ESCUELA DE ELECTRÓNICA Y
TELECOMUNICACIONES
MODALIDAD CLÁSICA
“ESTUDIO DE PROPIEDADES ELECTRÓNICAS DE
HEMATITA CON DEFECTOS PUNTUALES”
TESIS DE GRADO PREVIA A LA OBTENCIÓN DEL TITULO DE INGENIERO EN ELECTRÓNICA Y TELECOMUNICACIONES
AUTOR:
RICHARD AUGUSTO RIVERA ESCOBAR
DIRECTOR:
PH.D. ARVIDS STASHANS
CENTRO UNIVERSITARIO LOJA
Certificación:
Ph.D. Arvids Stashans
DIRECTOR DE TESIS
CERTIFICO:
Que el presente trabajo de investigación, titulado “Estudio de Propiedades
electrónicas de Hematita con defectos puntuales”, realizado por el egresado
Richard Augusto Rivera Escobar, ha sido cuidadosamente revisado, por lo
que he podido constatar que cumple con todos los requisitos de fondo y de
forma establecidos por la escuela de Electrónica y Telecomunicaciones de la
Universidad Técnica Particular de Loja para esta clase de trabajos, por lo
que autorizo su presentación.
Loja, Septiembre del 2009
Ph.D Arvids Stashans
Cesión de derechos:
Yo, Richard Augusto Rivera Escobar, declaro ser autor del presente trabajo y
eximo expresamente a la Universidad Técnica Particular de Loja y a sus
representantes legales de posibles reclamos o acciones legales.
Adicionalmente declaro conocer y aceptar la disposición del Art. 67 del
Estatuto Orgánico de la Universidad Técnica Particular de Loja, que su parte
pertinente textualmente dice: “Forman parte del patrimonio de la Universidad
la propiedad Intelectual de Investigaciones, Trabajos Científicos o Técnicos y
Tesis de Grado que se realicen a través, o con el apoyo financiero,
académico o Institucional (Operativo de la Universidad).”
Introducción
El desarrollo de la moderna tecnología se debe a la electrónica. Multitud de
dispositivos electrónicos nos rodean y se han vuelto algo cotidiano e irreemplazable
en nuestra vida diaria. No siempre fue así, pero en la actualidad es impensable
carecer de esta gama de artículos que están aquí para facilitarnos la vida.
La electrónica se basa en la transmisión de portadores de carga para transmitir
señales. Estos portadores de carga se desplazan a través de dispositivos que los
interpretan y procesan para cumplir determinadas tareas. A un nivel muy profundo,
estos dispositivos funcionan según la forma en que están construidos, pero más
importante, según el material del que están conformados.
Los materiales semiconductores son la base para todo dispositivo electrónico.
Algunos son buenos para ciertas tareas, otros para unas diferentes, etc. Sin
embargo, para poder determinar la mejor aplicación para la que son aptos, deben
realizarse estudios de sus propiedades en diferentes estados. Como es literalmente
imposible obtener materiales en los que no haya defectos (como impurezas o
vacancias), es necesario estudiar los materiales en presencia de estos.
El estudio de materiales con defectos puede hacerse experimentalmente, pero los
equipos y los procedimientos son muy costosos y complejos, y son llevados a cabo
por ciertos Centros de Investigación y Universidades de países del primer mundo,
con recursos suficientes para ello. Un método mucho más asequible es el de
realizar simulaciones de los materiales a través de herramientas computacionales
específicas, que se pueden realizar en cualquier país, independientemente de su
nivel de desarrollo. La naturaleza está descrita en lenguaje matemático, y por ende
es posible realizar modelos matemáticos de las estructuras que conforman los
diversos compuestos existentes, y estudiarlos a través de simulaciones que, como
ha sido demostrado en multitud de trabajos realizados alrededor del mundo, son
confiables, certeros y de bajo costo.
Con esto en mente, se ha desarrollado una investigación sobre un cristal a través
de una herramienta computacional. El material que se ha elegido tiene importancia
centrarnos en la importancia electrónica del mismo. De que manera es afectado
este material en presencia de defectos puntuales, para así poder encontrar una
aplicación acorde a las alteraciones inducidas en el mismo, y compartir estos
resultados con la comunidad científica para poder compartir criterios e ideas.
Objetivo General:
- Analizar las propiedades electrónicas del cristal de Hematita (-Fe2O3) sometido a la presencia de defectos puntuales.
Objetivos Específicos:
- Analizar los fundamentos de los métodos computacionales, a fin de conocer
en que se basan para describir las estructuras a estudiarse.
- Describir las principales propiedades del cristal a estudiarse, hematita, a fin
de poder analizar las posteriores alteraciones producidas por la presencia
de regiones defectuosas en el mismo.
- Introducir defectos puntuales en el material para poder determinar como se
ven alteradas su estructura y propiedades electrónicas en presencia de los
Dedicatoria
A mis Queridos Padres,
Hermanos, Tíos, Abuelitos, y
a toda mi familia y amigos
que me han llevado a ser la
Agradecimientos
En primer lugar gracias a Dios por haberme permitido vivir en esta tierra maravillosa
y conocer a tantas buenas personas en el camino de mi vida.
Gracias a mi Padre y a mi Madre por siempre estar apoyándome
incondicionalmente en todo lo que he hecho a lo largo de mi vida, por formarme
como soy y por brindarme su amor sin condiciones.
A mis hermanos, con quienes he compartido toda mi vida y con quienes he pasado
muchos momentos que perdurarán para siempre.
A mis Abuelitos, y en especial a mi Abuelita Luz, que siempre me aconsejaron con
esa sabiduría que solo la experiencia puede dar, y siempre tratando de guíarme por
la senda del bien.
Gracias a mis tíos y tías, primos, amigos y a toda esa gente que siempre estuvo ahí
para apoyarme.
Un agradecimiento especial para el Ph.D Arvids Stashans, que me inició en el
mundo de la investigación científica y me ha apoyado cuando ha sido necesario, en
Índice de contenidos
Capítulo 1: Metodología 1
1.1 Introducción 1
1.2 Fundamentos de Mecánica Cuántica 1
1.2.1 La Ecuación de Schrödinger 1
1.2.2 Propiedades de la función de onda 2
1.2.3 Aproximación Born-Oppenheimer 3
1.2.4 Método Variacional 4
1.3 Teoría de Orbitales Moleculares 4
1.3.1 Orbitales Atómicos 5
1.3.2 Orbitales moleculares 7
1.4 Energía de un sistema 8
1.5 Ecuaciones de Hartree-Fock 9
1.6 Método INDO 11
1.6.1 Parámetros Semi-empíricos 13
Capítulo 2: Hematita (-Fe2O3): Propiedades y Modelización 14
2.1 Introducción 14
2.2 Estructura 14
2.3 Propiedades electrónicas 16
2.4 Modelización 17
2.4.1 Modelo de supercelda 17
2.4.2 Procedimiento de cálculo 17
2.4.3 Parametrización 21
Capítulo 3: Hematita con defectos Puntuales 27
3.1 Introducción 27
3.2 Defectos puntuales en Hematita: F centros 27
3.2.1 Resultados 28
3.3 Defectos puntuales en Hematita: Impureza de Titanio 31
3.3.1 Resultados 32
Lista de Figuras
Figura 1.1: Forma del orbital s 5
Figura 1.2: Forma del orbital p 6
Figura 1.3: Forma del orbital d 6
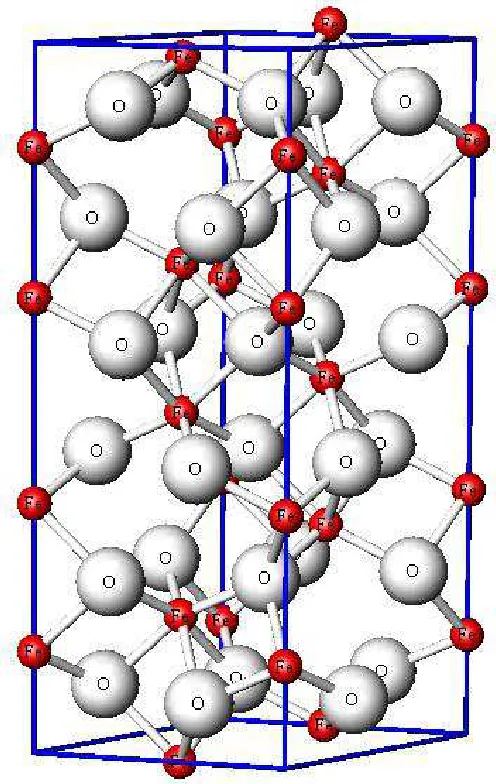
Figura 2.1: Estructura hexagonal de Hematita 15
Figura 2.2: Bandas de energía de la Hematita 16
Figura 2.3: Cabecera del archivo de entrada 18
Figura 2.4: Coordenadas de los átomos en la estructura 18 Figura 2.5: Aproximación inicial y vectores de traslación 19 Figura 2.6: Diagrama de flujo del procedimiento de cálculo 20 Figura 2.7: Esquema del parámetro de la red vs. La energía del sistema 22 Figura 2.8: Variación de la energía de la celda vs. Parámetro de la red
para un valor dado de alfa. 24
Figura 2.9: Gráfica de la Densidad de Estados de la Hematita. 25 Figura 3.1: Átomos en la vecindad del defecto y sus desplazamientos (F centro) 28 Figura 3.2: Densidad de estados del cristal con F centro. 30 Figura 3.3: Átomos en la vecindad de la impureza y sus desplazamientos (Ti) 33 Figura 3.4: Densidad de estados del cristal con F centro 35
Lista de Tablas
Capítulo 1
Metodología
1.1 Introducción
La mecánica cuántica se desarrolló a partir de la cuantización de la energía por el físico alemán Max Planck, y recibió importantísimos aportes, especialmente de Albert Einstein, con su explicación del efecto foto-eléctrico; y otros más de parte de físicos como Heisenberg, Pauli, Dirac, Schrödinger, entre otros. Los principios básicos de las teorías desarrolladas por estos científicos se emplearon en distintas teorías, y se desarrollaron otras nuevas basadas en estos aportes. Caso especial a recalcar es el de Schrödinger, cuyo legado principal, la ecuación que lleva su nombre, es capaz de describir todos los sistemas moleculares, aunque su solución para sistemas cristalinos es casi imposible.
Esto llevó al desarrollo de métodos de aproximación que otorgan resultados fiables y que empezaron a desarrollarse en la década de 50, propiciando el nacimiento de la Química Cuántica y el desarrollo de la Teoría de Orbitales Moleculares (TOM), y con ellas nuevos métodos de investigación de materiales y sus propiedades.
1.2 Fundamentos de Mecánica Cuántica 1.2.1 La ecuación de Schrödinger
Como se puede observar de [1-2], la Ecuación de Schrödinger sirve para representar la función de onda de un sistema. Esta función sirve para describir un sistema en general. Para un sistema atómico, la ecuación de Schrödinger es:
H = E (1.1)
núcleos se debe introducir en el Hamiltoniano del sistema, aparte de los operadores de energía cinética y potencial, términos coulombianos positivos para la interacción electrón-electrón y núcleo-núcleo, además de un término coulombiano negativo para la interacción electrón-núcleo, quedando el hamiltoniano de la forma:
B A AB B A N A A A total r Z Z e M h n N H 2 2 2 2 8 ) ,..., 2 , 1 ; ,..., 2 , 1 (
b a ab na A a Aa A a r e r Z e m
h 2 2 2
2 2
8
(1.2)Donde M se refiere a la masa de los núcleos, m y e se refieren a la masa y a la carga de los electrones, respectivamente; Z se refiere al número atómico, r es la distancia entre dos partículas. Las letras que se encuentran en mayúscula se refieren a los núcleos de los átomos, y aquellas en minúscula a los electrones.
Se debe tener en cuenta que la función de onda debe de ser continua, de valor único y tender a cero en el infinito. Además, uno de los preceptos básicos de la teoría cuántica es que la energía está cuantizada (o “discretizada”), por lo que la ecuación de Schrödinger se cumplirá solamente para ciertos valores de energía:
Hi= Ei (1.3)
1.2.2 Propiedades de la función de onda
- Sies una solución a la ecuación de Schrödinger, entonces c., (donde c
es una constante) es también una solución de la ecuación de Schrödinger. El valor de c puede hallarse a través de la condición de normalización. Para el caso de funciones de onda asociadas a un solo electrón que únicamente son dependientes de las coordenadas de este, se las denotará con . Este tipo de funciones, llamadas orbitales, son análogas a las órbitas planetarias, en el sentido de que pueden describir la posición del electrón alrededor del núcleo.
- Para un electrón con una función de onda estacionaria, i, la condición de
1 ) 1 ( 1
2
i d (1.4)Donde d1 es el elemento de volumen relacionado al electrón, y i2d
representa la probabilidad de encontrarlo en un elemento infinitesimal de
volumen d. La condición de normalización garantiza que el electrón se
encuentre en algún lugar del Universo.
- Para un sistema de partículas de función de onda i, la condición de
normalización será:
... i2(1,2,...)d1d2...1 (1.5)Donde d1, d2,... son los elementos volumétricos de las partículas
individuales y i 2
(1,2,…)d1d2… (que se abreviará i 2
d) es la probabilidad que la partícula 1 esté en el volumen d1, la partícula 2 esté en el volumen
d2, y así sucesivamente.
- Dos soluciones diferentes de una ecuación de onda son ortogonales entre
sí, es decir:
0 ... . 1
i jd (1.6)1.2.3 Aproximación Born – Oppenheimer
1.2.4 Método Variacional
El método variacional, como está descrito en [2], es una herramienta que nos permite encontrar una solución aproximada de la ecuación de Schrödinger. Este método nos permite trabajar en un sistema con muchas partículas, y con el podemos encontrar funciones de onda aproximadamente correctas a partir de funciones de prueba. Si nuestra función de onda depende de los parámetros c1,
c2,... de tal manera que = (c1, c2,...), entonces la energía dependerá de estos
parámetros, y tenemos que:
0 ... ,...) , ( ,...) , ( ,...) , ( 2 2 2 1 1 1 2 1 2
1
c c c c E c c c c E c c
E
(1.7)La forma en que se escoge la función de prueba se fundamenta en principios físicos. Por ejemplo, en el caso de un sistema molecular, es preferible utilizar como función de onda la combinación lineal de los orbitales atómicos.
1.3 Teoría de Orbitales Moleculares
La Teoría de Orbitales Moleculares [3], es un método para determinar la estructura molecular de un sistema, considerando que los electrones no se asignan a enlaces individuales entre átomos, sino que están bajo la influencia de los núcleos de toda la molécula. En esta teoría, cada molécula tiene un grupo de orbitales moleculares, y se asume que la función de onda del orbital molecular está escrita como una suma entre los n orbitales atómicos constituyentes, según la ecuación:
... 1 3 2 2 1
1
i ci c i ci (1.8)
Esta ecuación quiere decir que se considera a los orbitales moleculares , como una Combinación Lineal de Orbitales Atómicos , (LCAO: Llinear Combination of
Atomic Orbitals, por sus siglas en inglés) de los átomos que constituyen el sistema, como puede extraerse de [1].
En (1.8), ci son coeficientes numéricos, determinados a través de la ecuación de
El problema entonces se reduce a determinar cuánto influye un determinado electrón en el orbital molecular i. En un sistema de coordenadas esféricas, la
solución general de la ecuación de Schrödinger para un sistema atómico de un electrón y un núcleo de carga Ze, como el átomo de Hidrógeno, es de la forma:
) ( ) ( ) ( ) , ,
(
r iRnl r lm m (1.9)
Donde Rnl(r), lm() y m() son la función radial, Polar y Azimutal, respectivamente,
y están asociadas a la probabilidad de encontrar al electrón a lo largo del radio r, ángulo y en torno al azimut . Estas soluciones son funciones dependientes de los
números cuánticos n, l, y m, que representan el número cuántico principal, el
número cuántico azimutal y el número cuántico magnético, respectivamente. Usualmente, la notación se abrevia de la forma “nl” (el número cuántico “m” no se especifica, pues depende del valor de “l”), donde “n” toma valores enteros positivos y “l” se expresa a través de las siguientes letras: s, p, d, y f. Así, se encuentran los estados del sistema, como orbitales o estados 1s, 2s, 2p, 3d, etc.
1.3.1 Orbitales atómicos
Un orbital atómico, descrito en [4], es una representación de la posición de un electrón en el espacio. Sin embargo, dada la naturaleza mecano-cuántica de está partícula, un orbital atómico solamente representa una región del espacio en torno al núcleo atómico en la que la probabilidad de encontrar al electrón es elevada. El nombre de los orbitales se debe a sus líneas espectroscópicas (en inglés s sharp, p principal, d diffuse y f fundamental). La forma de los orbitales atómicos puede representarse de manera gráfica, teniendo que los orbitales s poseen simetría esférica alrededor del núcleo (lo que quiere decir que esta es el área en la que el electrón pasa la mayor parte del tiempo), como se puede observar en la figura 1.1.
La forma en que se presentan los orbitales p asemeja a la de dos esferas achatadas hacia el punto de contacto (que es el núcleo del átomo). Estas se encuentran orientadas según los ejes de coordenadas, presentándose tres orbitales: px, py, y pz. La forma de este se puede apreciar en la figura 1.2.
Fig. 1.2: Forma del orbital p
Los orbitales d tienen formas más diversas: cuatro de ellos tienen forma de 4 lóbulos en diferentes orientaciones espaciales, y el último es un doble lóbulo, a lo largo del eje z, rodeado por un anillo. En la figura 1.3 se puede apreciar su forma. La forma de los orbitales f es aún más compleja, pero como en el presente trabajo no se presentan átomos que contengan estos orbitales, no se los describirá.
[image:15.612.138.474.445.686.2]1.3.2 Orbitales moleculares
Los orbitales moleculares [3] pueden representarse a través de funciones matemáticas que describen el comportamiento ondulatorio que pueden tener los electrones en las moléculas. Con estas funciones se puede calcular propiedades tanto físicas como químicas, como la probabilidad de encontrar un electrón en una región del espacio. Utilizando métodos de cálculo de la estructura electrónica, como por ejemplo, el método de Hartree-Fock, es posible obtenerlos de forma cuantitativa.
En la Teoría de Orbitales Moleculares, los enlaces de las moléculas se forman por solapamiento de los orbitales atómicos, de modo que los nuevos orbitales moleculares pertenecen a la molécula entera y no a un solo átomo. Al formarse el enlace, los orbitales atómicos se acercan y comienzan a solaparse, liberando energía a medida que el electrón de cada átomo se ve atraído por la carga positiva del núcleo de otro átomo. Cuanto mayor sea el solapamiento, mayor será el desprendimiento de energía y, por lo tanto, menor será la energía del orbital molecular. Este proceso continúa hasta que los núcleos atómicos llegan a repelerse mutuamente, aumentando la energía. La máxima estabilidad (mínima energía) se alcanza cuando los núcleos se encuentran a una distancia determinada, llamada longitud de enlace.
Cuando se resuelve la ecuación de Schrödinger para el átomo de hidrógeno, la solución corresponde a un polinomio multiplicado por un factor exponencial de la forma e-r donde es conocido como exponente orbital, y su valor viene dado por la relación = Z/n, donde Z corresponde al número atómico y n al número cuántico principal.
en el cálculo de los orbitales moleculares son muy complicadas, por lo que el matemático Slater1 propuso una forma más sencilla de Rnl(r):
1/2 1 ( ) 2 / 1 )! 2 ( ) 2 ( )( n n r
nl r n r e
R (1.10)
Estos se denominan Orbitales de tipo Slater (STO) [1] y son funciones sin nodos, es decir, sin cambios de signo de un punto a otro.
1.4 Energía de un sistema
La función de onda corresponde a un sistema que posee energía, y para
encontrar el valor esperado de la energía del sistema, se procede separando el Hamiltoniano del sistema en 2 partes:
2 1 H H
H (1.11)
Donde H1 es el operador Hamiltoniano asociado a la suma de las interacciones de
cada uno de los electrones dentro de los campos debidos a la presencia de los núcleos desnudos, de un solo electrón a la vez, sin considerar el resto. El
Hamiltoniano H2 considera la interacción electrostática debido al resto de
electrones. Desarrollando los términos de la expresión anterior, se llega a la expresión de la energía de un sistema:
n i n i j ij ij n iii J K
H
E 2 (2 ) (1.12)
Donde Hii es el valor esperado del Hamiltoniano asociado a un electrón sobre su
correspondiente orbital molecular, es decir, representa la energía de un electrón en el orbital molecular (o estado) i bajo un campo de fuerzas electrostático debido
solamente a los núcleos. El factor 2 indica que pueden existir 2 electrones en cada orbital molecular i; Jij representa la energía de interacción de las distribuciones de
carga i*iy j*j y son conocidas como Integrales de Coulomb y Kij se denominan
1 Slater John Clark, Físico y químico teórico estadounidense, estudioso de la teoría cuántica,
Integrales de intercambio. Las integrales de intercambio tienen signo negativo debido a que estas reducen la energía de interacción entre electrones debido a efectos cuánticos (reducción de densidad alrededor del electrón. El efecto se denomina interacción de intercambio, o agujero de correlación, como puede verse en [5]) con espines paralelos en orbitales diferentes. Las integrales de Coulomb proporcionan el valor virtual de la energía de interacción entre los pares de electrones, como si los electrones se movieran en órbitas independientes.
1.5 Ecuaciones de Hartree-Fock
Utilizando la aproximación de un electrón (en la que se construye una función de onda multielectrónica a partir de una combinación de funciones que dependan solamente de las coordenadas de un electrón) para la función de onda; considerando los espín-orbitales (producto del orbital-electrón por su espín), en el método variacional se puede determinar cuánto contribuye cada orbital molecular para obtener una función de onda más próxima a la real, que debe tener la mínima energía total posible. Los orbitales que permiten este estado se denominan auto-consistentes, u Orbitales Hartree-Fock. [1-2]
En la ecuación de Schrödinger, el operador Hamiltoniano que hace uso de estos
orbitales se denomina Operador Hamiltoniano de Fock, F. La ecuación queda
entonces de la forma:
j j ij i
F
(1.13)El operador de Fock puede interpretarse como un operador asociado a un electrón dentro de una disposición molecular. El sistema de ecuaciones (1.13) tiene un conjunto de valores ij (una matriz, matriz de Fock), en lugar de un único valor
propio, debido que las soluciones del sistema de ecuaciones no son únicas.
A través de ciertas transformaciones ortogonales sobre los orbitales i, que son de
elevada complejidad y que no serán desarrollados, (1.13) se diagonaliza y puede reducirse a un sistema de ecuaciones de valores propios convencional (cada orbital
n i
c
F
i i
i, 1,2,... (1.14)Al sistema de ecuaciones (1.14) se les conoce como las Ecuaciones de Hartree-Fock2, y expresan que los orbitales moleculares óptimos son aquellos orbitales propios (funciones propias) del operador Hamiltoniano de Fock. De lo anterior, es posible deducir el procedimiento general para poder obtener los OMs:
1) Se elige un conjunto de funciones propias i´. con las que se define los
operadores de Coulomb y de Intercambio. 2) Se calcula Jj y Kj.
3) Las funciones propias encontradas i´´ son una primera aproximación, que
se usa para repetir el proceso anterior.
4) El procedimiento se repite hasta que los orbitales dejan de variar dentro de un cierto rango de tolerancia en las iteraciones sucesivas.
Por la naturaleza del procedimiento, este es conocido como Método-Autoconsistente [1-2]. Adicionalmente a los n orbitales ocupados, en la solución habrán orbitales adicionales desocupados con valores propios de energía i más
altos, llamados orbitales virtuales, aunque desde el punto de vista físico, perfectamente permitidos. Estos orbitales virtuales no se incluyen en las iteraciones del método Autoconsistente.
La ecuación (1.13) puede expresarse en forma de una combinación lineal:
u u ui i c
(1.15)Esta representación tiene la ventaja de facilitar la interpretación física en razón de que los orbitales atómicos mantienen su relación con los átomos constituyentes del sistema, y por consiguiente es más sencillo relacionar propiedades químicas de las moléculas con sus átomos constituyentes. Además, el problema se reduce a
2El Método de desarrolló originalmente por el físico Inglés Douglas Hartree, basándose en la
encontrar los coeficientes cui de la Combinación lineal de orbitales atómicos. No se
debe olvidar que los orbitales atómicos han de ser ortogonales.
La precisión con la que es posible obtener los orbitales moleculares es fundamentalmente dependiente del número de orbitales atómicos que se use como funciones base. Estas bases pueden ser de tres tipos [1]:
1) Bases mínimas: Están compuestas por todos los orbitales de los electrones del sistema, incluidos los electrones de valencia
2) Bases extendidas: Incluyen los orbitales de base mínima más un número de orbitales superiores a los ocupados por los electrones de valencia.
3) Bases de valencia: Únicamente comprende los orbitales de los electrones de valencia de cada átomo. Este incluye los orbitales virtuales.
Para obtener los orbitales atómicos se hace uso, nuevamente, del método Autoconsistente. Con la finalidad de expresar un orbital molecular en forma de Combinación Lineal de Orbitales Atómicos, se emplea el concepto de densidad de carga, la misma que se define como la densidad de probabilidad de encontrar un electrón en determinado estado energético multiplicada por la carga, donde la unidad de carga empleada es la Unidad Atómica (U.A.), donde e = 1. La integración de esta densidad de carga en todo el espacio resulta en la carga total, misma que corresponde al producto de las matrices de densidad (Puv) y de Solapamiento (Suv),
que permiten hacer un análisis detallado de la población de los orbitales atómicos.
1.6 Método INDO
de integrales que se debe resolver depende necesariamente del número de orbitales atómicos, dependientes a su vez de la cantidad de electrones del sistema.
Las ecuaciones y aproximaciones que se ha desarrollado hasta el momento constituyen lo que se conoce como método ab-initio, útiles para estudiar átomos y pequeños sistemas moleculares. Pero el sistema que se ha planteado resolver (hematita) es un sistema cristalino, y por ello el número de electrones del sistema es muy grande, lo que significa que el número de integrales es extremadamente elevado, de modo que es necesario simplificar el método aún más.
Las bases de valencia mencionadas anteriormente son una forma de simplificar el problema. Esta es una buena aproximación porque los electrones de los orbitales mas externos son los que poseen mayor interés en ser analizados ya que en un átomo son estos los que interactúan con los orbitales externos de otros átomos para formar los enlaces químicos, siendo así los responsables de la variación de la función de onda del sistema. Los orbitales internos no se involucran en estas interacciones, además de ser independientes del tiempo. Por lo tanto, solo serán considerados los orbitales mas externos, (es decir, los orbitales de valencia) como base de los orbitales moleculares del sistema, y estarán sujetos a variación por la presencia de reacciones químicas o defectos, como impurezas.
Sin embargo, a pesar de esta simplificación, las integrales continúan siendo complejas. Para resolver este inconveniente, se introducen parámetros para simplificar y/o reemplazar las integrales anteriores. Con estos parámetros se contribuye a que cada iteración se realice en un tiempo menor.
Estos parámetros cumplen además la función de coeficientes de corrección de las aproximaciones realizadas en la TOM. Estos parámetros se obtienen de modo empírico, de modo que los métodos que los utilizan se denominan métodos semi-empíricos [6]. En varios estudios realizados con métodos semi-semi-empíricos con
parametrización, se ha observado mayor precisión en relación a los métodos
ab-initio (que no utilizan parámetros de corrección).
Este método desprecia parcialmente la contribución del solapamiento entre orbitales a la energía final. Incluye algunas contribuciones de repulsión entre orbitales atómicos de un mismo átomo y puede ser utilizado para calcular efectos de espín. Sin embargo, este método debe ser adaptado para funcionar adecuadamente en sistemas cristalinos, surgiendo el método INDO modificado.
Existen 5 parámetros semi-empíricos que utiliza este método. Estos deben ser elegidos de modo que nos permitan modelar adecuadamente el cristal, es decir, que sean capaces de reproducir las propiedades del material en cuestión, para así reducir el error, debido a las aproximaciones empleadas, al mínimo.
1.6.1 Parámetros Semi-empíricos
Como ya se ha mencionado antes, el método INDO modificado para sistemas cristalinos usa 5 parámetros semiempíricos que son:
- : Es el exponente del orbital de Slater. Denota la parte radial de los OAs y depende del tipo de átomo, del orbital, y del número cuántico n.
- Es el parámetro de la integral de resonancia. Este parámetro define el ancho de las bandas de energía.
- Eneg: Es la electronegatividad.
- P0: Define la población electrónica de los orbitales atómicos.
- iB: Describe la interacción de un electrón en el orbital i de un átomo A con
el “core” de un átomo B. El “core” es el átomo sin tomar en cuenta los orbitales atómicos de valencia.
Capítulo 2
Hematita (-Fe2O3): Propiedades y Modelización
2.1 Introducción
Los óxidos de hierro son compuestos comunes dispersos en la naturaleza. Se los puede encontrar en casi todos los lugares del planeta: atmósfera, biósfera, hidrósfera, litósfera, etc. Se forman principalmente por aclimatación aeróbica de roca magmática en ambientes terrestres y marítimos. Esto involucra erosión causada por el viento, agua, etc. [7].
La hematita es el más antiguo mineral de óxido de hierro conocido, está disperso ampliamente en rocas y tierras. Su color es rojo sangre (su nombre se deriva del griego Haima = Sangre). Es un mineral extremadamente estable y usualmente es el producto final de la transformación de otros óxidos de hierro. Como la mayoría de los óxidos de hierro, tiene estructura cristalina. Posee variadas aplicaciones, como es su utilización en sensores de gas [8], baterías de litio [9], electrónica de alta temperatura [10], espintrónica [11], procesos de foto-oxidación de agua [12], etc.
2.2 Estructura
La estructura de los óxidos de hierro ha sido determinada principalmente a través de difracción de rayos X, o difracción de neutrones complementada con espectroscopia infrarroja, difracción de electrones y microscopía electrónica de alta resolución [7].
es igual al que le precede (en el ejemplo anterior no se menciona b, lo que significa que es igual al valor de a). Se ha determinado entonces que la celda unitaria de la hematita es de forma hexagonal, con a=0.5034nm y c=1.375nm, como se puede constatar en [7]. Existen 6 unidades de fórmula por celda unitaria. (Unidad de Fórmula: Fe2O3, 5 átomos). La celda unitaria es la mínima unidad del cristal, y el
material está conformado por un sinfín de celdas unitarias repetidas periódicamente. El cristal también puede ser descrito mediante el sistema romboédrico, en el cual arh=0.5427nm. (los valores de b y c son iguales al de a) y
[image:24.612.187.435.296.688.2]=55.3° (es la medida de la inclinación de los planos entre sí, ya que el sistema romboédrico es distorsionado), con 2 unidades de fórmula por celda. La estructura puede apreciarse en la figura 2.1.
2.3 Propiedades electrónicas
La hematita es un material semiconductor, y como en todos los semiconductores, la característica esencial es que la separación entre la Banda de Valencia Alta (UVB: Upper Valence Band, por sus siglas en inglés) y la Banda de Conducción (CB: Conduction Band, por sus siglas en inglés) sea menor a 5 electrón-voltios (eV). Además, en cualquier semiconductor, el nivel de Fermi (el nivel bajo el cual todos los niveles de energía están llenos) se encuentra en el último nivel de la banda de valencia alta.
La hematita es un semiconductor de tipo n. Esto significa que la conductividad en este es debido al movimiento de portadores de carga (electrones) en la banda de conducción. La configuración electrónica del átomo de hierro es: 1s2 2s2 2p6 3s2 3p6
[image:25.612.116.481.436.615.2]3d6 4s2; mientras que la del oxígeno es: 1s2 2s2 2p4. La banda de conducción del cristal está compuesta por orbitales d vacios de iones Fe3+, y la banda de valencia alta consiste de orbitales 3d llenos de Hierro con mezclas de los orbitales 2p del Oxígeno. El ancho de la banda prohibida (la separación entre la banda de valencia alta y la banda de conducción, conocida mas por su nombre en inglés, band gap) es comúnmente considerada en 2.2eV, como está descrito en [7]. Podemos apreciarlo en la Fig. 2.2.
Fig. 2.2: Bandas de energía de la Hematita
Banda de orbitales d vacios, de Fe
Banda ocupada, principalmente por orbitales Fe 3d
Banda ocupada principalmente de O 2p
2.2eV
Banda de Conducción
Banda Prohibida
2.4 Modelización
2.4.1 Modelo de Supercelda
El Modelo de Supercelda se refiere al hecho de trabajar con cristales en los que las celdas unitarias (mínima unidad que forma el sistema cristalino) se han extendido en determinada dirección. En el caso de la hematita, la celda unitaria es 30 átomos se ha extendido en la dirección (2x2x1), es decir, se duplica a lo largo del eje a y del eje b, formando un sistema de 120 átomos, que se considera como la celda primitiva. El modelo de Supercelda utiliza periodicidad infinita, es decir, que repite la estructura cristalina indefinidamente, por lo que no presenta efectos de borde.
2.4.2 Procedimiento de cálculo
Para el análisis de las propiedades del cristal se utiliza el software CLUSTERD, mismo que codifica el método Hartree-Fock con la aproximación INDO modificada para cristales, y emplea el modelo de Supercelda [13]. Así, hace posible realizar cálculos de la estructura geométrica y electrónica de moléculas, cristales, superficies y defectos puntuales sobre la geometría del cristal.
El procedimiento en base al cual se realizan los cálculos consiste en preparar archivos de entrada (formatos de texto con extensión ddp). Estos archivos contienen la información sobre el cristal: número de átomos y de especies atómicas (es decir, de elementos), coordenadas espaciales de los átomos que conforman el sistema, parámetros semi-empíricos, carga de la celda, multiplicidad, número de iteraciones, precisión deseada, etc. A continuación se analizará un ejemplar de un archivo, correspondiente al cristal Hematita.
Fig. 2.3: Cabecera del archivo de entrada
Luego de estos datos se tiene las coordenadas geométricas de los átomos del sistema. Los átomos se numeran como se muestra en la figura 2.4:
Fig. 2.4: Coordenadas de los átomos en la estructura
La segunda columna de la gráfica anterior representa el elemento al que pertenecen los átomos: 1 para los átomos de hierro, y 2 para los de oxígeno.
Finalmente, a continuación de las coordenadas de los átomos, se colocan los valores que constituyen la aproximación inicial, es decir, las funciones propias i´, o
funciones de prueba con las que se inician los cálculos. La elección de estos valores se fundamenta en principios físicos, siendo positivos para átomos con valencia positiva, como el hierro; y negativos para átomos con valencia negativa, como el oxígeno. Estos se pueden apreciar en la figura 2.5. Las tres últimas filas representan los vectores de traslación, es decir, los valores de a, b y c.
1 0 0 0 0 0 0 0 1 1
* supercelda de alpha-Fe2O3 (HEMATITA) * 120 0 1 4 2 0 0 0
0 0 0 0
100 109 1 0.000100 0.600000 0.001001 26 3 8 0.00 0.15
3 2 4 0 4 1 2.38 2.10 1.7
16.14 -8.6 12.4 -3.0 0.7 -3.0
0.82 0.82 0.82 0.82 0.82 1.6 0.0 0.0 0.0
8 2 6 -0.34 0.15 2 0 2 1
2.27 1.86
4.5 -16.0 -12.6 -16.0 1.974 1.96 1.96 1.96
1 1 0.000000 0.000000 0.967349 2 1 1.000000 0.000000 0.967349 3 1 0.000000 1.000000 0.967349
. . . . .
. . . . .
. . . . .
. . . . .
[image:27.612.213.401.353.468.2]Fig. 2.5: Aproximación inicial y vectores de traslación
A fin de obtener los parámetros más óptimos para los átomos, estos se deben estimar inicialmente. Al elaborar la Supercelda y realizar los cálculos se pueden obtener datos como la densidad de estados del sistema (DOS), composición química de las bandas de energía, densidad electrónica de los átomos constituyentes, parámetros de la red, etc., y estos se comparan con resultados experimentales. En caso de que los resultados se aproximen lo suficiente, estos son escogidos como adecuados. Si no es el caso, los parámetros se reemplazan y se repite el procedimiento.
Una vez que el cristal en su estado puro se ha modelado correctamente, y el modelo es consistente con el modelo físico real (experimental), se puede introducir los defectos que se desea analizar. Una vez que se ha hecho esto, es necesario recordar que todos los sistemas tienden a un estado de mínima energía, de modo que los átomos tienden a desplazarse dentro de la estructura para llegar a este estado. Con esto en mente, se procede a relajar (desplazar) manualmente los átomos circundantes al defecto: acercándolos o alejándolos de la impureza, o rotándolos con respecto al eje de la misma (el defecto debe introducirse lo más cerca posible al centro de la Supercelda), hasta que se obtiene el valor mínimo de energía del sistema. Esta información se obtiene a través de la generación de un archivo de salida (archivo de texto, de extensión .out), que contiene los datos sobre las diferentes iteraciones, la carga de los átomos, los valores propios, etc. En la figura 2.5 se puede apreciar un diagrama de flujo del procedimiento de cálculo que realiza el programa, como está descrito en [2].
0.17 0.17 0.17 0.17 0.17 1.48000 1.48000 1.48 1.48000
-1.90000 -1.30000 -1.30000 -1.30000 1 10 0 0 0 0 0 0 0 0 120 120
Fig. 2.6: Diagrama de flujo del procedimiento de cálculo Uso de valores de aproximación inicial para el
cálculo de los coeficientes de los Orbitales Moleculares (Matriz de Fock)
Autoconsistencia: ¿Se ha alcanzado la precisión deseada?
Electrones asignados en pares a cada orbital molecular con la menor energía. Se determina
orbitales ocupados
Cálculo de la matriz de densidad Puv a partir de
los coeficientes de los orbitales moleculares ocupados. El resultado se utiliza para calcular
una nueva matriz de Fock
Diagonalización de la matriz de Fock, obteniéndose valores y funciones propias (un
nuevo conjunto de cui y i)
Cálculo de la energía total del sistema Si
[image:29.612.180.446.70.655.2]La precisión se calcula a partir de las matrices de densidad de dos ciclos sucesivos, a través de:
uv
uv uv P
P ' (2.1)
Una vez que la energía del sistema es la menor, se dice que la geometría se ha optimizado. Es este momento es cuando se analiza los desplazamientos atómicos, las distancias inter-atómicas, la densidad electrónica, la composición de las bandas de energía, y sobre todo la presencia de los electrones o huecos resultantes de introducir el defecto en el cristal.
2.4.3 Parametrización
Con la ayuda de los parámetros de la red cristalina (a y c), se puede obtener la estructura geométrica del cristal. Una vez que esta se ha conformado, se acoplan los parámetros semi-empíricos para que puedan minimizar la energía del sistema en función de su geometría.
La celda se ha elaborado de modo que sea de simetría hexagonal, y los átomos se
han ubicado de manera que se han obtenido los valores de a=0.5035nm, y
c=1.372nm, los cuales concuerdan bien con los datos experimentales mencionados anteriormente, en [7].
atómicas de longitud, o radios de Bohr (1 Radio de Bohr = 0.052nm), que convertidas a nanómetros resultan en los valores de a y c.
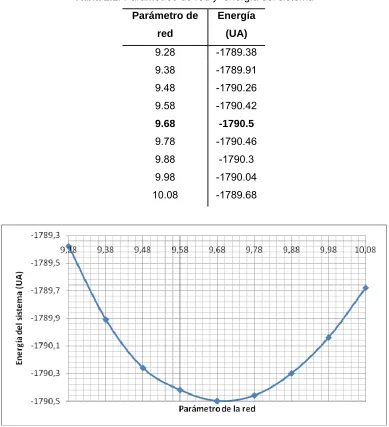
Tabla 2.1. Parámetros de red y energía del sistema Parámetro de
red
Energía (UA)
9.28 -1789.38 9.38 -1789.91 9.48 -1790.26 9.58 -1790.42 9.68 -1790.5 9.78 -1790.46 9.88 -1790.3 9.98 -1790.04 10.08 -1789.68
Fig. 2.7: Esquema del parámetro de la red vs. La energía del sistema
Una vez que el parámetro de la red es el óptimo, se procede a encontrar los otros parámetros que requiere el método. En el caso presente, correspondiente al cristal de -Fe2O3, se requiere los parámetros de los átomos de Hierro y Oxígeno, pero el
tomaremos los valores optimizados del mismo [14], de modo que se obtendrá los parámetros del átomo de Hierro. Estos parámetros se pueden apreciar en las tablas 2.2 y 2.3.
Tabla 2.2. Conjunto de parámetros semi-empíricos Eneg P0
Átomo AO (au) (eV) (e) (eV)
3d 2,38 16,14 0,82 -8,6
Fe 4s 2,1 12,4 1,6 -3
4p 1,7 0,7 0 -3
O 2s 2,27 4,5 1,974 -16
2p 1,86 -12,6 1,96 -16
Tabla 2.3. Conjunto de parámetros semi-empíricos para 2 centros αB (au-1)
Fe O
Fe 0 0,15
O -0,34 0,15
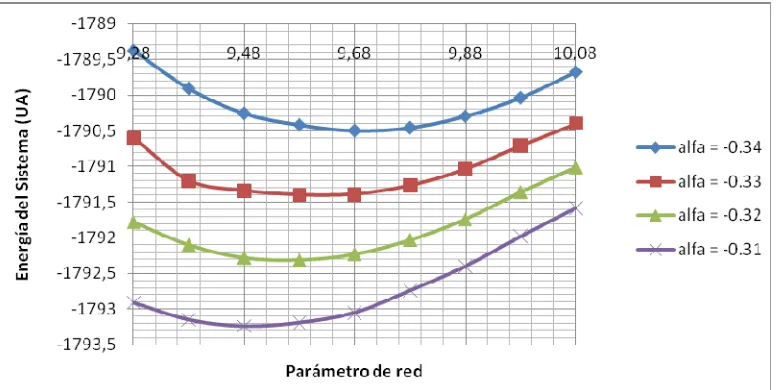
[image:32.612.224.399.354.433.2]Fig. 2.8: Variación de la energía de la celda vs. Parámetro de la red para un valor dado de alfa.
Como se ha mencionado anteriormente, estos parámetros deben reproducir en la mejor medida posible los datos experimentales sobre el material estudiado, especialmente en cuanto a la geometría del cristal y la composición de las bandas de energía. Para poder corroborar los datos obtenidos analíticamente con los datos experimentales, se recurre a la elaboración de una gráfica denominada Densidad de estados (Density of States, DOS, por sus siglas en inglés). Está gráfica nos muestra la composición de todas las bandas de energía del cristal y puede obtenerse mediante la utilización de los valores propios encontrados en los cálculos de la red cristalina.
A cada valor propio de energía i se le asigna un número D(i) que representa la
probabilidad de que un electrón del sistema se encuentre en el nivel de energía i,
que corresponde a su vez a un orbital molecular determinado, como puede verse en la ecuación (2.2), extraída de [1]. De este modo, se puede construir una gráfica como la que se puede apreciar en la figura 2.5.
j ij i c
Fig. 2.9: Gráfica de la Densidad de Estados de la Hematita. La gráfica ha sido suavizada.
La línea en el punto cero marca el último orbital ocupado de la banda de valencia (Nivel de Fermi), que es el punto donde inicia la banda prohibida. Está gráfica se ha “suavizado”, de modo que sea más sencillo el poder apreciar la composición de las bandas (por eso la línea cruza la banda Fe3d, cuando en realidad se encuentra en el borde).
La aproximación de 1 electrón que se ha empleado produce un efecto notorio en la parte electrónica del modelo. Como no se toman en cuenta la totalidad de las interacciones electrónicas, el ancho de la banda prohibida aparece mayor al real. En este caso, su valor es de 6.2eV. Es necesario recalcar sin embargo, que este ensanchamiento no afecta a la composición de las bandas (como puede verse de los datos antes expuestos), distorsiones de la red o energías de relajación. La energía de la banda prohibida se obtiene a través del método HOMO-LUMO, que consiste en la diferencia entre el más alto orbital molecular ocupado (Highest Occuped Molecular Orbital, HOMO, por sus siglas en inglés) y el más bajo orbital
molecular desocupado (Lowest Unoccupied Molecular Orbital, LUMO, por sus
excitar un electrón desde el último nivel ocupado de la banda de valencia al primer nivel desocupado de la banda de conducción, y calcular la energía necesaria para el salto (esta energía es la mínima que ha de haber para realizar dicho salto), como la diferencia entre el estado fundamental del cristal, y el estado excitado del mismo. A través de este método, el valor calculado es de 2.8eV, lo cual es muy cercano al valor experimental.
Capítulo 3
Hematita con defectos puntuales
3.1 Introducción
Los defectos puntuales en materiales son eventos comunes en la naturaleza. De hecho, muchos de ellos fueron descubiertos en minerales y a partir de allí se ha empezado su estudio. Cristales que corresponden a una misma estructura y composición, pero con ciertas propiedades alteradas (el color es la más reconocible) se deben a la presencia de estos defectos. Como es imposible obtener una sustancia ciento por ciento “pura”, el estudio de cómo son afectadas las propiedades de un material es imprescindible, no solo para saber cómo se comporta una sustancia con un defecto, sino para saber cual es este defecto y como se puede aprovechar.
Posteriormente, se encontró interés en introducir estos defectos intencionalmente en los materiales, con la finalidad de obtener sustancias con las propiedades deseadas. En la actualidad, los estudios se centran principalmente en los materiales semiconductores, siendo la hematita uno de los más estudiados.
3.2 Defectos puntuales en Hematita: F centros
Uno de los defectos más comunes en los cristales óxidos es el llamado F centro (centro F, o también centro de color). Este defecto se descubrió en Alemania, y por ello la letra “F” es por Farb, la palabra alemana para color, y significa que la presencia de este defecto afecta la coloración del cristal. Este defecto consiste en 2 electrones atrapados en una vacancia de oxígeno, afectando las propiedades estructurales (se reduce la cantidad de átomos), electrónicas (el átomo removido poseía carga) y ópticas (de ahí el cambio de color) del material.
3.2.1 Resultados
[image:37.612.121.509.239.540.2]La presencia del defecto en la estructura del cristal hace que los átomos que se encuentran en la vecindad de la región defectuosa (región en la que se encuentra el F centro) se desplacen de las posiciones que originalmente tenían. Como es posible apreciarse en la Fig. 3.1, los átomos que se encuentran en el mismo plano que la región defectuosa, presentan una tendencia a moverse hacia esta zona, al igual que la mayoría que los átomos de oxígeno que no se encuentran en el mismo plano.
Fig. 3.1: Átomos en la vecindad del defecto y sus desplazamientos
átomos O(13) y O(14) no se han movido. La explicación para estos fenómenos reside en una redistribución de la densidad electrónica en dichos átomos. Estos átomos acumulan una mayor concentración de carga en los enlaces que se encuentran en direcciones opuestas a la del defecto. Los movimientos de los átomos pueden apreciarse en la tabla 3.1.
Tabla 3.1: Carga de los átomos y la variación de su distancia respecto al defecto. La numeración corresponde a la de la Fig. 3.1. Los valores positivos en los
desplazamientos significan incremento, los negativos decremento.
Átomo Qo(e) Qf(e) R (Å)
Fe(1) 2,75 2,79 0,06 Fe(2) 2,75 2,36 0,11 Fe(3) 2,75 2,67 0,35 Fe(4) 2,81 2,80 -0,18 Fe(5) 2,76 2,79 0,10 Fe(6) 2,69 2,26 0,11 Fe(7) 2,74 2,71 -0,18 Fe(8) 2,74 2,71 0,39
O(9) -1,83 -1,83 -0,13
O(10) -1,83 -1,85 0,04
O(11) -1,83 -1,84 -0,09
O(12) -1,83 -1,85 -0,13
O(13) -1,83 -1,85 0,00
O(14) -1,83 -1,83 0,00
O(15) -1,83 -1,84 -0,17
O(16) -1,82 -1,85 -0,07
O(17) -1,84 -1,87 -0,15
O(18) -1,85 -1,88 -0,11
una redistribución electrónica en los átomos, en la cual, los átomos de hierro mencionados concentran una mayor densidad electrónica en la dirección hacia la que se encuentra el plano que contiene la región defectuosa, y si consideramos que los átomos de Oxígeno que se encuentran en el mismo plano se encuentran más cercanos a ellos que el F centro, se puede concluir que la atracción por la interacción de Coulomb entre los átomos de Hierro y Oxígeno es más fuerte que la repulsión entre el defecto y los átomos de Fe.
[image:39.612.123.503.381.621.2]La presencia de la región defectuosa en la estructura cristalina hace que se forme un nivel local (que podría describirse como una pequeña banda de energía), cerca de la Banda de Valencia Alta, lo que reduce la energía que necesitaría un electrón para ser excitado desde la Banda de Valencia Alta hasta la Banda de Conducción. La Figura 3.2, correspondiente al cálculo de la Densidad de Estados del material con el defecto, nos muestra con mayor claridad la presencia de este nivel local que se ha formado a continuación del último estado ocupado de la Banda de Valencia Alta (la gráfica ha sido suavizada para obtener una mejor interpretación).
Fig. 3.2: Densidad de estados del cristal con defecto. En el círculo rojo, se puede apreciar el nivel local, producto del F centro introducido.
energía de absorción del mismo, siendo menor en relación a la del cristal sin defectos. Las energías de absorción se determinaron en los valores de 0,9 y 3,6 eV, correspondiendo al salto desde la Banda de Valencia Alta hacia el nivel local, y desde el mismo hasta la banda de conducción. Estos valores se encontraron a través de la ecuación E=hf, que nos dice que la energía está en función de la frecuencia de la luz por la constante de Planck.
El valor de 0,9 eV, que corresponde a una longitud de onda de 1380nm, no ha sido reportado en otras investigaciones; pero en el caso de la energía de absorción de 3,6 eV, que corresponde a una longitud de onda de 345nm, se han divulgado resultados experimentales basados en métodos foto-electro-químicos realizados en películas de hematita [15-16] que dan como resultado longitudes de onda de 340nm y 350nm, que prácticamente coinciden con los resultados encontrados en esta investigación. En estos trabajos se atribuye estas energías de absorción a una reducción de portadores de carga (electrones), evitando que se dé una inmediata recombinación electrón-hueco en la hematita. Esta reducción puede deberse a vacancias positivas de oxígeno, que, como se ha visto en la presente investigación, puede producirse por la presencia de F centros.
La hematita tiene aplicaciones ópticas, y la presencia de F centros en el cristal puede favorecer a las mismas. Se debe notar que la longitud de onda en la que se presenta la energía de absorción corresponde a cierta región del espectro visible, próximo a la región del ultravioleta, lo que significa que las radiaciones de esta zona son absorbidas por el cristal, y esto puede ser útil en procesos de foto-oxidación de agua [12], que es una de las principales aplicaciones de la hematita en el campo óptico.
3.3 Defectos puntuales en Hematita: Impureza de Titanio
Para introducir una impureza, en este caso de Titanio, se elige un átomo de Hierro que se encuentre lo más cerca posible a la región central de la Supercelda. El Titanio es un átomo que posee la siguiente distribución electrónica: 1s2 2s2 2p6 3s2 3p6 3d2 4s2, y posee valencia +4, de modo que contiene un electrón de valencia más que el átomo de hierro que reemplaza. Una vez que se ha introducido el átomo en la red cristalina, este produce perturbaciones estructurales que influyen tanto en las propiedades electrónicas como en las ópticas del material. Los parámetros semi-empíricos del átomo de titanio están descritos en las tablas 3.2 y 3.3.
Tabla 3.2. Conjunto de parámetros semi-empíricos para interacción entre 2 centros
αB
Átomo Fe O Ti
Fe 0.0 0.15 0.0
O -0.34 0.15 0.0
Ti -0.4 0.15 0.0
Tabla 3.3. Conjunto de parámetros semi-empíricos
Átomo AO Eneg P0
3d 1.88 0.07 0.55 -8.0
Ti 4s 1.75 1.5 0.44 -0.8
4p 1.7 -2.0 0.22 -0.8
3.3.1 Resultados
[image:41.612.204.422.416.495.2]Fig. 3.3: Átomos en la vecindad de la impureza y sus desplazamientos
Como se puede apreciar de la gráfica, los átomos de Hierro Fe(1), Fe(2) y Fe(3) se mueven en dirección a la impureza, obviamente debido a la interacción de Coulomb. El átomo de Titanio introducido en el material posee una carga eléctrica menor a la que poseía el átomo de Hierro que ha sustituido, de modo que la fuerza de repulsión originada por las cargas eléctricas es menor. Sin embargo, es necesario recalcar que no todos los átomos de hierro cumplen con esta tendencia. El átomo Fe(5) no se ha movido de su posición original, debido a que su carga eléctrica se ha incrementado notablemente. Esto significa que la naturaleza de su enlace químico se ha vuelto mucho más iónica. En el caso del átomo Fe(4), se observa un interesante fenómeno: una redistribución de su densidad electrónica, que pasa desde el orbital atómico 3dyz, hacia el orbital 3dx2-y2, lo que produce que el
Tabla 3.4: Carga de los átomos y la variación de su distancia respecto a la impureza. La numeración corresponde a aquella de la Fig. 3.3. Los valores positivos
en las distancias significan incremento, los negativos decremento.
Átomo Qo(e) Qf(e) ∆R(Å)
Fe(1) 2,70 2,82 -0,13
Fe(2) 2,75 2,90 -0,13
Fe(3) 2,76 2,76 -0,13
Fe(4) 2,81 2,74 0
Fe(5) 2,82 3,54 0
O(6) -1,83 -1,83 0
O(7) -1,83 -1,59 -0,45
O(8) -1,83 -1,83 -0,15
O(9) -1,83 -1,80 0,06
O(10) -1,83 -1,68 -0,17
O(11) -1,83 -1,82 0,07
O(12) -1,83 -1,59 -0,09
O(13) -1,83 -1,82 0,16
O(14) -1,85 -1,79 0,07
O(15) -1,85 -1,86 0,22
O(16) -1,85 -1,68 0,1
O(17) -1,85 -1,85 0,06
Ti(18) - 0,89 -
Analizando la estructura de bandas del material, se puede apreciar que la presencia de la impureza origina un estrechamiento de la Banda de Energías Prohibidas, lo que significa que se podría esperar una mejora en la conductividad tipo n en el cristal dopado. La introducción de la impureza, que posee un electrón de valencia más que el átomo de Hierro, incrementa el número de electrones de valencia presentes en el material. Este electrón adicional se encuentra parcialmente deslocalizado (no se encuentra en un punto específico), y es posible observar estados 3d del Titanio en el fondo de la banda de conducción. La gráfica 3.4 nos muestra la Densidad de estados del cristal con la impureza de titanio.
Capítulo 4
Conclusiones y Recomendaciones
4.1 Conclusiones
Al realizar la modelización del cristal, se obtuvo que la geometría del mismo, así como la composición de las bandas de energía coinciden en gran medida con los datos experimentales. Sin embargo, el valor obtenido para la banda de energía prohibida, 2.8 eV, es mayor al valor experimental de 2.2ev, debido a la aproximación de un electrón utilizada en el método.
La presencia del F centro en la hematita afecta la geometría del cristal. La configuración de equilibrio espacial indica que la mayoría de átomos de hierro se mueven alejándose de la impureza, debido a la repulsión electrostática. En el caso de los átomos de oxígeno, la mayoría se mueve hacia la impureza, debido principalmente a la atracción por la interacción de Coulomb, mientras el resto se aleja o permanece inmóvil, debido a redistribuciones de la densidad electrónica.
El F centro genera la presencia de un nivel local en la banda de energías prohibidas, que lleva a una contribución en el espectro de absorción del cristal, en la banda de 345nm, estando de acuerdo con resultados experimentales foto-electroquímicos.
La presencia del nivel local hace que la energía que necesitaría un electrón para pasar desde la banda alta de valencia hasta la banda de conducción sea menor, lo que implica una mejora en la conductividad eléctrica del material.
alejándose de la impureza, principalmente, debido a una menor atracción por la interacción de Coulomb. Aquellos que no se acercan, presentan una redistribución de su densidad electrónica hacia la zona defectuosa, que explica su comportamiento.
El átomo de titanio en la red cristalina de la hematita afecta a la composición de las bandas de energías del mismo. Luego del dopaje se tiene un electrón extra en el sistema (el Ti tiene un electrón de valencia más que el Fe), estando el electrón extra parcialmente deslocalizado (no en un punto específico), y se pueden notar estados de Ti 3d en el fondo de la banda de conducción, lo que produce que la banda prohibida se estreche. Esto significa que la conductividad tipo n del material ha mejorado
El hierro posee características magnéticas, y así mismo la hematita. No se ha podido determinar si la presencia de los defectos afecta en alguna medida a las características magnéticas del cristal, debido a que el software utilizado no permite analizar dichas características.
No se ha considerado efectos debido a variaciones de temperatura en la
4.2 Recomendaciones
Impulsar el uso de herramientas computacionales para el estudio de
estructuras cristalinas de importancia tecnológica. Su uso es mucho más conveniente en esta Universidad debido a los costos, principalmente.
Los temas de investigación no deben limitarse a ciertos materiales. Existen gran cantidad de compuestos con características únicas que pueden estudiarse y que no cuentan con la cantidad de estudios adecuada para poder explicar satisfactoriamente sus propiedades, por lo que debe tomarse en cuenta a la hora de elegir un objetivo de estudio
Los defectos en los materiales son muy comunes. Por lo tanto, el estudio de la presencia en los mismos no debe limitarse a ciertos elementos, sino que debe investigarse materiales de otro tipo, como pueden ser aceptores (que poseen menor valencia que el elemento que reemplazan), y no donadores (como el ti), o elementos de igual valencia. Además, puede variarse su concentración, puede estudiarse la presencia de más de un tipo de defectos, etc.
Realizar investigaciones a través de métodos que consideren efectos que no se toman en cuenta con el software utilizado (como los efectos magnéticos o
térmicos). Un ejemplo puede ser el programa VASP, que si los toma en
Referencias
[1] PATIÑO Edgar, Estudio de los efectos físicos producidos por el dopaje de Nb y La en el BaTiO3 utilizando el método cuántico LUC. Tesis de grado de la Escuela Politécnica Nacional. 2000
[2] PINTO Henry, Estudio de las propiedades estructurales y ópticas de los polarones en el BaTiO3 utilizando el método INDO. Tesis de grado de la Escuela Politécnica Nacional. 1999.
[3] Wikipedia, la enciclopedia libre, Teoría de los orbitales moleculares [en línea]. [citado el 09 de Noviembre del 2009] disponible en la dirección: http://es.wikipedia.org/wiki/Teoria_de_orbitales_moleculares
[4] Wikipedia, la enciclopedia libre, Orbital atómico [en línea]. [citado el 09 de Noviembre del 2009] disponible en la dirección: http://es.wikipedia.org/wiki/Orbital_at%C3%B3mico
[5] R. M. Martin, Electronic Structure, University of Cambridge ,2004
[6] FONT. María, Mecánica Cuántica [en línea]. [citado el 29 de Septiembre del
2009] disponible en la dirección: http://www.unav.es/organica/docencia/modeling_d/mecanicacuantica/mecani
cacuantica.pdf.
[7] R.M Cornell, U. Schwertmann, The Iron oxides, Wiley, 2000
[8] K. Arshak, I. Gaidan, L. Cavanagh, “Screen-Printed Fe2O3/ZnO thick films for gas sensing applications” Journal of Microelectronic and Electronic
Packaging 2 (2005) 25.
[9] J. Chen, L. Xu, W. Li, X. Gou, “-Fe2O3 Nanotubes in Gas Sensor and
Lithium-Ion Battery Applications”, Advanced Materials, 17 (2005) 582.
[10] F. Zhou, S. Kotru, R.K. Pandey, “Pulsed laser deposited ilmenite-hematite films for application in high temperature electronics”, Thin Solid Films 408
(2002) 33.
[11] J. Dou, L. Navarrete, P. Kale, P. Padmini, R.K. Pandey, H. Guo, A. Gupta, R. Scha, “Preparation and characterization of epitaxial ilmenite-hematite films”,
Journal of Applied Physiscs, 101 (2007) 53908.
[12] G. Jinghua, “Hematite nanoarrays promise water photo-oxidation by solar irradiation”, Micro/Nano Lithography & Fabrication (2007). Disponible en la
[13] E.V. Stefanovich, E.K. Shidlovskaya, A.L. Shluger, M.A. Zakharov, “Modification of The INDO Calculation scheme and parametrization for ionic crystals” Phys. Status Solidi B 160 (1990) 529.
[14] H. Pinto, A. Stashans, “Quantum-chemical simulation of Al- and Sc-bound hole polarons in BaTiO3 crystals” Computer Material Science 17 (2000) 73. [15] N. Beermann, L. Vayssieres, S.-E. Lindquist, A. Hagfeldt,
”Photoelectrochemical Studies of Oriented Nanorod Thin Films of Hematite”,
Journal of the Electrochemical. Society. 147 (2000) 2456.
[16] A. Kay, I. Cesar, M. Grätzel, “New Benchmark for Water Photooxidation by Nanostructured -Fe2O3 Films”, Journal of American Chemical Society. 128
Resumen— Los efectos producidos en el cristal de hematita (α-Fe2O3) debido a la presencia de defectos puntuales han sido investigados a través de un método computacional químico-cuántico que hace uso de la metodología de Hartree-Fock para el procedimiento de cálculo. Para ello, se ha empleado una supercelda de 120 átomos. Primero, se determinó los parámetros numéricos del átomo de Hierro, reproduciendo las características principales del material puro. Luego, los defectos puntuales F centro y Titanio, se introducen en la estructura y se procede a determinar los efectos geométricos (movimientos) y relativos a las propiedades electrónicas que pueden generarse debido a la perturbación del cristal. La presencia del F centro produce un nivel local en la banda de energías prohibidas, alterando el espectro de absorción del cristal; mientras que el átomo de Titanio produce el estrechamiento de la mencionada banda, que lleva a una mejora en la conductividad del material.
.
Índice de Términos — Hematita, Hartree-Fock, F centro, Titanio, defecto puntual.
I. INTRODUCCIÓN
os óxidos de hierro son materiales abundantes en la corteza terrestre, y entre ellos, la hematita es uno de los más comunes. Este cristal puede encontrarse en estructura hexagonal con parámetros a = 5.03Å y c = 13.75 Å, componiéndose de 6 unidades de fórmula (Unidad de fórmula: Fe2O3, 5 átomos) por celda unitaria (30 átomos).
Este material posee una gran variedad de aplicaciones, entre las que se puede destacar su utilización como un sensor de gas [1], sus características semiconductoras han sido estudiadas en un amplio rango de temperaturas, y tiene un gran futuro para dispositivos electrónicos de alta temperatura [2]; espintrónica [3], procesos de foto-oxidación [4], además de que podría exhibir un efecto de memoria debido al ordenamiento de los momentos magnéticos internos de las partículas [5].
Las sustancias no existen en un estado libre de impurezas o defectos, y la existencia de dichos defectos produce diversas alteraciones en las propiedades de los materiales, mismas que pueden ser de tipo estructural, electrónico u óptico. Por esto, el
A. Stashans, Instituto de Química Aplicada (IQA), Universidad Técnica Particular de Loja (UTPL), Apartado 11-01-608 (e-mail: [email protected])
R. Rivera, Escuela de Electrónica y Telecomunicaciones (EET), Universida técnica Particular de Loja (UTPL) Apartado 11-01-608 (e-mail: [email protected]).
estudio de impurezas en los cristales es muy importante si se quiere conocer las características de los materiales reales. Con la finalidad de estudiar los efectos producidos por la presencia del F centro y el Titanio en la hematita, se ha aplicado una metodología químico-cuántica basada en la aproximación de Hartree-Fock y el modelo de supercelda, con lo que es posible tomar con precisión las peculiaridades cristalográficas del material dado. Con esto en mente, se ha empleado el código computacional CLUSTERD [6]. Cabe recalcar que este método ha probado ser muy confiable y ha dado resultados precisos en el estudio de cristales complejos
como PbTiO3 [7-8]; PbZrO3 [9]; y WO3 [10-11].
II. METODOLOGÍAYPROPIEDADESDE HEMATITAPURA
El modelo usado para la investigación se basa en la versión avanzada de la teoría de Hartree-Fock modificada para cálculos en cristales. El método, implementado en el software CLUSTERD [6], se ha desarrollado especialmente para sistemas periódicos (cristales) que contienen diversos defectos puntuales. Este programa resuelve la ecuación de Schrödinger y calcula la estructura electrónica de bandas y la energía total de un cristal a través de Orbitales Moleculares, como combinaciones lineales de Orbitales Atómicos, y considera las peculiaridades y periodicidad de un sistema a través del modelo de Supercelda. En el caso presente, se utilizado una supercelda de 120 átomos, resultado de extender cuatro veces (2x2x1) la celda primitiva de 30 átomos de la hematita.
Debido a que el método es de carácter semi-empírico, existe un número de aproximaciones y simplificaciones matemáticas que deben ser consideradas si se pretende estudiar un sistema real. Para ello debe obtenerse los parámetros numéricos para cada tipo de átomo en el sistema, acoplando las diferentes propiedades del cristal con los datos experimentales, principalmente el ancho de la banda de energías prohibidas, ancho y composición de las bandas de valencia y de conducción, estructura espacial, constantes de la celda, cargas atómicas, etc.
Se desarrolló la parametrización del átomo de Hierro. Aplicando el proceso de minimización de energía (todos los sistemas tienden al estado de mínima energía) para la supercelda, se obtuvo los parámetros de la red: a = 5.035 Å, y
c = 13.72 Å, que prácticamente coinciden con los datos experimentales [12]. Una vez que se obtuvo los parámetros de la estructura, se precedió a determinar los parámetros semi-empíricos del método, que se puede apreciar en las tablas 1 y 2.
Estudio de Propiedades Electrónicas de
Hematita con defectos Puntuales
Arvids Stashans, Richard Rivera, Universidad Técnica Particular de Loja