DESPOOL: un nuevo programa para estudios predictivos en informática química y biológica
77
0
0
Texto completo
(2) DICTAMEN. El que subscribe, Addel Arnaldo Goya Jorge, hago constar que el trabajo titulado “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica” fue realizado en la Universidad Central “Marta Abreu” de Las Villas como parte de la culminación de los estudios de la especialidad de Ciencia de la Computación, autorizando a que el mismo sea utilizado por la institución, para los fines que estime conveniente, tanto de forma parcial como total y que además no podrá ser presentado en eventos ni publicado sin la autorización de la Universidad.. ________________ Firma del Autor. Los abajo firmantes, certificamos que el presente trabajo ha sido realizado según acuerdos de la dirección de nuestro centro y el mismo cumple con los requisitos que debe tener un trabajo de esta envergadura referido a la temática señalada.. ______________________. ___________________________. Firma del Tutor. Firma del Jefe de Seminario. Dra. Gladys Casas Cardoso. Dr. Ricardo Grau Ábalo. _____________ Fecha. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(3) Tabla de contenido Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(4) ÍNDICE RESUMEN ABSTRACT INTRODUCCIÓN .............................................................................................................................................................................. 1 CAPÍTULO I: ESTRATEGIAS UTILIZADAS EN LA INFORMÁTICA QUÍMICA ............................................................................... 6 1.1. Consideraciones generales sobre estudios predictivos QSPR/QSAR ................................................................................. 6 1.2. Descriptores moleculares ..................................................................................................................................................... 9 1.2.1. Definición y Clasificación de los descriptores moleculares .......................................................................................... 9 1.2.2. Propiedades que debe poseer un nuevo descriptor molecular .................................................................................. 14 1.3. Programas para el cálculo de descriptores moleculares utilizados en la informática química ........................................... 17 1.3.1. Software comerciales ................................................................................................................................................. 17 1.3.2. Software libres ............................................................................................................................................................ 18 1.3.3. Bibliotecas utilizadas en la informática química ......................................................................................................... 20 1.4. Consideraciones parciales ................................................................................................................................................. 21 CAPÍTULO II: DISEÑO E IMPLEMENTACIÓN DEL PROGRAMA DESPOOL .............................................................................. 22 2.1.. Suite de trabajo, TOMOCOMD-CARDD ...................................................................................................................... 22. 2.2.. Uso de la biblioteca CDK ............................................................................................................................................. 23. 2.3.. Análisis, diseño e Implementación de la Herramienta ................................................................................................. 25. 2.3.1.. Diagrama de casos de uso ................................................................................................................................ 26. 2.3.2.. Diagrama de clases ........................................................................................................................................... 27. 2.3.3.. Diagrama de actividades ................................................................................................................................... 28. 2.3.4.. Artefactos del sistema ........................................................................................................................................ 29. 2.4 Consideraciones finales ...................................................................................................................................................... 30 CAPÍTULO III: DISEÑO TEÓRICO DE DESPOOL ........................................................................................................................ 31 3.1.. Manual de usuario ....................................................................................................................................................... 31. 3.1.1.. Requerimientos del software ............................................................................................................................. 32. 3.1.2.. Modo de entrada de fichero ............................................................................................................................... 32. 3.1.3.. Modo de salida de ficheros ................................................................................................................................ 33. 3.1.4.. Definición de Invariantes .................................................................................................................................... 34. 3.1.5.. Definición de Locales ......................................................................................................................................... 37. 3.1.6.. Definición de propiedades atómicas .................................................................................................................. 38. 3.1.7.. Elección de descriptores moleculares a calcular ............................................................................................... 39. 3.1.8.. Reportes Estadísticos ........................................................................................................................................ 41. 3.2.. ANÁLISIS DE VARIABILIDAD DE LOS DESCRIPTORES DEL DESPOOL ............................................................... 42. CONCLUSIONES ........................................................................................................................................................................... 46 RECOMENDACIONES ................................................................................................................................................................... 47 REFERENCIAS BIBLIOGRÁFICAS ............................................................................................................................................... 48 ANEXOS ......................................................................................................................................................................................... 51. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(5) Dedicatoria Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(6) A mis padres, que encuentran la felicidad cuando yo la encuentro. Cuando yo vivo algo hermoso, lo viven a través de mi experiencia. Gracias mami, Gracias papi.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(7) Agradecimientos Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(8) Con este trabajo culmina una etapa importante de mi vida y el momento insta a la reflexión y en mi memoria se dibujan las imágenes de todos aquellos contribuyeron de una forma u otra en mi educación y me ayudaron alcanzar una meta como esta: Así, que doy gracias: A mis padres por el esfuerzo y la dedicación que han tenido durante toda su vida, quienes les debo mucho porque se han sacrificado y lo han dado todo para que yo sea la persona que soy hoy, por haberme dado siempre fuerzas para seguir adelante y vencer todos los obstáculo y sobre todo por haberme regalado la vida. A mi familia por todo el apoyo que me dieron en esta etapa y durante toda mi vida, por estar pendiente siempre de mis éxitos y en especial a mi hermanita, mis abuelitos, tíos y mis primos, la confianza, el apoyo inigualable que siempre me brindaron, por darme un espacio de su tiempo, y un pedacito de su corazón. A mis tutores, la Dra. Gladys Casas Cardoso, M.Sc. Abdel Rodríguez Abed y el Dr. Yovani Marrero Ponce por su insuperable ayuda, su guía certera, por todos los conocimientos que pusieron a mi disposición y su confianza en que llegaríamos hasta aquí. Gracias a Yovani por haberme dado la posibilidad de incursionar en este tema, a la profesora Gladys por recibirme y a Abdel por dedicarme todo el tiempo de que dispuso no solo este año sino en investigaciones anteriores, profes son mi ejemplo a seguir en el futuro. Un agradecimiento muy especial a Stephen Jones y Yoan Martínez quienes a pesar de su juventud cubrieron la ausencia física de mis tutores dando muestras de consagración y dedicación, compartiendo conmigo largas horas de trabajo y brindándome todo su apoyo. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(9) A Ricardo Grau Ábalo y familia quienes han estado pendientes de mi formación desde sus inicios, dándome aliento y sabios consejos. A Ricardo agradezco el haberme recibido en el grupo de investigación de Bioinformática y la infinita amabilidad mostrada siempre que lo necesité. A mis compañeros y amigos del aula por permitirme compartir con ellos todos los momentos agradables y malos, entender la belleza de la vida y de esta forma perfeccionar la formación como profesional. En especial a Sergio, Sandro, Ledis, Yaidel Oscar, Carlos, Pepe… y otros tantos que soy injusto al no mencionar, a todos un abrazo. Al claustro de profesores de la carrera de Ciencias de la Computación que durante estos cinco años aportaron a mi formación y mi sed de aprender. En fin deseo agradecer la dedicación, la confianza, la ayuda desinteresada y la paciencia, de todo aquel que se mostró espontáneo por darme un espacio de su tiempo, un pedacito de su corazón. A los que me enseñaron poniendo en mí su esperanza, su confianza, su respeto para que sea en cada instante de mi existencia un mejor ser humano y en el futuro un mejor profesional, a todos muchas gracias.. “La gratitud es el más legítimo pago al esfuerzo ajeno, es reconocer que todo lo que somos, es la suma del sudor de los demás. Que un hombre solo no vale nada, y que la dependencia humana, además de necesaria, es hermosa.” José Martí Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(10) Pensamiento Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(11) “La única manera de realizar un buen trabajo es amando lo que haces. Si todavía no lo has encontrado sigue buscando y no te conformes.” Steve Jobs. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(12) Resumen Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(13) RESUMEN El empleo de métodos computacionales en combinación con las técnicas automatizadas de evaluación masiva de propiedades de las moléculas, debe convertirse en una norma para mejorar las oportunidades de desarrollar fármacos permitiendo la reducción de los costos en términos de recursos materiales, humanos y de tiempo, factores limitantes en este propósito. El presente trabajo, enmarcado en esta tendencia científica, se propuso diseñar un nuevo programa (DESPOOL) para el cálculo de descriptores moleculares para Estudios Predictivos en Informática Química y Biológica, el cual forma parte de la suite TOMOCOMD-CARDD, aplicación interactiva de código abierto para la caracterización y discriminación de estructuras moleculares, desarrollada por el grupo de investigaciones CAMD-BIR Unit (Unit of Computer-Aided Molecular “Biosilico” Discovery and Bioinformatic Research) de la Facultad de Química y Farmacia. Hasta el momento, la mayoría de los software conocidos en este campo, presentan limitaciones tales como; pocas ponderaciones, uso del operador suma de las partes como única vía para caracterizar las moléculas y tienen un carácter global, es decir, toman en cuenta la molécula como un todo y no permiten el cálculo para determinadas regiones o grupos de átomos. DESPOOL es un software libre y de código abierto capaz de realizar los cálculos de los descriptores moleculares clásicos y de superar las limitaciones descritas de los programas anteriores.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(14) Abstract Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(15) ABSTRACT The use of computational methods in combination with automatic methods for massive evaluation of molecular properties should become a norm for the improvement of the opportunities for drug development allowing cost reduction in terms of the material and human resources, and time, which are limiting factors for this goal. This work, framed in this scientific tendency, the design of new program (DESPOOL) for the calculation of molecular descriptors was proposed for Predictive Studies in Chemical and Biological Informatics, which forms part of the suite TOMOCOMD-CARDD, a open source interactive application, used for the characterization and discrimination of molecular structures, developed by the research group CAMD-BIR Unit (Unit of Computer-Aided Molecular “Biosilico” Discovery and Bioinformatic Research) of the Faculty of ChemistryPharmacy. Up to this moment, the majority of the software known in this field, present limitations such as: few weighting schemes, the use of the summation operator as the exclusive way of globally characterizing the molecule, that is to say, they only take into account the molecule as a whole and do not permit the computation of determined zones or group of atoms. DESPOOL is an open source software capable of performing the calculation of the classic molecular descriptors and overcome the limitations described in previous programmes.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(16) Introducción Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(17) Introducción INTRODUCCIÓN. 1. El lenguaje matemático, en toda su riqueza y diversidad, se ha convertido en una herramienta indispensable para la interpretación teórica del comportamiento de la materia. Sin embargo, la complejidad de los fenómenos aquí involucrados provoca que los modelos matemáticos utilizados para describirlos no puedan ser completamente desarrollados, en la mayoría de los casos, con el fin de obtener respuestas exactas. En el intento de mejorar la capacidad predictiva y determinar resultados tan fiables como sea posible, se emplean tanto cálculos aproximados como modelos teóricos del comportamiento de los sistemas. En la práctica, todas estas aplicaciones implican el uso extensivo de procedimientos matemáticos computarizados. Los estudios predictivos, tales como los denominados por sus siglas en ingles QSAR (siglas en inglés de Quantitative Structure-Activity Relationship), se han convertido en una importante área de investigación en la química computacional(1, 2). Este tipo de estudio tiene dos objetivos fundamentales: 1) el primero es brindar una vía para estimar la actividad/propiedad estudiada a nuevos compuestos con un aceptable grado de precisión y 2) en segundo lugar, pero no menos importante, es obtener una interpretación en términos estructurales de la actividad estudiada. El paradigma de los estudios predictivos en este campo está relacionado con el hecho de que las propiedades físicas, físico-químicas, químicas y biológicas de los compuestos orgánicos, dependen, en último término, de la estructura molecular (3, 4). Basado en este paradigma, Crum-Brown y Fraser publicaron en 1868 el primer estudio QSAR(5,. 6). .. Entonces, el reto consiste en desarrollar métodos que permitan expresar la estructura química mediante parámetros, reduciendo el problema a relaciones entre dos series de números, una representando la estructura química (denominados como descriptores moleculares) y la otra a las actividades(7). En la actualidad, existe un gran número de descriptores moleculares que pueden ser usados para los estudios predictivos, de tipo QSAR/QSPR y/o de similitud molecular. Los descriptores moleculares son “el resultado final de un procedimiento lógico y matemático que transforma información química codificada dentro de una representación simbólica de Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(18) Introducción una molécula en un número útil o el resultado de algún experimento estandarizado”(8). Según esta definición, los descriptores moleculares se dividen en dos categorías principales, en dependencia de cuál haya sido el proceder utilizado para su definición: a) medidas experimentales, tales como log P, refractividad molar, el momento dipolar, polarizabilidad, en general, propiedades físico-químicas y b) descriptores moleculares teóricos, los que se derivan a partir de una representación simbólica de la molécula, estos pueden tener en cuenta rasgos topológicos (2D), geométricos (3D), y/o electrónicos de las moléculas al igual que usar otras representaciones posibles. Este tipo de descriptores moleculares (en lo adelante DMs) son los más adecuados debido a que pueden ser calculados a cualquier molécula, incluso una hipotética, que no tenga aun existencia física. En cambio los DMs experimentales conllevan un gasto de recursos materiales en su determinación y de tiempo, además de no ser tan reproducibles dado que dependen de las condiciones usadas. En este sentido el número de índices teórico tiene un gasto mínimo de recursos y pueden ser obtenidos gran cantidad de ellos (con total reproducibilidad) en un tiempo razonable logrando una mejor representación de la estructura molecular. Para calcular los DMs a partir de la estructuras químicas es necesario disponer de las posibilidades de las representaciones de estructuras químicas que nos brinda CDK (siglas en inglés de Chemistry Development Kit)(9) y JOELib(10), que son bibliotecas implementadas en Java de código abierto para Quimioinformática y Bioinformática. Están disponibles para Windows, Unix, y Mac OS. Se distribuyen bajo licencia GNU LGPL. Con diversas aplicaciones en Quimioinformática para la generación de geometría 3D de moléculas, el soporte para muchos formatos de archivos químicos, cálculo de DMs diversos, la detección de farmacóforos, etc; y en la Bioinformática para la detección de sitios activos de proteínas, lectura de archivos DPB, etc. En la actualidad existe un número considerable de programas libres y comerciales para el cálculo de DMs, tales como el Dragón, Molconn-Z, Volsurf, PreADMET, CDK Descriptor Calculator, BlueDesc, MODEL, MOL2 y el PaDEL-Descriptor, por solo citar algunas.(11) Muchos de estos programas tienen implementado un número reducido y común de DMs y realmente pocos han sido evaluados exhaustivamente en la práctica. Además, muchos de estos software son comerciales y su acceso es limitado dado su alto costo y solo son Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 2.
(19) Introducción disponibles por línea de comandos, es decir no poseen una interfaz gráfica que le permita al usuario una adecuada interacción y configuración de los cálculos. Finalmente, es importante destacar que los descriptores incluidos en estos programas presentan varias y significativas limitaciones, tales como: 1) limitado número de ponderaciones y poco relevantes a la hora de discriminar los átomos, 2) la mayoría son DMs definidos globalmente y no se permite su cálculo para algunas regiones o determinados grupos de átomos (carencia de definición local), 3) en su mayoría son obtenidos usando el operador “suma de las partes” o al menos pueden ser expresados de esta forma, 4) no explotan la diversidad de representaciones de las moléculas y se restringen a usar las más tradicionales, 5) presentan alta co-linealidad entre ellos, 6) están basados en representaciones matriciales o invariantes muy relacionadas y 7) tienen poca o ninguna interpretación estructural y/o químico-física(12). El grupo de investigaciones CAMD-BIR Unit (Unit of Computer-Aided Molecular “Biosilico” Discovery and Bioinformatic Research) de la Facultad de Química y Farmacia, con la colaboración del grupo de Bioinformática del Centro de Estudio Informáticos (CEI) de la Facultad de Matemática, Física y Computación de la Universidad Central “Marta Abreu” de Las Villas, ambos con una vasta experiencia en el desarrollo de varias aplicaciones informáticas dirigidas a la predicción de principios activos en la industria farmacéutica, han estado trabajando con el objetivo de resolver las limitaciones citadas en los descriptores referidos. En el marco de dichas investigaciones, este trabajo de diploma propone “Un nuevo programa para estudios predictivos en Informática Química y Biológica (Despool). Problema Científico La mayoría de los descriptores moleculares que existen en la actualidad para realizar los estudios predictivos carecen de definición local, al no permitir su utilización en el cálculo de determinados fragmentos o átomos en la molécula. Además, estos DMs se han obtenido a partir del operador suma y/o no utilizan información química de relevancia para codificar los átomos en la molécula, por todo lo cual estos índices moleculares necesitan de ser extendidos-generalizados con el propósito de mejorar su desempeño en estudios de informática química y biológica.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 3.
(20) Introducción Objetivo general Mejorar el desempeño de los Descriptores Moleculares, superando las limitaciones actuales de los programas utilizados en la predicción de principios activos en la industria farmacéutica, mediante el desarrollo de una nueva plataforma de cálculo, y la adecuación y/o implementación de nuevos Descriptores Moleculares. Este objetivo general se desglosa en los siguientes objetivos específicos:. Objetivos específicos Adecuar a índices locales los descriptores moleculares reportados en CDK e implementados en PaDel, transformar los de JOELib al formato de la biblioteca CDK e implementar nuevos descriptores moleculares; utilizando invariantes, locales y ponderaciones. Demostrar la utilidad del programa DESPOOL en estudios de diversidad comparando los índices tradicionales con los nuevos propuestos, derivados de las extensiones y generalizaciones. El trabajo desarrollado tiene novedad, así como valor teórico, práctico y metodológico. Novedad Científica y Aportes: La principal novedad del trabajo radica en la definición e implementación computacional de nuevas familias de Descriptores Moleculares, y la redefinición y/o extensión-generalización de índices ya conocidos en el campo de los estudios predictivos. Valor teórico: Obtención de nuevos Descriptores Moleculares empleando una estrategia de uso de invariantes totales o locales sobre un vector de LOVI, local vertex invariants. Valor práctico: Creación de un nuevo módulo para el cálculo de índices de código abierto, acceso libre y multiplataforma amigable con el usuario (a partir del empleo de una GUI). Valor metodológico: Las extensiones y generalizaciones propuestas en el trabajo pueden ser empleadas por otros autores para la extensión-generalización de sus índices. El primer paso para la realización de este trabajo fue la confección del marco teórico. Para ello se realizó una amplia revisión de la literatura consultando libros, artículos y páginas de Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 4.
(21) Introducción internet, entre otras fuentes. Sus elementos esenciales se encuentran expuestos de manera resumida en el primer capítulo de la presente tesis. Como conclusión de la elaboración del marco teórico se enuncia la siguiente hipótesis de investigación: La creación de una plataforma de cálculo que permita integrar de forma sistémica los Descriptores Moleculares con las bibliotecas CDK y JOELib, y la modificación e implementación de nuevos descriptores, permiten superar las limitaciones actuales de las aplicaciones informáticas dirigidas a la predicción de principios activos en la práctica farmacéutica. Este trabajo se ha estructurado en tres capítulos. El primero de ellos trata sobre estrategias utilizadas en la informática química y sirve de marco teórico para nuestra investigación. En él se analizan y exponen las consideraciones generales sobre estudios predictivos QSPR/QSAR, las teorías, enfoques teóricos, investigaciones y antecedentes relacionados con los descriptores moleculares, así como se citan los aspectos más importantes relacionados con programas utilizados para el cálculo, que fueron tomados en consideración en el desarrollo del trabajo. Todo lo anterior es válido para el correcto encuadre del estudio. Como resultado se adoptan las herramientas necesarias para la consecución de los objetivos. El segundo capítulo está dedicado al desarrollo de nuevos descriptores moleculares y su implementación en la biblioteca seleccionada. Este capítulo incluye una aplicación práctica que permite comprobar la validez de la propuesta. En el tercer capítulo se aborda el diseño teórico del nuevo programa, lo cual incluye un manual de usuario con los requerimientos del software, los modos de entrada y salida de los ficheros, las definiciones de las invariantes, de locales y propiedades atómicas, así como la elección de los descriptores moleculares a calcular y sus reportes estadísticos. Además, en este capítulo se hace un análisis de la variabilidad de los descriptores moleculares de DESPOOL. Finalmente se presentan las conclusiones de la tesis, así como algunas recomendaciones que abren futuras líneas de investigación.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 5.
(22) Capítulo I: Estrategias utilizadas en la informática química. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(23) Capítulo I: Estrategias utilizadas en la informática química CAPÍTULO I: ESTRATEGIAS UTILIZADAS EN LA INFORMÁTICA QUÍMICA. 6. 1.1. Consideraciones generales sobre estudios predictivos QSPR/QSAR La búsqueda de nuevos fármacos en la terapia de procesos patológicos como el Síndrome de Inmunodeficiencia Adquirida (SIDA), las enfermedades cardiovasculares y neoplásicas, la enfermedad de Alzheimer, y una inmensa variedad de infecciones de naturaleza viral y parasitaria, se encuentra entre las prioridades de la industria farmacéutica en la actualidad(13) .Las estrategias actuales para el descubrimiento/diseño de fármacos están determinadas por el desarrollo de la química combinatoria, las técnicas HTS (acrónimo de High-Throughput bioinformática. Screening),. (14, 15). la. ingeniería. genética,. la. biología. molecular. y la. .. Estos avances han permitido que en el presente, el número de compuestos químicos conocidos supere los 50 millones(16).Una parte considerable de ellos (más de un millón) están disponibles a través de diferentes proveedores. Además, la mayoría de las compañías farmacéuticas cuentan con colecciones propias de compuestos que representan una fuente de mucho valor para el descubrimiento y optimización de compuestos líderes. Por otro lado, el empleo de la robótica y el desarrollo de las técnicas HTS han hecho posible la evaluación diaria de las propiedades biológicas de millones de compuestos contra cientos de dianas biológicas(17-19). Sin embargo, un estudio reciente, que incluyó aproximadamente cincuenta compañías e instituciones académicas, reveló que se invierten como promedio 880 millones de dólares y 15 años de investigación para el desarrollo de un nuevo fármaco, desde su descubrimiento hasta su uso terapéutico(20). Aproximadamente el 14% del costo se emplea en la evaluación de propiedades biológicas y farmacológicas durante la fase pre-clínica(21) . A pesar de estos avances y las inversiones millonarias, el número de nuevos fármacos introducidos en el mercado ha permanecido constante durante los últimos años(22, 23). En este contexto los métodos que emplean técnicas asistidas por computadoras usados en el descubrimiento, diseño, y optimización de compuestos con estructura y propiedades deseadas han desempeñado un rol importante en el desarrollo de fármacos que se encuentran actualmente en el mercado o en fase de estudios clínicos (6). El resultado de Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(24) Capítulo I: Estrategias utilizadas en la informática química este tipo de estudio in sílico puede aplicarse como estrategia de tamizaje virtual que pospone los costosos procesos de síntesis y bioensayos, que se llevan a cabo solo después que se exploran conceptos iniciales mediante modelos computacionales (24). Este proceso implica, a menudo, la obtención de modelos para estimar las relaciones cuantitativas de estructura-actividad (QSAR – según sus siglas del inglés Quantitative Structure-Activity Relationship), que se centran en predecir la actividad biológica de un compuesto a partir de una representación vectorial de la estructura molecular (18,. 25). .. Además de los estudios QSAR referidos a la descripción de la actividad, los estudios QSPR/QSTR (siglas en inglés acrónimos de Quantitative Structure Property/Toxicity Relationships) también se han convertido en una importante área de investigación en la química computacional(14). Este tipo de estudios se encuentra en la intersección entre la biología, la química y la computación y tienen dos objetivos fundamentales. El primero es brindar. una. vía. para. estimar,. con. un. aceptable. grado. de. precisión,. la. actividad/propiedad/toxicidad estudiada a nuevos compuestos. El segundo, pero no menos importante,. es. obtener. una. interpretación. en. términos. estructurales. de. la. actividad/propiedad/toxicidad estudiada. El desarrollo de modelos computacionales basados en la relación estructura/– actividad/propiedad requiere de la representación adecuada de la estructura molecular. Estas representaciones se logran mediante los descriptores moleculares (DMs) (también se empleará el término índices moleculares). Los DMs son términos que caracterizan un aspecto específico de la molécula(26) y se clasifican en experimentales y teóricos(8). Los DMs ocupan un lugar importante en el escenario del descubrimiento/diseño de fármacos, donde se emplean como herramientas en el desarrollo de modelos QSAR/QSPR y como instrumentos en estudios comparativos de similitud/disimilitud molecular(26). Los estudios QSPR/QSAR constituyen un enfoque que permite entender como la variación estructural afecta la propiedad/actividad biológica de un conjunto de compuestos. En estos estudios, los descriptores moleculares (X) se correlacionan con una variable respuesta (Y) (27-29). . Es decir, este análisis puede definirse como una aplicación de métodos matemáticos. y estadísticos al problema de encontrar una ecuación empírica de la forma Yi = fi(X1, X2, ...Xn), donde Yi son las propiedades y/o actividades biológicas de la molécula, y X1, X2, ...Xn Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 7.
(25) Capítulo I: Estrategias utilizadas en la informática química son propiedades estructurales experimentales o calculadas (descriptores moleculares) de los compuestos. En este sentido, cada compuesto puede representarse como un punto en un espacio multidimensional, en los cuales los descriptores X1, X2, ...Xn son coordenadas independientes del compuesto. El objetivo más usual de este análisis es incrementar el entendimiento del sistema biológico bajo investigación o predecir la propiedad estudiada a un objeto (compuesto) no utilizado en la obtención del modelo. Lo anterior se explica a través de la teoría de grafos, la cual ha sido ampliamente aplicada a diversos campos de la ciencia. Un grafo se expresa usualmente como vértices interconectados por aristas(25, 30). Donde cada vértice del grafo se representa un objeto y la arista que conecta dos vértices representa la relación entre estos dos objetos. En la química grafo-teórica los objetos del grafo pueden representar orbitales, átomos (o sus núcleos), enlaces, grupos de átomos, moléculas, o colecciones de moléculas. Las aristas de un grafo químico simbolizan las interacciones entre objetos químicos y se usan para definir enlaces químicos, reacciones, mecanismos de reacciones, modelos cinéticos, u otra relación o transformación de los objetos químicos. En la literatura existente sobre la química grafo-teórica y sus aplicaciones;(18, 19, 24, 31, 32) podemos encontrar la mayoría de las principales aplicaciones de los grafos químicos, como son: 1) los índices topológicos (ITs) y otros índices estructurales para los estudios QSAR(1, 3, 11, 14, 26, 33-36). ;. 2) el enfoque de orbitales moleculares de Hückel (37, 38); 3) la enumeración de isómeros, percepción de simetría estructural y codificación de compuestos químicos (39-41); 4) grafos cinéticos y de reacción(42) y 5) el diseño de síntesis asistida por computadora(43). De todas estas aplicaciones mencionadas, la de interés para el presente trabajo es aquella relacionada con la obtención de descriptores estructurales para el diseño molecular. Aunque hasta el momento el número de índices moleculares reportados supera el millar, el desempeño de estos en la predicción de determinadas propiedades no siempre es totalmente satisfactorio. Por esa razón, la definición de DMs se mantiene como un área de intensa actividad en el campo de la química computacional. La aplicación de conceptos de Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 8.
(26) Capítulo I: Estrategias utilizadas en la informática química la Teoría de Grafos (TG) en el desarrollo de métodos teóricos para la representación de estructuras químicas ha tenido un enorme impacto, entre las aplicaciones más importantes de la TG a la química se encuentra la caracterización numérica de la estructura molecular a partir de invariantes grafo-teóricas, que se emplean como DMs de compuestos químicos en estudios de estructura-propiedad (QSPR/QSAR). 1.2. Descriptores moleculares Los índices o descriptores moleculares (DMs) representan la vía por la cual la estructura química se transforma en números permitiendo el tratamiento matemático de la información química contenida en la molécula y son considerados representaciones matemáticas de las moléculas que se obtienen al aplicar algoritmos específicos sobre una representación molecular definida o a partir de procedimientos experimentales específicos. 1.2.1. Definición y Clasificación de los descriptores moleculares La química matemática, cuyo surgimiento data del siglo XVIII por los trabajos del científico alemán Georg Ferdinand Helm, tiene como objetivo central la deducción matemática de las propiedades moleculares a partir de rasgos estructurales de las moléculas. (25) Para esto, se hace referencia al término índice o descriptor molecular. Todeschini y Consonni definen un descriptor molecular como “el resultado final de un procedimiento lógico y matemático que transforma información química codificada dentro de una representación simbólica de una molécula en un número útil o el resultado de algún experimento estandarizado” (8). En una de las primeras publicaciones de estudios QSAR, realizada por Crum Brown y Frazer en 1868, los autores relacionan la acción fisiológica (f) como una función de la constitución química (C), según la ecuación [1.1]: f = f (C). [1.1]. Actualmente, el principal escollo en obtener una definición precisa de la función f reside en la caracterización de los cambios en la estructura química que producen una determinada respuesta. La información estructural y propiedades fisicoquímicas se representan numéricamente en descriptores que codifican a las moléculas. A pesar de la investigación teórica y experimental en este campo, no existe acuerdo acerca de aquel conjunto de descriptores óptimo, y dado que diferentes descriptores codifican distinta información, la Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 9.
(27) Capítulo I: Estrategias utilizadas en la informática química estrategia consiste en aplicar aquellos más relevantes según la particularidad del caso de estudio. La naturaleza de los descriptores, depende de cual haya sido el proceder utilizado para la definición de los mismos, pudiendo tener en cuenta rasgos topológicos(44), geométricos(45), y electrónicos de las moléculas. Algunos de estos descriptores sin embargo, tienen “más información” de propiedades físico-químicas que de los rasgos estructurales de la molécula. Estos incluyen los basados en la determinación experimental de propiedades físicoquímicas, tales como la mayoría de las constantes de los sustituyentes, hidrofobias, electrónicas y estéricas(46). En contraste, los llamados índices topológicos (IT) tienen la información estructural contenida en una representación bidimensional de las moléculas, generalmente el grafo molecular con los átomos de hidrógenos suprimidos, sin considerar ningún rasgo físico-químico de las moléculas. La mayoría de estos índices pueden considerarse como descriptores estructurales explícitos. Otro grupo de descriptores, llamados químico-cuánticos describen rasgos electrónicos de las moléculas basados en el uso de la función de onda molecular. Los descriptores geométricos tienen información de los rasgos estructurales 3D de las moléculas en una vía explícita, tales como distancia y ángulos de enlaces o en una vía implícita, en forma de descriptores topográficos. Además, se pueden clasificar en función de: La invariabilidad de sus propiedades, es decir, su capacidad para rendir un valor independiente de características particulares de la representación del compuesto. Estas propiedades son la invariabilidad química (tipos de átomos o enlaces), invariabilidad translacional y rotacional (en función del marco de referencia espacial) y la conformación de la representación geométrica. Los descriptores 3D que presentan invariabilidad de traslación y rotación son particularmente útiles, ya que no requieren el alineamiento previo de las moléculas, por lo que se ahorra tiempo de cálculo y se evitan problemas asociados con el alineamiento. Su degeneración o capacidad de evitar asignar valores idénticos a compuestos distintos. El tipo de propiedad que describen (estéricas, electrónicas, lipofílicas, de forma, descriptores farmacofóricos…). Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 10.
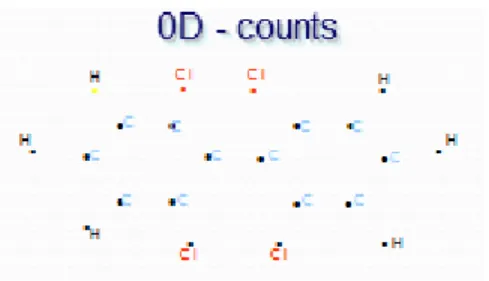
(28) Capítulo I: Estrategias utilizadas en la informática química En cualquier caso, no existe un único esquema de clasificación de los descriptores, aunque entre las propuestas más aceptadas destacan la de Todeschini (8), cuyo handbook se ha convertido en una de las referencias básicas del campo de descriptores. A continuación se detalla la clasificación realizada de acuerdo a la representación simbólica de la molécula teniendo en cuenta rasgos de la misma, por constituir la más utilizada en el presente trabajo: Descriptores 0D: Se derivan de la fórmula química de la molécula. Se puede decir que son independientes de la estructura molecular, describen solamente la constitución de la molécula, pero no dicen nada sobre la conformación ni tipo de conectividad presente. Los más simples son el número de átomos de un determinado tipo, el número de enlaces y el peso molecular, entre otros(8). La figura 1.1 muestra un ejemplo.. Figura 1.1. Descriptor 0D Descriptores 1D: La representación en una dimensión consiste en una lista de fragmentos estructurales en la molécula y no requiere un conocimiento completo de la estructura molecular. La lista puede estar compuesta por grupos funcionales, sustituyentes de interés presentes en la molécula. Los descriptores derivados de esta representación se denominan DMs 1D. Se aplican fundamentalmente en análisis y búsqueda subestructural(8). La figura 1.2 muestra un ejemplo.. Figura 1.2. Descriptor 1D Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 11.
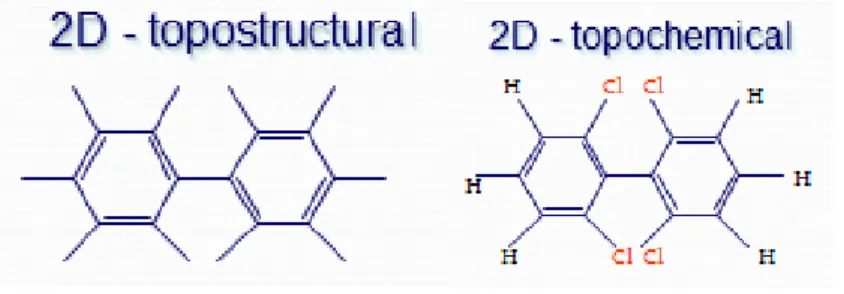
(29) Capítulo I: Estrategias utilizadas en la informática química Descriptores 2D: Basados en la representación en dos dimensiones de la molécula la cual se basa en el conocimiento de la conectividad entre los átomos que la forman. (47) Utilizan una función de autocorrelación bidimensional que contiene la topología del grafo, y además representa la distribución de una propiedad atómica determinada en la molécula. La propiedad atómica con la que se pesa/pondera al descriptor considera los átomos presentes en la molécula a través de la electronegatividad, masa atómica, polarizabilidad atómica, estado electrotopológico o volumen de Van der Waals, con lo cual se pueden seleccionar aquellos átomos que dan mayor peso a la variable. Estos descriptores tienen en cuenta las interacciones inter/intra-moleculares. Por ejemplo, los métodos basados en grafos moleculares permiten una representación 2D de la molécula, generalmente conocida como representación topológica. Los DMs obtenidos al aplicar algoritmos sobre la representación topológica se denominan DMs 2D(8). La figura 1.3 muestra un ejemplo.. Figura 1.3. Descriptores 2D Descriptores 3D: Basados en la representación tridimensional de la molécula como un objeto rígido. Permite la representación de la conectividad entre los átomos y de la configuración espacial de la molécula. Ejemplo de estos descriptores son los descriptores geométricos, estéricos y de talla(48). esta clase tiene en cuenta los aspectos conformacionales de la estructura molecular, considerando de esta manera las propiedades estereoquímicas de las moléculas. Para su cálculo se utilizan estructuras moleculares previamente optimizadas con métodos convenientes, tales como el Método de Campos de Fuerza de la Mecánica Molecular MM+, en combinación con métodos derivados de la Mecánica Cuántica, sean ab initio o Métodos de la Teoría de Orbitales Moleculares Semiempírica. Los descriptores geométricos, algunos estéricos y de talla constituyen ejemplos de DMs 3D(8). La figura 1.4 muestra un ejemplo. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 12.
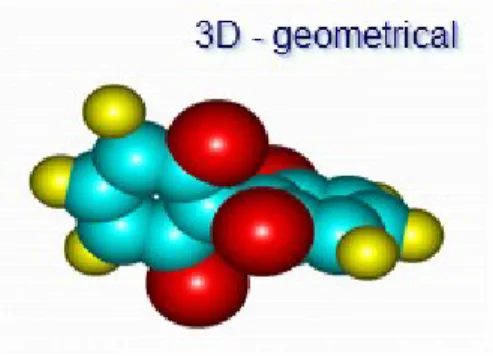
(30) Capítulo I: Estrategias utilizadas en la informática química 13. Figura 1.4. Descriptor 3D Descriptores 4D: Derivados de la representación tridimensional de la molécula y su interacción con una sonda que caracteriza el ambiente (campos de interacción molecular).(49) Descriptores 5D: Basados en representación estereodinámica (QSAR dinámico): es una representación dependiente del. tiempo que adiciona propiedades estructurales a las. representaciones 3D como flexibilidad, comportamiento conformacional, propiedades de transporte. La utilidad de un DMs debe analizarse con doble sentido: el número puede brindar una interpretación más profunda en términos estructurales de la propiedad molecular y/o es capaz de tomar parte en un modelo para la predicción de propiedades moleculares de interés.(8) Incluso si la interpretación del DM es débil o carente, este podría estar estrechamente correlacionado con algunas propiedades moleculares permitiendo obtener modelos con alta capacidad predictiva. Por otro lado, DMs con baja capacidad predictiva pueden ser mantenidos en el modelo cuando están correctamente fundamentados por la teoría y son. interpretables debido a su capacidad para codificar información de la química(8).. estructura. (aproximadamente (50). DRAGON. Actualmente. 500-1500,. , CODESSA. muchos. existen de. ellos. muchos. índices. implementados. o en. descriptores el. programa. (51). , etc.), la mayoría de ellos están relacionados por su definición. analítica. Incluso, índices tan diversos en su definición analítica como los índices MTI (índices de Schultz) y los índices de conectividad de Kier y Hall pueden ser expresados de igual forma a partir del procedimiento vector-matriz-vector transpuesto(8, 18).. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
(31) Capítulo I: Estrategias utilizadas en la informática química 1.2.2. Propiedades que debe poseer un nuevo descriptor molecular Randic propuso una lista de propiedades que deben poseer los nuevos descriptores moleculares,(52) considerada actualmente de un nivel de sofisticación muy alto, el cual deben alcanzar los nuevos descriptores moleculares diseñados. Estas propiedades son las siguientes: 1) interpretación estructural directa 2) buena correlación con al menos una propiedad 3) buena discriminación entre isómeros 4) localmente definidos 5) generalizables a análogos superiores 6) linealmente independientes 7) simplicidad 8) no basados en propiedades físico-químicas 9) no trivialmente relacionados con otros índices 10) eficiencia de construcción 11) basados en conceptos estructurales familiares 12) mostrar una dependencia correcta con el tamaño 13) tener cambios graduales con cambios graduales en la estructura. En realidad, muchos de estos atributos tienen cierto grado de interrelación. A continuación, se comentarán algunas de estas propiedades “deseables” a través, también, de algunos problemas o desventajas que presentan algunos descriptores moleculares. Al emplear los descriptores moleculares para la caracterización de una molécula intrínsecamente se tiene cierta pérdida de información, ya que se está tratando de representar un objeto tridimensional por un número simple. Esta cuestión es crítica, cuando se trata de una actividad biológica que depende de la interacción estereoespecífica del receptor con un sitio de la molécula. Sin embargo, los descriptores moleculares pueden contener una sorprendente información estructural sobre las moléculas, lo que los hace de gran utilidad en los fines prácticos de sus aplicaciones. Existen dos posibilidades de resolver la pérdida de información de los descriptores grafo-teóricos. La primera, consiste en la generalización de un descriptor simple a análogos “superiores” y segunda, la Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 14.
(32) Capítulo I: Estrategias utilizadas en la informática química generalización de la invariante grafo-teórica como secuencias de números. (52). . La. generalización de los índices es necesaria para salvar situaciones en las que un descriptor simple no es suficiente y la investigación de una familia de descriptores estructuralmente relacionados puede resolver el problema. Sucede que en la obtención de una familia de descriptores de diferentes órdenes (familia de descriptores relacionados), como generalización de un simple descriptor, se ha observado que muchos de estos son colineales (unos índices pueden ser expresados como combinación lineal de los restantes, por lo que puede existir “redundancia de la información”). La independencia lineal u ortogonalidad de los índices es uno de los atributos deseables, pues los descriptores colineales pueden afectar la “estabilidad del coeficiente de correlación” y dificulta la interpretación de los modelos obtenidos. En este sentido, es importante también que los descriptores sean ortogonales en relación a los restantes. Esta independencia lineal de los índices significa que los mismos conduzcan a una correlación con una propiedad que no es satisfactoriamente explicada por los otros descriptores existentes. Una de las principales deficiencias que aún presentan la mayoría de los descriptores está relacionada con su selectividad estructural; o sea, la capacidad de los mismos para diferenciar entre estructuras que no sean isómeras. La sensibilidad de la discriminación de isómeros, surge debido a que la diferenciación de isómeros garantiza que estos descriptores puedan ser utilizados en estudios donde las propiedades moleculares varían para diferentes isómeros y en general, en los estudios de aquellos aspectos de las propiedades moleculares que dependen del tamaño de la molécula. De forma general, la selectividad de los descriptores disminuye a medida que aumenta el número de vértices en el grafo que representa el esqueleto carbonado molecular, por ejemplo el andamiaje carbonado de un alcano. Se ha demostrado además que no existe una relación directa entre la discriminación de isómeros y la utilidad de un índice para correlacionar con propiedades físicas de interés(53). Este resultado, demuestra que una cuestión importante en el diseño de un nuevo descriptor, es que este contenga información estructural valiosa y no sólo que sea altamente discriminante para isómeros. Por otro lado, la mayor parte de los índices utilizan matrices que caracterizan la molécula son muy similares. Así por ejemplo, la mayoría de los descriptores parten de calcular Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 15.
(33) Capítulo I: Estrategias utilizadas en la informática química invariantes a las matrices de adyacencia o de distancia entre átomos o de combinaciones de estas dos, es decir; no existe gran diversidad en los índices definidos hasta el momento debido a que el número de matrices usadas para la representación de moléculas es reducida y las invariantes que se usan para extraer su información están muy relacionados entre sí. Además de estos dos factores, los índices definidos hasta el momento no permiten describir adecuadamente todas las propiedades químico-físicas y biológicas, así por ejemplo algunas propiedades físicas de octanos (moléculas sin heteroátomos, anillos e instauraciones) no pueden ser predichas adecuadamente por los índices definidos hasta el momento(45). Uno de los aspectos más importantes de un parámetro molecular, incluidos en la lista de los trece atributos para la definición de nuevos descriptores(18,. 24). , es su capacidad de. definición local; este término está referido al hecho de que estos no sean obtenidos de forma global para una estructura molecular, sino que puedan ser definidos sobre determinados fragmentos de la propia estructura. Esta propiedad resulta muy importante dado que muchas propiedades o actividad biológicas depende más de los rasgos estructurales de determinados zonas de la molécula que la molécula como un todo. De hecho, hay propiedades que se miden experimentalmente de forma local, tales como la reactividad de grupos nucleofílicos sobre determinados centros electrofílicos, los corrimientos químicos de protones, etc. Este requisito representa una debilidad importante para los índices definidos hasta el momento, puesto que todos son de definición global y no se emplea para la definición de fragmentos o determinadas zonas de las moléculas. Finalmente, es importante resaltar que todos los índices propuestos hasta el momento y que son definidos localmente son empleados para obtener índices globales (de forma invariante) usando el enfoque de que la suma de las partes hacen el total. Así por ejemplo, los índices del estado electro-topológico de una molécula o fragmento son calculados como la suma de los estados electro-topológicos de cada átomo, es decir; como una combinación lineal de los índices atómicos(18).. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 16.
(34) Capítulo I: Estrategias utilizadas en la informática química 1.3. Programas para el cálculo de descriptores moleculares utilizados en la informática química Búsquedas aplicadas en quimio-informática y la toxico informática cada vez más usan la representación de moléculas en forma de descriptores moleculares, que capturen tanto las características estructurales como las propiedades de la molécula. Estas representaciones son útiles para ADME/toxicidad, predicción, análisis diversos, diseño de bibliotecas, QSAR/QSPR, exploraciones virtuales, etc...Estos descriptores se pueden utilizar para evaluar la estructura molecular-actividad o estructura-propiedad, así como para el análisis de similitud y selección de alto rendimiento de bases de datos de moléculas. Una gran variedad de descriptores son usados cuantitativamente para describir estructuras moleculares y propiedades para muchas aplicaciones en química computacional y bioinformática, la selección apropiada de uno de ellos para cumplir una tarea computacional específica es rápida y en la mayoría de los casos obvia(54, 55). Existe en el mundo de la Química Computacional varios software para el cálculo de descriptores moleculares (Ver Tabla A, Anexos). A continuación se relacionan una selección de programas que incluyen grupos de descriptores moleculares utilizados en la Bioinformática y que fueron tomados en consideración para el desarrollo del presente trabajo por su representatividad, es decir se trata de que los software que se seleccionen tenga el menor solapamiento posible en cuanto a la variedad de sus descriptores. Además la selección fue realizada con preferencia para software de código abierto que permita la reutilización de su código fuente con vistas a resolver las debilidades presentes en su funcionamiento. 1.3.1. Software comerciales Software DRAGON: DRAGON es una aplicación para el cálculo de descriptores moleculares originalmente desarrollado por Milano Chemometrics and QSAR Research Group. Dragon calcula 1600 descriptores moleculares agrupados en 20 bloques lógicos. El software ha sido diseñado para trabajar tanto para Windows (DRAGON professional y DRAGON plus) como Linux (DRAGONx), con una interfaz gráfica y una de línea de comandos. El programa puede calcular no solo las propiedades más simples como tipo de átomos, grupo funcionales y conteo de fragmentos, sino también diversos descriptores topológicos y geométricos. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 17.
(35) Capítulo I: Estrategias utilizadas en la informática química Algunas propiedades moleculares como logP, refractividad molecular, número de enlaces rotables, H-donantes, H-aceptores y superficie de área topológica (TPSA) son además calculados usando modelos comunes tomados de la literatura. Los formatos de molécula que permite DRAGON son (MDL, Sybyl, HyperChem, MacroModel, SMILES, CML, HyperChem). Su interfaz gráfica para la importación de moléculas es flexible y permite la selección de las estructuras que se almacenan en diferentes formatos de archivo y ubicados en diferentes carpetas. Las estructuras moleculares almacenadas en formatos de diferentes tipos pueden cargarse y simultáneamente ser tratados en el mismo lote (50). El DRAGON permite unir el cálculo de descriptores moleculares con un conjunto de propiedades definidas por el usuario, ofreciendo un archivo de salida que será fácilmente cargado por cualquier aplicación de análisis de correlación. 1.3.2. Software libres Software MODEL Este programa calcula cerca de 3780 descriptores basados en la estructura 3D de la molécula. Los descriptores pueden ser de tipo constitucional, físico-químicos, topológicos, geométricos, electrónicos y de propiedades de la superficie de la molécula. El formato de los archivos de entrada puede ser en mol, mol2, pbd o cor. El software es libre para su utilización, aunque de código cerrado y está basado en su desarrollo en aplicaciones Web62. MODEL está accesible en http://jing.cz3.nus.edu.sg/cgi-bin/model/model.cgi, libre y sin costo para uso académico(56). Software BlueDesc El BlueDesc da la mayor importancia en su trabajo a la relación cuantitativa clásica de estructura- actividad o propiedad (QSAR/QSPR), fijando su objetivo en la modelación del compuesto químico dentro de un vector de descriptores numéricos. El software presenta una línea de comandos que transforma los archivos de entrada MDL SD y devuelve formatos ARFF (WEKA) y LIBSVM para aprendizaje automático y minería de datos. El software esta implementado en JAVA en su totalidad y debe incluir para su funcionamiento JOELib2 (GNU GPL) y el CDK (LGPL), en Multiplataforma (Windows, Linux, MacOS).. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 18.
(36) Capítulo I: Estrategias utilizadas en la informática química Brinda el cálculo de 174 descriptores tomados de ambas bibliotecas. Funciona únicamente con estructuras en 3D(44). Software MOLGEN Este descriptor fue desarrollado desde hace 17 años por el grupo de Bioinformática de la Universidad de Bayreuth, Alemania y es profundamente usado en la industria y la docencia. Este programa tienen varios componentes tale como: un generador para gráficos químicos, un generador para conectividad de isómeros, un editor gráfico de moléculas y un monitor 2D, etc. Una versión del programa MOLGEN-CID (MOLGEN-Chemical Identifier) fue desarrollado recientemente y está libre en internet (http://www.molgen.de/). Este trabaja con los gráficos suprimiendo el hidrógeno y usa la multiplicidad de enlace, por lo que la salida no contienen átomos de hidrógeno(57). Software Mold2 El software Mold2 fue desarrollado para trabajar sobre datas diversas aplicando sobre ellas descriptores codificados en una y dos-dimensiones únicamente. Análisis comparativos del Mold2 con otros software de cálculos como DRAGON y Molconn-Z con varias datas y usando un análisis de la entropía de Shannon demostraron que ofrece similares resultados. Sin embargo el software solamente funciona sobre la plataforma Windows y su código es cerrado. Todo el funcionamiento está basado en un interfaz de línea de comandos lo cual lo hace poco apetecible por los expertos en este campo(58). Software PaDel PaDEL-Descriptor es un software para calcular descriptores moleculares y de conteo de huellas. El software calcula 797 descriptores (663 descriptores 1D y 2D y 134 descriptores 3D) y 10 de tipo de conteo de huellas. Estos descriptores son calculados principalmente usando la biblioteca CDK (The Chemistry Development Kit). Algunos otros descriptores y conteos de huellas les fueron adicionados, tales como los de estado electrotopológico de tipo átomo, volumen de McGowan, conteo de anillos, conteo de subestructuras químicas identificadas por Laggner y conteo de huellas binarias. El PaDEL-Descriptor fue desarrollado usando lenguaje JAVA y consiste en un componente de biblioteca y uno de interfaz. El componente de biblioteca permite la integración de la relación estructura Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 19.
(37) Capítulo I: Estrategias utilizadas en la informática química actividad cuantitativa a través de las características de cálculo del descriptor, mientras que el componente de interfaz permite ser usado como un software autónomo. El software permite el procesamiento mediante múltiples núcleos de CPU, presentes en la mayoría de las computadoras modernas, para aumentar la velocidad de cálculos de los descriptores moleculares, además posee una interfaz de usuario y otra de línea de comando y trabaja sobre las plataformas más usadas en la actualidad (Windows, Linux, MacOS). Soporta 90 formatos de moléculas diferentes, y trabaja usando múltiples hilos(12). 1.3.3. Bibliotecas utilizadas en la informática química A medida que aumenta los problemas en la Química Computacional y en la Biología Computacional, tener herramientas potentes es una de las necesidades de estas ramas de la ciencia. Biblioteca CDK El Chemistry Development Kit es una biblioteca Java de código abierto para Quimioinformática y Bioinformática, no un programa usable.1 Está disponible para Windows, Unix, y Mac OS. Se distribuye bajo licencia GNU LGPL. El CDK fue creado por Christoph Steinbeck, Egon Willighagen y Dan Gezelter, quienes desarrollaron el Jmol y JChemPaint en aquella época para proveer una base común de código en la Universidad de Notre Dame. Desde entonces muchas personas han contribuido al proyecto, llevando al programa a altos niveles de funcionalidad, actualmente es desarrollada por más de 50 programadores, contribuyendo más de 10 universidades de todo el mundo con proyectos empresariales. Ha sido integrado en varios entornos para hacer que sus funciones estén disponibles (por ejemplo, R,2 CDK-Taverna,3 Bioclipse, y Cinfony.4 Además existe una extensión CDK para KNIME(9). Biblioteca JOELib JOELib es un sistema experto químico software libre que se utiliza principalmente para convertir formatos de archivo químico. Debido a su fuerte relación con la informática, este programa pertenece más a la categoría de quimioinformática que a la de modelado molecular. Se encuentra disponible para Windows, Unix y otros sistemas compatibles con Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 20.
(38) Capítulo I: Estrategias utilizadas en la informática química Java. Se distribuye bajo licencia GPL. El logotipo del proyecto es simplemente la palabra JOELib escrita en los caracteres del alfabeto tengwar creado por el escritor británico de fantasía J. R. R. Tolkien para escribir sus lenguas artísticas, particularmente el quenya y el sindarin. Las letras se agrupan como JO-E-Li-b. Las vocales normalmente se agrupan con una consonante, pero dos vocales seguidas deben estar separadas por una construcción auxiliar. Los Formatos de archivo químico son: Formato MDL Molfile, formato SD, SMILES, GAUSSIANO, Lenguaje de marcado químico (CML) y MOPAC(10). 1.4. Consideraciones parciales En el campo de la química computacional los programas especializados en apoyar las tareas cotidianas de los investigadores generalmente son comerciales y muy costosos, por tanto es poco probable que se puedan encontrar los códigos fuentes o versiones de código abierto. Por otra parte, los descriptores moleculares existentes no poseen definición local, lo cual resulta muy importante para trabajar sobre determinados fragmentos de la propia estructura, que en muchas ocasiones son los que definen el comportamiento de la misma. Lo anterior sumado a la necesidad de obtener descriptores que incluyan ponderaciones e invariantes para su generalización son razones suficientes para el desarrollo del presente trabajo, que se propone poner a disposición de la comunidad de investigadores un nuevo software, llamado DESPOOL, que resuelve las dificultades citadas anteriormente.. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”. 21.
(39) Capítulo II: Diseño e Implementación del Programa DESPOOL Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
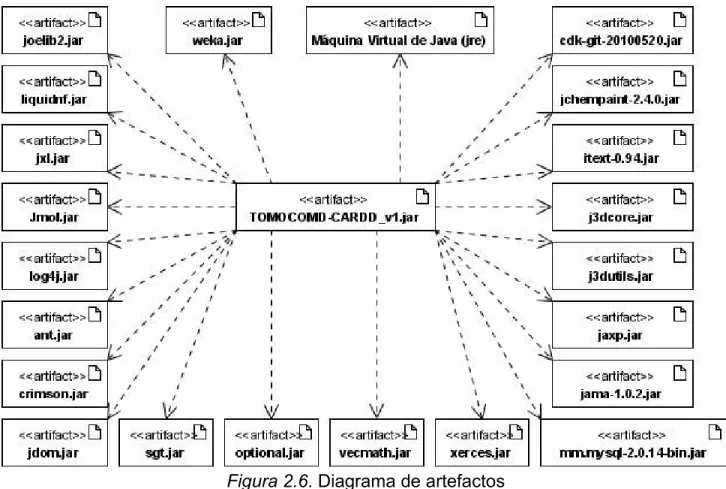
(40) Capítulo II: Diseño e implementación del programa DESPOOL CAPÍTULO II: DISEÑO E IMPLEMENTACIÓN DEL PROGRAMA DESPOOL. 22. Este capítulo está dedicado a explicar brevemente algunos aspectos relacionados con el análisis, diseño e implementación del producto de software elaborado 2.1. Suite de trabajo, TOMOCOMD-CARDD TOMOCOMD-CARDD es una aplicación interactiva, de código abierto, amigable para el usuario que calcula descriptores moleculares (o índices) para estructuras moleculares, con el objetivo de caracterizar o discriminar algunos de ellos. Está implementada en Java. Java actualmente es libre, y esto es muy conveniente para su uso en el desarrollo de aplicaciones en los países del tercer mundo. Este lenguaje fue creado para trabajar con objetos y es independiente de la plataforma. Al compilar un programa, Java genera un seudocódigo para una máquina genérica, que corre indistintamente en cualquiera de los ordenadores disponibles en el mercado, así funcionen sobre Windows, Linux, Mac u otro sistema operativo. Es un lenguaje robusto justamente por la forma en que está diseñado, no permite el manejo directo del hardware ni de la memoria, implementa mecanismos de seguridad que limitan el acceso a recursos de las máquinas donde se ejecuta. TOMOCOMD-CARDD consta de dos suites funcionalmente. La primera es una extensa colección de módulos para el cálculo de. descriptores. moleculares basados en las llamadas matrices de relación de frecuencia, conteo de huellas moleculares y el conjunto de los más relevantes descriptores reportados en la literatura. Dentro de sus módulos incluye: . DIVATI (siglas de DIscreteDeriVAtiveTypeIndices).. . GT-STAF (siglas de GraphTheoreticalThermodynamicSTAteFunctions).. . FREMESSA (siglas de FREquency-type Matrices Extended classical Algorithms).. . FREMXALF (siglas de FREquency – typeMatriX – based Algebraic Forms). . MOLFIP (siglas de MOLecular FInger Prints).. . DESPOOL (siglas de DEScriptorPOOLs).. Addel Arnaldo Goya Jorge “DESPOOL: Un Nuevo Programa para Estudios Predictivos en Informática Química y Biológica”.
Figure

+7



Documento similar
En el se especifican las condiciones de realización de la tesis, los derechos y deberes del doctorando, incluyendo los posibles derechos de propiedad intelectual y/o
Artículo 8. Las solicitudes de reconocimiento presentadas, en las que se aleguen créditos obtenidos en títulos universitarios oficiales de Graduado, para la convalidación de
Para eso, será necesario que el alumnado solicite autorización a la Comisión Académica del programa, en la que indicará las fechas de inicio y de fin de la estancia, junto con una
Habilidad para construir y criticar argumentos formales e informales en relación a problemas de las diversas ciencias, formales o empíricas, así como de problemas de lógica,
Como resultado de esta propuesta , el plan de estudios para el Grado en Química incluye una asignatura con el nombre de “Informática Aplicada a la Química” y presenta un
En cada antecedente debe considerarse como mínimo: Autor, Nombre de la Investigación, año de la investigación, objetivo, metodología de la investigación,
En el siguiente apartado de la entrevista, se trata la polémica entre Twitch y los periodistas de medios tradicionales, que ven una amenaza en la plataforma digital, ya
Algunos autores han sugerido que la hipoxia del tumor actúa como regulador del metabolismo energético y puede redirigir a las células cancerígenas a explotar la glucolisis como
