El primer capítulo describe la síntesis total estereoselectiva de aspergilida A (IV), un producto natural aislado del hongo marino Aspergillius ostianus y que ha demostrado tener una importante actividad citotóxica contra las células de leucemia linfocítica. En el segundo capítulo se presenta una aproximación a la síntesis total de citospolida Q (VI), un producto natural aislado del moho Cytospora sp, que ha mostrado actividad citotóxica contra el adenocarcinoma de pulmón.
INTRODUCCIÓN Y ANTECEDENTES
- TETRAHIDROPIRANOS EN PRODUCTOS NATURALES
- MÉTODOS SINTÉTICOS DE TETRAHIDROPIRANOS
- Reacción hetero Diels-Alder
- Reordenamiento de Petasis-Ferrier
- Ciclación radicalaria
- Ciclación de Prins
- Ciclación de 5-hidroxiepóxidos
- Ciclación mediada por metales
- Reacción intramolecular oxa-Michael
- Reducción de hemiacetales cíclicos
- SÍNTESIS PREVIAS DE LA ASPERGILLIDA A
- Síntesis total de la aspergillida A por el grupo de Marco
- Síntesis total de la aspergillida A por el grupo de Fuwa
- Síntesis total de la aspergillida A por el grupo de Shishido
- Síntesis formal de la aspergillida A por el grupo de Loh
- TRABAJO PREVIO REALIZADO EN EL GRUPO DE INVESTIGACIÓN
Uno de estos ejemplos lo vemos en la síntesis de (-)-kendomicina, trabajo de Smith y colaboradores, donde se lleva a cabo la condensación entre el hidroxiácido 13 y el aldehído 14 para generar dioxano 15 (Esquema 2). Un ejemplo de este protocolo es el reportado por el grupo Jennings en la síntesis de (-)-cianolida.

OBJETIVOS
PROPUESTA SINTÉTICA
RESULTADOS Y DISCUSIÓN
- Síntesis de la γ-hidroxi-δ-valerolactona 90
- Resolución cinética del alcohol 88
- Primera aproximación a la síntesis de la aspergillida A
- Síntesis del alqueno 134
- Síntesis del análogo 148 de la aspergillida A
- Síntesis total de la aspergillida A (1)
Para resolver este problema, la síntesis de la lactona 98 se llevó a cabo sin la función éster lateral. Es por ello que se llevó a cabo la síntesis de la hidroxilactona 138, la cual presenta grupos protectores con suficiente ortogonalidad reactiva.

CONCLUSIONES
CONSIDERACIONES EXPERIMENTALES GENERALES
- Consideraciones experimentales
- Abreviaturas
Se eliminó el THF a presión reducida y la reacción se extrajo con acetato de etilo (3 x 100 ml). Una vez que se completó la reacción, se añadió una solución saturada de NH4Cl y se extrajo con acetato de etilo (3 x 15 ml). Una vez que se completó la reacción, se añadió una solución saturada de NH4Cl y se extrajo con acetato de etilo (3 x 20 ml).
Una vez completada la reacción, se añadió agua, el disolvente se eliminó a presión reducida y se extrajo con acetato de etilo (3 x 15 ml). Una vez completada la reacción, se añadió una solución saturada de Na2CO3 (15 ml) y se extrajo con diclorometano (3 x 30 ml).
TETRAHIDROFURANOS EN PRODUCTOS NATURALES
Muchos productos naturales tienen un anillo de tetrahidrofurano (THF) en su estructura, incluidos macrólidos, poliéteres ionóforos, acetogeninas, etc.38 Este tipo de compuestos exhiben una amplia gama de actividades biológicas, incluyendo agentes antitumorales, antimicrobianos o antipalúdicos, entre otros. Figura 8).39. Debido a la importancia biológica de estos compuestos, la construcción de moléculas que contengan THF en su estructura ha despertado un gran interés a lo largo de los años. Estos incluyen el uso de ciclaciones de alcoholes β,γ y γ,δ-insaturados, ciclación de γ-hidroxicetonas, ciclación de 1,4-dioles, reacciones radicalarias o reducción de hemiacetales (sustitución en ion oxocarbenio).40.
MÉTODOS SINTÉTICOS DE TETRAHIDROFURANOS
- Síntesis de THFs mediante la formación de enlaces C–O
- Adición intramolecular de alcoholes sobre epóxidos
- Haloeterificación y sustitución de halógeno
- Ciclación de alcoholes insaturados mediada por metales
- Reacción intramolecular oxa-Michael
- Síntesis de THFs mediante la formación de enlaces C–C
- Formación radicalaria de enlaces C-C
- Reacción de cicloisomerización
- Reacción de metátesis de olefinas
- Síntesis de THFs mediante la formación simultánea de enlaces C–C y C-O
- Reacciones de cicloadición [3+2]
- Adición nucleofílica sobre un intermediario oxonio
La reacción se agitó a -60 °C hasta el consumo completo de la materia prima (control por CCF). Se dejó que la mezcla de reacción alcanzara temperatura ambiente y se agitó hasta que se consumió el material de partida (control por CCF). La mezcla de reacción se agitó hasta que se observó el consumo completo de la materia prima (control por CCF).
Se dejó que la mezcla de reacción alcanzara temperatura ambiente y se agitó hasta que se consumió el material bruto (control por CCF). La mezcla de reacción se dejó calentar hasta temperatura ambiente y se agitó hasta el consumo total de materia prima (control por CCF).
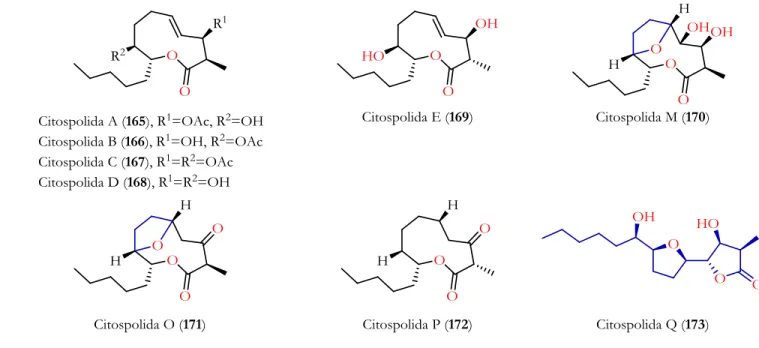
SÍNTESIS PREVIAS DEL CITOSPOLIDE Q
- Síntesis total del citospolide Q por el grupo de Stark
- Síntesis total del citospolido Q por el grupo de Goswami
PROSPECTIVAS
PARTE EXPERIMENTAL
Se dejó que la reacción alcanzara temperatura ambiente, se añadió agua y se extrajo con diclorometano (2 x 50 ml). Una vez completada la adición, la reacción se enfrió a temperatura ambiente y se diluyó con acetato de etilo. La reacción se agitó a -78 °C hasta el consumo total del material de partida (verificación por CCF).
Stark y colaboradores se basaron en la interconversión de citospolida M (170) para obtener citospolida Q (173) mediante transesterificación en un medio básico, siendo esta la primera síntesis de 173 (Esquema 56). Con las condiciones estandarizadas para la síntesis de la lactona 138, se sintetizó el alqueno 243. Una vez completada la reacción, se añadió agua, el disolvente se eliminó a presión reducida y se extrajo con acetato de etilo (3 x 5 ml).
El aceite bruto de reacción se extrajo con acetato de etilo (3 x 5 ml), la fase orgánica se secó con Na2SO4 y se concentró a presión reducida.
NAFTOPIRANONAS EN PRODUCTOS NATURALES
Las naftopiranonas son un pequeño grupo de metabolitos secundarios, principalmente extraídos de hongos, con importante actividad biológica como antibacteriana57 y antialérgica.60 Algunos de los compuestos más representativos son la viriditoxina (257), su monómero semiviriditoxina (258), su análogo estructural semivioxatina (259 ). ) y su dímero, vioxatina (260) (Figura 10). 104 Por otro lado, debido al amplio espectro biológico que presentan esta clase de compuestos y los pocos métodos para acceder a dicho núcleo, se ha puesto gran atención en la síntesis de compuestos que contienen naftopiranonas en su estructura.
MÉTODOS SINTÉTICOS DE NAFTOPIRANONAS
SÍNTESIS PREVIAS DE LA VIRIDITOXINA
La posterior desprotección selectiva del grupo EOM, seguida del acoplamiento mediado por vanadio, conduce al dímero 271, que contiene en su estructura la mayoría de los átomos presentes en el producto natural. Finalmente se realiza la protección de los fenoles libres, desprotección del grupo TIPS, oxidación al aldehído correspondiente seguida de una segunda oxidación al ácido en cuestión y su esterificación, y finalmente desprotección de los grupos isopropilo y metilo a viriditoxina (257) (Esquema 69 ).58.
TRABAJO PREVIO REAIZADO EN EL GRUPO DE INVESTIGACIÓN
Además, se han reportado reacciones intramoleculares, como el caso del halobenceno 280, que en presencia de un catalizador de paladio o n-BuLi (dependiendo del sustituyente carbonilo) y la posterior reducción conduce al benzociclobutenol 279.67 Asimismo, la apertura intramolecular del estireno óxidos mediados por n-BuLi y MgBr2 como es el caso del epóxido 281 (Esquema 70a).68. 108 El benzociclobutenol 290 se utilizó como intermediario clave en la síntesis de (-)-lasionectrina (295), para lo cual se logró la construcción del pentaciclo 294 mediante una reacción de Diels-Alder entre el dieno 291, derivado del benzociclobutenol 290, y la lactona 293. un enfoque para la síntesis total de viriditoxina (257) utilizando un benzociclobutenol como intermediario clave en una reacción de Diels-Alder.
Teniendo en cuenta los resultados previos de nuestro grupo de trabajo y debido a la importancia sintética y biológica que presentan las bisnaftopiranonas, en este proyecto se propuso la síntesis total de viriditoxina (257) mediante cicloadición de benzociclobutenoles y acoplamiento oxidativo de naftopiranonas. Por otro lado, el benzociclobutenol 296 se prepararía a partir del bromobenceno 298 y la lactona 297 surgiría de la yodolactonización y posterior olefinación entre el alcohol alílico 300 y el ácido yodoacético 299 (Esquema 72).
Síntesis de la lactona α,β-insaturada 305
Síntesis del acetato de benzociclobutilo 319
La posterior reducción y protección del alcohol formado condujo al acetato de benzociclobutilo 317 con excelentes rendimientos. Finalmente, la desprotección del grupo TBS y la oxidación del alcohol bencílico condujeron al aldehído 319, lo que permitió realizar una reacción tipo Baeyer-Villiger para acceder al ciclobuteno deseado. Desafortunadamente, cuando la reacción se llevó a cabo en condiciones típicas (m-CPBA en diclorometano), no se observó formación del producto deseado tras la recuperación del material de partida.
Además, se probaron algunas variantes de dicha reacción, como el uso de peróxido de hidrógeno en metanol con ácido sulfúrico catalítico, y peróxido de hidrógeno en THF con ácido bórico y ácido sulfúrico catalítico. Sin embargo, en ninguno de los casos anteriores fue posible acceder al producto deseado observando la materia prima o su descomposición (Esquema 75).
Síntesis del benzociclobutenol 327
Reacción de cicloadición 4+2
A la luz de los resultados obtenidos, se consideró realizar la protección del benzociclobutenol 327 para evitar la formación de los correspondientes aldehídos. Con la hipótesis de que el calentamiento en estas reacciones era la causa de la formación preferencial de los aldehídos antes mencionados, se decidió realizar algunos experimentos a baja temperatura.71 Para ello, primero se trató el benzociclobutenol 327 con n-BuLi a -35 °C para generar la especie reactiva 327a, que posteriormente se hizo reaccionar con la lactona 305, previamente activada con un Por otro lado, se hizo reaccionar benzociclobutenol 327 con LDA para formar de manera similar la especie reactiva 327a y se hizo reaccionar con la lactona 305 previamente activada con un ácido de Lewis (Et2AICI, Esquema 80).
En ambos casos, sin embargo, la mayor parte del producto resultó ser el aldehído 328, resultado que llevó a la conclusión de que el benzociclobutenol 327 efectivamente realizó la apertura electrocíclica antes mencionada, pero la lactona 305 no mostró reactividad hacia la reacción de cicloadición. Afortunadamente para nosotros, en las dos últimas pruebas se observó la formación de los productos de cicloadición 335 y 337, lo que confirma que el benzociclobutenol efectivamente es reactivo en la reacción de Diels-Alder, pero el problema en las pruebas anteriores fue la baja reactividad de la lactona 305.
Modificación de la ruta sintética
Con el alquino 344 en la mano, se realizó una reacción de cicloadición con el correspondiente benzociclobutenol 327.120 Para verificar si la geometría del alquino era responsable de la formación de las cetonas 347 y 349, decidieron probar una reacción de cicloadición intramolecular. formación de alcohol alílico. Con base en los resultados obtenidos en la reacción de cicloadición intramolecular, se decidió realizar la síntesis del alcohol alílico 358, el cual contendría los sustituyentes necesarios para continuar con la ruta sintética.
Para ello se realizaron algunas pruebas iniciales con n-BuLi y se encontró que la reacción no procedía, por lo que se decidió utilizar t-BuLi, donde se observó una mezcla compleja de productos. Se descubrió que la reacción de cicloadición intramolecular de benzociclobutenoles con alquenos era la ruta más viable para proceder con la síntesis de viriditoxina (257).
PATE EXPERIMENTAL
El disolvente se eliminó a presión reducida y el crudo de reacción se usó en la siguiente etapa. La fase orgánica se secó con Na2SO4, el disolvente se eliminó a presión reducida y la materia prima de reacción se usó en la siguiente reacción. La fase orgánica se secó con Na 2 SO 4 , el disolvente se eliminó a presión reducida y el crudo de reacción se usó en la siguiente etapa sin purificación adicional.
La fase orgánica se secó con Na2SO4, el disolvente se eliminó en el rotavapor y la mezcla de reacción cruda se usó en la siguiente reacción. La fase orgánica se secó con Na2SO4, el disolvente se eliminó a presión reducida y la mezcla de reacción cruda se usó en la siguiente reacción sin purificación adicional.
Stereoselective Total Synthesis of Aspergilide A: A Visible Light-Mediated Photoredox Access to the Trisubstituted Tetrahydropyran Core. In this work, we report the total synthesis of aspergillide A based on a photoredox free radical approach to construct the switch. The synthesis of the key alkene 19 was completed by a partial hydrogenation in near-quantitative yield using the Lindlar catalyst (Scheme 4).
In summary, we have completed a stereoselective total synthesis of aspergillide A in an overall yield of 0.8% by employing a visible light-mediated photoredox radical-ionic iodolactonization for the construction of the main tetrahydropyran core. After stirring for 3 hours at room temperature, the reaction was quenched with a saturated aqueous Na 2 CO 3 solution (15 ml) and extracted with dichloromethane (3 x 30 ml).







