SULEIMA CARPETA SANCHEZ
Trabajo de grado presentado como requisito para optar al título de MAESTRÍA EN CIENCIAS BIOLÓGICAS
CON ENFASIS EN GENÉTICA HUMANA
PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS
MAESTRÍA EN CIENCIAS BIOLÓGICAS BOGOTÁ. D.C
___________________________ DRA. INGRID SCHULLER
DECANA ACADEMICA FACULTAD DE CIENCIAS
_____________________________ DR. MANUEL FRANCO DIRECTOR DE POSGRADO
FACULTAD DE CIENCIAS
PONTIFICIA UNIVERSIDAD JAVERIANA FACULTAD DE CIENCIAS
MAESTRÍA EN CIENCIAS BIOLÓGICAS BOGOTÁ. D.C
NOTA DE ADVERTENCIA
Artículo 23 de la resolución No 13 de julio de 1946
“La universidad no se hace responsable por los conceptos emitidos por sus alumnos en sus trabajos de tesis. Solo velará porque no se publique nada contrario al dogma y la moral católica y porque las tesis no contengan ataques
A mi familia y en especial a mi hermana quien ha estado a mi lado brindándome su gran apoyo incondicional para poder alcanzar esta meta académica
AGRADECIMIENTOS
A Dios por darme fortaleza espiritual para enfrentar y superar todas las dificultades encontradas durante el camino.
A la Vicerrectora Académica de la Pontificia Universidad Javeriana quien aportó los recursos para desarrollar este estudio
A la Dra. Olga Moreno por compartir su conocimiento y coordinar el desarrollo de este trabajo.
Al Dr. Zarante por toda la orientación académica brindada.
A los pacientes por que fueron ellos quienes permitieron la realización del trabajo.
A todas las Instituciones y médicos Genetistas que apoyaron esta investigación.
A los residentes de Genética de la Pontificia Universidad Javeriana y en especial a Tatiana Pineda por su ayuda profesional en el seguimiento de los pacientes.
Al grupo de trabajo del Laboratorio Genetix por su apoyo profesional y personal para la realización de esta investigación.
RESUMEN ...16
SUMMARY ...17
1. FORMULACIÓN DEL PROBLEMA DE INVESTIGACIÓN ...18
1.1. Justificación ...19
2.1. Fisura labio y/o palatina ...21
2.2. Antecedentes históricos ...22
2.3. Epidemiologia. ...23
2.4. Embriología del labio y del paladar ...25
2.4.1. Patogénesis del Desarrollo de las fisuras de labio y paladar ...26
2.5. Clasificación de las Fisuras Labio y/o palatinas ...27
2.6. Etiología ...29
2.6.1. Genética de las fisuras orofaciales. ...30
2.6.1.1. Genética de las FL/P no sindrómicas ...30
2.6.1.2. Genética de las FL/P Sindrómicas. ...31
2.6.1.2.1. Síndrome de Van der Woude. ...32
2.6.1.2.2. Fisura Palatina ligada a X y anquiloglosia ...32
2.6.1.2.3. Síndrome de displasia ectodérmica con FL/P. ...32
2.7. Síndrome de deleción 22q11.2 ...33
2.7.1. Antecedentes históricos ...34
2.7.2. Epidemiología...35
2.7.3. Características Clínicas ...36
2.7.4. Descripción Clínica del síndrome de 22q11.2 ...37
2.7.4.1. Malformaciones cardíacas ...37
2.7.4.2. Alteraciones en el paladar. ...37
2.7.4.3. Alteraciones inmunes ...39
2.7.4.4. Función paratiroidea ...39
2.7.4.5. Alteraciones en la alimentación...39
2.7.4.6. Alteraciones craneofaciales ...39
2.7.4.7. Alteraciones en el crecimiento. ...40
2.7.4.8. Desarrollo sicosocial y función cognitiva ...40
2.7.4.9. Alteraciones psiquiátricas ...40
2.7.5 Abordaje diagnóstico. ...41
2.7.5.1 Pruebas diagnósticas ...42
2.7.6 Etiología Genética del Síndrome de deleción 22q11.2 ...43
2.7.7 Genes afectados por la deleción ...46
2.7.7.1 Gen TBX1 ...47
2.7.7.2 Gen HIRA ...48
2.7.7.3 Gen UFD1L...48
2.7.7.4 Gen CRKL...48
2.7.7.5 Genes COMT y PRODH ...48
2.7.8 Modificadores Genéticos del síndrome 22q11.2 ...49
2.7.8.1 Factor de crecimiento de fibroblastos 8 (Fgf8) ...49
2.7.8.2 Factor de crecimiento de fibroblastos 10 (Fgf10) ...49
2.7.8.3 Gen GBX2...50
2.7.8.4 Gen Pitx2 ...50
3 OBJETIVOS ...52
3.1 Objetivo General. ...52
3.2 Objetivos específicos ...52
4. METODOLOGÍA ...53
4.1 Población de estudio. ...53
4.1.1 Criterios de Selección de las muestras ...53
4.1.1.1 Criterios de inclusión ...53
4.1.1.2 Criterios de exclusión. ...53
4.2 Selección y recolección de Muestras ...54
4.3 Análisis citogenético...55
4.3.1 Cultivo de linfocitos y obtención de cromosomas de alta resolución. ...55
4.3.1.1 Siembra y sincronización de cultivos ...55
4.3.1.2. Cosecha ...55
4.3.1.3. Extendido en placa y bandeo G ...56
4.3.1.4. Análisis de cariotipo. ...56
4.4.1. Descripción de las regiones analizadas por MLPA ...58
4.4.2 Aislamiento de ADN ...58
5.4.2.1 Cuantificación de ADN ...58
4.4.3. Reacción de MLPA ...58
4.4.3.1. Electroforesis capilar de MLPA ...59
4.4.3.2. Análisis de de los resultados de MLPA ...61
4.5 Correlación del Fenotipo con los resultados citogenéticos y moleculares. ...63
5 RESULTADOS ...64
5.1. Selección de la muestra ...64
5.2. Descripción de la población. ...65
5.5 Resultados del análisis citogenético ...65
5.2.1. Resultado del caso con alteraciones en 21q ...66
5.2.2. Resultado del caso con cromosoma 4 derivado ...69
5.3. Análisis por MLPA de la región 22q11.2 ...74
5.4. Correlación con la clínica de los pacientes ...78
5.4.1. Tipo de Fisuras ...78
5.4.2. Características clínicas en los pacientes con Fisuras orales. ...80
5.5. Análisis citogenético y molecular en los padres de pacientes que presentaron alteraciones cromosómicas y deleción 22q11.2. ...81
6. DISCUSIÓN ...83
6.1 Caso con alteración en 21q ...85
6.2 Caso con cromosoma 4 derivado...87
6.3 Deleción 22q11.2 ...90
7. CONCLUSIONES ...99
8. PERSPECTIVAS Y APLICACIONES ...101
9. BIBLIOGRAFÍA ...102
INDICE DE TABLAS
Tabla 1. Prevalencias estimada de hendiduras orofaciales en diferentes regiones del mundo ..25
Tabla 2. Genes candidatos y loci implicados en la etiología de las FL/P no sindrómicas . ...31
Tabla 3. Frecuencia de la deleción 22q11.2en una población de Atlanta entre 1994-1999 . ...36
Tabla 4. Hallazgos cardiacos en pacientes con Síndromedeleción 22q11.2. ...38
Tabla 5. Hallazgos palatinos en pacientes con Síndrome deleción 22q11.2 ...38
Tabla 6. Genes y función conocida presentes dentro de la región cromosómica 22q11.2 ...51
Tabla 7. Programa del termociclador para la reacción y amplificación por MLPA. ...59
Tabla 8.Valores de corte de DQs para análisis de resultados de MLPA ...62
Tabla 9. Bases de datos e Instituciones participantes para la selección de pacientes. ...64
Tabla 10. Descripción de la población por edad y sexo. ...65
Tabla 11.Descripción de resultados del análisis cromosómico por micrroarray para el cromosoma 21 derivado. ...68
Tabla 12.Datos del resultado de análisis cromosómico por micrroarray para la región 4p16.3 y 8p23. ...71
Tabla 13.Tipo de delecion presente en los pacientes y total de pacientes sin delecion para la region 22q11.2 . ...75
Tabla 14. Resultados de MLPA de los siete pacientes con la deleción 22q11.2. Lo marcado en rojo señala las sondas comprometidas en la deleción. ...76
Tabla 15. Tipos de fisuras orales en los pacientes analizados ...79
Tabla 16.Características Fenotípicas Mayores de los pacientes con la delecion 22q11.2 ...82
Tabla 17.Descripción de otras anormalidades congénitas en los pacientes con presencia la delecion 22q11.2 ...82
INDICE DE FIGURAS
Figura 1. Recipiente de arcilla que representa un individuo afectado por labio leporino en la
cultura de los mochica. ...22
Figura 2.Origen embrionario de las estructuras faciales de línea media. ...26
Figura 3. Representacion en Y de los diferentes tipos de fisuras . ...27
Figura 4. Clasificación de las fisuras a nivel embriológico. ...28
Figura 5. Representación gráfica de recombinación homóloga no alelica (NAHR) ...45
Figura 6 Organización de los LCR. ...45
Figura 7. Representación esquemática de la región 22q11.2. ...46
Figura 8. Organización de Genes en la región 22q11.2.. ...46
Figura 9. Electroferograma de un individuo normal (control) analizado con la SALSA MLPA probemix P250-B1 DiGeorge(MRC-Holland)). ...60
Figura 10. Electroferograma de una muestra control positivo analizado con la SALSA MLPA probemix P250-B1 DiGeorge(MRC-Holland). ...61
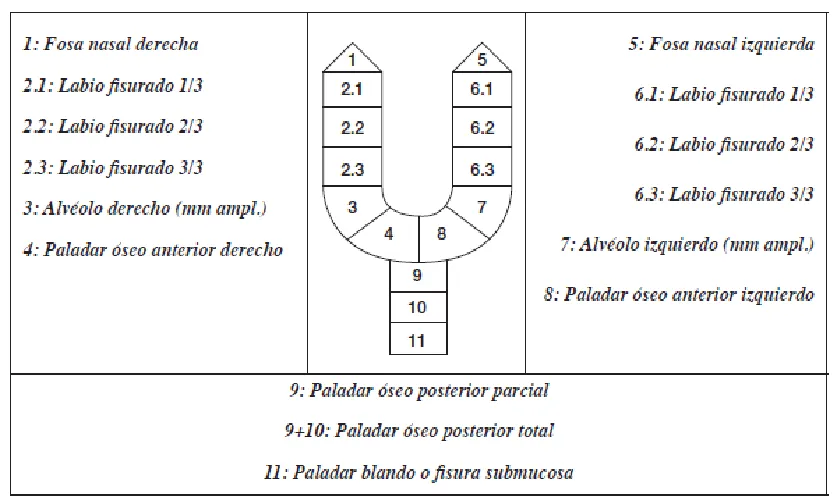
Figura 11. Clasificación de las Fisuras del labio y/o paladar ...63
Figura 12. Porcentaje de pacientes con cariotipo normal y anormal en el grupo de Fisuras labio y/o palatinas sindrómicas. ...65
Figura 13 Cariotipo femenino con bandeo G en donde se visualiza el cromosoma 21 derivado ...66
Figura 14. Ampliación del cromosoma 21 e ideograma del cromosoma normal y derivado en donde se señalan los puntos de rotura, inversión y perdida. ...67
Figura 15. Metafase con bandeo C en donde se muestra el cromosoma derivado 21 con la presencia de un solo centrómero ...67
Figura 16. Resultado de CGHa en donde se identifican los segmentos deletados (en rojo) y los segmentos duplicados (en verde) en 21q. ...68
Figura 18. Representación gráfica del análisis de MLPA en dónde se muestra la duplicación en
la región 8p23.1.. ...70
Figura 19. Ampliación e ideograma de los cromosomas normales 4 y 8 y del derivado 4. . ...71
Figura 20. Resultado de CGH array en donde se identifica la deleción en 4p16 ...72
Figura 21. Resultado de CGH array en donde se identifica la duplicación en 8p23 . ...72
Figura 22. Resultado final del CGH array en donde se identifican conjuntamente las dos alteraciones en las regiones 4p16 y 8p23. ...73
Figura 23. Características dismórficas de los pacientes con alteraciones cromosómicas. ...74
Figura 24. Porcentaje de pacientes con y sin presencia de la delecion 22q11.2 en el grupo de Fisuras labio y/o palatinas sindrómicas ...75
Figura 25. Representación gráfica del análisis de MLPA de un paciente (caso 2316) con deleción 22q11.2 en TDR (LCRA a D). . ...77
Figura 26. Representación grafica del análisis de MLPA de un paciente (caso 2341) con deleción distal en 22q11.2 (LCR D a F). Figura 27. Representación gráfica del análisis de MLPA de un paciente normal (caso 2365) de la región 22q11.2. ...78
Figura 28. Distribución por sexo de los diferentes tipos de FL/P ...79
Figura 29. Tipo de fisura y anormalidad palatina presente en los pacientes con delecion 22q11.2 ...80
TABLA DE ANEXOS
Anexo 1. Características clínicas asociadas con el síndrome de delecion 22q11.2. .. 120
Anexo 2. Encuesta de seguimiento telefónico ... 121
Anexo 3.Consentimiento informado para participar en proyecto de investigación ... 122
Anexo 4. Gráfica de reacción de MLPA y diseño de las sondas empleadas. ... 126
Anexo 5. Descripción del kit SALSA MLPA probemix P250-B1 DiGeorge ... 127
Anexo 6. Protocolo de aislamiento de ADN ... 128
Anexo 7. Protocolo de la reacción de MLPA ... 130
Anexo 8. Radios de muestras positivas y negativas para el análisis de la región 22q11.2 ... 131
Anexo 9. Datos del análisis de MLPA ... 132
Anexo 10. Cariotipo femenino de un caso sin alteraciones cromosómicas ... 133
Anexo 11. Cariotipo de un caso con deleción 22q11.2 (caso 2411) identificado por MLPA ... 134
Anexo 12. Descripción general y resultados citogenéticos y moleculares de los pacientes con fisura de labio y o de paladar enalizados en este estudio. ... 135
ABREVIATURAS
FL/P: Fisura del labio y/o del paladar FLP: Fisura labio palatina
FL: Fisura labial.
FP: Fisura palatina
IVF: Insuficiencia Velofaríngea
FLPS: Fisura del labio y paladar sindrómica SMVC: Síndrome Velocardiofacial.
SDG: Síndrome de DiGeorge. WHS: Síndrome Wolf–Hirschhorn. CIV: Comunicación interventricular CIA: Comunicación interauricular
LCR: Regiones de bajo número de copias (low copy repeats) SDS: Duplicaciones segmentarias
MLPA: Amplificación de Sondas Múltiples dependientes de Ligación. (Multiplex dependent Probe Amplification).
PCR: Reacción en Cadena de la Polimerasa (Polimerase Chain Reaction)
TBX1: Gen de la Proteína 1 T-box
NAHR: Recombinación homologa no alélica.
CNV: Variación de número de copias (Copy number variant ) TDR: Región típicamente deletada.
16 RESUMEN
Introducción: Las fisuras del labio y/o del paladar son defectos frecuentes del
nacimiento. Las formas sindrómicas de las fisuras se asocian a mutaciones en un solo gen y a alteraciones cromosómicas como la microdeleción 22q11.2, que incluye las fisuras dentro de su espectro fenotípico. En Colombia se desconoce la frecuencia con que la microdeleción 22q11.2 es causante del fenotipo sindrómico como parte de su etiología genética. EI objetivo de este estudio fue identificar anomalías cromosómicas, así como la deleción de la región 22q11.2 en pacientes con fisuras de labio y/o palatinas con sospecha de síndrome de deleción 22q11.2.
Metodología: Los pacientes analizados presentaron fisura de labio y/o de paladar
acompañadas de características asociadas al síndrome de deleción 22q11.2. Se realizó cariotipo BG de alta resolución, análisis por MLPA de la región 22q11.2 (SALSA MLPA KIT P250-B1 DiGeorge) y correlación con la clínica. Resultados: Se analizaron 49 pacientes, 7(14.3%) mostraron la deleción 22q11.2 y 2(4.1%) alteraciones en otros cromosomas: un cromosoma 21 derivado por inversión y un cromosoma derivado 4 por translocación. La anomalía orofacial con mayor representación en los pacientes con deleción 22q11.2 fue la fisura palatina 4(57%), acompañada de otras características representativas del síndrome. Los resultados sugieren un papel importante de la deleción 22q11.2 y otras anomalías cromosómicas en la etiología de la fisura labio palatina sindrómica en la población estudiada. Conclusión: Los estudios citogenéticos y moleculares realizados identificaron la deleción 22q11.2 en un alto porcentaje de casos con fisura palatina y alteraciones en regiones distintas a 22q11.2, contribuyendo así al conocimiento de la etiología genética de esta patología.
Palabras claves: Fisura labial, fisura palatina, fisura labio-palatina sindrómica,
17 SUMMARY
Introduction: The cleft lip and/or palate are common birth defects. The syndromic
forms are associated with mutations in single genes and chromosomal disorders such as 22q11.2 microdeletion syndrome. In Colombia, the frequency in which the 22q11 microdeletion is responsible for the phenotype is unknown. EI objective of this study was to identify chromosomal abnormalities, as well as the 22q11 microdeletion in patients with cleft lip and / or palate with suspected 22q11.2 deletion syndrome.
Methodology: The patients analyzed had cleft lip and / or palate accompanied by
characteristics associated with 22q11.2 deletion syndrome. High-resolution G-Banding karyotype was performed and 22q11.2 region analysis by MLPA (SALSA MLPA KIT P250 -B1 DiGeorge ) and correlated with the phenotype. Results: 49 patients were analyzed, 7 (14.3%) showed the 22q11 microdeletion and 2 (4.1%) abnormalities on other chromosomes: one derivative chromosome 21 from an inversion and one derivative chromosome 4 from a translocation. The most common orofacial abnormality in patients with 22q11.2 microdeletion syndrome was cleft palate (57%), accompanied by other representative characteristics of the syndrome. The results suggest an important role of the 22q11.2 deletion and other chromosomal abnormalities in the etiology of syndromic cleft lip cleft in studied population. Conclusion: Cytogenetic and molecular studies identified the 22q11.2 microdeletion in a high percentage of cases with cleft palate and in different chromosomal regions, contributing to the knowledge of the genetic etiology of this condition.
Key words: cleft lip, cleft palate, cleft lip and –palate syndromic, 22q11.2 deletion
18 1.
Las fisuras labiales y palatinas se producen durante el desarrollo embrionario por alteración en la fusión de los tejidos que darán origen al labio superior y al paladar, afectan a 1 de cada 700 nacidos vivos a nivel mundial. (1, 2)
Existen diferentes grados de severidad que comprenden fisuras del labio, fisuras de labio y paladar y fisuras del paladar. Estas anormalidades comúnmente están acompañadas de alteraciones en el desarrollo facial y dental, del habla y de la audición, así como de desajustes en el comportamiento psicosocial (3). Clínicamente las fisuras del labio y/o paladar pueden presentarse con otras malformaciones congénitas definiéndose como sindrómicas; si aparecen como un defecto aislado se clasifican como no sindrómicas. La etiología de las fisuras es causada por una interacción compleja entre factores ambientales y genéticos principalmente asociados a mutaciones de un solo gen y anormalidades cromosómicas como las duplicaciones y las deleciones. (1, 3,4). Esta anomalía está descrita en el síndrome de microdeleción 22q11.2 como parte de los 180 signos clínicos conocidos que contribuyen al amplio espectro fenotípico del Síndrome. Su origen genético es una deleción hemicigota en el brazo largo del cromosoma 22 que incluye los síndromes de DiGeorge, velocardiofacial y de anomalía facial conotruncal. Se ha estimado una prevalencia en la población de 1 en 4000 a 1 en 6000 nacimientos considerándose como la deleción intersticial más frecuente en humanos (5, 6,7). La deleción frecuentemente se presenta dentro de la región típicamente deletada con algunos casos de deleción adyacentes o solapadas a esta región (8,9). Se ha informado que la delecion puede ser heredada en 7% de los casos, pero la mayoría de los individuos tienen una deleción de novo, (6,7,9,10)
FORMULACIÓN DEL PROBLEMA DE INVESTIGACIÓN
19 embargo en algunos casos no hay una clara expresión fenotípica, por lo cual es necesario investigar la deleción como parte de la patología que puede presentarse con fisuras orales.
Investigaciones previas de la correlación genotipo fenotipo en pacientes con la deleción 22q11.2, reportan que hasta un 69% de estos pacientes tienen anomalías palatinas (10). En la población Chilena se ha reportado un 76.6% de anormalidades palatinas en pacientes con la deleción 22q11.2 (11). Dada la alta frecuencia reportada de esta anormalidad en el síndrome de deleción 22q11.2 resulta importante evaluar los pacientes con fisuras asociadas a otras anormalidades congénitas.
Varios genes han sido identificados en el origen de las fisuras. Los análisis sobre el contenido génico de la región 22q11.2 en ratones han identificado al gen TBX1 como posible causante de las principales alteraciones de las estructuras embrionarias que forman el arco faríngeo y que se relacionan con las características fenotípicas del síndrome de deleción 22q11.2, incluidas las anomalías palatinas (12). La identificación de la etiológica genética de las fisuras sindrómicas permiten una precisión diagnostica para el adecuado seguimiento y asesoramiento genético por la implicaciones clínicas, genéticas y familiares que estos pacientes pueden tener.
1.1. Justificación
20 de los síndromes en el que se incluyen las anormalidades palatinas como una de sus principales características es el síndrome de deleción 22q11.2; los individuos con este síndrome pueden manifestar un amplio rango de malformaciones estructurales del paladar como las fisuras palatinas y la insuficiencia velopalatina con repercusiones estéticas y funcionales como las dificultades de alimentación, respiración, audición y comunicación asociadas también con infecciones a repetición y problemas sicosociales (3,10).
Los estudios de fisuras palatinas y deleción 22q11.2 se han realizado principalmente en fisuras aisladas; los resultados han sugerido la posibilidad de evaluación de la deleción 22q11.2 en fisuras labio/palatinas asociadas a otras características clínicas.(7.6) A nivel general los estudios genéticos de las fisuras sindrómicas en nuestra población no han sido reportados; en población colombiana se desconoce la frecuencia con que se presenta el síndrome de deleción 22q11.2 y de otras anormalidades cromosómicas como causa genética del fenotipo sindrómico de las hendiduras orofaciales. La identificación del origen genético de esta condición permite realizar un adecuado asesoramiento a los padres y/o a los individuos afectados.
El fenotipo del Síndrome de delecion 22q11.2 es clínicamente variable puede oscilar de leve a severo, donde pueden presentarse casos con rasgos típicos como casos sin un fenotipo característico; este comportamiento plantea la importancia de evaluar las diferencias en la presentación clínica para orientar hacia un posible diagnóstico de deleción 22q11.2.
21 2. MARCO TEORICO
2.1. Fisura labio y/o palatina
Las fisuras labiales y palatinas representan un grupo heterogéneo de desordenes que afectan el labio y la cavidad oral. La formación de estas estructuras se originan por una serie compleja de eventos que se producen a nivel embrionario y que requieren de una estrecha coordinación de los programas para la migración celular, el crecimiento, la diferenciación y la apoptosis que se dan a nivel embrionario (3). Las alteraciones en estos procesos de crecimiento y fusión de tejidos, originan las hendiduras labiales y/o palatinas (FL/P). (15). El labio fisurado (FL) y el paladar hendido (FP) son las deformidades congénitas más comunes que afectan las estructuras orofaciales en humanos y constituyen cerca del 13% de todas las anomalías reportadas. (14). Estos defectos se presentan a nivel general en 1.7 por 1000 nacidos vivos con algunas variaciones étnicas y geográficas. (2,3)
Las estructuras afectadas comúnmente son el labio superior, el paladar duro, el paladar blando y la cresta alveolar. Como consecuencia de estas deformidades los individuos afectados presentan alteraciones físicas en el desarrollo de la cara, la oclusión maxilodental, la audición, la calidad del habla, y pueden ser la causa de trastornos psicológicos o del comportamiento. Estos pacientes requieren atención multidisciplinaria del nacimiento hasta la edad adulta y tienen una mayor morbilidad y mortalidad a lo largo de la vida que las personas no afectadas. (3,16)
Las FP/L son desordenes complejos multifactoriales en los que los factores genéticos y/o ambientales son responsables de su etiología. Estos factores inhiben el flujo de células de la cresta neural o disminuyen su número, imposibilitando el adecuado
contacto de las prominencias faciales y formando así las hendiduras. (14).
22 están asociados con alguna anomalía reconocible, el 70% son no sindrómicos y ocurren como una condición aislada. (18,19)
2.2. Antecedentes históricos
[image:22.612.241.410.380.569.2]Desde la antigüedad se conocen las malformaciones congénitas en grabados y figurillas, testigos de la malformación en civilizaciones antiguas de diversas partes del mundo. Los primeros datos reportados sobre fisuras labio palatinas aparecen en el año 200 a. C. (20). Su primera documentación está representada en una estatuilla griega del siglo IV a. C., que muestra con gran detalle los caracteres de la fisura labial (20,21). Evidencia de esta alteración también ha sido encontrada en un cráneo del siglo I a.C presente en una cerámica de la cultura mochica del siglo IV al VI a.C (Figura 1) y en momias egipcias en las que se identificó un caso de FLP que correspondió al año 2500 a.C. (21)
23 Las FLP han recibido diferentes denominaciones en cada civilización de acuerdo a su cultura y tradición. En las sociedades primitivas, se definieron estas condiciones humanas mediante comparaciones con características encontradas en animales. Es así como el término “labio leporino” viene del latín“Lepore”: (liebre en latín) y constituye la denominación más frecuente de las fisuras primarias del labio y paladar, debido a la similitud anatómica del labio superior de estos animales con una ”V” invertida, y por lo tanto, con una fisura. En la literatura médica francesa se ha utilizado el termino bec-de –Lievre usado por primera vez en 1970 por Pare (22).
En 1869 Dursy propone que la fisura se forma por la falla en la fusión de los elementos ectodérmicos y mesodérmicos en el área de la fisura. En la década de 1940, Poul Fogh-Andersen proporcionó la primera evidencia que el labio y/ o paladar hendido tenía un fuerte componente genético y 1943 Josef Warkan postuló que anomalías craneofaciales podrían también ser causadas por la exposición ambiental. (21)
2.3. Epidemiologia.
La prevalencia reportada de todas las hendiduras orofaciales en el mundo es de aproximadamente 1:700 nacidos vivos con variación étnica y geográfica; la variación reportada para las fisuras labiales con o sin paladar es de 0.34 a 2.2 por 1000 nacidos vivos y para las fisuras palatinas aisladas de 0.13 a 2.5 por 1000 nacidos vivos. (23,24)
Las tasas más altas de FL/P han sido reportadas en población Indígena Norte Americana y en población asiática con 1 afectado por cada 500, mientras que para la población caucásica se han reportado tasas intermedias de 1 afectado por cada 1000 nacidos vivos y la población africana tiene las más bajas tasas de prevalencia reportadas de 1 afectado por cada 2500 (25). Para la población de Latino América se ha reportado alta prevalencia de esta condición, 1.67 afectados por 1000 nacidos vivos (2,26).
24 1.4 por 1000 nacidos vivos, la más alta frecuencia calculada por cada mil habitantes en Bolivia (2.3), seguido por Ecuador (1.49) y Paraguay (1.33); las tasas más bajas fueron observadas en Venezuela (0.72), seguido por Perú (0.84), Uruguay (0.97) y Brasil (0.1) (20).
En cuanto a la fisura palatina se ha reportado alta prevalencia en Canadá y Norte de Europa y bajas prevalencias en Latino América y Norte de Europa (Tabla 1)(2). La fisura palatina aislada (FP), es menos común, con una prevalencia de 1/1500-2000 en población caucásica (24). También se han informado diferencias en la presentación de FL/P en cuanto al género de los pacientes, con más varones afectados por FL/P, mientras que más mujeres son afectadas por FP (24,26)
En Colombia, los estudios del ECLAMC informan para el periodo de 2001 y el 2008, una frecuencia de fisura labio palatina de 1.5 afectados por 1000 nacidos vivos y en 2012 0.6 afectados por cada 1000 nacidos vivos (13,27)
GBD Región Total HOFs*/1,000 (no-chr)
FP**/1,000 (non-chr)
FL(P) ***1,000
(no-chr) FP, % Total
Latinoamérica, meridional 2.39 0.72 1.67 30
Latinoamérica, tropical 2.39 0.72 1.67 30
Australasia 2.01 1.02 0.98 51
Norteamérica, 2.00 0.83 1.17 41
Oceanía 1.85 1.13 0.72 61
Europa occidental 1.66 0.59 1.07 35
Asia Pacifico 1.65 0.64 1.00 39
Sur de Asia 1.60 0.30 1.30 19
Centroamérica 1.54 0.39 1.15 25
Europa central 1.45 0.67 0.77 47
Sudeste Asiático 1.36 0.28 1.08 20
Región Andina 1.29 0.17 1.12 13
Este Asiático 1.28 0.27 1.01 21
25 *HOFs Hendiduras orofaciales ** FP Fisura Palatina *** FL(P) Fisura labial con o sin paladar
[image:25.612.113.563.84.276.2]Se excluyen anomalías cromosómicas
Tabla 1. Prevalencias estimada de hendiduras orofaciales en diferentes regiones del mundo: (2)
2.4. Embriología del labio y del paladar
Durante el desarrollo embrionario los arcos branquiales primero, segundo y tercero desempeñan un papel crítico en el desarrollo de la cara, la boca y la lengua (28). La formación de la cara ocurre principalmente entre la cuarta y octava semanas del desarrollo embrionario: las estructuras que formarán la cara en el ser humano están compuestas por primordios que aparecen alrededor del estomodeo (boca) embrionario tempranamente en la cuarta semana de desarrollo (29).
La boca primitiva inicia su formación hacia la quinta semana de gestación con la migración de células desde la cresta neural hacia la región anterior de la cara. El labio se forma entre la quinta y sexta semana de gestación, cuando el proceso frontonasal se va fusionando con los procesos maxilares (figura 2a) (29,30). Posteriormente se produce la formación del paladar con la fusión de los procesos palatinos, entre la séptima y octava semana del desarrollo embrionario (figura 2b) (29,30). Los procesos palatinos derivados de los respectivos procesos maxilares inicialmente se encuentran orientados verticalmente hacia abajo a cada lado de la cavidad oral primitiva. Para la adecuada formación del paladar estos procesos palatinos deben migrar hacia una
Asia Central 1.19 0.62 0.57 52
Medio Oriente 10.2 0.30 0.72 29
Caribe 0.93 0.31 0.62 34
África Central 0.54 0.04 0.51 7
África Occidental 0.54 0.08 0.46 15
África Meridional 0.45 0.15 0.30 33
Norte de África 0.44 0.12 0.29 35
África Oriental subsahariana 0.38 0.12 0.27 31
26 posición horizontal para finalmente fusionarse en la línea media con el septum nasal, la cual se lleva a cabo desde la parte anterior (foramen incisivo) a la posterior (úvula). (31,32).
Figura 2.Origen embrionario de las estructuras faciales de línea media. (a y b) Procesos embrionarios que forman el paladar primario, secundario, nariz y labio superior (29)
2.4.1. Patogénesis del Desarrollo de las fisuras de labio y paladar
Las fisuras de labio y/o paladar embrionariamente se deben a un trastorno en la fusión de los procesos embrionarios que constituyen la región facial, por una falta de cierre del labio y del paladar entre la 4ª y 12ª semanas de vida intrauterina durante el desarrollo de los paladares primario y secundario del embrión (33).El mecanismo que se ha propuesto para la formación de la patología es una ausencia de la mesodermización e irrigación del tejido epitelial que se reabsorbe secundariamente, provocada por una disminución en la fuerza dinámica del desarrollo y crecimiento de las masas mesodérmicas separadas inicialmente por el muro epitelial. (3,33). La fusión de las
placas del paladar secundario forman el techo de la boca y el suelo nasal entre la semana séptima y duodécima gestacional, las fallas en este proceso pueden dar lugar a hendiduras en el paladar posterior al foramen incisivo (3)
27 2.5. Clasificación de las Fisuras Labio y/o palatinas
Existe variación en la morfología de las fisuras labio-alveolo-palatinas debido a que puede implicar cuatro estructuras diferentes: el labio, el proceso alveolar, el paladar duro y el paladar blando, además de la posibilidad de presentación unilateral o bilateral (20).
Varios investigadores a través del tiempo, han propuesto varios sistemas de clasificación basados en diferentes criterios : embriológico, anatómico, odontológico y quirúrgico. Una aplicada clínicamente es la Kernahan quien propone una clasificación sencilla que abarca todos los tipos de fisuras de paladar primario y secundario, representados en un figura en Y la cual ha sido modificada en los últimos años (30) (ver figura 3)
Figura 3. Representacion en Y de los diferentes tipos de fisuras (30).
La clasificación embriológica es una de la más utilizada, la cual contempla 4 clases de fisuras (ver fig 4):
Fisura labial uni o bilateral que afecta exclusivamente al labio; puede incluir el
[image:27.612.92.508.318.567.2]28
Fisura labio palatina unilateral completa: afecta al labio paladar primario y paladar
secundario.
Fisura labio palatina bilateral completa que afecta al labio paladar primario y
[image:28.612.177.452.203.541.2]paladar secundario en ambos lados. (30)
Figura 4. Clasificación de las fisuras a nivel embriológico. (30)
Aproximadamente el 80% de FLP son unilaterales y 20 % bilaterales, la fisura labio palatina bilateral es rara con el 10 % de los casos. (2).(30)
29 sindrómicas de causa no conocida (3); la base de datos del Online Mendelian
Inheritance In Man (OMIM) contiene más de 400 entidades que se asocian con hendiduras (34).
Una hendidura es no sindrómica, cuando se presenta esta malformación o múltiples anomalías originadas únicamente de solo un evento primario (1). Gran parte de las hendiduras se presentan como defectos aislados o no sindrómicos, cuyo origen es multifactorial en donde se combinan factores genéticos y factores ambientales.(35,36).
El 15 % de todas la hendiduras son sindrómicas (12 % para las FLP y 25 % para las FP) con más de 400 síndrome conocidos asociados, la fisura labial y la fisura palatina aisladas se presentan en 20 y 25 % de los casos respectivamente (3,4)
2.6. Etiología
Las FL, FP, FLP son de naturaleza multifactorial y son el resultado de diferentes procesos bioquímicos y de desarrollo (37). Las fisuras faciales se producen por múltiples causas, pueden ser genéticas, infecciosas, ambientales o de origen multifactorial que actúan sincrónicamente (16)
Durante el desarrollo embrionario de la región oral intervienen diferentes tipos celulares provenientes del ectodermo y endodermo y factores que actúan sobre la diferenciación celular, el crecimiento, la apoptosis, la adhesión celular y la señalización inter e intra celular; de esta manera, la etiología de las hendiduras puede deberse a alteración en genes que controlan estos factores, a la inhibición de la función celular por teratógenos ambientales o a la combinación de estos dos (1).
30 teratógenos en el lugar de trabajo y en el hogar al inicio del embarazo han sido investigados como factores de riesgo (38,39). Se ha demostrado que muchos factores ambientales que inhiben la cadena transportadora de electrones son inductores de FL/FP en mamíferos (40).Las deficiencias nutricionales en las madres gestantes han sido consideradas como un factor de riesgo muy importantes en la etiología de las FL/P; también la deficiencia de ácido fólico y zinc ha sido asociada con la presencia de fisuras de labio y/o de paladar tanto en humanos como en animales (1,3)
2.6.1. Genética de las fisuras orofaciales.
Dentro de las causas genéticas que pueden generar labio y paladar fisurado se han considerado tres categorías:
1. Herencia monogenética
2. Herencia poligénica o multifactorial 3. Aberraciones cromosómicas
Los análisis de la etiopatogenia de la fisura labio-palatina la asocian fundamentalmente a la herencia poligénica o multifactorial, debido a que su origen se relaciona al resultado de interacciones complejas entre un número variable de genes "menores" que actúan por acción aditiva (poligénica); a estos genes se les ha denominado factores genéticos predisponentes cuando actúan con factores ambientales usualmente desconocidos. Este modo particular de herencia se ha denominado multifactorial, y no sigue los patrones básicos de las leyes mendelianas (20)
2.6.1.1. Genética de las FL/P no sindrómicas
La FL/P no sindrómica se ha considerado como un rasgo genéticamente complejo debido a que la mayoría de los pacientes afectados no tienen antecedentes familiares y en los casos familiares no se observa un modelo simple de herencia Mendeliana (41).
31 Estudios de ligamiento en todo el genoma humano han identificado regiones del genoma que probablemente contienen genes que cuando presentan mutaciones causan hendidura orofaciales (17,43), los genes candidatos para FL/P se muestran en la tabla 2 y algunos de los cuales están relacionados con vías del desarrollo.
Tabla 2. Genes candidatos y loci implicados en la etiología de las FL/P no sindrómicas. No se reporta
valores de LOD y OR (17).
2.6.1.2. Genética de las FL/P Sindrómicas.
Se ha descrito que las mutaciones de un solo gen y las alteraciones cromosómicas son los principales mecanismos en la etiología de las FL/P sindrómicas. La base de datos de OMIN (Mendelian Inheritance in Man) describe más de 500 Síndromes con FL/P como parte del fenotipo. Las trisomías de los cromosomas 13,18 y 21, han sido
Cromosoma Gen* Evidencia
Mutación identificada
Desequilibrio de ligamiento
Asociación
1p36 MTHFR
1q32 IRF6
2p13 TGFA
2q32 SATB2
4p26 MSX1
4p21 ACOD4
6p23 ---
11q23 PVRL1
14q24 TGFB3
19q13 CLPTM1
Xq12 TBX22
*MTHRF-5.10-metilenotetrahidrofolato reducatasa, IRF6 interferón factor regulador -6., TGFA
32 asociadas con FL/P, así como deleciones y duplicaciones parciales de otros cromosomas(44). Estas observaciones han sugerido la existencia de loci en diferentes regiones genómicas, que en exceso o insuficiencia pueden conducir a FL/P. Se ha informado que aproximadamente el 5% de deleciones y duplicaciones autosómicas producen defectos congénitos con FL/P como parte de los hallazgos clínicos (37)
Dentro de los muchos síndromes genéticos en los que la fisura de labio y/o paladar es una característica común se describen algunos a continuación:
2.6.1.2.1. Síndrome de Van der Woude.
Es un desorden autosómico dominante con una prevalencia de 1 en 34000 nacidos vivos y representa la forma más frecuente de fisura labial y palatina sindrómica (43). En
los pacientes se han identificado mutaciones en el gen del factor 6 de regulación del interferon (IRF6), que generan una proteína no funcional por haploinsuficiencia de la misma.(45,46)
2.6.1.2.2. Fisura Palatina ligada a X y anquiloglosia
Este desorden se ha reportado como una condición recesiva ligada al cromosoma X. (47). El gen involucrado se ha localizado en el brazo largo del cromosoma X(Xq21)y fue
identificado como TBX22; éste gen codifica una proteína de la familia T-box que actúa como factor de transcripción durante el desarrollo temprano del embrión, y de manera particular se ha detectado su expresión en la lengua y el paladar en desarrollo. (48)
2.6.1.2.3. Síndrome de displasia ectodérmica con FL/P.
Este síndrome tiene un patrón de herencia autosómica recesiva y se caracteriza por presentar hendidura labial y/o palatina acompañada de otras anomalías físicas (49). El
33 PVRL1 fue encontrado expresado en el epitelio del ángulo medial de la superficie palatina y en el epitelio de la piel (50).
Dentro del Grupo de Fisuras labio Palatinas Sindrómicas se ha incluido también el Síndrome Velocardiofacial (SVCF), este síndrome es parte del espectro fenotípico del Síndrome de deleción 22q11.2 que a continuación se describe.
2.7. Síndrome de deleción 22q11.2
Este es un desorden autosómico dominante que se caracteriza por presentar un amplio espectro fenotípico en donde se han descrito más de 180 características (51). Este Síndrome incluye el síndrome de DiGeorge (SDG), el Síndrome Velocardiofacial, el síndrome de anomalía facial conotruncal y el síndrome Cayler, todos causados por la misma alteración, una microdeleción en hemicigosis del cromosoma 22 en la banda q11.2 (51).
Este síndrome de expresión altamente variable presenta dentro de sus principales características alteraciones congénitas del corazón, facies características, anomalías palatinas, deficiencia inmune y alteraciones cognitivas o de comportamiento, entre otras (52). El 93% de los pacientes tienen la microdeleción de novo y el 7% es heredada de los padres. (10)
34 2.7.1. Antecedentes históricos
La primera descripción médica de un caso probable de deleción 22q11.2 fue realizada en 1955 por Sedlackova (54) quien describió una condición de habla hipernasal con alteraciones faciales. (51)
En 1968 el cardiólogo pediatra William Strong, informó una asociación de anormalidades cardíacas, alteraciones de aprendizaje y apariencia facial característica, en cuatro miembros de una familia. Poco después el endocrinólogo pediátrica Angelo DiGeorge, describió tres niños con una deficiencia inmune congénita de células T acompañada de hipoplasia de la glándula paratiroidea descrito posteriormente como Síndrome de DiGeorge (55). En 1969, Cayler describe una serie de casos con anomalías cardíacas conotruncales acompañado de facies asimétricas. (56) En 1976, Kinouchi, un médico en Japón, reportó hallazgos faciales típicos vistos en pacientes con problemas de corazón (anomalías conotruncales) y lo denominó síndrome de facies y anomalías conotruncal (CTAF) (57)
En 1978, Robert Shprintzen, describió 12 individuos que tenían la asociación de defectos congénitos del corazón, habla hipernasal con anomalías palatinas, apariencia facial característica, y discapacidad de aprendizaje, Shprintzen el denominó a esta asociación “Síndrome Velocardiofacial”(58).
En 1981 Shprintzen sugirió que los pacientes con CTAF tenían en realidad el síndrome velocardiofacial. En 1982 Richard Kelley y Elaine Zackai, encontraron que los pacientes con síndrome de DiGeorge tenían una alteración en el cromosoma 22, identificado como una pequeña deleción de material cromosómico en q11.2. (59)
35 hallazgo explicó la coincidencia en los resultados clínicos entre los dos diagnósticos, por lo que apoya la idea de que se trataba, del mismo síndrome. (59)
En 1993 Wilson et al. propusieron el acrónimo CATCH 22CATCH 22, por C: Cardiac Malformations (malformaciones cardíacas), A: Abnormal Face (facie anormal), T: Timic Hipolas (hipoplasia del timo), C: Cleft Palate (fisura palatina); H: Hypocalcemia (hipocalcemia) y 22: deleción 22q11 (60). Este término fue rechazado por los genetistas clínicos y hoy se ha propuesto el nombre de síndrome de 22q11.2 (10)
2.7.2. Epidemiología
Aunque el síndrome de deleción 22q11.2 es considerado como un síndrome común, se conoce muy poco a cerca de su prevalencia; la literatura relacionada con el tema ha estimado una prevalencia general de 1 afectado en 4000 a 1 afectado en 7000 nacidos vivos (52).Reportes a nivel local han informado en la población Belga una incidencia anual de 1 en 6395 nacidos vivos para los años 1992 y 1996 (61) y en población de Atlanta entre los años informan una prevalencia de 1 por 5950 para los 1994 y 1999 (62).
36
Tabla 3. Frecuencia de la deleción 22q11.2 en una población de Atlanta entre 1994-1999 (62).
2.7.3. Características Clínicas
La deleción 22q11.2 es uno de los síndromes más variables clínicamente. El espectro fenotípico incluye un amplio rango de anormalidades (anexo 1), con severidad variable, que han llevado a describir varios síndromes dentro de la misma condición (51). Una de las variantes descritas dentro de este síndrome es el síndrome velocardiofacial (SVCF) que presenta alteraciones cardiacas, principalmente malformaciones conotruncales, anomalías palatinas, deficiencia inmune, facies características y alteraciones cognitivas y comportamentales (51). El síndrome de DiGeorge es otra de estas variantes
Grupo Nacimientos Casos Tasa Riesgo relativo
Por 10000
nacimientos Nacimientos por caso Estimación puntual 95% CI
Total 255 849 43 1.7 1 en 5950
Por Raza
Blanco 116 459 18 1.5 1 en 6470 1.0 Referenci
a
Negro 103 247 17 1.6 1 en 6073 1.1 0.7-1.4
Hispano 22 584 6 2.7 1 en 3764 1.7 0.7-2.7
Asiático 12 747 2 1.6 1 en 6374 1.0 0.2-2.8
Por genero
Masculino 130 577 21 1.6 1 en 6218 1.0 Referenci
a
Femenino 125 272 22 1.8 1 en 5694 1.1 0.8-1.3
Por año
1994 39 837 9 2.3 1 en 4426
1995 40 258 6 1.5 1 en 6710
1996 40 900 9 2.2 1 en 4544
1997 42 903 6 1.4 1 en 7151
1998 44 936 6 1.3 1 en 7489
1999 47 015 7 1.5 1 en 6716
1994-1996 120 995 24 2.0 1 en 5041
37 caracterizado por anormalidad o ausencia congénita del timo, paratiroides, hipocalcemia, defecto del tracto de salida del corazón, facies dismórficas, infecciones frecuentes y problemas del paladar (51,65)
2.7.4. Descripción Clínica del síndrome de 22q11.2
2.7.4.1. Malformaciones cardíacas
Se presentan con alta frecuencia en este síndrome hasta en un 75 a 78% de los pacientes (Tabla 5), afectan la anatomía derivada del tronco y son la principal causa de mortalidad en estos pacientes; más del 90% de todas las muertes de estos pacientes son causadas por el defecto cardiaco (10,66). El defecto cardiaco incluye principalmente cardiopatías conotruncales como la tetralogía de fallot (TF), atresia pulmonar con comunicación interventricular (AP+CIV), Tronco arterioso(TA), interrupción del arco aórtico (IAA), comunicación interventricular (CIV) y otras anomalías asociadas al arco aórtico (AAA) como anillos vasculares, arco aórtico derecho y subclavia aberrante. (66)
2.7.4.2. Alteraciones en el paladar.
38
[image:38.612.115.399.102.321.2]Tabla 4. Hallazgos cardiacos en pacientes con Síndrome deleción 22q11.2.( 10)
Tabla 5. Hallazgos palatinos en pacientes con Síndrome deleción 22q11.2 (10) Alteraciones cardiacas % de individuos afectados
Tetralogía de Fallot (TOF) 20%
Arco aórtico interrumpido (IAA) 13%
Defecto septal ventricular (VSD) 14%
Tronco arterioso (TA) 6%
Anillo vascular 5.5%
Defecto septal auricular 3.5%
VSD; ASD 4%
Otro1 10%
Normal 24%
Alteraciones palatinas % de individuos afectados
Insuficiencia Velofaríngea (VPI) 27%
Paladar Hendido Submucoso 16%
Paladar Hendido abierto 11%
Úvula Bífida 5%
Fisura labial/Fisura labio y paladar 2%
VPI Infantil 8%
39 2.7.4.3. Alteraciones inmunes
Las deficiencias inmunes se presentan en 77% de los casos, con un grado de compromiso del sistema inmune muy variable. La mayoría de los pacientes presentan disminución en el número de linfocitos T en grado leve a moderado como consecuencia de hipoplasia tímica (68).
2.7.4.4. Función paratiroidea
El Síndrome del 22q11 afecta el desarrollo de estructuras derivada del tercer y cuarto arco faríngeo y por lo tanto se ha informado que hasta en un 50 a 60% de los pacientes con hipoplasia o aplasia de las glándulas paratiroides pueden desarrollar hipoparatiroidismo (69,70). Usualmente se manifiesta como hipocalcemia sintomática en el período neonatal lo aparecer en adolescentes y adultos por un hipoparatiroidismo latente. (69)
2.7.4.5. Alteraciones en la alimentación.
Alrededor del 36% de los niños tienen dificultades en la alimentación, con frecuencia se presenta disfagia grave que requiere la alimentación por sonda nasogástrica y/o la colocación del tubo de gastrostomía. (10)
2.7.4.6. Alteraciones craneofaciales
40 2.7.4.7. Alteraciones en el crecimiento.
La incidencia de baja talla en pacientes de este síndrome ha sido estimada en un 39 a 67%. La baja estatura puede ser secundaria al retardo de crecimiento intrauterino, retardo constitucional del desarrollo, desnutrición por enfermedad concomitante como cardiopatía, hipotiroidismo y deficiencia de hormona de crecimiento y a la dificultad en la alimentación (71).
2.7.4.8. Desarrollo sicosocial y función cognitiva
De manera general, los niños con la deleción 22q11.2 tienen retraso en el desarrollo motor (caminan en promedio a los 18 meses de edad), retraso en el lenguaje, y tienen trastornos del espectro autista en aproximadamente el 20% de los casos (72).
2.7.4.9. Alteraciones psiquiátricas
En los pacientes con deleción 22q11.2 se ha descrito que con frecuencia sufren de déficit de atención, ansiedad, dificultad en la interacción social, junto con autismo y trastornos del espectro autista (73). Se ha planteado que hasta un 60% de los adultos llegan a tener trastornos psiquiátricos y aproximadamente un 25% esquizofrenia; también se ha descrito en los pacientes trastorno bipolar, ansiedad y depresión (73,74).
41
2.7.5 Abordaje diagnóstico.
El síndrome de deleción 22q11.2 se sospecha en individuos con una serie de hallazgos que incluyen:
Cardiopatías congénitas (principalmente defecto conotruncal)
Anomalías palatinas (especialmente insuficiencia velofaríngea)
Hipocalacemia
Inmunodeficiencia
Dificultades de aprendizaje (especialmente problemas de aprendizaje no verbal, con una división mayor de diez puntos entre el coeficiente intelectual verbal y el coeficiente intelectual de rendimiento).
Anomalías faciales características. Nariz prominente, de punta ancha y bulbosa e hipoplasia de narinas, fisuras palpebrales estrechas, comisuras labiales descendidas.facies de respirador oral, con mejillas hipotónicas y labios entreabiertos, orejas en asa o dismórficas (10,51,52,54,56)
Con menos frecuencia se observan alteraciones funcionales como:
Disfagia severa
Deficiencia de hormona del crecimiento
Enfermedad autoinmune (trombocitopenia , artritis reumatoide juvenil, enfermedad de Grave, vitiligo , neutropenia , anemia hemolítica)
Pérdida de audición ( neurosensorial y conductiva)
Enfermedad psiquiátrica
Autismo. (10)
Otras anomalías estructurales importantes que contribuyen al diagnóstico incluyen :
42 poliquísticos / displásicos , duplican riñón, riñón en herradura , ausencia de útero , hipospadias , hernia inguinal y criptorquidia
Anomalías laringotraqueoesofágica: anillo vascular, membrana laríngea, malacia laringotraqueal y estenosis subglótica.
Hallazgos oftalmológicos : vasos retinianos tortuosos , ptosis , coloboma , cataratas, anoftalmia y estrabismo.
Anomalías del sistema nervioso central: atrofia cerebelosa, defectos del tubo neural , médula anclada , convulsiones no provocadas , y facies de llanto asimétricas
Anomalías gastrointestinales: ano imperforado , atresia esofágica, atresia yeyunal , bazos accesorios , hernia umbilical , hernia diafragmática , mal rotación intestinal y la enfermedad de Hirschsprung. (10)
2.7.5.1 Pruebas diagnósticas
La prueba diagnóstica para la deleción 22q11.2 comúnmente se realiza por FISH. Las dos sondas disponibles en el mercado para el análisis FISH 22q11.2 son TUPLE1 y N25 ubicadas en la parte proximal del inicio de la deleción entre el LCR A y B. El FISH no es lo suficientemente sensible para detectar deleciones más pequeñas (< 40 kb) en 22q11.2. La mayoría de las deleciones ahora están siendo identificados por el uso de microarreglos cromosómicos y MLPA (Multiplex ligation-dependent probe amplification). Estos estudios no detectan mutaciones en cualquiera de los genes dentro de la región deletada (10).
43 del paciente. El análisis CMA utilizando matrices de BAC , oligonucleótidos, o SNPs (polimorfismos de un solo nucleótido ) puede detectar la deleción de 3 Mb en un individuo afectado . La capacidad de identificación del tamaño de la deleción depende del tipo de microarrays utilizado y de la densidad de sondas en la región 22q11.2. (10) Los métodos como MLPA , se pueden utilizar para el diagnóstico rápido ante la la sospecha clínica del síndrome. Se ha demostrado que esta técnica es un costo efectivo, rápida, y un método altamente sensible y específico para la detección de deleciones y duplicaciones en la región proximal 22q11 y de variantes distales o solapadas que frecuentemente se pueden presentar dentro de esta región. La identificación de estas variantes es de particular interés, ya que puede dar una idea de qué genes o regiones genómicas son cruciales para manifestaciones fenotípicas (10, 137,138)
2.7.6 Etiología Genética del Síndrome de deleción 22q11.2
La causa genética específica del síndrome fue descrita por Driscollen et al :en 1992, al encontrar una microdeleción de la banda q11.2 en el cromosoma 22 en pacientes con el SVCF que no difería de la observada en los pacientes con SDG (59).
El síndrome resulta de una deleción comúnmente de tres megabases en la región cromosómica 22q11.2 que está flanqueado por repeticiones de bajo número de copias (LCR, Low copy repeats). Se planteado que esta deleción es causada por un evento de recombinación meiótica no alélica durante la espermatogénesis u ovogénesis (figura 5). (75)
44 Los cuatro LCR proximales han sido descritos como LCR-A a LCR-D posicionados en dirección centrómero telómero y han sido ampliamente caracterizados por estar involucrados en los reordenamientos recurrentes de 22q11.2 que conducen a SDG/ SVCF (76). Los LCR A a D tienen un alto nivel de identidad (> 98%)y son los que con más frecuencia están involucrados en la generación de reordenamientos. Los cuatro LCR distales, LCR-E a LCR-H, son más pequeños, y rara vez se han asociado con deleciones (9,76).
Se han propuesto dos modelos para explicar el origen de la deleción promovido por la presencia de los LCR en el cromosoma 22. (77) El primer modelo propone un alineamiento intracromosómico errado durante la Meiosis I entre los dos cromosomas 22 homólogos. Este alineamiento errado puede ser mediado por los módulos de repetición dentro de los LCRs separados que están en orientación directa uno respecto al otro y por crossing-over conduciendo a eventos de deleción y duplicación reciproca (Figura 5) (79). El segundo modelo propone la formación de la deleción del DNA que está dentro del loop intercromosomico formado por la recombinación entre los módulos duplicado (77).
La deleción más común de 3 Mb, ha sido descrita en el 85% de los individuos; esta deleción se extiende desde la LCRA a LCRD e incluye TBX1 (Figura 7), un gen considerado responsable de características típicamente asociadas, en particular anomalías cardíacas conotruncales. El 15% restante de los individuos afectados tienen deleciones atípicas. (10 ,51,52,58)
45
Figura 5. Representación gráfica de recombinación homóloga no alelica (NAHR). Duplicaciones segmentarias LCR (cajas azules y blancas) proporcionan un sustrato para la recombinación homóloga no alélica (NAHR) originado por crossing over intracromosómico entre las repeticiones directamente orientadas (a) causa deleción (b) o duplicación (c). (78)
[image:45.612.133.470.353.643.2]46
Figura 7. Representación esquemática de la región 22q11.2. Ubicación de los LCR y de las deleciones descritas en la región 22q11.2 (10)
2.7.7 Genes afectados por la deleción
Las deleciones en la región 22q11.2 afectan la carga génica de cerca de 40 genes cuando la pérdida es de 3Mb y de aproximadamente 30 genes cuando esta es de 1.5 Mb (Figura 8). Se ha propuesto que la deleción genera haploinsuficiencia de todos los genes afectados y que esta condición en uno o más genes presentes en esta región son responsables de la etiología del síndrome (tabla 6); estos genes podrían tener su efecto en la regulación del desarrollo del aparato faríngeo y de sus estructuras derivadas. (78,79,80)
47 Los modelos animales y el análisis funcional de los genes que se expresan del arco branquial y cresta neural han identificado algunos genes candidatos en la patología del Síndrome (79). Sin embargo, no se conoce mucho acerca de la función de los genes en
la región implicada. A continuación se describen algunos cuya función ya ha sido establecido
2.7.7.1 Gen TBX1
El gen TBX1 codifica un factor de transcripción, involucrado en el desarrollo embrionario temprano incluida la formación de mesodermo y endodermo y la morfogénesis del corazón y de las extremidades (81). En investigaciones hechas en ratones se ha demostrado que la homocigosis para la mutación en TBX1 muestran características similares a las del síndrome de deleción 22q11.2 y mueren perinatalmente; estos animales presentan defecto conotruncal, características faciales anormales, hendidura palatina, hipoplasia del timo y alteraciones de glándula paratiroidea. Por el contrario, los ratones heterocigotos para Tbx1, solo tienen defectos del arco aórtico con un fenotipo menos penetrante, y no revelan el espectro fenotípico completo del síndrome de deleción 22q11.2. Estos resultados dan evidencia probable para el TBX1 como el mayor determinante genético en la etiología del 22q11.2. (81,82)
La deleción de TBX1 contribuye a las anomalías del corazón, pero no es la causa putativa en el Síndrome de deleción 22q11 (81).TBX1 está implicado en un mecanismo más complejo del desarrollo del corazón que implica regulación corriente abajo de otros genes o complejas interacciones genéticas de múltiples genes. Hay evidencia de que TBX1 interactúa con varios genes importantes para la embriogénesis incluyendo genes homeobox que están muy involucrados en el desarrollo regional de los arcos faríngeos y bolsas que forman las estructuras de la cabeza y el cuello y las porciones del corazón y su sistema vascular (83). Se ha confirmado que la hemizigocidad para el gen TBX1 es responsable del fenotipo cardiaco en SVCF (83,84).
48 2.7.7.2 Gen HIRA
Este gen es un regulador transcripcional. Estudios en ratones han evidenciado su expresión embrionaria durante el desarrollo del tubo neural (85). Se ha propuesto que este gen puede contribuir con el defecto cardiaco ya que experimentos por atenuación antisentido de la expresión de Hira en la cresta neural cardiaca durante la embriogénesis han demostrado la persistencia del tronco arterioso en ratones (79).
2.7.7.3 Gen UFD1L
Este gen está relacionado con la ubiquitinación; fue propuesto inicialmente como candidato para el Síndrome de deleción 22q11.2 por los hallazgos vistos en modelos animales. Mediante hibridación in situ se ha localizado la expresión de UFD1L en los arcos faríngeos y el cuarto arco branquial (79,83) Fue identificado como un gen corriente abajo del factor de transcripción Hand2 en embriones de ratones Hand2-/-. Ratones con mutaciones de estos genes tienen defectos del desarrollo del corazón (79).
2.7.7.4 Gen CRKL
Codifica factores de crecimiento y señalización de adhesión celular y es expresado en tejidos derivados de células de la cresta neural durante el desarrollo embrionario. La homocigosis de Crkol en ratones es letal y el fenotipo es similar al de la delecion 22q11.2, incluyendo defecto conotruncal y del arco aórtico, sugiriendo un efecto potencial de CRKL en el fenotipo del síndrome de deleción 22q11.2 (86).
2.7.7.5 Genes COMT y PRODH
49 pacientes con síndrome 22q11.2. Se ha demostrado que las mutaciones en el gen
COMT provocan alteraciones en el comportamiento emocional en ratones (87)
2.7.8 Modificadores Genéticos del síndrome 22q11.2
Una elevada variabilidad en la expresión del fenotipo se observa en pacientes no relacionados y entre los miembros de una misma familia de afectados que tienen deleciones idénticos presuntamente heredadas (88). Esta variación observada no
parece ser atribuible al tamaño de la deleción o al origen parental del cromosoma deletado (88,89). Se han propuesto varios mecanismos que explicarían esta variabilidad: la presencia de genes modificadores que residen fuera de la región deletada, variación alélica de los genes dentro de la región 22q11.2 del cromosoma no deletado; mutaciones somáticas, fenómenos epigenéticos, características individuales (raza o sexo), factores ambientales o eventos al azar (88,89)
Algunos genes modificadores están en la misma vía genética, corriente arriba de TBX1 como se describen a continuación de acuerdo a diferentes vías de interacción propuesta para TBX1 (89).
2.7.8.1 Factor de crecimiento de fibroblastos 8 (Fgf8)
El gen Fgf8 pertenece a la familia del factor de crecimiento de fibroblastos (FGF) asociado con procesos del desarrollo como la proliferación, migración y diferenciación celular (90). Los embriones de ratones con alteraciones en Fgf8 muestran fenotipos observados en SVCF/SDG como alteraciones craneofaciales, tímicas, paratiroides y cardiovasculares (91).
2.7.8.2 Factor de crecimiento de fibroblastos 10 (Fgf10)
50
2.7.8.3 Gen GBX2
El gen Gbx2, es un factor de transcripción homeobox. Los embriones de ratones nulisómicos para Gbx2 tienen defectos cardiovasculares asociados con el desarrollo anormal del cuarto arco faríngeo incluyendo arteria aorta interrumpida alteración retroesofágica de la arteria subclavia (93). Estos defectos están relacionados con SVCF/SDG, por lo que se cree que este gen puede ser un modificador del locus para estos Síndromes (89).
2.7.8.4 Gen Pitx2
51
Gen Función/homología Expresión Referencia
DGCR6
Similar a lamina de cadena-g & proteína gonadal de Drosophila
Ampliamente
expresado Demosuk et al 1996
LAN/DGCR2/IDD Similar a receptor LDL & C-tipo lectina Ampliamente expresado
Budarf et al 1995, Wadey et al 1995, Demosuk et al 1996 TSK2
(serine/threonine kinase
Serina/ treonina quinasa Testículo Gong et al 1996, Goldmuntz et al 1997
DGSI/ES2
Similitud a la proteína de función desconocida C. elegans
Ampliamente expresado
Gong et al
1996,1997, Rizzu et al 1996
GSCL (goosecoid-like)
Proteína Homeobox de
pares-como la clase Testículo; cerebro Gottlieb et al 1997
CPT ( citrate transport protein)
cambio de la membrana mitocondrial interna electroneutro
Ampliamente expresado
Heisterkamp et al 1995, Goldmuntz et al 1996
CLTCL (cathrin heavy chain-like)
Similitud de la cadena
pesada de clatrina Mas abundante Sk músculo
Gong et al 1996, Kedra et al 1996, Sirotkin et al 1996 Holmes et al 1997
HIRA Homología con la levadura HIR1 y HIR2
Ampliamente expresado
Halford. Wilson et al 1993, Lorain et al 1996
NLVCF Desconocido Ampliamente expresado Funke et al 1998
UFDIL
similitud a la levadura proteína de fusión de ubiquitina 1 degradación
Ampliamente
expresado Pizzuti et al 1997
CDC45L Similitud con otras proteínas CDC45 Ampliamente expresado
Mckie et al 1998, Saha et al 1998, Shaikh et al 1999
TMVCF
Similitud con proteína de rata de función
desconocida
Ampliamente expresado; más alto en el pulmón
Sirotkin, Odonell, et al 1997
hCDCrel-1(human cdc-related
Similitud con proteínas
de unión GTP Desconocido Zieger et al 1997
GPIBB
(glycoprotein Ibβ)
Subunidad plaquetaria del receptor del factor von
Willebrand Plaquetas Budarf et al 1995
TBX-1
Miembro de la familia de factores transcripción de ADN T-box
Adulto Sk; Musculo y testículos; tejidos
fetales Chieffo et al 1997
COMT (cathecol-
O-methyltransferase
Metabolismo
Catecolamina Ampliamente expresado Grossman et al 1992
ARVCF Miembro de subfamilia de catenina Ampliamente expresado Sirotkin, Morrow et al 1997 T10 Desconocido Ampliamente expresado Halford, Wadey, et
al 1993
N41 cDNA Desconocido Ampliamente expresado Emanuel et al 1993
[image:51.612.91.505.89.661.2]LZTR-1 similitud con leucina dominio superior Ampliamente expresado Kurahashi et al 1995
52
3 OBJETIVOS
3.1 Objetivo General.
Identificar anomalías cromosómicas numéricas y estructurales, así como la longitud de la deleción y el número de copias de la región 22q11.2 en pacientes con fisuras labio palatinas con sospecha de síndrome de deleción 22q11.2 evaluando la correlación genotipo-fenotipo.
3.2 Objetivos específicos
1. Identificar la presencia de la microdeleción 22q11.2 y/o otras alteraciones cromosómicas mediante bandeo G de alta resolución.
2. Determinar la longitud de la deleción y la variación en el número de copias presentes en la región 22q11.2 mediante la técnica de citogenética molecular de MLPA.
3. Correlacionar el fenotipo clínico de los pacientes con los hallazgos citogenéticos y moleculares obtenidos.
53 4. METODOLOGÍA
4.1 Población de estudio.
Se realizó un estudio descriptivo, en donde se evaluaron 49 pacientes con fisura de labio y/o de paladar acompañados de otras alteraciones congénitas que revelaran una condición sindrómica. Los pacientes fueron seleccionados de la base de datos del programa de vigilancia de malformaciones congénitas de Bogotá, de la consulta de Genética del Instituto de Genética Humana (IGH), de la clínica de labio y paladar hendido de la Facultad de Odontología de la Pontificia Universidad Javeriana, de la consulta pediátrica del Hospital Universitario San Ignacio (HUS), de Brigadas de Salud de labio y paladar hendido en Neiva y de otras de Instituciones como Hospital de la Victoria, Laboratorio López Correa (Pereira), FISULAB, Hospital Simón Bolívar e Higuera Escalante (Bucaramanga).
4.1.1 Criterios de Selección de las muestras
4.1.1.1 Criterios de inclusión
1. Los pacientes incluidos en el estudio debieron tener cualquier tipo de fisura en condición sindrómica: fisura labial, fisura de labio y de paladar, fisura de paladar, anormalidades palatinas como insuficiencia velo faríngea y úvula bífida, acompañadas de al menos otra característica fenotípica que estuviera asociada al síndrome 22q11.2 como facies dismórficas, anomalías cardiacas, alteraciones del desarrollo y cognitivas, anomalías del sistema inmune, malformaciones esqueléticas, entre otras.
2. Pacientes de ambos sexos y de cualquier edad
54 1. Pacientes con cualquier tipo de Fisura de labio y/o paladar en condición aislada
(no sindrómica).
2. Pacientes con fisuras de labio y/o de paladar que se asocien a un diagnóstico especifico de un síndrome diferente a la deleción 22q11.2.
4.2 Selección y recolección de Muestras
La selección de los pacientes candidatos de la base de datos del programa de vigilancia de malformaciones congénitas se realizó mediante la selección de los pacientes por código del ECLAMC para las fisuras del labio y/o paladar e identificación posterior de los casos como unimalformadoso y polimalformados. Mediante el trabajo coordinado con el grupo de seguimiento de malformaciones congénitas se ubicaron todos los casos con fisuras de labio y/o de paladar en condición sindrómica, y por llamada telefónica se contactó a la persona a cargo del paciente para invitarlo a participar del estudio.
Los pacientes de la base de datos de la consulta de genética del Instituto de Genética Humana (IGH) de la consulta de otorrinolaringología y de consulta Externa del Hospital Universitario San Ignacio (HUSI) fueron filtrados por diagnostico presuntivo que incluyó todos los tipos de fisuras. A todos los casos seleccionados en estas bases de datos se les contactó telefónicamente, se les explicó el motivo de la misma, se les invitó a participar del estudio y luego de dar su aceptación se les realizó una encuesta dirigida a establecer la presencia de alguna anormalidad congénita adicional a las fisuras (ver anexo 2). Los pacientes participantes de la jornada de salud de Healing the Children en Neiva fueron seleccionados por un Genetista.