuales se observó también una variación en la morfología de los individuos, posiblemente a causa del nivel de ploidía. Recientemente, en CORPOICA, en el C.I. Tibaitatá, se evaluaron 11 genotipos de uchuva, a partir de los cuales se determinó el arreglo cromosómico 2n=4x=48 para accesiones de P. peruviana y 2n=2x=24, así como 2n=2x=48 para el taxon relacionado P. floridana. Estos resultados fueron altamente relacionados con la cantidad de ADN de las accesiones evaluadas, determinada por citometría de flujo (Liberato, 2012).
Genes - RCGs), se realizaron evaluaciones en 60 accesiones del banco de germoplasma de CORPOICA-Tibaitatá. En este estudio, se obtuvo un promedio de 7 y 4 alelos por locus para COSII y RCGs, con un índice de contenido polimórfico (PIC) de 0,46 y 0,36 respectivamente. Se determinaron valores de heterocigosidad altos (Heterocigosidad observada: 0.54) e índices de fijación negativos (FIS = -60), indicando un exceso de individuos heterocigotos, baja endogamia y fijación alélica (Delgadillo, 2012), lo cual podría estar relacionado con la forma de reproducción mixta de la especie sugerida hasta el momento (Lagos et al., 2003). Recientemente, Simbaqueba et al. (2011) desarrollaron y evaluaron 162 marcadores SSRs provenientes de ESTs de uchuva, obteniendo un 83% de amplificación con una tasa de polimorfismo del 22%, útiles para estudios de diversidad y estructura poblacional.
Sin embargo, P. peruviana aún carece de suficiente información genómica que permita el desarrollo de estudios a nivel molecular con miras a mejoramiento genético. En la base de datos Taxonomy del NCBI (National Center for Biotechnology Information), para Mayo de 2011, se registran menos de 50 secuencias a nivel nucleotídico y protéico. La mayoría de estas secuencias han sido generadas a partir de estudios filogenéticos del síndrome del cáliz inflado (Inflated-Calyx Sindrome - ICS), característica novedosa de algunos géneros la familia Solanaceae, como el género Physalis. El ICS se encuentra asociado a la expresión del gen MPF2, así como la presencia de hormonas giberelinas y citoquininas, dando como resultado el control de la división celular y la elongación de los sépalos (He & Saedler, 2005; Hu & Saedler, 2007).
3.3 Técnicas de secuenciación de siguiente generación
Tabla 1: Técnicas de secuenciación y sus aplicaciones
Técnica Química de
secuenciación Método de amplificación Longitud de las lecturas Rendimiento
por corrida Ventajas Desventajas Aplicaciones
Sanger
Secuenciación automática por la técnica de Sanger
Clonación-amplificación 330-700pb NA
Lecturas más largas Tiempo de preparación de muestras más largos, mayor costo Secuenciacón de genomas
Pirosecuenciación Secuenciación por
síntesis
PCR en
emulsión 600pb 0.45Gb
Lecturas más largas que otras
plataformas de segunda generación; menor tiempo de corrida Homopolimero, inserciones y deleciones Genómica y transcriptómica de
novo
Illumina Secuenciación por síntesis PCR en puente 75-100pb 18-35Gb
Alta cobertura, plataforma comúnmente utilizada actualmente Lecturas cortas; baja capacidad en multiplexado de muestras, sustituciones Resecuenciación; descubrimiento de variantes alélicas; descubrimiento de genes en metagenómica
ABI SOLiD Secuenciación por
ligación
PCR en
emulsión 50pb 7-14Gb
Código de dos bases genera mayor exactitud
en las lecturas
Tiempos de corrida más largos, sustituciones Resecuenciación; descubrimiento de variantes alélicas; descubrimiento de genes en metagenómica Helicos Heliscope Secuenciación de molécula única – Secuenciación por
síntesis-
NA 25-35pb 37Gb No requiere
PCR
Baja calidad de la secuencias
RNA-seq; secuenciación directa de RNA
Pacific Biosciences
Secuenciación de molécula única en
tiempo real
NA 964pb NA
No requiere PCR; Lecturas más largas que
otras plataformas
Baja calidad de las secuencias
Secuenciación de transcriptomas; resecuenciación
hidrógeno tras la incorporación de un nucleótido en la cadena de ADN a secuenciar (Thudi et al., 2012).
Estas nuevas técnicas tienen en común, la eliminación de la clonación de los fragmentos de ADN y la paralelización en una sola corrida de cientos de millones de reacciones, lo cual tiene como ventaja un alto rendimiento, dando como resultado la generación de millones de lecturas de secuencia, a bajo costo y en menor tiempo, en comparación con tecnologías clásicas como la de Sanger (Deschamps & Campbell, 2010). Además, reducen la taza de falsos positivos del 6% al 1% en el descubrimiento de polimorfismos de un solo nucleótido (SNPs), en comparación con datos generados por microarreglos, y permite realizar genotipado de alta densidad. Esto permite un aumento en la eficiencia en estudios de mapeo de loci de caracteres cuantitativos (por sus siglas en inglés, QTL) y la identificación de genes asociados con características de interés en especies con genomas aún sin secuenciar (Schneeberger & Weigel, 2011).
No obstante, una de las mayores limitaciones en las técnicas de secuenciación de siguiente generación es la precisión en la determinación de las secuencias, teniendo en cuenta que las lecturas generadas son más cortas y la exactitud en pares de bases (pb) para cada lectura es menor comparada con el método de Sanger. Pese a esto, esta limitación ha sido contrarrestada con el aumento en la cobertura (redundancia de las lecturas en una determinada posición nucleotídica), que puede ser usada para discernir entre errores de secuenciación y una verdadera variación genética (Deschamps & Campbell, 2010; Schneeberger & Weigel, 2011).
De esta manera, gracias a los avances y beneficios de estas nuevas tecnologías, el número de genomas secuenciados ha crecido exponencialmente en los últimos años. En la base de datos GOLD (The Genomes Online Database, www.genomesonline.org) se registran un total de 19.060 proyectos genómicos para el año 2012, que incluyen 2.555 proyectos en Eucariotas, de los cuales 175 ya han sido terminados. Así mismo, estás técnicas tienen hoy en día numerosas aplicaciones como por ejemplo, proyectos de ensamblajes de novo, resecuenciación de genomas, transcriptómica, descubrimiento de polimorfismos y ARN no codificante, análisis epigenéticos, mapeo por asociación, entre otras (Mardis, 2008; Thudi et al., 2012).
3.3.1 Pirosecuenciación ó 454
generación, que ofrecen mayor rendimiento pero lecturas más cortas. De esta manera, el sistema de secuenciación 454 es frecuentemente utilizado para proyectos de ensamblaje de novo o secuenciación de grandes genomas (Morozova et al., 2009).
Esta técnica emplea PCR en emulsión sobre la base de cientos de microesferas, que poseen secuencias complementarias a los adaptadores de la cola terminal, ligados a cada uno de los fragmentos a secuenciar durante la construcción de la librería (Figura 1A). Posteriormente, cada microesfera es aislada en micelas individuales de agua y aceite, con los reactivos necesarios para llevar a cabo la PCR. Estas micelas permiten la captura de una única molécula o fragmento de ADN (Figura 1B), como molde para la PCR en emulsión. De esta forma, después del enriquecimiento de cada microesfera, éstas son depositadas en una placa PicoTiterPlate (PTP), en la cual se disponen en pozos individuales, seguido de las enzimas necesarias para la reacción, tales como, el fragmento Klenow de la ADN polimerasa, ATP sulfurilasa, luciferasa y apirasa (Figura 1 C); además de los sustratos, adenosina fosfosulfato (APS) y D-luciferina (Ahmadian et al., 2006). La corrida, se realiza en un equipo GS-FLX Titanium de Roche. Este, permite la liberación de los 4 nucleótidos, uno a la vez, de forma consecutiva, de tal manera que si hay incorporación del nucleótido en la cadena, se libera pirofosfato inorgánico (PPi). El equipo posee un sistema de detección bioluminiscente, que mide la liberación de pirofosfato inorgánico, convirtiéndolo a luz visible, mediante una serie de reacciones enzimáticas.
Figura 1: Procedimiento general de la técnica de secuenciación 454 de Roche. Modificado de: Mardis (2008) y Metzker (2010).
3.3.2 Aplicaciones de la pirosecuenciación en transcriptómica de novo en plantas
particulares (Alagna et al., 2009; Rosero et al., 2011), anotación y descubrimiento de genes y rutas metabólicas (Alagna et al., 2009; Cheung et al., 2006; Li et al., 2010; Lin et al., 2011; Natarajan & Parani, 2011; Sun et al., 2010; Wang et al., 2009; Zeng et al., 2010), relaciones filogenéticas (Zeng et al., 2010), descubrimiento de ARN no codificante (Paul et al., 2011), anotación de genomas (Kalavacharla et al., 2011) y desarrollo de marcadores moleculares (Blanca et al., 2011; Dutta et al., 2011; Novaes et al., 2008; Parchman et al., 2010; Sloan et al., 2012; Zeng et al., 2010).
Alagna et al. (2009) secuenciaron cuatro librerías de ADNc de 2 cultivares de Olea europea caracterizados por patrones de acumulación de diferentes tipos de fenoles. De esta forma, los análisis de expresión diferencial realizados, permitieron la identificación de genes asociados a diferentes rutas metabólicas relacionadas con características de interés de la oliva. Parchman et al. (2010), mediante 454, realizaron la secuenciación de novo de Pinus concorta, un árbol ecológica y económicamente importante. Obtuvieron un total de 586.732 ESTs, ensamblados en 63.657 contigs; los cuales fueron anotados contra las bases de datos UniRef50 y TAIR, encontrando una alta presencia de retrotransposones. Además, identificaron y desarrollaron 15.084 marcadores SSRs, así como 3.707 marcadores tipo SNP. Sun et al. (2010) generaron la primer colección de ESTs del ginseng americano (Panax quinquefolius), una de las especies más usadas en el mundo por sus propiedades medicinales. Mediante la secuenciación y el ensamblaje de los ESTs, se identificaron diferentes enzimas asociadas a la biosíntesis de diferentes metabolitos secundarios, entre ellos, la biosíntesis de ginsenosides, principal componente bioactivo de la planta. Kalavacharla et al. (2011) secuenciaron y ensamblaron de novo, librerías de ADNc de Phaseolus vulgaris, a partir de las cuales obtuvieron nuevo transcritos (53.4%) en la especie útiles en genómica funcional, que permitieron el desarrollo de marcadores moleculares para futura identificación de QTLs y apoyo a programas de mejoramiento. Por otro lado, Dutta et al. (2011) generaron los primeros marcadores SSRs de Cajanus cajan, una de las principales leguminosas de regiones tropicales y subtropicales. De estos, 20 marcadores identificados como polimórficos fueron evaluados en 22 variedades de C. cajan. Los resultados mostraron que los marcadores generados son de gran valor en estudios de diversidad y mapeo genético en la especie. Lin et al. (2011) sugieren que la secuenciación por tecnologías de siguiente generación, permiten el descubrimiento de genes y la rápida caracterización funcional de especies no modelo, tal como el Ginkgo biloba; encontrándose diferentes transcritos asociados con rutas metabólicas de gran importancia en la especie, como la síntesis de flavonoides y ginkgolide/bilobalide, así como transcritos relacionados con desarrollo, respuesta a enfermedades y defensa.
de marcadores moleculares específicos para especies con limitado número de información genómica, como lo es la uchuva.
3.4 Análisis computacionales de ESTs
A partir del uso de las técnicas NGS, miles de lecturas o ESTs, especialmente de organismos no modelo están siendo generados. Estas lecturas deben ser procesadas con la estrategia más conveniente que ayude a cumplir con el objetivo biológico propuesto. Usualmente, el análisis de ESTs comienza con la asignación de un valor de calidad a cada base, mediante un software de llamado de bases que permite identificar errores de secuenciación. A su vez, es posible realizar limpieza para secuencias contaminantes, como vectores, adaptadores, cebadores u otros xenocontaminantes (Bouck & Vision, 2007). Posteriormente, las lecturas pueden ser alineadas a una secuencia de referencia o ensambladas de novo, formando secuencias consenso o contigs (Metzker, 2010). Y, finalmente, los transcritos ensamblados pueden ser anotados, con el fin de predecir su función, con la ayuda de diferentes bases de datos públicas disponibles.
3.4.1 Ensamblaje de ESTs
El ensamblaje de lecturas pequeñas, generadas a partir de NGS, es considerado como uno de los procesos más desafiantes en genómica, especialmente de genomas largos y complejos; entre los cuales encontramos los de algunas plantas, caracterizados por su frecuente poliploidía, elementos repetitivos y duplicación génica (Imelfort & Edwards, 2009). Es por esto, que en los últimos años la secuenciación y ensamblaje de transcriptomas es una alternativa que no sólo sobrepasa el principal obstáculo de un ensamblaje, como lo es la cantidad de ADN repetitivo de regiones no codificantes, sino además es una alternativa para la generación de información génica de organismos con limitada o nula información a nivel genómico (Pop & Salzberg, 2008).
Según Martin & Wang (2011) el ensamblaje de ESTs puede involucrar tres estrategias diferentes (Figura 2): ensamblaje basado en un genoma de referencia, ensamblaje de novo o una estrategia combinada de las dos anteriores.
abundancia. Sin embargo, el correcto ensamblaje depende de la calidad del genoma de referencia a usar y la disponibilidad del mismo (Martin & Wang, 2011).
La segunda estrategia, la cual ha sido usada en este estudio, es el ensamblaje de novo de las lecturas obtenidas. Este ensamblaje, se basa en la alta redundancia de las lecturas, que comparten bases entre ellas, para formar transcritos. Esta estrategia, permite en comparación con el ensamblaje comparativo, la identificación de transcritos faltantes en los genomas, así como el dar a conocer un primer set de transcritos cuando el genoma no está aún disponible. Además, tiene como ventaja, la no dependencia en el correcto alineamiento de las lecturas para identificar sitios de splicing. No obstante, los requerimientos computacionales para este tipo de ensamblaje son mayores y necesitan de un alto nivel de cobertura de las lecturas (Martin & Wang, 2011; Miller et al., 2010).
Finalmente, la estrategia combinada, se basa en la sensibilidad en el uso de un genoma de referencia y la capacidad del ensamblaje de novo de detectar nuevos transcritos. Sin embargo, aún no existen algoritmos automatizados para llevar a cabo esta estrategia de manera conjunta (Martin & Wang, 2011).
Actualmente, existen diferentes ensambladores comúnmente usados en estudios genómicos y transcriptómicos en plantas. Algunos de los ensambladores usados en estudios transcriptómicos de novo en plantas son Newbler (Bai et al., 2011; Dutta et al., 2011; Ying Li et al., 2010; Natarajan & Parani, 2011; Novaes et al., 2008), Seqman Ngen (DNAstar, Inc.) (Parchman et al., 2010), CAP3 (W. Wang et al., 2009), entre otros pipelines (Alagna et al., 2009).
Figura 2: Estrategias de ensamblaje de lecturas generadas a partir de técnicas de secuenciación de siguiente generación. A. Ensamblaje comparativo B. Ensamblaje de novo
[image:30.612.114.525.492.679.2]La mayoría de estos ensambladores, usan tres tipos de algoritmos para la generación de contigs e isoformas. Una de las estrategias es el uso de algoritmos de avance rápido (greedy), basado en la extensión de los contigs por solapamientos continuos de las lecturas hasta que no exista una posible unión (Miller et al., 2010). Otra estrategia, usada para lecturas cortas, por ensambladores como Euler, Velvet, SOAPdenovo, Trinity, son los grafos de Bruijn; el cual consiste en el uso de secuencias de una longitud k (k-mers) de cada lectura, las cuales representan un nodo. Posteriormente, cada par de nodos es conectado, de tal manera que se mueve una base (k-1) entre cada par de k-mers. Finalmente, los nodos son unidos en uno solo, formando transcritos con sus respectivas isoformas (Figura 2), teniendo en cuenta que la unión de nodos puede generar diferentes caminos a través del grafo (Martin & Wang, 2011). Ensambladores como Celera, Arachne, CAP3, Newbler, utilizan otra estrategia llamada OLC (Overlap/Layout/Consensus). Este método usa un grafo de solapamiento que involucra el uso de k-mers y un alineamiento pareado de todos contra todos, que lleva a la construcción de un grafo de solapamientos y un posterior alineamiento múltiple para formar la secuencia consenso (Figura 3) (Miller et al., 2010).
Una selección del ensamblador correcto para estudios en transcriptómica y aún más en genómica es difícil de realizar, basado en el hecho de que la precisión de un ensamblador es difícil de medir. No obstante, usualmente su exactitud, se puede llegar a determinar por el tamaño y precisión de los contigs ensamblados; teniendo en cuenta la longitud máxima y promedio, y el N50, definido como la longitud del contig a partir del cual se encuentran el 50% de las bases usadas en el ensamblaje (Miller et al., 2010).
3.4.1.1 GS de novo Assembler o Newbler
Versiones actuales de newbler, poseen la opción cDNA para proyectos transcriptómicos. Básicamente, usa la misma metodología para la construcción de grafos de contigs, pero crea subgrafos que posiblemente representan un gen, que tienen como nombre isogrupos. Luego newbler, empieza a atravesar o converger los diferentes caminos de los contigs en los subgrafos de cada isogrupo, generando variantes alternativas, llamadas isotigs (Figura 3) (Nederbragt, 2011). Los parámetros por defecto, especifican un número máximo de 100 isotigs por isogrupo con 500 contigs.
Como ventaja, newbler tiene la capacidad de corregir grandes secuencias de homopolímeros, haciendo uso de la cobertura de las lecturas para detectar errores en las secuencias (Martin & Wang, 2011; Miller et al., 2010). Además, en comparación con otros ensambladores comúnmente usados, se caracteriza por construir contigs más largos, de manera rápida, siendo a su vez, fácil de usar (Kumar & Blaxter, 2010).
Figura 3: Ensamblaje basado en el algoritmo OLC en Newbler, con la opción cDNA para ensamblaje de transcriptomas. A. Solapamiento (Overlap), B. Diseño (Layout), C. Consenso (Consensus). Tomado de: Nederbragt (2011).
3.4.2 Anotación de secuencias consenso
el fin de generar información biológica, mediante la integración de análisis computacionales, datos biológicos y criterio biológico; siendo éste, el proceso en el cual se une el vacío que hay entre el secuenciamiento y la biología de un organismo (Stein, 2001). No obstante, el proceso de anotación es el cuello de botella en la genómica hoy día, teniendo en cuenta el gran volumen de información a nivel nucleotídico generado a partir de las nuevas tecnologías de secuenciación. De esta manera, actualmente, existen diferentes algoritmos para anotación, que tienen como meta facilitar el proceso de análisis de secuencias, de una forma rápida y menos laboriosa.
La anotación de secuencias generalmente involucra la anotación a nivel estructural y funcional. La anotación estructural está relacionada con la identificación de genes, en conjunto con regiones génicas como codones de iniciación y terminación, regiones no traducidas (UTR), elementos transponibles, sitios de splicing, regiones promotoras y reguladoras, islas CpG, sitios poliA, pseudogenes, entre otros elementos biológicamente importantes (Kawaji & Hayashizaki, 2008).
Existen diferentes algoritmos usados para predicción génica, basados en diferentes métodos computacionales. El método ab initio, consiste en el uso de ciertas características de la secuencias de ADN (v.g regiones ricas Guanina+Citocina) para la identificación de un gen. Un segundo método, utiliza la homología de secuencias entre organismos relacionados. Mientras que la última generación de algoritmos en predicción génica, utilizan en conjunto los dos métodos anteriores (Stein, 2001).
Por otro lado, la anotación funcional es el proceso más importante en estudios genómicos y transcriptómicos. Este proceso tiene como objetivo asignar una función putativa a cada uno de los genes identificados, generalmente mediante la identificación de dominios y motivos con base en similaridad de secuencias nucleotídicas traducidas, con secuencias de aminoácidos de proteínas ya caracterizadas. A este proceso, se suma la identificación del rol biológico de los genes y proteínas identificadas, en base al vocabulario controlado para describir las funciones génicas, llamado Gene Ontology (GO) (Stein, 2001).
Entre las herramientas más utilizadas para anotación tanto estructural como funcional, se encuentran algunos predictores ab initio como Augustus (Stanke et al., 2006) y Genmark (Besemer & Borodovsky, 2005) y predictores por similaridad como BLAST (Altschul et al., 1990), PSI-BLAST, RPS-BLAST (Altschul et al., 1997) y HMMER (Johnson et al., 2010), así como herramientas basadas en GO como Blast2Go.
generada mediante una intervención manual, donde los expertos o curadores manuales juegan un papel importante, a la hora de producir información más confiable y refinada de las estructuras y funciones génicas predichas (Ashurst & Collins, 2003).
3.4.2.1 Bases de datos para anotación
Con el fin de llevar a cabo el proceso de anotación por homología de secuencias, existen a nivel mundial, diferentes tipos de bases de datos con información periódicamente actualizada, de secuencias de nucleótidos, proteínas, dominios, rutas metabólicas, entre otros. A continuación se hace una breve descripción de las bases de datos usadas en este estudio.
UniprotKB/Swiss-prot
Hace parte de la base de datos central curada Uniprot Knowledgebase, perteneciente al consorcio Uniprot, la cual está dividida en dos secciones, UniprotKB/Swiss-prot y UniprotKB/TrEMBL. Swiss-prot es una base de datos de proteínas, con entradas curadas y revisadas manualmente, con un mínimo de redudancia; mientras que TrEMBL es una colección de entradas que provienen de otras bases de datos (DDB/EMBL/GenBank), las cuales son posterioremente anotadas automáticamente (Consortium Uniprot, 2011).
RefSeq
La base de datos RefSeq pertenece a los repositorios de Centro Nacional para la Información Biotecnológica (NCBI). Esta base de datos contiene datos de secuencias genómicas, transcriptómicas y proteicas no redundantes y curadas (Pruitt et al., 2012).
Pfam
Pfam es una base de datos de familias de proteínas, generada a partir de alineamientos múltiples y construcción de perfiles de modelos ocultos de Markov (HMMs) que permiten la identificación de familias proteicas. Se encuentra dividida en dos secciones: Pfam – A con familias curadas manualmente de alta calidad y Pfam-B con familias automáticamente generadas (Punta et al., 2012).
KEGG PATHWAY
metabólicas, generados a partir de evidencia experimental reportada en la literatura (Kanehisa et al., 2012).
4.4.2.2 Herramientas bioinformáticas para anotación funcional
En bioinformática diferentes herramientas son usadas para el proceso de anotación estructural y funcional, con el fin de la identificación de genes, proteínas y función biológica de transcritos o ADN de manera más eficiente, teniendo en cuenta el gran volumen de datos a nivel de secuencia generados por las últimas tecnologías de secuenciación. A continuación se presentan algunos programas usados en este estudio.
BLAST (Basic Local Alignment Search Tool)
La herramienta BLAST (Altschul et al., 1990), es un algoritmo comúnmente usado en bioinformática, que tiene como fin la búsqueda por similaridad de secuencias, mediante alineamientos locales, de una entrada problema contra las bases de datos disponibles (Camacho et al., 2009). Existen diferentes tipos de BLAST, que permiten personalizar la búsqueda teniendo en cuenta el tipo de entrada usado. Algunos de los tipos de BLAST se encuentran en la tabla 2.
Tabla 2: Tipos de BLAST comúnmente usados para anotación. BD=Base de datos
Programa Descripción
blastn Compara secuencias de nucleótidos contra una BD de nucleótidos blastp Compara secuencias e aminoácidos contra una BD de proteínas
blastx Compara secuencias de nucleótidos traducida en sus seis posibles marcos de lectura contra una BD de proteínas
tblastx Compara una secuencia de nucleótidos traducida contra una BD de nucleótidos traducida en sus seis posibles marcos de lectura cada una tblastn Compara una secuencia de proteínas contra una BD de nucleótidos
traducida
rpsblast Busca una secuencia de proteínas contra una BD de matrices de puntaje de posición específica (PSSM, dominios conservados)
Blast2GO
vocabulario estructurado y controlado de los productos génicos, mediante una estructura jerárquica de padres e hijos, representada por nodos (términos ontológicos o GO terms) con relaciones entre sí, a más de un término ontológico (Figura 4). Agrupa las ontologías en tres grandes grupos; función molecular, proceso biológico y componente celular (The Gene Ontology Consortium, 2012). La categoría proceso biológico está relacionada con el objetivo biológico que lleva a cabo el producto génico, como por ejemplo el término “transducción de señales” o “fotosíntesis”. Función molecular se asocia con la actividad bioquímica del producto génico, como “actividad transportadora” o “regulación enzimática”. Y finalmente, la ontología componente celular corresponde al lugar donde el producto génico lleva a cabo su función, como por ejemplo la “membrana nuclear” o el “ribosoma” (Ashburner et al., 2000).
Figura 4: Estructura jerárquica de padres e hijos, de las ontologías génicas, donde cada nodo o término GO puede estar relacionado con más de un término GO. A. Grafo de la ontología génica “regulación de la fotosíntesis, reacciones lumínicas”. En este caso, un nodo puede tener más de un padre, por ejemplo “regulación de la fotosíntesis” tiene dos padres, “fotosíntesis” y “regulación de procesos metabólicos celulares”. Por otro lado, el proyecto GO usa diferentes
tipos de términos relacionales con el fin de unir término ontológicos; por ejemplo es_un (por sus
siglas en inglés is_a) (líneas azules), parte_de (por sus siglas en inglés part_of) (líneas azul
claro), regula (por sus siglas en inglés regulates) (líneas negras), entre otros. El término
“regulación de la fotosíntesis, reacciones lumínicas” es_un regulador de la “fotosíntesis” y es_un
regulador de la “generación de metabolitos precursores y energía”; además regula las
“reacciones lumínicas de la fotosíntesis”, término que hace parte_de la “fotosíntesis” y es_un “generador de metabolitos precursores y de energía”. B. Grafo de la ontología génica “plastidio”.
Este término, es_una “parte citoplasmática” y es_un “organelo intracelular limitado por una
membrana”. Fuente: http://amigo.geneontology.org/cgi-bin/amigo/go.cgi
Blast2GO (B2G) es una herramienta catalogada como un anotador universal basado en GO, especialmente diseñada para organismos no modelo, capaz de asociar secuencias génicas a términos ontológicos. B2G usa el algoritmo BLAST para encontrar homólogos de las secuencias problema, extrae las ontologías asociadas para cada uno de los hits obtenidos y evalúa y asigna la mejor ontología a la secuencia problema (Conesa & Gotz, 2008). Esta herramienta realiza 3 pasos en el proceso de anotación: BLASTing, mapeo y anotación.
1. BLASTing: consiste en el uso de BLAST para encontrar secuencias similares a la secuencia pregunta.
2. Mapeo: obtiene las ontologías asociadas a los hits obtenidos.
3. Anotación: consiste en la selección de ontologías más confiables. a partir de aquellas anteriormente identificadas. La selección, se realiza con base en un puntaje de anotación generado por el porcentaje de similaridad entre la secuencia pregunta y el hit de BLAST, la calidad de la fuente o código de evidencia (siglas en inglés-EC) de la asignación del término ontológico (v.g “inferido por ensayo directo” (por sus siglas en inglés IDA), “inferido por anotación electrónica” (por sus siglas en inglés IEA), entre otros) y la estructura del grafo..
Finalmente, B2G ofrece la posibilidad de visualizar los resultados obtenidos, mediante gráficas de barras y tablas que facilitan el análisis de datos.
Bases de datos
Existen diferentes herramientas de libre acceso para la construcción de este tipo de bases de datos, tales como PostgreSQL (http://www.postgresql.org), MySQL
(http://www.mysql.com/), GMOD (O’Connor et al., 2008), Tripal
4. MATERIALES Y MÉTODOS
4.1 Localización
El estudio se llevó a cabo en el Laboratorio de Genética Molecular Vegetal, en la Unidad de Bioinformática y Biología Computacional del Centro de Biotecnología y Bioindustria CBB, de la Corporación Colombiana de Investigación Agropecuaria (CORPOICA), sede Tibaitatá. Esta tesis, formó parte del proyecto “Evaluación de resistencia o tolerancia de germoplasma de uchuva (Physalis peruviana) a Fusarium oxysporum como contribución al control integrado de la enfermedad” financiado por el Ministerio de Agricultura y Desarrollo Rural (MADR), código 072-2008L4787-3281.
4.2 Material vegetal
Para este estudio, se usó material foliar, de la accesión 09U216-6, perteneciente a la colección del Banco de Germoplasma de uchuva, administrado por CORPOICA. Según datos de pasaporte, este material pertenece al ecotipo Colombia, seleccionado a partir de cultivos comerciales del departamento de Nariño. En estudios previos, la accesión usada fue catalogada como susceptible a F. oxysporum en condiciones de invernadero y campo (González & Barrero, 2011). Adicionalmente, el ecotipo fue caracterizado a nivel citogenético, mediante conteo cromosómico y citometría de flujo, dando como resultado un nivel de ploidía de 2n=4x=48, con un tamaño de genoma haploide o valor C de 1.93pg de ADN (1.8Gpb) (Liberato, 2012).
4.3 Extracción de ARN y construcción de la librería normalizada de ADNc
doble cadena y el uso del oligonucleótido SMART IV (en inglés - Switching Mechanism At 5' end of RNA Transcript) para producir ADNc completo (Zhu et al., 2001).
Una vez sintetizado el ADNc, se realizó la normalización de la librería, usando el método de normalización modificado de Patanjali et al. (1991) y Soares et al. (1994). Este proceso, tuvo como fin igualar el número de copias de genes en la librería, reduciendo la prevalencia de transcritos abundantes, equiparándolos a la concentración de transcritos que se expresan en un bajo nivel (Bogdanova et al., 2011). El procedimiento general, consistió en la síntesis de ADNc completo ligado a dos pares de adaptadores tester3/tester5 fosforilado y driver3/driver5 fosforilado (Anexo 1) para la muestra prueba (tester) y la muestra de referencia (driver). El ADNc de cadena sencilla fue usado para la hibridación en lugar del ADNc de cadena doble. De esta manera, se realizó una digestión con la enzima exonucleasa lambda (), la cual permitió la degradación de las cadenas con el extremo 5’ fosforilado. Posteriormente, las hebras sencillas tanto del tester como driver, fueron sometidas a hibridación; así, los transcritos que se presentaron en exceso fueron hibridados entre sí. Después, las moléculas con cadena doble fueron separadas de la sencilla usando cromatografía en hidroxiapatita. Las moléculas de ADNc normalizadas, de cadena sencilla, fueron amplificadas con el cebador L4N, específico para el tester (FailSafeTm PCR system, Epicentre Biotechnologies, USA). Finalmente, se realizaron cortes de los fragmentos de ADNc amplificados en geles de agarosa al 1% y se seleccionaron fragmentos mayores a 0.5kb, los cuales fueron purificados por electro-elución y almacenados en 80% de EtOH a -80ºC.
4.4 Secuenciación y ensamblaje
La librería de ADNc normalizada, fue secuenciada mediante la técnica de secuenciación de segunda generación pirosecuenciación ó 454 - GS FLX Titanium de Roche, en el Centro Genómico de Emory (Atlanta, GA, USA). Aproximadamente, 5 μg de ADNc purificado fue fragmentado mediante el sistema Covaris E210, y posteriormente secuenciado en tres cuartos de placa de 454. Mediante esta técnica se generaron archivos tipo SFF (Standard Flowgram File), que contienen las secuencias con sus respectivos valores de calidad, los cuales fueron sometidos al NCBI Sequence Read Archive (SRA) (número de accesión SRP005904).
generaron nuevamente archivos tipo SFF, los cuales fueron usados para realizar un ensamblaje de novo, mediante el software Newbler, GS de novo Assembler (Roche, versión 2.5.3), usando los parámetros por defecto. Adicionalmente, se llevó a cabo un ensamblaje comparativo con el genoma borrador de papa (Solanum tuberosum – versión PGSC_DM_v3) (X. Xu et al., 2011) y el genoma borrador de tomate (Solanum
lycopersicum – versión ITAG2.3)
(http://solgenomics.net/organism/solanum_lycopersicum/genome), utilizando GS Reference Mapper de Newbler.
En el ensamblaje de novo, newbler ensambló las lecturas en contigs, los cuales fueron posteriormente ensamblados en isotigs. Los isotigs, representan transcritos alternativos putativos de un gen, agrupados en isogrupos si comparten contigs entre ellos. Las lecturas que no ensamblaron dentro de un contig se definieron como singletons. El ensamblaje fue depositado en la base de datos Transcriptome Shotgun Assembly (por sus siglas en inglés, TSA) del NCBI, con números de accesión JO124085-JO157957.
4.5 Anotación Funcional
A partir del ensamblaje de novo, se realizó un BLASTX de los isotigs y singletons (unigenes) contra la base de datos UniProtKB/Swiss-Prot (versión Abril-2011). Se usó un valor esperado “e-value” de 1e-5 y se recuperó el mejor alineamiento para cada isotig, mediante el uso de los parámetros -num_descriptions y -num_alignments, a partir del formato de salida en XML. Aquellos unigenes que no presentaron homología con una proteína de la base de datos de UniProtKB/Swiss-Prot, fueron comparados contra la base de datos RefSeq del NCBI (versión 47).
4.6 Identificación de marcadores moleculares
Se identificaron marcadores tipo microsatélite (SSRs) en la colección de contigs obtenida a partir del ensamblaje de novo realizado, mediante el programa Phobos (versión 3.3.10) (Mayer, 2006-2010). Se identificaron marcadores tipo perfecto e imperfecto, con una longitud mínima de 18pb para dinucleótidos, 24pb para trinucleótidos y tetranucleótidos, 30pb para pentanucleótidos y 36pb para hexanucleótidos. El anexo 2 representa gráficamente el flujo de trabajo realizado en este estudio.
4.7 Organización de la información en una base de datos
5. RESULTADOS Y DISCUSIÓN
5.1 Síntesis de ADNc y normalización
A partir del ARN de 6 clones del ecotipo usado, se sintetizó una librería de ADNc normalizada. Este tipo de librerías ha sido ampliamente usada en diferentes estudios transcriptómicos (Blanca et al., 2011; Natarajan & Parani, 2011; Parchman et al., 2010; Sloan et al., 2012), permitiendo sobrepasar las diferencias que existen en la abundancia de transcritos en células de diferentes tejidos. Por medio de la librería normalizada, se equiparó el número de transcritos, lo cual probablemente permitió la secuenciación de ESTs expresados tanto a alto como a bajo nivel, haciendo más eficiente el análisis de ARNm. De esta manera, probablemente fue posible la identificación de un mayor número de genes, y por ende una anotación funcional más completa de la primera colección de ESTs en uchuva.
5.2 Secuenciación de ESTs y ensamblaje
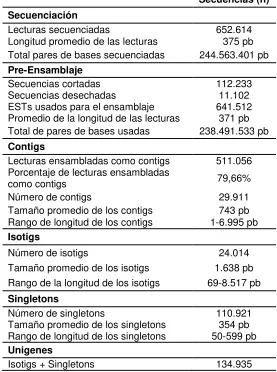
Tabla 3: Estadísticas de la secuenciación y ensamblaje
Secuencias (n) Secuenciación
Lecturas secuenciadas 652.614
Longitud promedio de las lecturas 375 pb
Total pares de bases secuenciadas 244.563.401 pb
Pre-Ensamblaje
Secuencias cortadas 112.233
Secuencias desechadas 11.102
ESTs usados para el ensamblaje 641.512
Promedio de la longitud de las lecturas 371 pb
Total de pares de bases usadas 238.491.533 pb
Contigs
Lecturas ensambladas como contigs 511.056
Porcentaje de lecturas ensambladas
como contigs 79,66%
Número de contigs 29.911
Tamaño promedio de los contigs 743 pb
Rango de longitud de los contigs 1-6.995 pb
Isotigs
Número de isotigs 24.014
Tamaño promedio de los isotigs 1.638 pb
Rango de la longitud de los isotigs 69-8.517 pb
Singletons
Número de singletons 110.921
Tamaño promedio de los singletons 354 pb
Rango de longitud de los singletons 50-599 pb
Unigenes
Isotigs + Singletons 134.935
Parani, 2011), Silene vulgaris (86%) (Sloan et al., 2012) y Fraxinus spp. (79%) (Bai et al., 2011). La mayoría de contigs fueron ensamblados con un promedio de 17 lecturas y cerca del 32% de los contigs tienen más de 20 lecturas, mientras que solo un 10% de los contigs tiene más de 50 lecturas. Según Blanca et al. (2011) está baja redundancia en el número de ESTs por contig se deba probablemente al proceso de normalización de la librería que suprimen los transcritos de mayor abundancia, permitiendo el aumento en el número de contigs.
Figura 5: Distribución de las lecturas secuenciadas. A. (ESTs) de P. peruviana, B. contigs
ensamblados y C. isotigs.
Posteriormente, los contigs generados fueron ensamblados entre sí, generando 24.014 isotigs con un promedio de 1.638pb (Figura 5C). La longitud N50 de los isotigs fue de 2.504pb. Aquellos isotigs que comparten contigs en común pertenecen al mismo isogrupo, para un total de 14.049 isogrupos, equivalentes a un locus génico con múltiples transcritos alternativos (promedio de 1,7 isotigs por isogrupo), donde la mayoría de los isogrupos (68%) contiene un sólo isotig.
El grupo restante de secuencias (20,3%) pertenecen a lecturas no sobrelapantes denominadas singletons, secuencias cortas de nucleótidos similares (semillas) en más de 70 lecturas las cuales son categorizadas como repeticiones, falsos positivos o lecturas problemáticas en el ensamblaje (v.g secuencias quiméricas) y secuencias pequeñas. La cobertura promedio de los isotigs ensamblados fue de 9.1X, la cual disminuye a 3.1X si se incluyen los singletons como porción transcrita del genoma, teniendo en cuenta que corresponden a un único EST. El porcentaje de singletons encontrados fue de un 17% de las lecturas usadas para el ensamblaje. Este alto porcentaje de lecturas, puede corresponder a secuencias poco abundantes, incapaces de ser ensambladas en contigs. Algunas de ellas, podrían representar secuencias contaminantes, producto del proceso de construcción de la librería normalizada o
[image:45.612.63.576.206.383.2]posiblemente estan relacionados con errores de secuenciación o regiones altamente polimórficas. Aunque Newbler se basa en el algoritmo OLC, el cual según Li et al. (2012) puede tolerar cierto número de errores en la fase de solapamiento, la presencia de lecturas de regiones con alta heterocigocidad o aquellas con errores de secuenciación podrían ser algunas de las principales limitaciones a la hora de realizar ensamblajes de novo, como el realizado en este trabajo. El anexo 3 muestra la tabla correspondiente a las estadísticas del ensamblaje generadas por Newbler. Los 24.014 isotigs y 110.921 singletons usados para análisis posteriores, se denominaron unigenes.
5.2.1 Ensamblaje comparativo con papa y tomate
Teniendo en cuenta, que el genoma borrador de la papa (Solanum tuberosum - versión PGSC_DM_v3) (Xu et al., 2011) y el tomate (Solanum lycopersicum –versión ITAG2.3) (http://solgenomics.net/organism/solanum_lycopersicum/genome) se encuentran disponibles, y son especies que pertenecen a la misma familia de P. peruviana, fue posible realizar un ensamblaje de referencia con estas dos especies. Éste, permitió realizar un control de calidad del ensamblaje de novo, basados en valores estadísticos estándar usados en otros estudios (Kumar & Blaxter, 2010).
[image:46.612.99.543.662.726.2]Los ensamblajes de referencia o comparativos, dependen de la calidad del genoma de referencia usado, siendo posible el uso de genomas de especies relacionadas (Martin & Wang, 2011), como en este caso papa y tomate. Sin embargo, como se observa en la tabla 4, el porcentaje de lecturas ensambladas como contigs es similar para las tres aproximaciones; pero el número, tamaño promedio y N50 de los contigs mayores a 500pb es superior para el ensamblaje de novo en comparación con los ensamblajes de referencia. Esta situación posiblemente se presente porque las lecturas no son mapeadas a lo largo del genoma de referencia, teniendo en cuenta la probable variabilidad a nivel nucleotídico entre las 2 especies (uchuva vs papa, tomate vs uchuva) y la presencia de intrones, situaciones que pueden llegar a producir una fragmentación de los contigs en tamaños más pequeños.
Tabla 4: Estadísticas del ensamblaje comparativo con papa y tomate
Ensamblaje de
novo
Ensamblaje comparativo papa
Ensamblaje comparativo tomate
Lecturas ensambladas
como contigs 511.056 531.386 504.515
ensambladas como contigs
Número total de contigs 29.911 72.077 65.245
Número de contigs
mayores a 500pb 15.124 6.181 5.173
Tamaño promedio de los
contigs 1.222 pb 917 pb 921 pb
N50 Contigs 1.438 pb 950 pb 957 pb
De ésta manera, el uso de ensamblajes de novo, tiene como ventaja el ensamblaje de regiones transcritas que no aparecen en el genoma de referencia, ya sea el caso por regiones incompletas en el genoma o regiones divergentes entre las 2 especies relacionadas (vg. papa-uchuva, tomate-uchuva). Aunque no existe un método exacto para medir la precisión y la calidad de un ensamblador y ensamblaje, en base a lo anterior y al hecho de que el ensamblaje de novo presentó tamaños y promedios superiores, éste último fue escogido para su uso en la anotación funcional de la primera colección de transcritos de uchuva.
5.3 Anotación funcional
5.3.1 Anotación por homología de secuencias BLASTX
Tabla 5: Número de isotigs, singletons y unigenes con hits de BLAST contra las bases de datos datos UniProtKB/Swiss-Prot y RefSeq
Isotigs (24.014) Singletons (110.921) Unigenes (134.935)
Swissprot
Hits de BLAST 14.720 (61,2%) 20.906 (18,8%) 35.626 (15,1%)
Proteínas únicas 5.953 7.702 11.232
RefSeq Isotigs (9.294*) Singletons (90.015*) Unigenes (99.309)
Hits de BLAST 4.442 (47,7%) 14.522 (16,3%) 18.964 (19,09%)
Proteínas únicas 2.768 7.490 9.411
Total Hits de Blast 19.162 (79,7%) 35.428 (31,9%) 54.590 (40,45%)
* Número de isotigs y singletons sin hits de BLAST con UniProtKB/Swiss-Prot, usados en la anotación por BLAST
contra RefSeq.